

The transcriptional profiles of related pathogens and their responses to host-induced stresses underpin their pathogenicity. Expression differences between related pathogens during host interaction can indicate when and how these genes contribute to virulence, ultimately informing new and improved treatment strategies for those diseases. In this paper, we compare the transcriptional profiles of five isolates representing four lineages of C. gattii in rich media. Our analyses identified key processes, including those involving cell capsule, ergosterol production, and melanin, that are differentially expressed between lineages, and we found that VGII has the most distinct profile in terms of numbers of differentially expressed genes. All lineages have also undergone subfunctionalization for several paralogs, including capsule biosynthesis and attachment genes. Most genes appeared downregulated during coincubation with macrophages, with the largest decrease observed for capsule attachment genes, which appeared to be coordinated with a stress response, as all lineages also upregulated oxidative stress response genes. Furthermore, VGII upregulated many genes that are linked to ergosterol biosynthesis and switched from expression of the laccase LAC1 to expression of LAC2 ex vivo. Finally, we saw a pronounced increase in the FosB/Jun/Egr1 regulatory proteins at early time points in bone marrow-derived macrophages, marking a role in the host response to C. gattii. This work highlights the dynamic roles of key C. gattii virulence genes in response to macrophages.
KEYWORDS: Cryptococcus, capsule, ergosterol, host response, host-pathogen interactions, laccase, mRNA
ABSTRACT
Cryptococcus gattii is a pathogenic yeast of humans and other animals which causes disease predominantly in immunocompetent hosts. Infection begins when aerosolized yeast or spores enter the body, triggering an immune response, including engulfment by macrophages. To understand the early transcriptional signals in both the yeast and its mammalian host, we performed a time-course dual-transcriptome sequencing (RNA-seq) experiment for four lineages of C. gattii (lineages VGI to IV) interacting with mouse macrophages at 1, 3, and 6 h postinfection. Comparisons of in vitro to ex vivo gene expression levels indicated that lineage VGII is transcriptionally divergent from non-VGII lineages, including differential expression of genes involved in capsule synthesis, capsule attachment, and ergosterol production. Several paralogous genes demonstrated subfunctionalization between lineages, including upregulation of capsule biosynthesis-related gene CAP2 and downregulation of CAP1 in VGIII. Isolates also compensate for lineage-specific gene losses by overexpression of genetically similar paralogs, including overexpression of capsule gene CAS3 in VGIV, which have lost the CAS31 gene. Differential expression of one in five C. gattii genes was detected following coincubation with mouse macrophages; all isolates showed high induction of oxidative-reduction functions and downregulation of capsule attachment genes. We also found that VGII switches expression of two laccase paralogs (from LAC1 to LAC2) during coincubation of macrophages. Finally, we found that mouse macrophages respond to all four lineages of C. gattii by upregulating FosB/Jun/Egr1 regulatory proteins at early time points. This report highlights the evolutionary breadth of expression profiles among the lineages of C. gattii and the diversity of transcriptional responses at this host-pathogen interface.
IMPORTANCE The transcriptional profiles of related pathogens and their responses to host-induced stresses underpin their pathogenicity. Expression differences between related pathogens during host interaction can indicate when and how these genes contribute to virulence, ultimately informing new and improved treatment strategies for those diseases. In this paper, we compare the transcriptional profiles of five isolates representing four lineages of C. gattii in rich media. Our analyses identified key processes, including those involving cell capsule, ergosterol production, and melanin, that are differentially expressed between lineages, and we found that VGII has the most distinct profile in terms of numbers of differentially expressed genes. All lineages have also undergone subfunctionalization for several paralogs, including capsule biosynthesis and attachment genes. Most genes appeared downregulated during coincubation with macrophages, with the largest decrease observed for capsule attachment genes, which appeared to be coordinated with a stress response, as all lineages also upregulated oxidative stress response genes. Furthermore, VGII upregulated many genes that are linked to ergosterol biosynthesis and switched from expression of the laccase LAC1 to expression of LAC2 ex vivo. Finally, we saw a pronounced increase in the FosB/Jun/Egr1 regulatory proteins at early time points in bone marrow-derived macrophages, marking a role in the host response to C. gattii. This work highlights the dynamic roles of key C. gattii virulence genes in response to macrophages.
INTRODUCTION
Infectious diseases impose a huge burden on human society. In recent years, fungi have gained widespread attention for their ability to threaten both animal and plant species on a global scale (1, 2). However, many features of fungal genomes and transcriptomes that enable the infection of diverse hosts and ecological niches remain largely unexplored, especially for emerging pathogens (3, 4). One such example is the basidiomycete yeast Cryptococcus gattii, which can cause pneumonia and meningoencephalitis in humans (5). While C. gattii causes less overall global morbidity than its sibling species C. neoformans, lineages have emerged with hypervirulent clinical phenotypes, a process that was most strikingly observed in the Pacific Northwest of the United States in the late 1990s (6). C. gattii is comprised of four genetically distinct lineages designated the VGI to VGIV molecular types, among which lineage VGII has been found to be associated with the highly virulent subtypes seen in the United States. These lineages are sufficiently divergent to include 737 lineage-specific gains and/or losses of genes across all four lineages (approximately 4% of the genes in any given isolate), including DNA transposons and genes involved in the response to oxidative stress and import into mitochondrial inner membrane (7).
In addition to genetic differences between lineages, changes in gene regulation underlie important morphological and physiological traits in fungal pathogens (8, 9). However, the mechanisms of gene regulation, evolution of gene networks, and rewiring of transcriptional modules within lineages remain largely uncharacterized for many infectious diseases, including those caused by C. gattii. New virulent lineages can emerge through mutation and/or recombination events (10), which in turn lead to different transcriptional profiles and enhanced virulence profiles, such as that of the hypervirulent C. gattii VGIIa sublineage in the Pacific Northwest (11), which descends from two alpha mating-type parents (6) and is characterized by enhanced intracellular parasitism (11).
Previous studies have demonstrated unique expression profiles among different isolates of Cryptococcus under various conditions. For example, in C. neoformans reference isolate H99, genes encoding membrane transporters for nutrients, general metabolism, and oxidative stress response have been shown to be upregulated in the presence of macrophages (12) and amoebae (13). Janbon et al. also characterized differential expression of transporters, transcription factors, and genes involved in lipid metabolism in C. neoformans in comparisons between rich media, limited/starved media, and pigeon guano (14). In addition to analyzing protein-coding gene expression, Janbon et al. also identified nearly 1,200 miscellaneous RNAs that may perform a range of functions that include morphogenesis via small open reading frames (ORFs) or noncoding RNA (ncRNA) with structural or regulatory roles. Concurrently, Chen et al. described an increase in genes involved in metabolic processes, alkaline response, salt tolerance, and oxidative stress for two C. neoformans isolates grown in cerebral spinal fluid compared to growth in rich media (15).
Few studies have focused on gene expression in C. gattii, although, notably, Ngamskulrungroj et al. identified an upregulation of laccase genes involved in melanin formation, and of other genes involved in cell wall assembly and metabolism in C. gattii VGIIa outbreak strain R265, relative to the nonoutbreak C. gattii VGIIb strain in minimal medium (16). In this report, we extend the description of C. gattii gene expression levels to characterize variation across all four major lineages of C. gattii, including the outbreak strain VGIIa R265 and a nonoutbreak isolate, ENV152, also from the VGIIa subgroup, both in vitro and at three early time points (1, 3, and 6 h) following coincubation and engulfment by murine bone marrow-derived macrophages (BMDMs). Furthermore, we describe concurrent expression changes in mouse macrophages during C. gattii coincubation, demonstrating the potential for the simultaneous profiling of both host and pathogen responses.
RESULTS
To identify mammalian and Cryptococcus gattii genes that are activated during infection, we performed transcriptome sequencing (RNA-seq) across five C. gattii isolates representing four lineages (VGI, VGII, VGIII, and VGIV), including an environmental and clinical isolate from VGII, both in vitro and at time points 1, 3, and 6 h postcoincubation with BMDMs (ex vivo). Each of the sequencing runs (n = 40) yielded between 904,770 and 17.6 million 30mer reads (10.9 gigabases of total sequence) (see Table S1, tab 1, and Fig. S1, S2, and S3 in the supplemental material), except for VGIV at h 6 (t6), which failed to yield sequencing data. Most of the sequences derived from mouse (84% to 99% for each data set), while C. gattii reads ranged from up to 56.97% (in vitro without macrophages; see Materials and Methods for mapping description) to between 0.02% and 1.44% reads ex vivo (mean of 38,949 reads) (Fig. S2). The C. gattii data sets formed two main clusters: (i) t0 (in vitro) for all isolates, probably owing to the large difference in read counts between samples, and (ii) VGII isolates at any time point (Fig. 1a). In contrast, gene expression in mouse macrophages clustered by time point (Fig. 1b).
FIG 1.

Heat maps of the Log2 fold change in the trimmed mean of M-values (TMM) normalized fragments per kilobase of transcript per million mapped reads (FPKM) of C. gattii (a) and mouse BMDM (b) transcripts per sample.
Alignments of C. gattii and mouse transcripts to sequences of the core eukaryotic genes (CEGs), suggesting mostly complete C. gattii gene-sets and the high quality of Mouse gene-set mm10 p4. Only VGIIa R265 and mouse mm10p4 were used in this study. Download FIG S1, PDF file, 0.1 MB (137.3KB, pdf) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
RNA was extracted from all four lineages of C. gattii in mouse macrophages at 1 h, 3 h, and 6 h postinfection, as well as C. gattii and macrophages in vitro (t0). Data represent percentages of sequenced reads deriving from the mouse macrophage (top) and C. gattii (bottom). Download FIG S2, PDF file, 0.2 MB (213KB, pdf) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Heat maps of all differentially expressed genes (FDR P value of <0.001 and greater-than-4-fold change of trimmed mean of M-values [TMM] expressed in normalized fragments per kilobase of transcript per million mapped reads [FPKM]) of C. gattii transcripts in vitro (t0) and during infection of mouse macrophages at 1 h, 3 h, and 6 h. Download FIG S3, PDF file, 0.8 MB (812.4KB, pdf) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(Tab 1) Contamination from E. coli K-12 MG1655, suicide vector pCD-RAsl1, and cloning vector pMJ016c identified by BLASTn searches of the nonredundant (NR) database. (Tab 2) Reads aligning either to mouse GRCm38 p4 mm10 gene sets or genome or to R265 updated gene set or genome. ARD, average read depth across genes. Download Table S1, XLSX file, 0.0 MB (48KB, xlsx) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differential expression was computed using the quantile-adjusted conditional maximum likelihood (qCML) method implemented in EdgeR (17), requiring a false-discovery-rate (FDR) P value of <1e−3 and at least 4-fold change in trimmed mean of M-values (TMM) normalized fragments per kilobase of transcript per million mapped reads (FPKM) to be considered a significantly differentially expressed gene (DEG). To check for the effect of different read depths on DEG prediction, subsets were created and reanalyzed. This analysis identified consistent numbers of DEGs in vitro and more variable numbers between infection time points (Fig. S4).
Subsets (75%, 50%, and 25%) of C. gattii data were used to recall differential expression data and for comparisons with the full dataset. x-axis categories for each bar chart correspond to data from each of the five isolates (combining t1, t3, and t6) and to in vitro data (combining all isolates at t0). (a) The total number of differentially expressed gene changes (either up- or downregulation) for isolates ex vivo but not in vitro. (b) The percentages of genes found in the full dataset (i.e., representing a proxy for true positives). Data are represented as >75% for all categories using 75% subsets, >50% for all categories using 50% subsets, and >25% for all categories using 25% subsets. (c) The number of genes found only in the full dataset (i.e., representing a proxy for false negatives). VGI, VGIII, and VGIV reidentified most of the genes also found with their full datasets, while VGII and in vitro conditions recovered fewer genes found in the full dataset as the subset became smaller. (d) The number of genes not found in the full dataset (i.e., representing a proxy for false positives). Values corresponding to previously unidentified genes either increased or decreased as the subset size decreased, with VGIV giving the most robust results and VGIII the least. Download FIG S4, PDF file, 0.2 MB (223.9KB, pdf) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
VGII is transcriptionally divergent in vitro from other C. gattii lineages.
Pairwise expression values for all C. gattii isolates in vitro (t0) revealed 524 DEGs between one or more isolates, indicating that nearly 1 in 10 (8.1%) of all C. gattii protein-coding genes situated throughout the genome (n = 6,456) are uniquely differentially regulated among the four divergent lineages, despite being all cultured in the same rich media at 37°C with 5% CO2 (Fig. 2a and e) (see also Data Set S1 in the supplemental material). A G-test of goodness of fit based on the numbers of in vitro DEGs per lineage (not considering specific genes) suggested differences (G = 328.52, X-squared df = 4, P value < 2.2e−16), and pairwise comparisons using G-tests with Bonferroni multiple correction also showed differences in VGII isolates compared with VGI, VGII ENV152, and VGIV isolates but not VGII R265 (P value < 2.2e−16). Results of comparisons of VGIV to VGI or VGIII were also highly distinct for numbers of DEGs (P value < 2.2e−16).
FIG 2.

The number of Cryptococcus gattii genes up- and downregulated between lineages in vitro (a) and between time points (b) and principal-component analysis (PCA) of differentially expressed genes in vitro (c) and between time points (d) and the locations of those genes in their genomes (synteny plotted using Synima [48]) alongside a phylogenetic tree constructed with RAxML (GTRCAT) with 1,000-bootstrap support (shown as asterisks) (e). Genes are considered differentially expressed where the FDR P value is <0.001 and the TMM FPKM is greater than 4-fold. Colored boxes show the genomic locations of differentially expressed genes (DEGs) with colors corresponding to panels a and b: in vitro, yellow; t0 versus another time point, blue; t1 versus another time point, red; t3 versus another time point, green; t6 versus another time point, purple.
(Tab 1 to 4) All genes differentially expressed among five C. gattii isolates in vitro. (Tab 1) Details of every differentially expressed gene. (Tab 2) Gene annotation, position in genome, and associated PFAM. Orthogroup numbers refer to ortholog groups from 15 C. gattii and C. neoformans H99 genome comparisons (7). (Tab 3) Gene ontology terms assigned to differentially expressed genes. (Tab 4) Differentially expressed genes were found in multiple pairwise comparisons. (Tab 5 to 8) All genes differentially expressed by each of the five C. gattii isolates at different time points (t0, t3, and t6) in coincubation with mouse macrophages. (Tab 5) Details of every differentially expressed gene. (Tab 6) Gene annotation, position in genome, and associated PFAM. Orthogroup numbers refer to ortholog groups from 15 C. gattii and C. neoformans H99 genome comparisons (7). (Tab 7) Gene ontology terms assigned to differentially expressed genes. (Tab 8) Differentially expressed genes were found in multiple pairwise comparisons. (Tab 9 and 10) Expression values and overlap of previously identified differentially expressed genes in Acanthamoeba castellanii and macrophages (13). (Tab 9) C. neoformans genes with similar modulation patterns after interaction of the fungus with amoebae and with murine macrophages. (Tab 10) C. neoformans genes with different modulation patterns after interaction of the fungus with amoebae and with murine macrophages. (Tab 11) Genes differentially expressed by mouse macrophages. LogFc, log fold change; LogCPM, log counts per million. C1, VGIV CBS10101; C2, VGII R265; C3, VGII ENV152; C4, CA1873; C5, VGI WM276. “A” and “B” represent replicates. Download Data Set S1, XLSX file, 2.1 MB (2.1MB, xlsx) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Of the 524 in vitro DEGs, 203 were differentially regulated in both VGII isolates, 245 additional genes were differentially regulated only for an individual VGII isolate, and 50 were differentially regulated inconsistently across VGII (e.g., upregulated in VGIV versus VGII R265 and upregulated in VGI versus both VGII isolates). While some of these genes were also differentially expressed in other lineages, only 5% (n = 27) of in vitro DEGs were unique to VGI, VGIII, or VGIV isolates, suggesting that VGII harbors expression profiles distinct from those seen with the other lineages. Furthermore, the greater number of genes uniquely differentially expressed within isolates of the VGII isolates suggests that substantial differences exist even within the VGIIa sublineage. Furthermore, VGII and VGIII isolates were distinct from each other by principal-component analysis (PCA) (Fig. 2c), while VGI and VGIV were not distinct from each other.
To understand the role that differential expression may play in each isolate in vitro, we opted to use both a targeted approach (looking at known genes of interest [18], including 35 capsule biosynthesis genes, 40 capsule attachment genes and cell-wall remodeling genes, and 20 ergosterol genes based on their orthology to C. neoformans H99 [7]) and a nontargeted approach (gene ontology term [GO-term] and PFAM enrichment). Of the 524 unique DEGs, 8/35 were involved in capsule biosynthesis, 10/40 in capsule attachment and cell-wall remodeling, and 4/20 in ergosterol production, and both laccase genes were differentially expressed in at least one pairwise comparison (Fig. 3). These gene categories are therefore enriched for DEGs based on the results of a hypergeometric test (P[X > x] = P = 1.207e−07).
FIG 3.

Bar charts showing mean expression (TMM FPKM) of ergosterol, capsular biosynthesis, capsular attachment, and laccase genes differentially expressed between the five isolates of Cryptococcus gattii in vitro. Red, significantly (Sig.) upregulated; blue, Sig. downregulated. Genes are considered differentially expressed (D.E.) where the FDR P value is <0.001 and the TMM FPKM is greater than 4-fold. Error bars show the range of TMM FPKM values in comparisons between the two replicates.
In vitro C. gattii lineages have distinct expression for capsule biosynthesis and attachment genes.
Capsule biosynthesis genes were differentially expressed between lineages in vitro (Fig. 3), including CAS3, which was upregulated in VGIV compared with all the other isolates. CAS3 mutants have a reduced capsule under conditions of combination with cas31Δ, cas32Δ, or cas33Δ mutants and have a partial defect in O-acetylation, leading to reduced overall levels of this modification (18, 19). Meanwhile, the closely related paralog CAS31 is absent in VGIV and VGIIIb (but present in the VGI, VGII, and VGIIIa lineages) (Fig. S5) and as such has no detectable expression (zero TMM FPKM for both “replicates”)—manifesting as downregulated compared with each of the other lineages. Similarly, CAP1 is upregulated in VGII and VGIV isolates relative to VGI and VGIII isolates, while the close paralog CAP2 is upregulated in VGIII compared with VGII. Other differentially expressed capsule biosynthesis genes may not be fully compensated by genetically similar paralogs, such as the hexose transporter HXT1 downregulated by both VGII isolates.
A paralogous cluster of Cas3 and Cas31 from a previously described study of 16 isolates (7) had their sequences aligned using MUSCLE v3.8.31 (46), and a neighbor-joining tree was constructed using PAUP version 4.0b10 (47) to decipher orthologs. Cn, Cryptococcus neoformans; Cg, Cryptococcus gattii. Note that VGIV has no Cas31 gene. Expression levels (TMM FPKM) for five isolates are shown below the dendrogram. Download FIG S5, PDF file, 0.2 MB (159.4KB, pdf) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Capsule attachment and cell-wall remodeling genes were also differentially expressed between lineages of C. gattii (Fig. 3). For example, chitin synthases CHI4 and CHS2 were upregulated in both VGII isolates. Chitinase CHI2 was upregulated in VGIV. Chitin deacetylase 1 (CDA1) was highly expressed by all isolates in vitro (TMM FPKM ranging from 932 to 2,265 across all replicates) and was not identified as differentially expressed. However, CDA2 was upregulated in VGI and VGII isolates compared with VGIII and VGIV. Meanwhile, CDA3 was upregulated in VGIV compared with VGII and VGIII (perhaps again showing a change in paralog expression regulation). Both CDA2 and CDA3 mutants have increased capsule size when combined with cda1Δ mutants (18), however, CDA1 was not found to be differentially expressed.
Differential expression of known drug targets and virulence genes.
In common with most fungi, ergosterol is found in the membrane of C. gattii and is a key target for numerous antifungal drugs including fluconazole and amphotericin B (20). The VGII lineage had significantly higher expression for several genes involved in ergosterol production, including NADPH-ferrihemoprotein reductase genes, ERG1 (squalene monooxygenase), ERG25 (methylsterol monooxygenase), and SRE1. Each of these genes are linked to drug resistance or the oxidative stress response; for example, ERG1 mutants have increased fluconazole susceptibility in Candida glabrata (21), ERG25 has a moderate susceptibility to hypoxia and the endoplasmic reticulum (ER) stress-inducing agent dithiothreitol (DTT) in Aspergillus fumigatus (22, 23), and SRE1 is required for hypoxic induction of genes coding for oxygen-dependent enzymes involved in ergosterol synthesis in C. neoformans (24).
Laccases are cell wall enzymes that catalyze melanin to protect Cryptococcus from various stresses, including oxidative stress, and are therefore considered important virulence factors (25). Laccase production in both C. gattii and C. neoformans is controlled by two cell wall enzymes that control melanin production (25). VGIV significantly upregulates both copies (LAC1 and LAC2) in vitro compared with VGIII and all the other isolates, respectively. Whereas LAC1 and LAC2 flank each other in C. neoformans, C. gattii has an additional gene (CNBG_2145) encoding a hypothetical protein with no functional annotation (PFAM, GO, KEGG) in the middle. This gene, unlike LAC1 and LAC2, was not consistently expressed by any of the lineages under any conditions.
Differential expression levels among isolates in vitro were also enriched (two-tailed Fisher’s exact test with false-discovery-rate [FDR {q-value}] analysis) for several GO-terms compared with the remaining genes (Table 1). Enriched terms included “oxidative reduction” (q = 1.85E−07) and oxidoreductase activity (q = 0.011). Genes with the oxidative reduction GO-term, including those coding for ferric reductases, metalloreductases, nitric oxide dioxygenases, acidic laccases, oxidoreductases, and various dehydrogenases, were both up- and downregulated by each isolate. VGII isolates had the greatest number of upregulated genes assigned an oxidative reduction function: VGII ENV152 (n = 39), VGII R265 (n = 23), compared with VGIII (n = 18), VGIV (n = 11), and VGI (n = 7). All R265 genes, apart from the single gene CNBG_2804 (encoding a hypothetical protein with a DUF455 domain), were also found in ENV152. The remaining 17 uniquely upregulated genes in ENV152 included genes encoding five dehydrogenases (methylmalonate-semialdehyde, 3-hydroxyacyl-coenzyme A [hydroxyacyl-CoA], glutaryl-CoA, glyceraldehyde-3-phosphate, and glutamate), the Fe-Mn family superoxide dismutase, and ferric reductase transmembrane component 4. Only a single PFAM, a PF00083.19 sugar (and other molecule) transporter, was found to be enriched in the in vitro comparisons. The ability of C. gattii to respond to host- and environment-derived oxidative stress is well described (22, 26, 27), and it is noteworthy that differences in expression levels between isolates and lineages were found even under in vitro conditions.
TABLE 1.
Enrichment of functional annotation for in vitro and ex vivo comparisonsa
| GO/PFAM designation | Isolate | Count 1 | Count 2 | Fisher P | q value | Rel. prop | GO description |
|---|---|---|---|---|---|---|---|
| Sig. enriched terms from in vitro comparisons |
|||||||
| GO:0055114 | 64 | 256 | 1.13E−10 | 1.85E−07 | 2.54 | Oxidation reduction | |
| GO:0016638 | 5 | 2 | 1.01E−04 | 1.11E−02 | 25.41 | Oxidoreductase activity, acting on the CH-NH2 group of donors |
|
| GO:0050660 | 13 | 32 | 9.79E−05 | 1.11E−02 | 4.13 | FAD binding | |
| PF00083.19 | 18 | 54 | 2.26E−05 | 2.66E−02 | 3.56 | Sugar (and other) transporter | |
| GO:0004553 | 12 | 33 | 4.15E−04 | 3.25E−02 | 3.7 | Hydrolase activity, hydrolyzing O-glycosyl compounds |
|
| GO:0005975 | 25 | 116 | 7.41E−04 | 4.86E−02 | 2.19 | Carbohydrate metabolic process | |
| Sig. depleted terms from in vitro comparisons |
|||||||
| GO:0043234 | 3 | 211 | 3.35E−06 | 6.14E−04 | 0.14 | Protein complex | |
| GO:0034645 | 11 | 316 | 6.33E−05 | 9.42E−03 | 0.35 | Cellular macromolecule biosynthetic process |
|
| GO:0006396 | 2 | 142 | 2.60E−04 | 2.25E−02 | 0.14 | RNA processing | |
| GO:0003723 | 2 | 137 | 3.78E−04 | 3.11E−02 | 0.15 | RNA binding | |
| GO:0005737 | 35 | 597 | 7.23E−04 | 4.86E−02 | 0.6 | Cytoplasm | |
| GO:0015031 | 2 | 126 | 7.96E−04 | 4.86E−02 | 0.16 | Protein transport | |
| Sig. enriched terms from ex vivo comparisons |
|||||||
| GO:0055114 | VGI WM276 | 18 | 302 | 3.56E−05 | 4.63 | Oxidation reduction | |
| GO:0016491 | VGI WM276 | 18 | 376 | 4.52E−04 | 3.72 | Oxidoreductase activity | |
| GO:0006364 | VGII ENV152 | 34 | 13 | 1.93E−12 | 10.64 | rRNA processing | |
| GO:0005730 | VGII ENV152 | 25 | 6 | 7.74E−11 | 16.95 | Nucleolus | |
| GO:0003735 | VGII ENV152 | 54 | 58 | 7.37E−10 | 3.79 | Structural constituent of ribosome | |
| GO:0006412 | VGII ENV152 | 71 | 117 | 4.27E−07 | 2.47 | Translation | |
| GO:0003723 | VGII ENV152 | 51 | 88 | 1.32E−04 | 2.36 | RNA binding | |
| GO:0005506 | VGII ENV152 | 35 | 51 | 3.66E−04 | 2.79 | Iron ion binding | |
| GO:0055114 | VGII ENV152 | 95 | 225 | 5.04E−04 | 1.72 | Oxidation reduction | |
| GO:0016627 | VGII ENV152 | 13 | 8 | 1.32E−03 | 6.61 | Oxidoreductase activity, on CH-CH | |
| GO:0008026 | VGII ENV152 | 24 | 32 | 2.60E−03 | 3.05 | ATP-dependent helicase activity | |
| GO:0016627 | VGII R265 | 11 | 10 | 4.25E−04 | 9.6 | Oxidoreductase activity, on CH-CH | |
| GO:0022900 | VGII R265 | 8 | 7 | 8.59E−03 | 9.97 | Electron transport chain | |
| GO:0020037 | VGII R265 | 11 | 17 | 8.59E−03 | 5.64 | Heme binding | |
| GO:0016491 | VGIII CA1873 | 33 | 361 | 9.41E−04 | 2.57 | Oxidoreductase activity | |
| GO:0055114 | VGIII CA1874 | 28 | 292 | 1.77E−03 | 2.69 | Oxidation reduction | |
| Sig. depleted terms from ex vivo comparisons |
|||||||
| GO:0043234 | VGII ENV152 | 20 | 194 | 1.39E−03 | 0.42 | Protein complex | |
| GO:0006508 | VGII ENV152 | 12 | 132 | 9.48E−03 | 0.37 | Proteolysis |
Enrichment of functional annotation for in vitro and ex vivo comparisons was determined by a two-tailed Fisher exact test with a q-value (Storey-Tibshirani) FDR, where count 1 data represent numbers of terms and parent terms in the differentially expressed set and count 2 data represent the remaining terms and parent terms in the non-differentially expressed set. The uncorrected Fisher P values, corrected q values, relative-proportion (Rel. Prop) values, and descriptions are provided for each term (requiring q values of <0.05 for each). FAD, flavin adenine dinucleotide; Sig., significantly.
One in five C. gattii genes is differentially expressed under in vitro and ex vivo conditions among the lineages.
Pairwise comparisons of expression values for each lineage at different time points (t0 versus t1, t3, and t6) identified 1,193 unique DEGs, the majority of which were upregulated in vitro versus ex vivo (Fig. 2b) (see also Data Set S1, tab 2). About 1/3 (60%) (n = 309) of these genes were also differentially expressed in an interlineage in vitro comparison, leaving 215 genes that were uniquely differentially expressed in vitro. PCA showed large differences between t0 and ex vivo time points in both VGII isolates (Fig. 2d).
Genes of interest (capsule, ergosterol, and laccases) were enriched for DEGs at different time points based on the results of a hypergeometric test (P[X > x]) = P = 0.007), including nine capsule biosynthesis genes (all VGII), 11 capsule attachment and cell-wall remodeling genes (all downregulated ex vivo), 14 ergosterol genes, and both laccase genes (Fig. 4). Separately, many (n = 40/97; 41%) C. neoformans genes that were previously found to be differentially expressed via microarray in the presence of amoebae and macrophages (13) were similarly modulated in C. gattii (111 genes with similar or different modulation results between amoebae and macrophages, 97 of which had a C. gattii ortholog, 56 of which were also C. gattii DEGs between time points, and 40 of which had the same directionality) (Data Set S1, tab 3).
FIG 4.

Bar charts showing mean expression (TMM FPKM) of differentially expressed ergosterol, capsular biosynthesis, capsular attachment and laccase genes between the five isolates of Cryptococcus gattii at each time point (t0 = in vitro, t1 = 1-h w/BMDMs, t3 = 3 h w/BMDMs, and t6 = 6 h w/BMDMs). Red, significantly upregulated; blue, significantly downregulated. Genes are considered differentially expressed where FDR P value < 0.001 and greater than 4-fold TMM FPKM. Error bars show the range of TMM FPKM values in comparisons between the two replicates. Glucan, glucan 1,3-β-glucosidase.
Genes differentially expressed ex vivo were statistically significantly enriched (two-tailed Fisher’s exact test with q-value FDR) for 18 GO-terms and no PFAM terms (Table 1). Strikingly, the oxidoreductase activity term was enriched in each lineage and isolate (apart from VGIV, which had only 20 differentially expressed genes and thus no enriched terms). The ability of C. gattii to respond to host- and environment-derived oxidative stress is thus a significant feature of genes that are differentially expressed (both between lineages in vitro and under in vitro versus ex vivo conditions). Additional terms included iron ion binding and terms related to ribosomes (i.e., processing of rRNA, a structural constituent of ribosome) for VGII ENV152, perhaps indicating an increase in translational activity.
Capsule biosynthesis genes in VGII were differentially expressed in the presence of macrophages (Fig. 4). For example, CAP1 and UGT1 were downregulated by both VGII isolates, while CAP4, CAS3, and UGD1 were all upregulated at one or more of the ex vivo time points. Other capsule biosynthesis DEGs included CAS35 (the CAS35Δ mutant has a decreased capsule [19]), which was upregulated in R265 at t3 versus t0; UXS1 (the uxs1Δ capsule is missing xylose [28]), which was upregulated in R265 at t1 and t3 versus t0; and CAS1 (the cas1Δ mutant has a defect in capsule O-acetylation and reactivity to GXM antibodies [29]), which was upregulated in VGII ENV152 at t3.
Capsule attachment and cell-wall remodeling genes were also differentially expressed ex vivo, predominantly by VGII isolates, and were all downregulated ex vivo (Fig. 4). For example, chitin synthase genes CHS4 and CHS8 were upregulated in vitro in VGII compared with any other time points ex vivo. Chitins generated by such chitin synthases are converted into chitosan by the chitin deacetylase genes CDA1, CDA2, and CDA3, where it constitutes an important component of the cell wall of Cryptococcus (30). Both CDA1 and CDA2 are downregulated ex vivo in one or more of the lineages of C. gattii, apart from VGIV.
The four ergosterol genes (CNBG0583/CNAG01003, ERF1, SRE1, and ERG25) that were upregulated in VGII compared with VGIII and VGIV in vitro were also upregulated in VGII in vitro compared with the three ex vivo time points, again suggesting that these are downregulated during infection. However, nine additional ergosterol biosynthesis genes, including ERG2, ERG6, ERG7, ERG13, ERG20, and ERG26, were each upregulated in the VGII isolates at 3 h postinfection compared with the levels seen in vitro, suggesting that these genes are activated between 1 and 6 h after coincubation with macrophages.
The laccase genes that produce melanin and that are upregulated in VGIV in vitro compared with the other lineages are also differentially expressed in VGII between in vitro and ex vivo conditions. Specifically, LAC1 (CNBG2144) in VGI and VGII is downregulated ex vivo. In contrast, LAC2 (CNB2146) is upregulated in both VGII isolates at t3 compared with t0—demonstrating that during infection, VGII isolates switch expression from LAC1 to LAC2.
Mouse macrophage response to C. gattii.
Mouse BMDM expression for each of 58,716 annotated mouse transcripts (including protein-coding genes and pseudogenes and other non-protein-coding genes) was highly consistent under in vitro conditions and conditions of coincubation with any of the four lineages of C. gattii at any of the three time points (Fig. 5), perhaps indicating low rates of yeast engulfment or the general low immunogenicity of cryptococci. In this case, differential expression of C. gattii ex vivo (see previous section) may be caused by indirect effects of macrophage coincubation. Only 24 upregulated DEGs and 42 downregulated DEGs were identified in total (Data Set S1, tab 4). Of those 24 upregulated genes, five separate/unique FBJ osteosarcoma viral oncogenes (specifically, FOSB and the truncated splice form Δfosb2) belonging to the eight most highly differentially expressed genes (log fold change = 3.58 to 7.78) were found at t1 for VGII R265 only. FOS genes encode a leucine zipper protein that dimerizes with the Jun family (among which JunB is also upregulated at t3 in VGII ENV152), forming the AP-1 transcription factor, which in turn regulates diverse functions, including cell proliferation, differentiation, and transformation, following the primary growth factor response. Perhaps it is therefore unsurprising that 20% of upregulated genes (n = 11/52 redundant upregulated genes), including EGR1-201 and EGR1-202, at t3 for all five C. gattii isolates tested and EGR3 at t1 for VGII R265 only, belonged to the early growth response protein 1 family genes.
FIG 5.
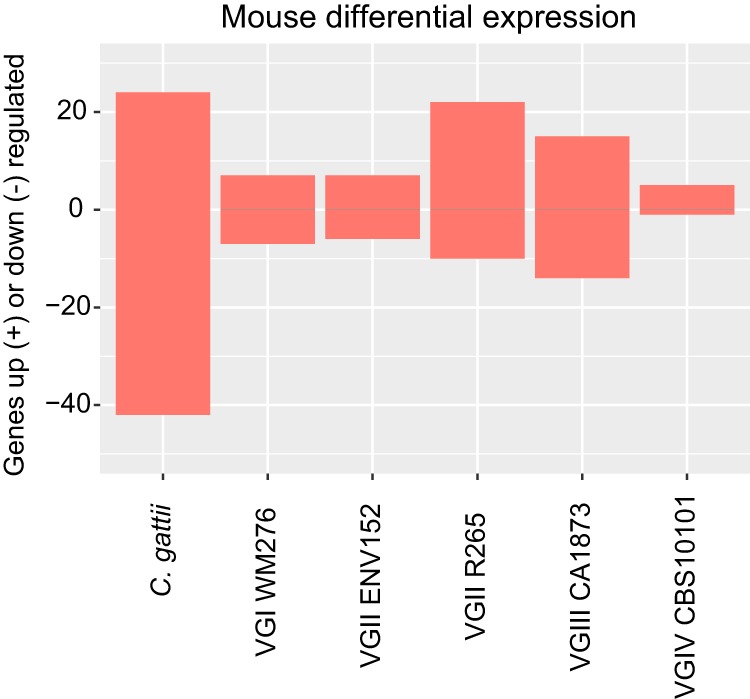
A bar chart showing the number of mouse genes up- and downregulated by each of the five isolates of C. gattii compared with growth without yeast. Where the same gene was found in multiple pairwise comparisons, it is included only once in the redundant categories (applicable only for the grouped category). Positive values indicate genes that were upregulated in that isolate/lineage compared with others, while negative values (below zero) indicate genes that were downregulated in that isolate/lineage compared with others. Genes are considered differentially expressed where FDR P value are <0.001 and TMM FPKM values are greater than 4-fold.
Most of the genes downregulated in mouse macrophages during infection appear to be signaling molecules and transcription factors, including ETV5 (ETS transcription factor variant 5), which is downregulated at t6 in all C. gattii isolates except VGIV. Other downregulated genes include those encoding the DENND2C proteins at t3 in VGII ENV152 and at t1 in VGII R265. DENND2C proteins act in diverse intracellular signaling pathways via GDP/GTP exchange, with many potential downstream targets. Additionally, DNA-binding protein ID1 (which inhibits basic helix-loop-helix [bHLH] transcription factors) was downregulated at t6 in VGI, VGII ENV152, and VGIII, as well as t1 in VGII R265. Similarly, dual specific protein phosphatase 6 (DUSP6), involved in mitogen-activated protein kinase (MAPK) signaling, was downregulated at t6 in VGI, VGII ENV152, and VGIII, while DUSP4 was also downregulated at t6 in VGI only.
DISCUSSION
The transcriptional responses of host and pathogen during infection can reveal key insights into their interactions and the molecular basis of pathogenicity. Transcriptional differences between distantly related isolates in culture can also reveal the impact of their genetic divergence and explain epidemiological differences. In this study, we compared the expression profiles of five isolates belonging to the four lineages of C. gattii in vitro between three early time points during coincubation with mouse bone marrow-derived macrophages (BMDM) and, in parallel, characterized the host response. Both C. gattii comparisons suggested that lineage VGII is transcriptionally divergent from non-VGII lineages in terms of number of genes differentially expressed. For example, only 5% of in vitro DEGs belonged to non-VGII isolates, despite these isolates accounting for 30% of all pairwise comparisons made. It is likely that the loss of RNA interference (RNAi) functionality in VGII (7, 31, 32) is partially responsible indirectly (rewiring of gene regulatory responses in result to loss of Argonaut proteins) and perhaps even directly (mRNA not being degraded).
VGII is responsible for nearly all C. gattii infections in the Pacific Northwest (PNW) (33), which is the location of the worst recorded outbreak of C. gattii infections worldwide. In this study, we found that two subgroup VGIIa isolates had distinct expression profiles, possibly owing to genetic variation generated from their different sources of origin (environmental or clinical). VGII isolates also show a high degree (albeit across a range) of virulence traits, including rate of intracellular proliferation within macrophages (R265 = 1.8 ± 0.1 and ENV152 = 2.3 ± 0.2 compared with VGI WM276 = 0.98 ± 0.2 and VGIV CBS10101= 1.23 ± 0.3), average mitochondrial tubularization percentage (R265 = 58.4% and ENV152 = 44.5% compared with VGI WM276 = 14.3% and VGIV CBS10101 = 21.6%), average phagocytosis percentage (R265 = 31.4% and ENV152 = 20.5% compared with VGI WM276 = 11.1% and VGIV CBS10101 = 8.6%), and macrophage cell death percentage (R265 = 15.2% and ENV152 = 12.3% compared with VGI WM276 = 16.8% and VGIV CBS10101 = 12%) (34, 35). VGIII isolate CA1873 was not evaluated for these phenotypic traits in those two papers. Therefore, isolates within lineages demonstrate high phenotypic variability. Although the sequenced isolates in this study are representative of each lineage, comparing multiple isolates from each lineage would help delineate the intralineage versus interlineage variations in gene expression.
The capsule biosynthesis pathway in C. gattii constitutes a complex trait controlled by numerous genes. Many of these genes presented various degrees of expression among the four lineages in vitro, suggesting the presence of diverse mechanisms operating to maintain and perhaps even diversify the properties of the capsule. However, non-VGII lineages did not differentially express capsule synthesis genes ex vivo, suggesting that they are perhaps less rigorously regulated (expressed under more diverse conditions) or expressed less abundantly or are perhaps less sensitive to host-derived stresses and stimuli. One such gene, CAS3, is upregulated by VGII isolates at multiple time points ex vivo compared with in vitro. CAS3 has previously been identified as upregulated in VGII R265 compared with low-virulence VGII isolate R272 under conditions of carbon and nitrogen starvation (16). However, we found no evidence of differential expression of CAS3 between VGII R265 and VGII ENV152—possibly indicating a uniqueness of R272 expression or experimental differences between studies. However, we found that CAS3 is upregulated by VGIV CBS10101 in vitro compared with other isolates, while close paralog CAS31 is a lineage-specific gene missing in VGIV (7). It is therefore possible that VGIV is overexpressing CAS3 to compensate for its CAS31 deletion or disruption and demonstrating subfunctionalization of these paralogs. Furthermore, cas31Δ mutants have previously been shown to manifest minor differences in GXM composition in C. neoformans (18, 19), which may also manifest in C. gattii VGIV as well as in VGIIIb. Separately, the capsule biosynthesis gene CAP2 is upregulated by VGIII, while CAP1 is downregulated by VGI and VGIII, suggesting a lineage transition from expressing one gene to expressing another. Capsule attachment DEGs (in comparison to capsule biosynthesis genes) were all downregulated ex vivo, suggesting that these do not play an active role during infection.
Ergosterol in the membrane of C. gattii is a key target for numerous antifungal drugs, including fluconazole and amphotericin B (20). VGII presented higher expression for genes involved in ergosterol production in vitro, including upregulation of ERG1, ERG25, SRE1, and an NADPH-ferrihemoprotein reductase gene. Mutants for each of these genes show a range of defects in the presence of antifungals or hypoxia (21–24). ERG1, ERG25, and SRE1 were also upregulated in VGII in vitro compared with the three ex vivo time points, suggesting that these genes can be switched off during non-drug-related stresses. However, a further nine ergosterol genes were upregulated between 1 and 6 h postinfection, including ERG2, ERG6, ERG7, ERG13, ERG20, and ERG26, suggesting that this pathway is active during infection. The biological significance of these expression differences are unclear but could manifest in lineage-specific drug resistance variations.
Laccase production in both C. gattii and C. neoformans is controlled by two cell wall enzymes (encoded by LAC1 and LAC2) that possess a broad spectrum of activity, oxidizing both polyphenolic compounds and iron (25). VGIV upregulates both genes compared with other lineages in vitro. LAC1 is downregulated in VGI and VGII ex vivo compared with in vitro, and LAC2 is instead upregulated in both VGII isolates at t3 compared with t0—suggesting that C. gattii VGII switches expression from one laccase gene to the other during infection, perhaps due to changing concentrations or requirements for metabolism of lactose and galactose or for production of melanin. In C. neoformans, LAC1 and LAC2 (along with capsule genes) are part of the Gpa1-cAMP pathway, which regulates capsule and melanin production using l-3,4-dihydroxyphenylalanine (l-dopa) as a substrate (36). We hypothesize that VGII uses LAC2 to regulate growth and glucose responses (and potentially virulence) instead of LAC1. In C. neoformans, LAC1 is localized to the cell wall, whereas LAC2 is cytoplasmic but is capable of localizing to the cell wall (37); therefore, C. gattii LAC2 could behave similarly to its C. neoformans counterpart, which may have increased versatility during infection of macrophages.
We also identified differential expression levels in VGII lineage-specific genes such as those encoding an MFS transporter and an oxidoreductase that may have contributed to the enrichment of oxidative reduction, which was identified in both in vitro comparisons and in vitro versus ex vivo comparisons. This term includes a wide range of functions, pathways, and genes, including those encoding ferric reductases, metalloreductases, nitric oxide dioxygenases, acidic laccases, oxidoreductases, and various dehydrogenases. Previously, an upregulation of genes involved in oxidative stress has been identified in C. neoformans isolate H99 coincubated with the J774A macrophage-like cell line for 16 h versus in vitro conditions (12) and in C. gattii VGII isolate R265 versus isolate R272 under carbon and nitrogen starvation conditions (16). The ability of C. gattii to respond to host- and environment-derived oxidative stress is well described—and it is consistent that genes involved in these processes should also be enriched between isolates and lineages at the expression level in vitro and among lineages at different time points ex vivo.
Finally, we found that mouse macrophages respond to C. gattii by upregulating FosB/Jun/Egr1 regulatory proteins at early time points, which may trigger differentiation and cell division of macrophages. We found little evidence for differential expression induced by different C. gattii lineages, suggesting that the macrophage responses to C. gattii are the same across lineages. Our report highlights the breadth of expression profiles among the lineages of C. gattii and the diversity of transcriptional responses at this host-pathogen interface, some of which may be the cause of the differences in phenotypic and clinical manifestations noted between lineages (38).
MATERIALS AND METHODS
Macrophage and Cryptococcus gattii infection assay.
C. gattii was grown in accordance with methods described in previous work (26). Briefly, five strains of C. gattii were grown in RPMI/C10 media with heat-inactivated (HI) fetal bovine serum (FBS) media and were preincubated at 37°C in C10 media for 1.5 h before infection. Bone marrow-derived macrophages (BMDM) were derived from bone marrow cells collected from the femur and tibia of C57BL/6 female mice. All mouse work was performed in accordance with the Institutional Animal Care and Use Committees (IACUC) guidelines and with relevant guidelines at the Broad Institute and Massachusetts Institute of Technology, with protocol 0615-058-1. Primary bone marrow cells were grown in C10 media as previously described (39) and supplemented with macrophage colony-stimulating factor (M-CSF) (Thermo Fisher Scientific) at final concentration of 10 ng/ml to promote differentiation into macrophages.
For the infection experiment, dilutions of 7.5E+5 cells were made for BMDM and C. gattii strains. BMDM were spun at 500 relative centrifugal force (rcf) at 37°C for 2 min to adhere to plates, and inoculated with C. gattii strains using a multiplicity of infection (MOI) of 1 macrophage to 2 C. gattii. Macrophages with C. gattii cells were centrifuged at 500rcf at 37°C for 2 min. Next, cells were incubated at 37°C with 5% CO2 for 1 h, 3 h, and 6 h postcentrifugation. In addition, we grew C. gattii in vitro, and BMDM without C. gattii, in duplicate with the same C10 media. At the end of the time points, the medium was removed, lysis buffer was added, and samples were placed in a −80°C freezer for RNA extraction.
RNA was extracted from population samples using a Qiagen RNeasy minikit. All samples were subjected to 3 min of bead beating with 0.5-mm-diameter zirconia glass beads (BioSpec Products) in a bead mill. Libraries were generated usinga TruSeq Stranded mRNA Library Prep kit (Illumina).
RNA-seq and data analysis.
All samples were sequenced on an Illumina HiSeq 2500 system to generate strand-specific paired-end reads that were 38 nucleotides (nt) in length. Raw reads from each sample were sequenced on multiple lanes and so were merged into individual replicates. Using BLAST searches of the nr database, we identified the following contaminants in each of the samples: Escherichia coli K-12 MG1655, suicide vector pCD-RAsl1, and cloning vector pMJ016c (see Table S1, tab 1, in the supplemental material). These contaminants were excluded from all of the read sets on the basis of Bowtie2 (40) alignments. We next aligned all reads to mouse GRCm38 p4 mm10 transcript sets (including protein-coding genes and pseudogenes and other categories of non-protein-coding genes) and genomes (41) using Bowtie2 (40). We took all reads that aligned to neither the mouse genome nor the gene sets and aligned those reads to the R265 updated gene set (7) as well as the R265 genome using Bowtie2 (Table S1, tab 2). Bowtie2 alignments to mouse or C. gattii were run though the Trinity version r20140413p1 (42) pipeline, using the following default parameters: max insert size 800, no-mixed (no unpaired reads), no-discordant (does not satisfy the paired-end constraints), gbar = 1,000 (disallow gaps within 1,000 bases), and end-to-end (entire read must align from one end to other without any trimming or soft clipping). To reveal the extent of the effect that this had on alignment data, we aligned R265 RNA in vitro replicate A to the genome of R265 with Bowtie2 using nonstringent parameters. The overall alignment rate increased from 56.97% to 68.62%. We also found that some reads that were left unaligned by Bowtie2 were aligned by the separate tool BWA v0.7.4.
Other factors that were likely to reduce alignment coverage were the draft quality of the C. gattii R265 genome and the gene sets used (see Fig. S1 in the supplemental material for evaluation of Core Eukaroyotic Gene coverage)—as a consequence of the effects of those limiting factors, genuine C. gattii mRNA would be unalignable. Another factor was that non-VGII isolates were aligned to VGII and that certain differences, including lineage-specific gene differences, would reduce gene coverage. However, the alignment strategy employed was deemed more suitable than aligning to individual lineage reference sets and relying on orthologs, which would reduce the number of genes subjected to comparisons and add errors caused by incorrect assignment of orthologs.
All reads from in vitro C. gattii experiments that were unaligned were assembled de novo using Trinity (5,730 sequences of total length 2.1 Mb), genes predicted by Transdecoder (521 sequences of 270 kb), and BLASTn searches of the NCBI nr database. Only 90 of these sequences (37 kb) did not align to either the mouse genome or that of C. gattii R265. The remainder of the unaligned sequence that did not map to mouse or C. gattii was highly repetitive and was enriched for Illumina adapter and control sequences. Indeed, FASTQC v0.11.4 identified large fractions of the unaligned unprocessed reads derived from Process Controls for TruSeq kits, which including >10% CTA and CTL as overrepresented sequences.
Reads that aligned only to coding sequence (CDS) from C. gattii or mm10 had their transcript abundances estimated using RSEM (43) and differential expression levels (FDR P value < 0.001 and >4-fold change of TMM-normalized FPKM) predicted using EdgeR (17) through the use of the Trinity version r20140413p1 (46) pipeline. Specifically, the pipeline uses the EdgeR quantile-adjusted conditional maximum likelihood (qCML) method after estimating common dispersion and tagwise dispersions using the Cox-Reid profile-adjusted likelihood method (also implemented in EdgeR). For Cryptococcus strains, we estimated transcript abundance and differential expression in the same way after (i) merged time points (i.e., calculations performed for each isolate only) and (ii) merged isolates (i.e., calculations performed for each time point only). Merging of isolate data resulted in far weaker correlations between replicates than merging of time point data, indicating that in vivo conditions were more influential on overall expression values than lineage/strain-specific differences. Principal-component analysis (PCA) was performed using SmartPCA from EIGENSOFT v4.0 (44), where each gene was given a value of 0 for non-DEG and a value of 1 for DEG (upregulated).
Orthologs to genes of known function in C. neoformans H99 were identified in C. gattii R265 using OrthoMCL (45) across 16 isolates as previously described (7). Genes of interest (capsule biosynthesis, capsule attachment, and ergosterol genes) that fell within paralogous clusters had the sequences of 16 isolates described previously (7) aligned using MUSCLE v3.8.31 (46), and a neighbor-joining tree was constructed using PAUP version 4.0b10 (47) to decipher orthologs. Synteny was visualized using Synima (48).
Of the 58,716 annotated mouse transcripts, 39,229 (67%) had evidence of expression in one or more samples (TMM FPKM > 1). Prior to differential expression analysis, we excluded 3,596 mouse transcripts that any C. gattii in vitro RNA (i.e., no macrophage present) aligned to them. Surprisingly, some mouse transcripts were very highly expressed in C. gattii-only samples (e.g., glutamate receptor interacting protein 2 [Grip2-205] had a TMM FPKM value of >15,000 for every C. gattii isolate and values between only 10 and 874 for t1, t3, and t6, which were time points when mouse macrophages were actually present), perhaps owing to sequence similarity of some 30mers or to inaccuracies in the mouse gene set. Applying the same threshold FDR P value of <0.001 and >4-fold change of TMM normalized FPKM using EdgeR (17) identified 24 genes that were upregulated during infection by 1 or more isolates of C. gattii and 42 genes that were downregulated during infection by 1 or more isolates of C. gattii.
Enrichment analyses for PFAM and GO terms previously assigned (7) were conducted using two-tailed Fisher’s exact test with q-value FDR. Multiple testing corrections were performed with the Storey-Tibshirani (49) method (requiring a q value of <0.05). For enrichment tests, we excluded PFAM and GO terms related to transposable elements and domains of unknown function.
Impact of read depth on differential expression.
On the basis of data quantity/transcriptome coverage alone, the host response and in vitro C. gattii lineage expression differences should be the most robust, while C. gattii ex vivo expression changes should be less robust. To explore the coverage and sensitivity of our C. gattii RNA-seq data and pipeline, we first made subsets (75%, 50%, and 25%) of our C. gattii data and recalibrated the differential expression data (Fig. S3). The number of genes differentially expressed between isolates under in vitro conditions remained most consistent in terms of number of differentially expressed genes following subsetting (100% = 1,208 genes, 75% = 1,205, 50% = 1,147, 25% = 800) (Fig. S3A).
For all C. gattii data sets (those corresponding to in vitro conditions and individual isolates corresponding to comparisons between conditions), the proportion of genes reidentified in the 75% subset was between 79% to 100% (VGIV CBS10101 had the same 20 genes corresponding to both data set sizes) (Fig. S3B). In contrast, the number of genes identified only in the full data sets increased in the remaining subsets, with the most pronounced increase being that seen with VGIIa ENV152, which lost the identification of 476 genes in the 75% subset, possibly indicating a lack of data for this isolate (Fig. S3C). We also assessed each subset for genes that were not found in the full set (i.e., genes that were unique to the subsets; Fig. S3D). VGII and VGIV and the in vitro data sets had fewer unique genes as the subsets became smaller, while VGI and VGIII had increases as the subsets became smaller.
Under in vitro conditions, between 10% to 15% of genes were unique in their subsets, while for VGII ENV152, the proportion was between 1% and 5%. For isolates with very few identified differentially expressed genes (such as VGIV CBS10101 and VGIII CA1873), the numbers of unique genes approached or even exceeded the number found in the full data set. Given this variation, it is therefore likely that most of the ex vivo C. gattii data sets would benefit from deeper RNA-seq data and that the in vitro expression values are more robust than the ex vivo data. Nevertheless, the majority of differential expressed genes were consistent between the larger subsets of data.
Accession number(s).
All RNA-seq data for mouse macrophages and C. gattii have been deposited in the Short Read Achieve under accession no. PRJNA428946.
ACKNOWLEDGMENTS
We thank Jose Munoz for his input on the analysis of the mouse RNA-seq enrichment.
R.A.F. was supported by a Wellcome Trust-Massachusetts Institute of Technology (MIT) Postdoctoral Fellowship. M.C.F. and J.R. were supported by Medical Research Council grant MR/K000373/1. R.C.M. is supported by a Wolfson Royal Society Research Merit Award and by funding from the European Research Council under the European Union’s Seventh Framework Program (FP/2007-2013)/ERC (grant agreement no. 614562). This work was funded in part by NIAID grant U19AI110818 to the Broad Institute.
REFERENCES
- 1.Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, Gurr SJ. 2012. Emerging fungal threats to animal, plant and ecosystem health. Nature 484:186–194. doi: 10.1038/nature10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farrer RA, Fisher MC. 2017. Describing genomic and epigenomic traits underpinning emerging fungal pathogens. Adv Genet 100:73–140. doi: 10.1016/bs.adgen.2017.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Farrer RA, Weinert LA, Bielby J, Garner TWJ, Balloux F, Clare F, Bosch J, Cunningham AA, Weldon C, Preez LH, Du Anderson L, Pond SLK, Shahar-Golan R, Henk DA, Fisher MC. 2011. Multiple emergences of genetically diverse amphibian-infecting chytrids include a globalized hypervirulent recombinant lineage. Proc Natl Acad Sci 108:18732–18736. doi: 10.1073/pnas.1111915108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farrer RA, Henk DA, Garner TWJ, Balloux F, Woodhams DC, Fisher MC. 2013. Chromosomal copy number variation, selection and uneven rates of recombination reveal cryptic genome diversity linked to pathogenicity. PLoS Genet 9:e1003703. doi: 10.1371/journal.pgen.1003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Springer DJ, Phadke S, Billmyre B, Heitman J. 2012. Cryptococcus gattii, no longer an accidental pathogen? Curr Fungal Infect Rep 6:245–256. doi: 10.1007/s12281-012-0111-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fraser JA, Giles SS, Wenink EC, Geunes-Boyer SG, Wright JR, Diezmann S, Allen A, Stajich JE, Dietrich FS, Perfect JR, Heitman J. 2005. Same-sex mating and the origin of the Vancouver Island Cryptococcus gattii outbreak. Nature 437:1360–1364. doi: 10.1038/nature04220. [DOI] [PubMed] [Google Scholar]
- 7.Farrer RA, Desjardins CA, Sakthikumar S, Gujja S, Saif S, Zeng Q, Chen Y, Voelz K, Heitman J, May RC, Fisher MC, Cuomo CA. 2015. Genome evolution and innovation across the four major lineages of Cryptococcus gattii. mBio 6:e00868-15. doi: 10.1128/mBio.00868-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H, Johnson AD. 2010. Evolution of transcription networks — lessons from yeasts. Curr Biol 20:R746–R753. doi: 10.1016/j.cub.2010.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fraser HB, Levy S, Chavan A, Shah HB, Perez JC, Zhou Y, Siegal ML, Sinha H. 2012. Polygenic cis-regulatory adaptation in the evolution of yeast pathogenicity. Genome Res 22:1930–1939. doi: 10.1101/gr.134080.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooke DEL, Cano LM, Raffaele S, Bain RA, Cooke LR, Etherington GJ, Deahl KL, Farrer RA, Gilroy EM, Goss EM, Grünwald NJ, Hein I, MacLean D, McNicol JW, Randall E, Oliva RF, Pel MA, Shaw DS, Squires JN, Taylor MC, Vleeshouwers VGAA, Birch PRJ, Lees AK, Kamoun S. 2012. Genome analyses of an aggressive and invasive lineage of the Irish potato famine pathogen. PLoS Pathog 8:e1002940. doi: 10.1371/journal.ppat.1002940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma H, Hagen F, Stekel DJ, Johnston SA, Sionov E, Falk R, Polacheck I, Boekhout T, May RC. 2009. The fatal fungal outbreak on Vancouver Island is characterized by enhanced intracellular parasitism driven by mitochondrial regulation. Proc Natl Acad Sci U S A 106:12980–12985. doi: 10.1073/pnas.0902963106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan W, Kraus PR, Boily M-J, Heitman J. 2005. Cryptococcus neoformans gene expression during murine macrophage infection. Eukaryot Cell 4:1420–1433. doi: 10.1128/EC.4.8.1420-1433.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Derengowski L da S, Paes HC, Albuquerque P, Tavares AHFP, Fernandes L, Silva-Pereira I, Casadevall A. 2013. The transcriptional response of Cryptococcus neoformans to ingestion by Acanthamoeba castellanii and macrophages provides insights into the evolutionary adaptation to the mammalian host. Eukaryot Cell 12:761–774. doi: 10.1128/EC.00073-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janbon G, Ormerod KL, Paulet D, Byrnes EJ, Yadav V, Chatterjee G, Mullapudi N, Hon C-C, Billmyre RB, Brunel F, Bahn Y-S, Chen W, Chen Y, Chow EWL, Coppée J-Y, Floyd-Averette A, Gaillardin C, Gerik KJ, Goldberg J, Gonzalez-Hilarion S, Gujja S, Hamlin JL, Hsueh Y-P, Ianiri G, Jones S, Kodira CD, Kozubowski L, Lam W, Marra M, Mesner LD, Mieczkowski PA, Moyrand F, Nielsen K, Proux C, Rossignol T, Schein JE, Sun S, Wollschlaeger C, Wood IA, Zeng Q, Neuvéglise C, Newlon CS, Perfect JR, Lodge JK, Idnurm A, Stajich JE, Kronstad JW, Sanyal K, Heitman J, Fraser JA, Cuomo CA, Dietrich FS. 2014. Analysis of the genome and transcriptome of Cryptococcus neoformans var. grubii reveals complex RNA expression and microevolution leading to virulence attenuation. PLoS Genet 10:e1004261. doi: 10.1371/journal.pgen.1004261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Toffaletti DL, Tenor JL, Litvintseva AP, Fang C, Mitchell TG, McDonald TR, Nielsen K, Boulware DR, Bicanic T, Perfect JR. 2014. The Cryptococcus neoformans transcriptome at the site of human meningitis. mBio 5:e01087-13. doi: 10.1128/mBio.01087-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ngamskulrungroj P, Price J, Sorrell T, Perfect JR, Meyer W. 2011. Cryptococcus gattii virulence composite: candidate genes revealed by microarray analysis of high and less virulent Vancouver Island outbreak strains. PLoS One 6:e16076. doi: 10.1371/journal.pone.0016076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson MD, McCarthy DJ, Smyth GK. 2010. EdgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Meara TR, Alspaugh JA. 2012. The Cryptococcus neoformans capsule: a sword and a shield. Clin Microbiol Rev 25:387–408. doi: 10.1128/CMR.00001-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moyrand F, Chang YC, Himmelreich U, Kwon-Chung KJ, Janbon G. 2004. Cas3p belongs to a seven-member family of capsule structure designer proteins. Eukaryot Cell 3:1513–1524. doi: 10.1128/EC.3.6.1513-1524.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santos JRA, Gouveia LF, Taylor ELS, Resende-Stoianoff MA, Pianetti GA, César IC, Santos DA. 2012. Dynamic interaction between fluconazole and amphotericin B against Cryptococcus gattii. Antimicrob Agents Chemother 56:2553–2558. doi: 10.1128/AAC.06098-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai H-F, Bard M, Izumikawa K, Krol AA, Sturm AM, Culbertson NT, Pierson CA, Bennett JE. 2004. Candida glabrata erg1 mutant with increased sensitivity to azoles and to low oxygen tension. Antimicrob Agents Chemother 48:2483–2489. doi: 10.1128/AAC.48.7.2483-2489.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J, Cho Y-J, Do E, Choi J, Hu G, Cadieux B, Chun J, Lee Y, Kronstad JW, Jung WH. 2012. A defect in iron uptake enhances the susceptibility of Cryptococcus neoformans to azole antifungal drugs. Fungal Genet Biol 49:955–966. doi: 10.1016/j.fgb.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blosser SJ, Merriman B, Grahl N, Chung D, Cramer RA. 2014. Two C4-sterol methyl oxidases (Erg25) catalyse ergosterol intermediate demethylation and impact environmental stress adaptation in Aspergillus fumigatus. Microbiology 160:2492–2506. doi: 10.1099/mic.0.080440-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chun CD, Liu OW, Madhani HD. 2007. A link between virulence and homeostatic responses to hypoxia during infection by the human fungal pathogen Cryptococcus neoformans. PLoS Pathog 3:e22. doi: 10.1371/journal.ppat.0030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu X, Williamson PR. 2004. Role of laccase in the biology and virulence of Cryptococcus neoformans. FEMS Yeast Res 5:1–10. doi: 10.1016/j.femsyr.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 26.Voelz K, Johnston SA, Smith LM, Hall RA, Idnurm A, May RC. 2014. ‘Division of labour’ in response to host oxidative burst drives a fatal Cryptococcus gattii outbreak. Nat Commun 5:5194. doi: 10.1038/ncomms6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Casadevall A. 1994. Susceptibility of melanized and nonmelanized Cryptococcus neoformans to nitrogen- and oxygen-derived oxidants. Infect Immun 62:3004–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozel TR, Levitz SM, Dromer F, Gates MA, Thorkildson P, Janbon G. 2003. Antigenic and biological characteristics of mutant strains of Cryptococcus neoformans lacking capsular O acetylation or xylosyl side chains. Infect Immun 71:2868–2875. doi: 10.1128/IAI.71.5.2868-2875.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moyrand F, Klaproth B, Himmelreich U, Dromer F, Janbon G. 2002. Isolation and characterization of capsule structure mutant strains of Cryptococcus neoformans. Mol Microbiol 45:837–849. doi: 10.1046/j.1365-2958.2002.03059.x. [DOI] [PubMed] [Google Scholar]
- 30.Baker LG, Specht CA, Donlin MJ, Lodge JK. 2007. Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot Cell 6:855–867. doi: 10.1128/EC.00399-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D’Souza CA, Kronstad JW, Taylor G, Warren R, Yuen M, Hu G, Jung WH, Sham A, Kidd SE, Tangen K, Lee N, Zeilmaker T, Sawkins J, McVicker G, Shah S, Gnerre S, Griggs A, Zeng Q, Bartlett K, Li W, Wang X, Heitman J, Stajich JE, Fraser JA, Meyer W, Carter D, Schein J, Krzywinski M, Kwon-Chung KJ, Varma A, Wang J, Brunham R, Fyfe M, Ouellette BFF, Siddiqui A, Marra M, Jones S, Holt R, Birren BW, Galagan JE, Cuomo CA. 2011. Genome variation in Cryptococcus gattii, an emerging pathogen of immunocompetent hosts. mBio 2:e00342-10. doi: 10.1128/mBio.00342-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, Hsueh Y-P, Li W, Floyd A, Skalsky R, Heitman J. 2010. Sex-induced silencing defends the genome of Cryptococcus neoformans via RNAi. Genes Dev 24:2566–2582. doi: 10.1101/gad.1970910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Byrnes EJ, Li W, Lewit Y, Ma H, Voelz K, Ren P, Carter DA, Chaturvedi V, Bildfell RJ, May RC, Heitman J. 2010. Emergence and pathogenicity of highly virulent Cryptococcus gattii genotypes in the northwest United States. PLoS Pathog 6:e1000850. doi: 10.1371/journal.ppat.1000850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farrer RA, Voelz K, Henk DA, Johnston SA, Fisher MC, May RC, Cuomo CA. 2016. Microevolutionary traits and comparative population genomics of the emerging pathogenic fungus Cryptococcus gattii. Philos Trans R Soc Lond B Biol Sci 371:20160021. doi: 10.1098/rstb.2016.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voelz K, Ma H, Phadke S, Byrnes EJ, Zhu P, Mueller O, Farrer RA, Henk DA, Lewit Y, Hsueh Y-P, Fisher MC, Idnurm A, Heitman J, May RC. 2013. Transmission of hypervirulence traits via sexual reproduction within and between lineages of the human fungal pathogen Cryptococcus gattii. PLoS Genet 9:e1003771. doi: 10.1371/journal.pgen.1003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pukkila-Worley R, Gerrald QD, Kraus PR, Boily M-J, Davis MJ, Giles SS, Cox GM, Heitman J, Alspaugh JA. 2005. Transcriptional network of multiple capsule and melanin genes governed by the Cryptococcus neoformans cyclic AMP cascade. Eukaryot Cell 4:190–201. doi: 10.1128/EC.4.1.190-201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Missall TA, Moran JM, Corbett JA, Lodge JK. 2005. Distinct stress responses of two functional laccases in Cryptococcus neoformans are revealed in the absence of the thiol-specific antioxidant Tsa1. Eukaryot Cell 4:202–208. doi: 10.1128/EC.4.1.202-208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen SC-A, Slavin MA, Heath CH, Playford EG, Byth K, Marriott D, Kidd SE, Bak N, Currie B, Hajkowicz K, Korman TM, McBride WJH, Meyer W, Murray R, Sorrell TC; Australia and New Zealand Mycoses Interest Group (ANZMIG)-Cryptococcus Study . 2012. Clinical manifestations of Cryptococcus gattii infection: determinants of neurological sequelae and death. Clin Infect Dis 55:789–798. doi: 10.1093/cid/cis529. [DOI] [PubMed] [Google Scholar]
- 39.Stubbs MC, Kim YM, Krivtsov AV, Wright RD, Feng Z, Agarwal J, Kung AL, Armstrong SA. 2008. MLL-AF9 and FLT3 cooperation in acute myelogenous leukemia: development of a model for rapid therapeutic assessment. Leukemia 22:66–77. doi: 10.1038/sj.leu.2404951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE, Group TMGD. 2014. The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res 42:D810–D817. doi: 10.1093/nar/gkt1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patterson N, Price AL, Reich D. 2006. Population structure and eigenanalysis. PLoS Genet 2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li L, Stoeckert CJ, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 48.Farrer RA. 2017. Synima: a Synteny imaging tool for annotated genome assemblies. BMC Bioinformatics 18:507. doi: 10.1186/s12859-017-1939-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Alignments of C. gattii and mouse transcripts to sequences of the core eukaryotic genes (CEGs), suggesting mostly complete C. gattii gene-sets and the high quality of Mouse gene-set mm10 p4. Only VGIIa R265 and mouse mm10p4 were used in this study. Download FIG S1, PDF file, 0.1 MB (137.3KB, pdf) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
RNA was extracted from all four lineages of C. gattii in mouse macrophages at 1 h, 3 h, and 6 h postinfection, as well as C. gattii and macrophages in vitro (t0). Data represent percentages of sequenced reads deriving from the mouse macrophage (top) and C. gattii (bottom). Download FIG S2, PDF file, 0.2 MB (213KB, pdf) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Heat maps of all differentially expressed genes (FDR P value of <0.001 and greater-than-4-fold change of trimmed mean of M-values [TMM] expressed in normalized fragments per kilobase of transcript per million mapped reads [FPKM]) of C. gattii transcripts in vitro (t0) and during infection of mouse macrophages at 1 h, 3 h, and 6 h. Download FIG S3, PDF file, 0.8 MB (812.4KB, pdf) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(Tab 1) Contamination from E. coli K-12 MG1655, suicide vector pCD-RAsl1, and cloning vector pMJ016c identified by BLASTn searches of the nonredundant (NR) database. (Tab 2) Reads aligning either to mouse GRCm38 p4 mm10 gene sets or genome or to R265 updated gene set or genome. ARD, average read depth across genes. Download Table S1, XLSX file, 0.0 MB (48KB, xlsx) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Subsets (75%, 50%, and 25%) of C. gattii data were used to recall differential expression data and for comparisons with the full dataset. x-axis categories for each bar chart correspond to data from each of the five isolates (combining t1, t3, and t6) and to in vitro data (combining all isolates at t0). (a) The total number of differentially expressed gene changes (either up- or downregulation) for isolates ex vivo but not in vitro. (b) The percentages of genes found in the full dataset (i.e., representing a proxy for true positives). Data are represented as >75% for all categories using 75% subsets, >50% for all categories using 50% subsets, and >25% for all categories using 25% subsets. (c) The number of genes found only in the full dataset (i.e., representing a proxy for false negatives). VGI, VGIII, and VGIV reidentified most of the genes also found with their full datasets, while VGII and in vitro conditions recovered fewer genes found in the full dataset as the subset became smaller. (d) The number of genes not found in the full dataset (i.e., representing a proxy for false positives). Values corresponding to previously unidentified genes either increased or decreased as the subset size decreased, with VGIV giving the most robust results and VGIII the least. Download FIG S4, PDF file, 0.2 MB (223.9KB, pdf) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(Tab 1 to 4) All genes differentially expressed among five C. gattii isolates in vitro. (Tab 1) Details of every differentially expressed gene. (Tab 2) Gene annotation, position in genome, and associated PFAM. Orthogroup numbers refer to ortholog groups from 15 C. gattii and C. neoformans H99 genome comparisons (7). (Tab 3) Gene ontology terms assigned to differentially expressed genes. (Tab 4) Differentially expressed genes were found in multiple pairwise comparisons. (Tab 5 to 8) All genes differentially expressed by each of the five C. gattii isolates at different time points (t0, t3, and t6) in coincubation with mouse macrophages. (Tab 5) Details of every differentially expressed gene. (Tab 6) Gene annotation, position in genome, and associated PFAM. Orthogroup numbers refer to ortholog groups from 15 C. gattii and C. neoformans H99 genome comparisons (7). (Tab 7) Gene ontology terms assigned to differentially expressed genes. (Tab 8) Differentially expressed genes were found in multiple pairwise comparisons. (Tab 9 and 10) Expression values and overlap of previously identified differentially expressed genes in Acanthamoeba castellanii and macrophages (13). (Tab 9) C. neoformans genes with similar modulation patterns after interaction of the fungus with amoebae and with murine macrophages. (Tab 10) C. neoformans genes with different modulation patterns after interaction of the fungus with amoebae and with murine macrophages. (Tab 11) Genes differentially expressed by mouse macrophages. LogFc, log fold change; LogCPM, log counts per million. C1, VGIV CBS10101; C2, VGII R265; C3, VGII ENV152; C4, CA1873; C5, VGI WM276. “A” and “B” represent replicates. Download Data Set S1, XLSX file, 2.1 MB (2.1MB, xlsx) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A paralogous cluster of Cas3 and Cas31 from a previously described study of 16 isolates (7) had their sequences aligned using MUSCLE v3.8.31 (46), and a neighbor-joining tree was constructed using PAUP version 4.0b10 (47) to decipher orthologs. Cn, Cryptococcus neoformans; Cg, Cryptococcus gattii. Note that VGIV has no Cas31 gene. Expression levels (TMM FPKM) for five isolates are shown below the dendrogram. Download FIG S5, PDF file, 0.2 MB (159.4KB, pdf) .
Copyright © 2018 Farrer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.