

Toxoplasma gondii is an obligate intracellular parasite that has infected one-third of the population. Upon infection of warm-blooded vertebrates, the replicating form of the parasite (tachyzoite) converts into a latent form (bradyzoite) present in tissue cysts.
KEYWORDS: protozoa, parasite, drugs, stress response, translation, antiparasitics, Apicomplexa
ABSTRACT
Toxoplasma gondii is an obligate intracellular parasite that has infected one-third of the population. Upon infection of warm-blooded vertebrates, the replicating form of the parasite (tachyzoite) converts into a latent form (bradyzoite) present in tissue cysts. During immune deficiency, bradyzoites can reconvert into tachyzoites and cause life-threatening toxoplasmosis. We previously reported that translational control through phosphorylation of the α subunit of T. gondii eukaryotic initiation factor 2 (eIF2α) (TgIF2α) is a critical component of the parasite stress response. Diverse stresses can induce the conversion of tachyzoites to bradyzoites, including those disrupting the parasite's endoplasmic reticulum (ER) (ER stress). Toxoplasma possesses four eIF2α kinases, one of which (TgIF2K-A) localizes to the parasite ER analogously to protein kinase R-like endoplasmic reticulum kinase (PERK), the eIF2α kinase that responds to ER stress in mammalian cells. Here, we investigated the effects of a PERK inhibitor (PERKi) on Toxoplasma. Our results show that the PERKi GSK2606414 blocks the enzymatic activity of TgIF2K-A and reduces TgIF2α phosphorylation specifically in response to ER stress. PERKi also significantly impeded multiple steps of the tachyzoite lytic cycle and sharply lowered the frequency of bradyzoite differentiation in vitro. Pretreatment of host cells with PERKi prior to infection did not affect parasite infectivity, and PERKi still impaired parasite replication in host cells lacking PERK. In mice, PERKi conferred modest protection from a lethal dose of Toxoplasma. Our findings represent the first pharmacological evidence supporting TgIF2K-A as an attractive new target for the treatment of toxoplasmosis.
INTRODUCTION
Toxoplasmosis is caused by the intracellular parasite Toxoplasma gondii, an apicomplexan parasite that has infected about a third of the global population (1). While the definitive host is the cat, Toxoplasma is capable of infecting any warm-blooded vertebrate (2). Toxoplasma invades nucleated cells and establishes a parasitophorous vacuole, a compartment separating the replicating parasites from the host cell. Proliferating parasites (tachyzoites) can differentiate into latent tissue cysts (bradyzoites) that persist in the host and afford a means of transmission through predation (3). In immunocompromised patients, bradyzoites can reconvert into tachyzoites, causing reactivation of acute disease (4). Due to the lack of the immune response, the tachyzoites continue to replicate and disseminate in the host, leading to serious complications and morbidity. During pregnancy, tachyzoites can cross the placental barrier and cause abortion or congenital birth defects (5). Antifolates are the primary drugs used to treat acute toxoplasmosis; however, in addition to toxic adverse effects, antifolates lack efficacy against tissue cysts (6). There is a dire need for new treatments that target both the acute and latent stages of Toxoplasma.
We previously demonstrated that translational control mediated by eukaryotic initiation factor 2 (eIF2) is a potential new drug target in apicomplexan parasites (7). The eIF2 complex delivers the initiator tRNA to ribosomes during protein synthesis. Upon cellular stress, the α subunit of eIF2 (eIF2α) is phosphorylated, reducing eIF2 activity and sharply lowering translation initiation. Lowered protein synthesis allows cells to conserve resources and reconfigure gene expression to optimize adaptation to the stress insult (8). We previously reported that translational control directed by phosphorylation of the Toxoplasma gondii α subunit of eIF2 (TgIF2α) is critical during both the acute and latent stages of infection (9). Phosphorylation of TgIF2α has a protective effect on extracellular tachyzoites or intracellular tachyzoites deprived of amino acids (10–12). Phosphorylation of TgIF2α is also associated with the formation and maintenance of bradyzoites (13).
There are multiple mammalian eIF2α kinases that are activated by stress conditions, including protein kinase R-like endoplasmic reticulum kinase (PERK) and GCN2, which are induced by endoplasmic reticulum (ER) stress and depletion of nutrients, respectively. Toxoplasma possesses four eIF2α kinases: a PERK-related protein kinase (TgIF2K-A) that responds to ER stress, two GCN2-like homologs (TgIF2K-C and TgIF2K-D) that respond to nutrient deprivation, and TgIF2K-B, which appears to be unique to this apicomplexan parasite (11–14). TgIF2K-A is situated in the parasite ER and upon activation by ER stress is released from its association with the molecular chaperone BiP (HSPA5, GRP78) (13). These key regulatory features suggest that TgIF2K-A is regulated analogously to mammalian PERK (15, 16).
Despite multiple attempts using a variety of approaches, TgIF2K-A has been refractory to genomic ablation in Toxoplasma; consistent with our inability to obtain a TgIF2K-A knockout parasite, a genome-wide loss-of-function screen using CRISPR determined a fitness score of −3.35 for TgIF2K-A (13, 17). These findings suggest that TgIF2K-A is essential, making this TgIF2 kinase an attractive target for small-molecule inhibitors. Small-molecule inhibitors of mammalian PERK which prevent autophosphorylation and eIF2α phosphorylation have been reported. For example, GSK2606414 (a PERK inhibitor [PERKi]) inhibits PERK activation in different cell types and has anticancer activity in mice (18). Recently, we reported that activation of the PERK-like eIF2α kinase in the apicomplexan parasite Plasmodium falciparum, PK4 (PfPK4), increases phosphorylation of P. falciparum eIF2α, leading to artemisinin-induced latency and treatment failure in humans (19). Administration of PERKi with artemisinin blocked P. falciparum recrudescence in vitro and in vivo (19).
In this study, we investigated the effect of PERKi on Toxoplasma. Our results show that PERKi blocks TgIF2K-A kinase activity in vitro, prevents TgIF2α phosphorylation in response to ER stress, and impairs tachyzoite replication. The pharmacological data presented in this study, considered with genetic evidence, support the idea that this eIF2α kinase is an attractive target for future antiparasitic drug design.
RESULTS
PERK inhibitor GSK2606414 is active against TgIF2K-A.
TgIF2K-A shares critical functional features with mammalian PERK, including localization to the ER, a dynamic association with BiP/GRP78, and efficient phosphorylation of eIF2α in vitro (13). We sought to determine whether the human PERK inhibitor (PERKi) GSK2606414 could inhibit TgIF2K-A kinase activity. Purified recombinant protein containing the kinase domain (KD) of TgIF2K-A fused to glutathione S-transferase (GST) at its N terminus (TgIFK-A-KD) phosphorylated TgIF2α with saturation after 60 min of reaction incubation (Fig. 1A). The addition of PERKi sharply decreased the TgIF2K-A-mediated phosphorylation of TgIF2α in a dose-dependent manner with a 50% inhibitory concentration (IC50) of 5 nM (Fig. 1B).
FIG 1.

PERKi impairs TgIF2K-A kinase activity. (A) Histidine-tagged TgIF2α was purified from E. coli and incubated with GST-TgIF2K-A-KD for the times shown. The amount of ATP consumed during the reaction time was measured using the ADP-Glo kinase assay and is expressed in relative light units (RLU). Error bars represent the standard deviation (n = 3). (B) TgIF2α was incubated with TgIF2K-A-KD for 30 min in kinase reaction buffer containing different concentrations of PERKi (0 to 2 μM). The amount of ATP consumed was measured using the ADP-Glo kinase assay, and the IC50 was calculated (IC50 = 5 nM). Error bars represent the standard deviation (n = 3). (C) Extracellular tachyzoites were treated with 1 μM thapsigargin in the presence of different concentrations of PERKi, as indicated, or incubated with the vehicle (DMSO) for 1 h. Parasite lysates were resolved by SDS-PAGE for immunoblotting with antibodies to total and phosphorylated TgIF2α. (D) Extracellular tachyzoites were treated with 1 μM thapsigargin (TG) or 50 nM halofuginone (HF) or exposed to extracellular stress for 8 h in the presence or absence of PERKi (1 μM), as indicated, and vehicle (DMSO). Samples were processed for immunoblotting as described in the legend to panel C.
Inhibition of TgIF2K-A would be predicted to impair TgIF2α phosphorylation in Toxoplasma during ER stress. We previously showed that thapsigargin (TG) elicits ER stress and induces TgIF2α phosphorylation in extracellular tachyzoites (13). Inclusion of PERKi with parasites subjected to ER stress in this manner sharply reduced the phosphorylation of TgIF2α (Fig. 1C).
Toxoplasma expresses four TgIF2α kinases, each of which is activated by distinct stress conditions (9). To determine if PERKi prevents TgIF2α phosphorylation specifically during ER stress, tachyzoites were subjected to other stress conditions that should not involve TgIF2K-A. We treated parasites with halofuginone (HF), which thwarts the aminoacylation of tRNAPro and is a potent inducer of the eIF2α kinase GCN2 (20). We also exposed parasites to extracellular stress for 8 h, which we have shown activates TgIF2K-D (11). While PERKi prevented TgIF2α phosphorylation during ER stress, it did not alter TgIF2α phosphorylation levels in response to halofuginone or extracellular stress (Fig. 1D). These findings indicate that PERKi subverts TgIF2α phosphorylation mediated by TgIF2K-A but not that mediated by GCN2-like TgIF2Ks.
Tachyzoite replication is impaired by PERKi independently of host cell PERK.
To determine the effects of PERKi on parasite replication in vitro, we infected human foreskin fibroblast (HFF) cells with tachyzoites stably expressing a β-galactosidase reporter (RHβ1) (21) in the presence of increasing doses of PERKi. We determined that 0.62 μM and 2.7 μM PERKi inhibited 50% and 90% of parasite replication, respectively (Fig. 2A).
FIG 2.

PERKi impairs replication of tachyzoites in vitro. (A) A colorimetric microtiter assay was performed to assess parasite growth in a transgenic RH parasite line expressing β-galactosidase. PERKi was used over a concentration range of 0 to 20 μM and reduced the replication of tachyzoites with an IC50 of 0.62 μM and an IC90 of 2.7 μM. The viability curve shows the means for three biological replicates. The y axis shows the percent viability relative to vehicle treatment, and the x axis shows the log of the concentrations of PERKi. (B) Plaque assays for wild-type RH strain parasite cultures in the presence of various concentrations of PERKi or vehicle. The uninfected HFF monolayer was treated with 5 μM PERKi. After 5 days, the monolayers were stained to measure the area of host cell lysis. Treated samples with results that were significantly different from those for vehicle-treated samples are indicated by asterisks (***, P < 0.005). (C) Parasite counting assay. At the indicated time points, the number of parasites in 250 random vacuoles was plotted as a percentage of the total number of vacuoles examined. Significant differences between PERKi-treated and vehicle-treated cells are indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.005. (D) Tachyzoites were allowed to infect MEF cells (wild-type [WT], PERK−/−, or GCN2−/− cells) in the presence or absence of 1 μM PERKi. At 48 h postinfection, parasite replication was assessed using the PCR-based assay for B1. Significant differences between the WT and knockout cells are indicated. *, P < 0.05. Error bars for all graphs show the standard deviation.
We also determined the effect of PERKi on Toxoplasma infection using a plaque assay. HFF host cells infected with RH strain tachyzoites were treated with PERKi or vehicle; 5 days later, the cultures were fixed and stained to measure the extent of host cell monolayer lysis. Uninfected HFFs treated with PERKi (5 μM) were also assayed in this fashion to assess if any host cell lysis occurred in the presence of drug alone. As shown in Fig. 2B, PERKi greatly reduced Toxoplasma viability in a dose-dependent fashion; note that PERKi treatment had no effect on the uninfected HFF monolayer. Parasite doubling assays confirmed the detrimental effect of PERKi on the tachyzoite replication rate, appearing with as little as 0.5 μM and as early as 24 h postinfection (Fig. 2C).
To address whether interference with host cell PERK also contributes to PERKi-mediated inhibition of Toxoplasma proliferation, we infected wild-type (WT), PERK−/−, and GCN2−/− mouse embryonic fibroblasts (MEF) (22) and treated these infected host cells with 1 μM PERKi or vehicle. After 30 h, we measured the number of tachyzoites inside the MEF host cells by quantitative PCR for the parasite-specific gene region B1. PERKi impaired the replication of tachyzoites at similar levels independently of the presence of host PERK or GCN2 protein kinases (Fig. 2D). These data suggest that the potential effect of PERKi on host cells at these concentrations is not a significant contributor to its efficacy in reducing tachyzoite replication. We note that the absence of PERK or GCN2 protein kinases in the MEF host cells led to a decreased number of tachyzoites, suggesting that host eIF2α kinases are required for optimal parasite replication.
Effects of PERKi on attachment, invasion, and egress.
We next assessed whether PERKi affects parasite attachment and invasion. We employed a standard attachment/invasion assay to determine if PERKi affects parasite entry into host cells (23). Pretreatment of extracellular tachyzoites with PERKi for up to 6 h prior to invasion diminished the parasite's capacity to attach and invade host cells in both a time- and dose-dependent fashion (Fig. 3A and B). In contrast, pretreatment of uninfected host cells with PERKi did not affect Toxoplasma infection, bolstering support for the suggestion that the drug directly affects the parasites (Fig. 3C).
FIG 3.

Effects of PERKi on the tachyzoite lytic cycle and differentiation. (A and B) Attachment (A) and invasion (B) assays. Extracellular RH strain tachyzoites were treated with the indicated concentrations of PERKi for 2, 4, or 6 h prior to infection of HFF host cells, and dual staining allowed determination of the percentage that attached or invaded. Significant differences between the vehicle-treated and PERKi-treated cells are indicated. ***, P < 0.005. (C) HFF monolayers were pretreated with the indicated concentrations of PERKi for 2, 4, or 6 h prior to infection. At 5 days postinfection, monolayers were stained to determine the percentage of host cell lysis. (D) Infected HFF monolayers were treated with the indicated concentrations of PERKi or vehicle for 10 min. Parasite egress was stimulated by the addition of 1 μM A23187. The percentage of egressed vacuoles in each treatment group was determined by scoring at least 100 randomly chosen vacuoles. Significant differences between the vehicle-treated and PERKi-treated cells are indicated. ***, P < 0.005. (E) The indicated concentration of PERKi (or vehicle) was included in cultures during the in vitro differentiation of ME49 type II strain parasites. At day 5, differentiated cultures were incubated with lectin stain to visualize tissue cyst walls. Significant differences between the vehicle-treated and PERKi-treated cells are indicated. **, P < 0.01. The error bars on each graph show the standard deviation.
We next examined whether PERKi affects parasite egress from the host cells. One day after infecting HFF cells with Toxoplasma, egress was induced by adding the calcium ionophore A23187. We found that PERKi, which does not induce egress alone, reduced ionophore-induced egress more than 50% in a dose-dependent manner (Fig. 3D).
Effect of PERKi on parasite differentiation.
We previously demonstrated that induction of ER stress in Toxoplasma triggers bradyzoite gene expression and formation of tissue cyst walls (13). We reasoned that inhibition of TgIF2K-A by PERKi would decrease tissue cyst formation in infected host cells. To test this hypothesis, type II ME49 strain parasites were used to infect HFF monolayers; at 4 h postinfection, bradyzoite conversion was induced by alkaline stress and CO2 deprivation for 5 days. The results showed that inclusion of PERKi during differentiation significantly decreased the number of tissue cysts generated, supporting the idea that TgIF2K-A plays a critical role in the conversion of tachyzoites into bradyzoites (Fig. 3E).
PERKi prolongs survival of mice given a lethal dose of Toxoplasma.
We examined whether PERKi protects mice from acute toxoplasmosis. Female BALB/c mice were infected intraperitoneally (i.p.) with a lethal dose of 100 RH strain tachyzoites and treated i.p. with 25 or 50 mg/kg of body weight PERKi once or twice daily. As expected, infected mice treated with the vehicle control died after 9 days; increasing amounts of PERKi conferred modest protection from lethal acute infection (Fig. 4). Once-a-day treatments with 25 or 50 mg/kg delayed death by 12 h. Administration of 25 or 50 mg/kg PERKi twice a day prolonged the life of some of the mice within the group for 30 h.
FIG 4.
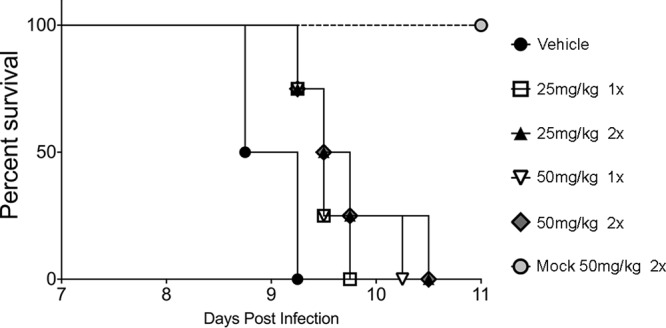
PERKi prolongs the survival of mice with acute Toxoplasma infection. Female BALB/c mice were infected i.p. with 100 RH strain tachyzoites (or were mock infected). At 12 h postinfection, the mice were given vehicle or the indicated dose of PERKi i.p. once or twice a day (indicated by 1× or 2×, respectively). Mice (4 per group) were monitored at least three times daily, and the time to death was recorded.
DISCUSSION
Our previous work has established that translational control through the phosphorylation of TgIF2α is a critical process as Toxoplasma progresses through its lytic cycle as well as transitions to its latent stage (9). Here, we present pharmacological evidence that the ER-resident TgIF2α kinase TgIF2K-A, a distal orthologue of mammalian PERK, operates at multiple points in the progression of infection. Our findings using the PERKi GSK2606414 bolster earlier genetic work that suggested that TgIF2K-A is an attractive new drug target. With an IC50 of 0.6 μM, PERKi is a potent inhibitor of tachyzoite replication in vitro (Fig. 2). Further characterization of the mechanism of action shows that PERKi interferes with numerous points of the lytic cycle, including attachment/invasion, replication, and egress. Consistent with our finding that ER stress and, hence, TgIF2K-A can induce tachyzoite-to-bradyzoite conversion in vitro, inclusion of PERKi significantly diminished parasite differentiation. PERKi is likely to interfere with any process in the parasite that requires the production of new proteins, which would tax the ER and activate TgIF2K-A to manage the added stress.
When examining the activities of drugs against intracellular parasites, it is always a concern whether detrimental effects occur indirectly through action on the host cell. Several lines of evidence suggest that the effect of PERKi on host cells appears to be minimal compared to its direct activity against the parasites. First, we were able to establish that PERKi impairs TgIF2K-A enzymatic activity in an in vitro kinase assay and reduces TgIF2α phosphorylation in parasites specifically in response to ER stress. Pretreatment of host cells with PERKi prior to infection did not impact Toxoplasma replication or viability. Moreover, PERKi adversely affected Toxoplasma replication in MEF cells independently of the status of the host PERK. Finally, concentrations as high as 5 μM (>5 times the 50% effective concentration [EC50] for Toxoplasma) had no overt detrimental effect on uninfected host cell monolayers.
PERKi conferred a modest degree of protection against a lethal dose of RH strain tachyzoites in a mouse model of acute infection. Our observation that better protection occurred in mice that received two doses per day is consistent with the findings of other studies showing that PERKi is quickly metabolized by the liver and has a limited half-life of 2.5 h in vivo (18). Alternative routes of administration of PERKi may increase its bioavailability and improve its efficacy against toxoplasmosis; presently, the high cost of PERKi makes it difficult to perform comprehensive analyses in mouse models of infection.
These data suggest that chemical derivatives of PERKi with increased in vivo activity against Toxoplasma are worthy of development. We previously showed that the TgIF2K-A protein sequence is highly divergent from the sequence of its human counterpart (14); however, the finding that PERKi selectively inhibits TgIF2K-A suggests that it has some shared structural and functional properties with human PERK. There are significant sequence similarities between the protein kinase domains of human PERK and the orthologues expressed in the apicomplexan parasites Toxoplasma and P. falciparum (PfPK4) (24). Further scrutiny of the kinase domain sequences of TgIF2K-A and human PERK reveals conservation of many of the residues involved in a back pocket critical for the binding of PERKi: in human PERK, the substituted phenyl ring of PERKi makes hydrophobic contacts with residues in the conserved αC helix, including L642, A643, Y653, and I885, and several of the amino acid residues that line the back pocket are conserved between PERK and TgIF2K-A (Fig. 5). Given the selectivity of PERKi for TgIF2K-A versus other Toxoplasma eIF2α kinases and the aforementioned sequence similarities between human PERK and TgIF2K-A in residues lining the binding pocket, we favor a mode for PERKi binding to TgIF2K-A that is similar to that described for human PERK. Along these lines, it might be possible to take advantage of differences in the predicted αC face of the pocket in TgIF2K-A (such as Asn4062 and Arg4063), exploiting the polar residues in this region of the pocket to design parasite-selective inhibitors. Additional strategies to achieve selectivity might include the design of irreversible inhibitors of TgIF2K-A, taking advantage of a cysteine in the hinge region (Cys4703) or in the vicinity of the back pocket (Cys4078) (Fig. 5). However, a formal description of the precise mode of PERKi binding to TgIF2K-A and the accessibility and orientation of sulfhydryl groups will require structural analysis.
FIG 5.

Alignments of ER-resident eIF2α kinase domains. The sequences of the kinase domains of Toxoplasma gondii TgIF2K-A, Plasmodium falciparum PfPK4, mouse PERK, and human PERK were aligned using the ClustalW program. Residues highlighted in black are identical or similar among all four species, and those displaying sequence divergence among individuals of a single species are highlighted in gray. Motifs comprising the kinase domain are designated I to XI. The catalytic lysine (K) in subdomain II and the DFG motif in subdomain VII, which plays an important role in the regulation of kinase activity, are enclosed in black boxes. The methionine (M) gatekeeper residue inside subdomain V is indicated with a black arrow. Amino acid residues in the hinge are indicated with asterisks, and back-pocket regions of the active site critical for binding of PERKi are boxed in gray. The sequences of TgIF2K-A (ToxoDB accession number TGGT1_229630), PfPK4 (PlasmoDB accession number PF3D7_0628200), mouse PERK (E2AK3_MOUSE-Mus musculus, gene ID 13666), and human PERK (E2AK3_HUMAN-Homo sapiens, gene ID 9451) were obtained from the indicated databases.
An unexpected finding during the course of these studies was that tachyzoites were less efficient at replicating in MEF cells lacking PERK or GCN2 (Fig. 2D), suggesting that host eIF2α phosphorylation is beneficial for the parasite's lytic cycle. Alternatively, these cells are less fit to support infection due to the lack of these eIF2α kinases. The role of translational control in the host cell during infection is an unexplored area, but this finding warrants its further investigation.
In conclusion, our studies show the first pharmacological evidence that translational control mediated by TgIF2K-A is a promising target for the development of future antiparasitics. Such inhibitors could have broad-spectrum utility against other apicomplexan parasites, such as the malaria parasite, which also rely on their PERK-like eIF2α kinase, PK4. PK4 not only is essential for the development of the erythrocytic cycle in malaria, but it also mediates drug-induced latency that leads to treatment failure (19, 24).
MATERIALS AND METHODS
Chemicals.
The PERK inhibitor (PERKi) GSK2606414 was purchased from Calbiochem (catalog no. 1337531-89-1) and MedChemExpress (catalog no. 133753136-8). The inhibitor was dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C. PERKi from the two vendors displayed equivalent effects against Toxoplasma growth in vitro (data not shown). Thapsigargin and halofuginone were purchased from Sigma-Aldrich.
Parasite culture and growth assays.
Tachyzoites of the RH strain were cultivated in human foreskin fibroblast (HFF) monolayers in Dulbecco modified Eagle medium (DMEM) supplemented with 1% heat-inactivated fetal bovine serum (FBS) (Gibco/Invitrogen) (25). Uninfected HFF and MEF cells were cultivated in DMEM supplemented with 10% FBS. The cultures were maintained in a humidified incubator at 37°C with 5% CO2.
The EC50s were determined using RHβ1, a transgenic line of RH that stably expresses β-galactosidase, to quantify parasite growth by colorimetric microtiter assays as previously described (21). Tachyzoites were allowed to invade an HFF host cell monolayer for 2 h; uninvaded parasites were then removed and medium was replaced with medium supplemented with the concentration of PERKi or vehicle (DMSO) indicated above and in the figures. After 96 h postinfection (hpi), chlorophenol red-β-d-galactopyranoside (CPRG) was added to a final concentration of 100 μM. The plates were incubated at 37°C with 5% CO2 for an additional 24 h, and then β-galactosidase activity was read at 570 nm using a BioTek microtiter plate reader.
For plaque assays, 500 parasites were used to infect HFF monolayers in 12-well plates. After 2 h, the medium was replaced with medium supplemented with the designated concentration of PERKi or vehicle. At 5 days postinfection, host cell lysis was determined by crystal violet staining as described previously (25). For doubling assays, 104 tachyzoites were allowed to invade an HFF host cell monolayer for 2 h, at which point the uninvaded parasites were removed by replacing the medium with medium containing the concentrations of PERKi or vehicle indicated above and in the figures. At 12, 24, and 36 hpi, cells were fixed with 4% paraformaldehyde for 20 min and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). The number of parasites in 250 randomly selected vacuoles was then counted.
As parasites are difficult to visualize in MEF cells, we used a PCR-based assay to determine parasite numbers (26). Briefly, wild-type (WT), GCN2−/−, and PERK−/− MEF cells (22) were infected with 104 parasites; after 2 h, the medium was replaced with fresh medium containing PERKi or vehicle, as indicated above and in the figures. At 30 hpi, genomic DNA was isolated and measured by quantitative PCR using primers specific for a parasite-specific gene region called B1 (27).
Attachment, invasion, and egress assays.
Parasite attachment and invasion efficiency were measured using a dual-staining technique as previously described (23). Briefly, tachyzoites were allowed to infect HFF monolayers for 1 h; the cells were then washed three times to remove unattached parasites. The cells were fixed with 4% paraformaldehyde for 20 min and blocked with phosphate-buffered saline (PBS) supplemented with 2% bovine serum albumin (BSA). Before permeabilizing the cells, extracellular parasites were labeled with a mouse anti-Sag1 monoclonal antibody (Invitrogen) for 1 h in blocking buffer. After the cells were permeabilized with blocking solution containing 0.2% Triton X-100 for 30 min, they were incubated with rabbit anti-TgIF2α for 1 h (14). Secondary goat anti-rabbit immunoglobulin-Alexa Fluor 488 and goat anti-mouse immunoglobulin-Alexa Fluor 594 (Invitrogen) were then for added for 1 h and visualized with Vectashield mounting medium and DAPI stain (Vector Laboratories). Intracellular and extracellular parasites were then distinguishable for counting: attached parasites appeared yellow, as they stained with both Alexa Fluor 488 (green) and Alexa Fluor 594 (red), whereas intracellular parasites were green, as they stained only with Alexa Fluor 488 (green). The number of attached parasites in 100 random fields was determined and compared to the 100% attachment for the vehicle-treated control parasites. The efficiency of invasion was measured as the percentage of intracellular parasites out of the total number of parasites.
To measure egress, tachyzoites were allowed to infect HFF monolayers for 24 h. The infected HFF cells were then washed and incubated with Hanks' buffered saline solution (HBSS) in the presence of PERKi or vehicle, with 1 μM the calcium ionophore A23187 being added where indicated for 2 min at 37°C (28). The infected HFF monolayers were then fixed with methanol and stained with differential Quik stain (Thermo Fisher Scientific). The percentage of egressed parasites was determined by counting at least 250 vacuoles per condition.
Bradyzoite differentiation assays.
Type II strain Toxoplasma ME49 tachyzoites were allowed to infect HFF monolayers for 2 h. After infection, the cells were washed in DMEM, and alkaline medium (pH 8.2) was added (29). After 4 h, PERKi or vehicle was added to the culture medium. Infected HFFs were cultured at 37°C in ambient CO2, and the alkaline medium containing PERKi or vehicle was replaced every other day. To visualize tissue cyst walls, infected monolayers were fixed with 4% paraformaldehyde and stained with rhodamine-conjugated Dolichos biflorus agglutinin.
Biochemical assays for eIF2α kinase activity.
A cDNA encoding TgIF2α was amplified by PCR from a previously reported plasmid (14) using the following primers: TgIF2PET19-forward (5′-ACGACGACAAGCATATGGAGGGCGAGAGACGGC-3′) and TgIF2PET19-reverse (5′-GTTAGCAGCCGGATCCTCACGCATTTCCATCATCGTTATCCT-3′). The amplified DNA was purified and inserted into the expression vector pET19b (Addgene) in the BamHI and NdeI sites. The resulting plasmid (pET19b-TgIF2α) encodes TgIF2α fused in frame to an amino-terminal polyhistidine tag. The plasmid was transformed into Escherichia coli Rosetta(DE3) cells, and expression of the recombinant TgIF2α protein was induced for 12 h at 37°C in the presence of 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). TgIF2α was purified from bacteria using a HisTALON kit (Clontech).
A cDNA encoding the protein kinase domain of TgIF2K-A (TgIF2K-A-KD) from amino acid residues 4002 to 5072 was amplified by PCR (forward primer, 5′-GAAGGTCGTGGGATCCGCTTCGAAAGA-3′; reverse primer, 5′-GGGAATTCGGGGATCTCAGTCTCTTTCATTCTTCCCC-3′) from previously described plasmid pYES-GST-TgIF2K-A (14) and inserted into plasmid pGEX-5X1 in the BamHI site. The resulting plasmid (pGEX-5xGST-TgIF2K-A-KD) carries the catalytic domain of TgIF2K-A-KD fused in frame with an amino-terminal GST. The plasmid pGEX-5xGST-TgIF2K-A-KD was introduced into Rosetta(DE3) cells, and expression was induced with IPTG, as described above. The bacterial cells were collected by centrifugation and then resuspended in a PBS solution supplemented with the protease inhibitor cocktail cOmplete and an EDTA-free protease inhibitor cocktail (Sigma-Aldrich). Cells were lysed by sonication. The TgIF2K-A-KD protein was purified from the lysate using GST-Bind resin (Novagen) following the manufacturer's instructions. After purification of the recombinant TgIF2α and GST-TgIF2K-A-KD proteins, each was dialyzed in a solution containing 50 mM NaCl and 15 mM Tris-HCl (pH 7. 4) at 4°C for 20 h. The eIF2α kinase reaction was performed in a 50 mM NaCl and 15 mM Tris-HCl (pH 7.4) solution supplemented with 5 μg/ml TgIF2α, 1 μg/ml TgIFK-A-KD, 10 mM MgCl2, and 0.5 mM ATP at 37°C for the times indicated above and in the figure legends. Phosphorylation of TgIF2α was measured using the ADP-Glo kinase assay (Promega Corporation) as described by the manufacturer.
Inhibition of TgIF2α phosphorylation.
Tachyzoites were purified from infected HFFs by syringe passage and filtration and then incubated in DMEM supplemented with 1% FBS in the presence or absence of a stress agent, 1 μM thapsigargin (TG; Sigma-Aldrich) or 50 nM halofuginone (HF; Sigma-Aldrich), for 1 h at 37°C in 5% CO2. Another stress condition involved depriving extracellular tachyzoites of host cells for 8 h (10). For each condition, the amount of PERKi indicated above and in the figure legends was added at the initiation of each stress. Tachyzoites were lysed in PBS containing 1% Triton X-100 supplemented with the protease inhibitor cocktail cOmplete and an EDTA-free protease inhibitor cocktail (Sigma-Aldrich). Total protein levels were quantified using the Bradford assay (Sigma-Aldrich). Equal amounts of total protein were separated by electrophoresis in NuPAGE 4 to 12% bis-Tris gels (Thermo Fisher Scientific). The proteins were transferred to nitrocellulose membranes, and Western blot analyses were performed using TgIF2α total or TgIF2α-phosphorylated (TgIF2α-P) antibodies diluted 1:20,000 and 1:2,000, respectively, in blocking solution (Tris-buffered saline with Tween 20 and 2% BSA) (13, 14). Secondary rabbit antibodies were used at a 1:5,000 dilution. After washing, the membranes were incubated with a Pierce enhanced chemiluminescence Western blot substrate to visualize the proteins.
Treatment of acute toxoplasmosis in a mouse model.
To test the efficacy of PERKi against acute Toxoplasma infection in vivo, 24 5- to 6-week-old female BALB/c mice were purchased from Envigo Laboratories, group housed with 4 per cage, and allowed to acclimate for 1 week on a 7 a.m. to 7 p.m. light/dark cycle. Following acclimation, the mice were randomized on the basis of weight into six groups (n = 4). Five of the six groups were infected intraperitoneally (i.p.) with 100 RH strain tachyzoites in sterile PBS, isolated by syringe lysis and filtration of an infected HFF monolayer. The sixth group was mock infected with sterile PBS alone. All groups were dosed twice per day with either vehicle (10% DMSO in sterile PBS) or PERKi (25 mg/kg or 50 mg/kg) i.p. starting at 12 hpi. Mice were observed at 7 a.m., noon, and 7 p.m.; percent survival was recorded at each time point. All animal research was conducted in accordance with the animal welfare act, and all protocols were approved by the institutional animal care and use committees at the Indiana University School of Medicine (approved protocol no. 10852).
Statistical analyses.
Quantitative data are presented as the mean and standard deviation for biological replicates (n = 3). Statistical analyses were performed using one-way analysis of variance with Dunnett's or Tukey's multiple-comparison tests in Prism (version 7) software (GraphPad Software, Inc.). The P values are indicated in the legend of each figure. For immunoblots, the reported images are representative of those from at least three independent experiments.
ACKNOWLEDGMENTS
This research was supported by the National Institutes of Health (grant AI124723 to W.J.S. and R.C.W.).
We thank the members of the Biology of Intracellular Pathogens group at the Indiana University School of Medicine for helpful discussions during the course of this work.
We have declared that no competing interests exist.
REFERENCES
- 1.Montoya JG, Liesenfeld O. 2004. Toxoplasmosis. Lancet 363:1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- 2.Hill DE, Chirukandoth S, Dubey JP. 2005. Biology and epidemiology of Toxoplasma gondii in man and animals. Anim Health Res Rev 6:41–61. doi: 10.1079/AHR2005100. [DOI] [PubMed] [Google Scholar]
- 3.Sullivan WJ Jr, Jeffers V. 2012. Mechanisms of Toxoplasma gondii persistence and latency. FEMS Microbiol Rev 36:725–733. doi: 10.1111/j.1574-6976.2011.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luft BJ, Remington JS. 1992. Toxoplasmic encephalitis in AIDS. Clin Infect Dis 15:211–222. doi: 10.1093/clinids/15.2.211. [DOI] [PubMed] [Google Scholar]
- 5.Havelaar AH, Kemmeren JM, Kortbeek LM. 2007. Disease burden of congenital toxoplasmosis. Clin Infect Dis 44:1467–1474. doi: 10.1086/517511. [DOI] [PubMed] [Google Scholar]
- 6.Halonen SK, Weiss LM. 2013. Toxoplasmosis. Handb Clin Neurol 114:125–145. doi: 10.1016/B978-0-444-53490-3.00008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmes MJ, Augusto LDS, Zhang M, Wek RC, Sullivan WJ Jr. 2017. Translational control in the latency of apicomplexan parasites. Trends Parasitol 33:947–960. doi: 10.1016/j.pt.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. 2016. The integrated stress response. EMBO Rep 17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joyce BR, Konrad C, Wek RC, Sullivan WJ Jr. 2011. Translation control is critical during acute and chronic stages of toxoplasmosis infection. Expert Rev Anti Infect Ther 9:1–3. doi: 10.1586/eri.10.146. [DOI] [PubMed] [Google Scholar]
- 10.Joyce BR, Queener SF, Wek RC, Sullivan WJ Jr. 2010. Phosphorylation of eukaryotic initiation factor-2{alpha} promotes the extracellular survival of obligate intracellular parasite Toxoplasma gondii. Proc Natl Acad Sci U S A 107:17200–17205. doi: 10.1073/pnas.1007610107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konrad C, Wek RC, Sullivan WJ Jr. 2011. A GCN2-like eukaryotic initiation factor-2 kinase increases the viability of extracellular Toxoplasma gondii parasites. Eukaryot Cell 10:1403–1412. doi: 10.1128/EC.05117-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konrad C, Wek RC, Sullivan WJ Jr. 2014. GCN2-like eIF2alpha kinase manages the amino acid starvation response in Toxoplasma gondii. Int J Parasitol 44:139–146. doi: 10.1016/j.ijpara.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narasimhan J, Joyce BR, Naguleswaran A, Smith AT, Livingston MR, Dixon SE, Coppens I, Wek RC, Sullivan WJ Jr. 2008. Translation regulation by eukaryotic initiation factor-2 kinases in the development of latent cysts in Toxoplasma gondii. J Biol Chem 283:16591–16601. doi: 10.1074/jbc.M800681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sullivan WJ Jr, Narasimhan J, Bhatti MM, Wek RC. 2004. Parasite-specific eukaryotic initiation factor-2 (eIF2) kinase required for stress-induced translation control. Biochem J 380:523–531. doi: 10.1042/bj20040262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. 2000. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 16.Ma K, Vattem KM, Wek RC. 2002. Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor-2 kinase in response to endoplasmic reticulum stress. J Biol Chem 277:18728–18735. doi: 10.1074/jbc.M200903200. [DOI] [PubMed] [Google Scholar]
- 17.Sidik SM, Huet D, Ganesan SM, Huynh MH, Wang T, Nasamu AS, Thiru P, Saeij JPJ, Carruthers VB, Niles JC, Lourido S. 2016. A genome-wide CRISPR screen in Toxoplasma identifies essential apicomplexan genes. Cell 166:1423–1435.e12. doi: 10.1016/j.cell.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, Atkins C, Liu Q, Rabindran S, Kumar R, Hong X, Goetz A, Stanley T, Taylor JD, Sigethy SD, Tomberlin GH, Hassell AM, Kahler KM, Shewchuk LM, Gampe RT. 2012. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 55:7193–7207. doi: 10.1021/jm300713s. [DOI] [PubMed] [Google Scholar]
- 19.Zhang M, Gallego-Delgado J, Fernandez-Arias C, Waters NC, Rodriguez A, Tsuji M, Wek RC, Nussenzweig V, Sullivan WJ Jr. 2017. Inhibiting the Plasmodium eIF2alpha kinase PK4 prevents artemisinin-induced latency. Cell Host Microbe 22:766–776.e4. doi: 10.1016/j.chom.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, Kim Y-J, Lee H-K, Cortese JF, Wirth DF, Dignam JD, Rao A, Yeo C-Y, Mazitschek R, Whitman M. 2012. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol 12:311–317. doi: 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McFadden DC, Seeber F, Boothroyd JC. 1997. Use of Toxoplasma gondii expressing beta-galactosidase for colorimetric assessment of drug activity in vitro. Antimicrob Agents Chemother 41:1849–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC. 2004. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol 24:1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huynh MH, Rabenau KE, Harper JM, Beatty WL, Sibley LD, Carruthers VB. 2003. Rapid invasion of host cells by Toxoplasma requires secretion of the MIC2-M2AP adhesive protein complex. EMBO J 22:2082–2090. doi: 10.1093/emboj/cdg217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang M, Mishra S, Sakthivel R, Rojas M, Ranjan R, Sullivan WJ Jr, Fontoura BM, Menard R, Dever TE, Nussenzweig V. 2012. PK4, a eukaryotic initiation factor 2alpha(eIF2alpha) kinase, is essential for the development of the erythrocytic cycle of Plasmodium. Proc Natl Acad Sci U S A 109:3956–3961. doi: 10.1073/pnas.1121567109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roos DS, Donald RG, Morrissette NS, Moulton AL. 1994. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol 45:27–63. doi: 10.1016/S0091-679X(08)61845-2. [DOI] [PubMed] [Google Scholar]
- 26.Konrad C, Queener SF, Wek RC, Sullivan WJ Jr. 2013. Inhibitors of eIF2alpha dephosphorylation slow replication and stabilize latency in Toxoplasma gondii. Antimicrob Agents Chemother 57:1815–1822. doi: 10.1128/AAC.01899-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kompalic-Cristo A, Frotta C, Suarez-Mutis M, Fernandes O, Britto C. 2007. Evaluation of a real-time PCR assay based on the repetitive B1 gene for the detection of Toxoplasma gondii in human peripheral blood. Parasitol Res 101:619–625. doi: 10.1007/s00436-007-0524-9. [DOI] [PubMed] [Google Scholar]
- 28.Black MW, Arrizabalaga G, Boothroyd JC. 2000. Ionophore-resistant mutants of Toxoplasma gondii reveal host cell permeabilization as an early event in egress. Mol Cell Biol 20:9399–9408. doi: 10.1128/MCB.20.24.9399-9408.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soete M, Camus D, Dubremetz JF. 1994. Experimental induction of bradyzoite-specific antigen expression and cyst formation by the RH strain of Toxoplasma gondii in vitro. Exp Parasitol 78:361–370. doi: 10.1006/expr.1994.1039. [DOI] [PubMed] [Google Scholar]