

Targeting lanosterol 14α-demethylase (LDM) with azole drugs provides prophylaxis and treatments for superficial and disseminated fungal infections, but cure rates are not optimal for immunocompromised patients and individuals with comorbidities. The efficacy of azole drugs has also been reduced due to the emergence of drug-resistant fungal pathogens.
KEYWORDS: antifungal, cytochrome P450, lanosterol 14α-demethylase, Saccharomyces cerevisiae expression, crystal structure, fungal pathogen, Candida albicans, Candida glabrata
ABSTRACT
Targeting lanosterol 14α-demethylase (LDM) with azole drugs provides prophylaxis and treatments for superficial and disseminated fungal infections, but cure rates are not optimal for immunocompromised patients and individuals with comorbidities. The efficacy of azole drugs has also been reduced due to the emergence of drug-resistant fungal pathogens. We have addressed the need to improve the potency, spectrum, and specificity for azoles by expressing in Saccharomyces cerevisiae functional, recombinant, hexahistidine-tagged, full-length Candida albicans LDM (CaLDM6×His) and Candida glabrata LDM (CgLDM6×His) and determining their X-ray crystal structures. The crystal structures of CaLDM6×His, CgLDM6×His, and ScLDM6×His have the same fold and bind itraconazole in nearly identical conformations. The catalytic domains of the full-length LDMs have the same fold as the CaLDM6×His catalytic domain in complex with posaconazole, with minor structural differences within the ligand binding pocket. Our structures give insight into the LDM reaction mechanism and phenotypes of single-site CaLDM mutations. This study provides a practical basis for the structure-directed discovery of novel antifungals that target LDMs of fungal pathogens.
INTRODUCTION
The eukaryotic CYP51 proteins of the endoplasmic reticulum are bitopic, membrane-monospanning, cytochrome P450 monooxygenases. Lanosterol 14α-demethylase (LDM) is a key enzyme in the ergosterol biosynthetic pathway of fungi and in the cholesterol biosynthetic pathway of humans (1). LDM is the target of the azole drugs used widely to prevent or treat fungal infections in humans. It is therefore important to ensure a broad window of clinical efficacy by identifying drugs that bind to fungal LDM without affecting drug metabolism in the host mediated by cytochrome P450 enzymes. Such drugs also should not be susceptible to known mechanisms that confer target-based resistance. Although mutations that confer azole resistance in clinical isolates have not been detected in C. glabrata LDM (CgLDM) thus far, more than 140 mutations have been found in Candida albicans LDM (CaLDM). The mutations shown experimentally to confer azole resistance have been located within three distinct hot spots in LDM (2–4). The acquisition of multiple mutations in the LDMs of several fungal pathogens is thought to ensure enzyme activity is sufficient in response to the threat posed by xenobiotics such as the azole drugs. For example, azole-based agrochemical protection of major crops against fungal pathogens seems to have selected for at least two distinct mutant forms of Aspergillus fumigatus LDM in the CYP51A isoform that have been reported worldwide (5–7). Both have a tandem repeat (TR) modification in the CYP51A promoter as well as at least one mutation in the open reading frame (ORF; TR36/L96H and TR46/Y121F/T289A). These environmentally acquired mutations confer significant resistance to voriconazole (VCZ), the front-line treatment of human A. fumigatus infections.
Until recently the further development of fungal LDM as a drug target was limited by a paucity of high-resolution structures that could visualize the effects of mutations and enable computer-based techniques to identify compounds that bind specifically to LDM. We have made significant progress toward implementing structure-based discovery of LDM inhibitors by overexpressing constitutively full-length Saccharomyces cerevisiae LDM with a C-terminal hexahistidine tag (ScLDM6×His) from the PDR5 locus of hypersusceptible S. cerevisiae strains deleted of multiple drug efflux pumps (8). The functional overexpression of ScLDM6×His has provided robust phenotypic screens not confounded by the presence of drug efflux pumps. Purified ScLDM6×His has yielded high-resolution structures of the full-length enzyme in complex with its substrate lanosterol, the pseudosubstrate estriol, several azole drugs, and, most recently, a range of azole agrochemicals (8–11). It has also provided the first structures of full-length fungal mutant enzymes, including ScLDM6×His Y140F/H mutants in complex with fluconazole (FLC), itraconazole (ITC), posaconazole (PCZ), and VCZ (8, 10, 12). Based on the X-ray crystal structure of ScLDM, mutations shown experimentally to confer azole resistance are located within the internal active site around the substrate entry channel and in the surface of the enzyme (4, 8).
Crystal structures have recently been obtained for the catalytic domains of LDMs from two pathogenic fungi, A. fumigatus and C. albicans. These structures used recombinant enzymes truncated of the N-terminal membrane-associated and transmembrane helices. The first structures published were of A. fumigatus CYP51B in complex with VCZ and the experimental inhibitor (R)-N-(1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide (VNI) (13). A. fumigatus CYP51B is thought to be responsible for the bulk of ergosterol production in this mold and is susceptible to the azole drugs, unlike its innately FLC-resistant paralog, CYP51A. The crystal structures of C. albicans LDM (N-truncCaLDM6×His) have been determined for complexes with PCZ and VT-1161 (14). The crystal structure of the Trypanosoma cruzi CYP51B catalytic domain in complex with VT-1161 has also been determined (15). VT-1161 shows tight type II binding to CaLDM and C. neoformans LDM and binds weakly to HsCYP51 (16, 17). Most recently, a crystal structure of the A. fumigatus CYP51B catalytic domain in complex with the investigational drug candidate VT-1598 was published (18). This broad-spectrum azole has the same tetrazole head group as VT-1161 but has a much longer tail that includes an additional aromatic ring, an alkyne, and a nitrile.
Our companion paper describes the biochemical and physiological characterization of functional, full-length CaLDM6×His or CgLDM6×His overexpressed from the PDR5 locus of S. cerevisiae deleted of 7 ABC transporters (19). Here, we report the full-length crystal structures of CaLDM6×His and CgLDM6×His and show their catalytic domains have the same fold and bind ITC in conformations nearly identical to that of full-length ScLDM6×His. The catalytic domains of the full-length LDMs have the same fold as the published structures of the CaLDM6×His catalytic domain in complex with PCZ or VT-1161. There are minor structural differences within the ligand binding pocket (LBP) comprised of the substrate entry channel (SEC), the heme-containing active site, and a putative product exit channel (PPEC). This detailed exploration of the LBP provides structure-based insight into the LDM reaction mechanism and suggests explanations of the phenotypes of several single-site mutations in CaLDM found in clinical isolates. Most importantly, it provides a practical basis for the rational design and discovery of antifungals. Computer-based, phenotypic, and biochemical screens can now be used effectively to identify broad-spectrum fungicides that selectively target the LDMs of fungal pathogens.
RESULTS
Enzyme purification and crystallization.
Full-length C. glabrata and C. albicans LDM6×His were extracted from crude membranes with the detergent n-decyl-β-d-maltoside (DM) and purified by nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography, using imidazole as the eluent, followed by size exclusion chromatography (8). Both preparations gave a single size exclusion peak with a relative migration of ∼100 kDa, as shown previously with ScLDM6×His (data not shown). This result is consistent with partial immersion of single LDM6×His molecules in 40-kDa vesicles of DM. The purified enzymes were concentrated in the presence of 40 μM ITC by centrifugal filtration and subjected to crystal trials using conditions similar to those used previously with ScLDM6×His (10, 11). CgLDM6×His (PDB entry 5JLC) and CaLDM6×His (PDB entry 5V5Z) crystallized in space groups I 4 3 2 and P 2 21 2, and their structures were refined to resolutions of 2.4 and 2.9 Å, respectively. Data collection parameters and refinement statistics are given in Table S1 in the supplemental material. The asymmetric unit in both structures is a monomer with the characteristic P450 fold seen previously in the related ScLDM6×His structure (PDB entry 5EQB). CgLDM6×His was resolved from L20-G534, revealing a segment of the amphipathic membrane-associated helix (MH) commencing at L20, leading into the transmembrane helix (TMH; L28-L52), followed by the catalytic domain (Fig. 1). The CaLDM6×His structure is comprised of amino acid residues I25 to E524. The resolved structure, assuming identity to ScLDM, lacks the N-terminal amino acid residues that contribute the MH, the turn between MH and TMH, and a single turn of the TMH.
FIG 1.
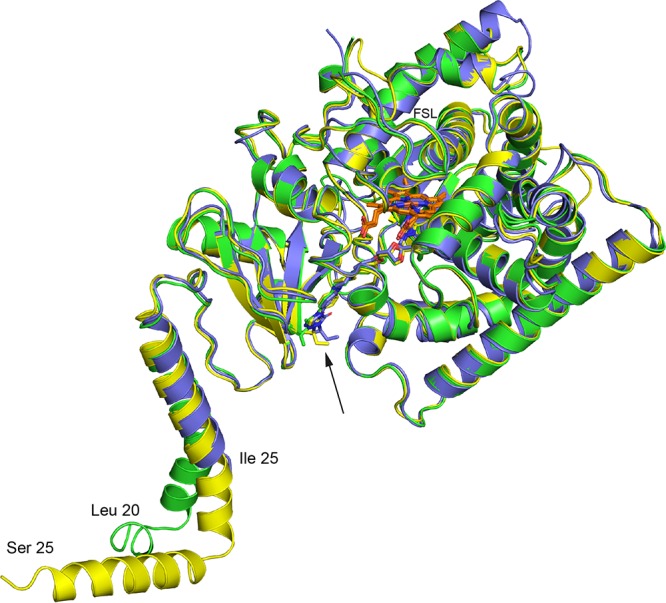
Structural comparison of C. glabrata, C. albicans, and S. cerevisiae LDM6×His in complex with ITC. The cartoons show the aligned polypeptide backbones of the 3 structures with the heme and ITC (arrow) shown as sticks. The polypeptide chains and the carbons of the bound ITC are shown in green for CgLDM, purple for CaLDM, and yellow for ScLDM. Heme carbons are shown in orange, nitrogen in blue, and oxygen in red. The most amino-terminal amino acid residue detected in each crystal structure is indicated. FSL, fungus-specific loop. For more detailed information about differences in surface features detected in these structures, see Fig. S2 and S3.
The CgLDM and CaLDM primary sequences of the external fungus-specific loop (FSL) are 1 and 4 amino acids longer, respectively, than the ScLDM sequence (Fig. S1). The crystal structures of both CgLDM6×His and CaLDM6×His lack electron density at the surface of the enzyme for the N-terminal third of the FSL (ΔD435-G443 of CgLDM6×His, FSL G434-G466; ΔA430-S441 of CaLDM6×His, FSL T429-G464; Fig. S2A), whereas the complete FSL (D434-G464) was modeled for ScLDM6×His.
Three additional differences were detected in the surface loop structures of the LDM catalytic domain. The length and composition of the N-terminal portion of the β5-β6 surface loop differed between the S. cerevisiae and fungal pathogen LDMs (Fig. S1 and S2B). This part of the surface loop contains 6 amino acids in CaLDM6×His (T494IDGYK499), 8 amino acids in CgLDM6×His (R496YPTEGET503), and 7 amino acids in ScLDM6×His (H494YPEGKT500). The polar and negatively charged G-H surface loop of CaLDM6×His differs from the other two structures with the insertion of a proline residue (Fig. S1 and S2A), which projects the loop closer to the C terminus of helix C and the N terminus of helix D. We speculate that these differences are involved in species-specific interactions with other partner molecules, as the β5-β6 loop should lie close to the endoplasmic reticulum surface while the G-H loop lies near the proposed NADPH-cytochrome P450 reductase binding site. There is also a 2-amino-acid extension of the C-terminal end of helix J in CaLDM6×His and a single-amino-acid insertion into the J-K loop compared with the aligned sequences in CgLDM6×His and ScLDM6×His (Fig. S1 and S2C). This allows formation of nonconserved hydrogen bonds between the main chain and side chain carbonyls of N349 and the R163 side chain guanidine in helix D of CaLDM.
The catalytic domains of the LDMs have comparable folds and bind ITC similarly.
There are no major structural deviations between the structures for the two full-length Candida LDMs (root mean squared deviation [RMSD] of 0.71 Å over a total of 434 αC atoms) (Table 1). Further comparisons made with other published structures, including the CaLDM catalytic domain, show the structures to be similar with only minor deviations (Table 1). In agreement with sequence and phylogenetic similarity, these and other data in Table 1 confirm that C. glabrata LDM is more closely related structurally to the S. cerevisiae enzyme than the C. albicans enzyme and that the catalytic domains of full-length and N-terminally truncated CaLDM6×His have very similar folds. The CgLDM and CaLDM structures contain a single ITC within a deep LBP coordinated to the iron of the heme cofactor in a manner identical to that of their S. cerevisiae counterpart. The major interactions between the drug and the enzyme are the coordinate bond between the heme iron and the N4 triazole nitrogen along with extensive van der Waals interactions. The conformation of ITC in each of the three full-length structures is essentially identical, with the terminal alkyl group differing slightly in orientation (Fig. 1). A water-mediated hydrogen bond exists between the piperazine ring of ITC and the main chain of the enzyme (S382) in the S. cerevisiae structure (PDB entry 5EQB). While no corresponding water molecule was modeled in the CgLDM6×His or CgLDM6×His structures, a water molecule that hydrogen bonds with the main chain of S378 has been assigned to a similar location in the N-truncated CaLDM6×His-PCZ structure. A total of 85 water molecules were modeled into the 2.5-Å-resolution CgLDM6×His + ITC crystal structure compared with 124 water molecules in the 2.1-Å-resolution ScLDM6×His + ITC structure (PDB entry 5EQB) and 61 water molecules in the 2.8-Å-resolution N-truncated CaLDM structure. We consider the 2.9-Å-resolution CaLDM6×His crystal structure to be insufficiently resolved to model water molecules.
TABLE 1.
Conservation of protein fold in full-length and truncated fungal CYP51 structures
| Structure | PDB entry | Resolution (Å) | No. of Cα atoms compared | RMSD (Å) |
|---|---|---|---|---|
| CgLDM6×His +ITC | 5JLC | 2.4 | 434 | 0.71 |
| CaLDM6×His +ITC | 5V5Z | 2.9 | ||
| CaLDM6×His +ITC | 5V5Z | 2.9 | 440 | 0.67 |
| ScLDM6×His +ITC | 5EQB | 2.19 | ||
| CgLDM6×His +ITC | 5JLC | 2.4 | 430 | 0.41 |
| ScLDM6×His +ITC | 5EQB | 2.19 | ||
| CaLDM6×His +ITC | 5V5Z | 2.9 | 403 | 0.58 |
| N-truncCaLDM6×His + PCZ | 5FSA | 2.86 | ||
| CgLDM6×His +ITC | 5JLC | 2.4 | 415 | 0.70 |
| N-truncCaLDM6×His + PCZ | 5FSA | 2.86 | ||
| CaLDM6×His +ITC | 5V5Z | 2.9 | 404 | 0.52 |
| N-truncCaLDM6×His + VT-1161 | 5TZ1 | 2.0 | ||
| ScLDM6×His +ITC | 5EQB | 2.19 | 419 | 0.67 |
| N-truncCaLDM6×His + PCZ | 5FSA | 2.86 | ||
| ScLDM6×His +VCZ | 5HS1 | 2.1 | 395 | 0.81 |
| N-truncAfCYP51B6×His +VCZ | 4UYM | 2.55 | ||
| ScLDM6×His +ITC | 5EQB | 2.19 | 401 | 0.79 |
| N-truncAfCYP51B6×His +VCZ | 4UYM | 2.55 | ||
| ScLDM6×His +ITC | 5EQB | 2.19 | 416 | 0.93 |
| N-truncAfCYP51B6×His + VNI | 4UYI | 2.81 | ||
| ScLDM6×His +ITC | 5EQB | 2.19 | 432 | 0.23 |
| ScLDM6×His +VCZ | 5HS1 | 2.1 | ||
| ScLDM6×His +ITC | 5EQB | 2.19 | 425 | 0.24 |
| ScLDM6×His + VT1161 | 5UL0 | 2.2 | ||
| ScLDM6×His + VT1161 | 5UL0 | 2.2 | 400 | 0.58 |
| N-truncCaLDM6×His + VT-1161 | 5TZ1 | 2.0 |
Interactions between the transmembrane domain and the catalytic domain.
The TMH appears similar in all three structures. However, there is a kink in the CgLDM6×His TMH (Fig. 1). The effect may be due to the substitution of T43 in CgLDM6×His for N42 in ScLDM and N34 in CaLDM6×His, combined with the impact of crystal contacts (see Discussion). In contrast, multiple polar contacts between one face of the C-terminal half of the TMH, its C-terminal turn, and the catalytic domain are conserved or involve nearly equivalent interactions in the crystal structures obtained for the full-length LDMs of the two fungal pathogens and S. cerevisiae (Table 2). The close packing of the one face of the C-terminal half of the TMH with a face of the catalytic domain in all three structures is mediated primarily through an extensive network of interactions with the adjacent long intervening loop, the β1β2 sheet, and a short loop that includes P401 and Y404 in S. cerevisiae LDM (Table 2). The most important of the conserved interactions is the hydrogen bond between the TMH Q46/Q47/Q38 side chain amide and the main chain carbonyl of Y61/Y62/Y53 in the adjacent long intervening loop of the ScLDM, CgLDM, and CaLDM structures. This is supported by hydrogen bonds between the Y49 hydroxyl and the Y404 hydroxyl in ScLDM, between the Y50 hydroxyl with the Y405 hydroxyl in CaLDM, and via a possible water-mediated interaction between the Y41 hydroxyl and the H400 imidazole group in CaLDM. In addition, the CgLDM structure has a water-mediated hydrogen bond network between the main chain carbonyls of V60 and S69 and the main chain amide of M95. Water-mediated hydrogen bond networks are also seen between the main chain carbonyl of F61 and the hydroxyl S69 and between the side chain hydroxyl of Y62 and the main chain amide of W66 of CgLDM. There is a sufficient spatial distribution for similar networks to exist in both CaLDM and ScLDM.
TABLE 2.
Polar interactions involving the transmembrane helix and neighboring elements of the catalytic domains in S. cerevisiae, C. albicans, and C. glabrata LDMa

Conserved interactions are highlighted in yellow, and a hypothetical semiconserved interaction is highlighted in green. Main chain interactions involving chemically similar amino acids are highlighted in light blue. Polar interactions: --, hydrogen bond; **, ionic interaction. The α-helix n + 4 hydrogen bonds are not shown.
Full-length and N-terminally truncated fungal CYP51 have similar LBPs.
The crystal structures of ScLDM6×His in complex with ITC, VCZ, and VT-1161 are essentially identical (RMSD of 0.23 to 0.24 Å over 432 atoms) (Table 2), consistent with a relatively rigid structure for this surrogate drug target that is likely applicable to CYP51 proteins, now including CgLDM and CaLDM. The crystal structures of the catalytic domain of A. fumigatus CYP51B (AfCYP51B) in complex with VCZ or VNI have folds similar to those of full-length CaLDM, CgLDM, and ScLDM in complex with ITC. However, the conformation of AfCYP51B within the drug binding pocket and efflux channel is clearly influenced by the ligand (13). For example, VCZ is bound within the active site, leaving the SEC and PPEC unoccupied, while VNI blocks the SEC and penetrates into the PPEC. The conformation/position of helix F′ at the nexus of the SEC and PPEC in the yeast structures (CgLDM, CaLDM, and ScLDM) differs from that of the mold enzyme by ∼2.0 Å. This is exemplified by the position of a conserved phenylalanine (F241 in ScLDM6×His) that projects beside the channel of the yeast enzymes but occupies the position of the tail of VNI in the mold enzyme. The conformation of this phenylalanine is predicted to obstruct the binding of VNI but not VCZ in the yeast enzymes. Comparison of the folds of complexes of ScLDM6×His and AfCYP51B-6×His with VCZ (RMSD of 0.81 Å over 395 atoms) highlights additional differences in the structures of the LBPs of the full-length yeast and the N-terminally truncated mold enzyme. For example, N-truncated AfCYP51B in complex with VCZ appears to have a slightly narrower mouth to the SEC and a much larger PPEC than the liganded full-length LDMs from S. cerevisiae (in complex with VCZ and ITC) and the two Candida LDMs (in complex with ITC). This is primarily due to differences in conformation of the AfCYP51B catalytic domains K94 and F234 compared with the structurally aligned residues in the LDMs (e.g., ScLDM R98 and F241).
The LBPs in S. cerevisiae, C. glabrata, and C. albicans LDMs are structurally and chemically similar.
Comparison of the full-length fungal LDM crystal structures (Table 3) revealed that only a fraction of amino acid residues identified previously as part of the fungal substrate recognition site (SRS) (20) contribute to the LBP interior surface we have defined using the exterior surface function in PyMOL, with the ITC ligand extracted. These include residues contributing to the active site distal to the heme, SEC, and PPEC, i.e., 9 to 10 of the 20 SRS1 residues, 7 to 8 of the 18 SRS4 residues, 6 of the 7 SRS5 residues, and 6 of the 11 SRS6 residues but none of the 9 SRS2 residues or the 11 SRS3 residues. In the three full-length structures, the 22 to 23 residues within 4 Å of ITC all contribute to the LBP. Residues in SRS2 and SRS3 contribute to enzyme function, but because they are part of the enzyme surface outside the LBP, we propose they should no longer be defined as part of substrate recognition sites. The putative solvent (S) channel (21) approaches the LBP from the enzyme surface adjacent to the membrane. The channel appears to be gated by an ionic interaction between two residues conserved in all CYP51s, i.e., ScLDM D233 in SRS2 and the LBP residue H317 in helix I. This interaction is conserved in all CaLDM structures (14). SRS3 is located at the N-terminal end of helix G in proximity to the membrane surface and may contribute to enzyme function by interacting with its cognate NADPH-cytochrome P450 reductase.
TABLE 3.
Amino acid residues contributing to the LBPa

SRS1 to SRS6 are substrate recognition sites as defined by Warrilow et al. (20). Residues in italics contribute to the interior surface of the LBP, i.e., the active site, the SEC (tunnel 2f [21]), and the PPEC (8). Residues not contributing to the LBP are in normal text. Residues within 4 Å of ITC are in boldface. Residues differing either chemically or in conformation compared with the reference structurally aligned S. cerevisiae residues are highlighted in yellow if within the LBP and in green if not. Nonidentical structurally aligned residues contributing to the LBP within 4 Å of ITC are shown in brackets. Y64 in CaLDM is within 4.1 Å of ITC. We find 48 residues contribute the interior surface of the LBP of CgLDM and ScLDM. CaLDM K118 at the external edge of the PPEC and P68 beside the water-containing pocket in the SEC may also contribute small areas to the LBP, while a further 5 residues may artifactually extend the LBP. Among the residues contributing to the LBP, single mutations at 8 residues (highlighted in purple) have been demonstrated to contribute to azole resistance in C. albicans. No such mutations have been reported for CgLDM. The 6 residues underlined are found in major ascomycete and basidiomycete fungal pathogens but not in their human or plant hosts and have yet to be shown to confer resistance to azole drugs. LBP, ligand binding pocket. SEC, substrate entry channel. PPEC, putative product exit channel.
The 48 amino acid residues that contribute to the LBP interior surface (shown in italics in Table 3) are almost absolutely conserved between CgLDM6×His and ScLDM6×His, except for the ScLDM/CgLDM V71/I70 and M75/T74 pairs in helix A′ at the mouth of the SEC (Fig. S3). The latter residues also differ chemically from CaLDM Q66.
There are 10 further amino acid substitutions in CaLDM LBP relative to the S. cerevisiae and C. glabrata enzymes. Each substitution within the LBP involves a chemically similar or isostructural contribution. For example, CaLDM V510 contributes a surface essentially identical to that generated by the α, β, and γ carbons of ScLDM T511 and CgLDM T514. Furthermore, none of the 12 LBP residues specific to CaLDM is within 4 Å of ITC apart from the main chain carbonyls of Y505 and S506 and main chain components and the β-carbon of I304. Similar relationships are expected for complexes with FLC, VCZ, VT-1161, and PCZ, consistent with the efficient binding by S. cerevisiae, C. glabrata, and C. albicans LDMs of the short-, medium-, and long-tailed azole drugs described in the companion paper (19).
Due to the modest (∼2.9-Å) resolution of the CaLDM structure, some apparent differences in the LBP surface are probably artifacts detected by PyMOL. For example, the active site in CaLDM6×His only appears to extend into a small pocket surrounded by A149, L150, Y156, L204, and L276. Slight changes in the conformation of surrounding side chains readily pinch off this pocket from the LBP. Similarly, P68 contributes a very small area to the surface of the LBP in both the full-length and truncated CaLDM structures but not to full-length ScLDM or CgLDM.
Polarity is conferred on the otherwise hydrophobic CaLDM LBP by a group of 10 hydrogen bond-accepting main chain carbonyls (Fig. 2). These are located around the mouth of the substrate entry channel (G65, Y505, and S506) and then track toward the active site (S507, M508, and P375) and are close to the kink in helix I (G303, I304, M306, and G307). A second group of residues on the opposite side of the LBP projects hydroxyl groups of 3 tyrosines into the LBP, with Y64 forming a hydrogen bond with S378 and Y118 and Y132 forming hydrogen bonds with the heme propionates (Fig. 2). The main chain carbonyl of S378, the heme propionate carboxyls, and the side chain carbonyl of the conserved Q142 residue also contribute polarity to the second group. The two groups of polarity-contributing residues are found in the LBPs of all full-length and N-terminally truncated LDM structures, including ScLDM in complex with lanosterol. Two broad hydrophobic strips, the PPEC and the heme, separate the two groups of residues. Opposite the nexus of the SEC and the PPEC, the groups of polarity are separated by the main chain hydrogen bond donating amide of a histidine (CaLDM H377, CgLDM H382, and ScLDM H381) and its ionizable imidazole. Proposals for the role of the polar groupings in substrate and drug binding, the three monooxygenase reactions of the enzyme, and product exit are discussed in the next section.
FIG 2.

Amino acid residues contributing polarity or charge to the LBP of CaLDM in complex with ITC. The ITC ligand and heme are shown in gray and the associated iron in orange. Selected residues of CaLDM6×His are shown with green carbons, blue nitrogens, and red oxygens. The surface of the LBP is shown in yellow (gray represents the back of the surface). The images are rotated in the horizontal plane ∼120° to each other and were generated in PyMOL for residues contributing to the LBP of PDB entry 5V5Z. SEC, substrate entry channel. PPEC, putative product exit channel.
DISCUSSION
Crystallization of functional full-length CaLDM and CgLDM.
As described in the companion paper (19), the type II binding of azole drugs showed detergent-solubilized, affinity-purified, full-length CgLDM6×His and CaLDM6×His are functional and stable in vitro. The structures of CgLDM6×His and CaLDM6×His in complex with ITC obtained with the purified enzyme are the first crystal structures obtained using full-length LDMs from fungal pathogens. Both structures could be refined for over 90% of their full-length primary sequence. They reiterate key features found for full-length ScLDM and N-terminally truncated CaLDM and provide additional insight, even though part of the N-terminal MH for CgLDM6×His and a portion of TMH plus the linking turn of TMH for CaLDM6×His could not be modeled despite the importance of MH-mediated crystal contacts in the ScLDM6×His structure.
The S. cerevisiae and fungal pathogen full-length LDMs have the same overall fold (Table 1), with the TMH interacting with the catalytic domain via a range of conserved or similar polar interactions and van der Waals interactions that bury one face of the C-terminal half of the TMH against the catalytic domain (Table 2). The CgLDM TMH veers away from highly similar structures obtained for the S. cerevisiae and C. albicans LDMs (Fig. 1). This difference may be due to CgLDM T43 substituting for an otherwise extensively conserved N43-Y62 or N43-H62 (C. glabrata numbering) polar interaction between TMH and the catalytic domain among fungal pathogens (Table 2 and Fig. S4). The deviation of the TMH may also be affected by intermonomer crystal lattice contacts between the C-terminal end of MH and the catalytic domain, i.e., adjacent CgLDM6×His monomers have an intermolecular electrostatic interaction between R31 and E79, as well as a water-mediated intermolecular hydrogen bond network between the L28 main chain amide and the carboxylates of D83 and E79 (PDB entry 5JLC).
The pose of each LDM with respect to the endoplasmic reticulum lipid bilayer appears to be set primarily by TMH plus the loop and F′ helix connecting the F and G helices (Fig. S1), as proposed previously for ScLDM6×His (8). This places the mouth of the SEC (tunnel 2f in human and Trypanosoma brucei CYP51 [21]) at the surface of the lipid bilayer. In addition, the S channel from the surface of the enzyme adjacent to the membrane is conserved across all structures, both full-length and truncated, and seems to be gated by an ionic interaction between the conserved LBP residue H317 of helix I and the conserved D233 in ScLDM SRS2 (14). The LBP (Table 3) is lined predominantly with hydrophobic residues, and in addition to an SEC, it comprises a heme-containing active site and a PPEC, which may provide a template to display demethylated lanosterol for attack by the Erg24-Erg27 enzymes (8). The pose of the enzyme places the mouth of the PPEC near the membrane surface and accessible to downstream metabolism by Erg24-Erg27.
The 2.9-Å-resolution structure of CaLDM6×His in complex with ITC (PDB entry 5V5Z) is insufficient to model resident water molecules, while the 2.4-Å-resolution C. glabrata structure in complex with ITC (PDB entry 5JLC) detects about 2/3 the number of water molecules found for the 2.1-Å-resolution structure of ScLDM6×His in complex with ITC (PDB entry 5EQB). Two water molecules in a pocket associated with the SEC are detected in the LBP of ScLDM6×His, one of which hydrogen bonds with N1 of the ITC piperazine ring. A similarly located water molecule is detected in the structure of N-truncated CaLDM6×His in complex with PCZ (PDB entry 5FSA) but not in full-length CgLDM6×His in complex with ITC, despite there being sufficient space for it to occur in the latter structure. The water-mediated hydrogen bond network between G467 and the heme ring D propionate in the CgLDM6×His structure is conserved among ScLDM and all published truncated CaLDM structures and appears likely to be conserved in the full-length CaLDM structure. In CgLDM, the heme ring D propionate forms a hydrogen bond with Y127 and an ionic bond with R386, while the heme ring C propionate forms a hydrogen bond with Y141 and an ionic bond with K152. These interactions are conserved in ScLDM and all of the CaLDM structures.
Binding of ITC and the similarity of CgLDM, CaLDM, and ScLDM ligand binding pockets.
All 3 full-length fungal LDM structures bind ITC in conformations close to that for N-truncated CaLDM6×His with PCZ. Slight differences in the conformation of the ITC and surrounding amino acid residues (CgLDM I71, T75, and I240; CaLDM A62, Q66, and I231; ScLDM V70, M74, and I239) have been modeled at the 1,2,4-triazlin-3-one end of the inhibitor (Fig. S3). These differences may not be significant due to modest electron densities for this region. The conformations of LBP residues that contribute to the active site, SEC, and PPEC appear very similar for all three full-length structures. Furthermore, the X-ray structures show FLC (PDB entry 4WMZ) or ITC is bound within the active sites of essentially identical protein structures (9).
Our crystal structures emphasize the identity of the ScLDM and CgLDM LBPs and their close chemical similarity to CaLDM. Apart from I71 and T75 at the mouth of the substrate entry channel of CgLDM, all 48 structurally aligned residues in the S. cerevisiae and C. glabrata LDM LBPs were identical. The ScLDM/CgLDM/CaLDM LBPs differ due to only 10 structurally aligned amino acid substitutions, 3 nonconservative (A124/A125/D116, M74/T75/Q66, and T511/T514/V510) and 7 conservative (V70/I71/A62, R98/R99/K90, V154/V155/A146, V311/V312/I303, L383/L384/I379, H405/H406/Y401, and F506T507/F509S510/Y505S506). Of these substitutions in CaLDM, only the F506T507/F509S510/Y505S506 substitution is within 4 Å of ITC. It is located at the mouth of the SEC, and only its main chain carbonyls contribute to the LBP. Two additional structurally aligned residues make minor contributions to the CaLDM LBP only, K119 at the external end of the PPEC and P68 in the SEC. Collectively, the Candida and S. cerevisiae LBP surfaces appear chemically similar with respect to the binding of the azole drugs ITC and PCZ. As previously described for ScLDM, N-truncated CaLDM, and trypanosomal CYP51s, these enzymes have a relatively rigid structure, with an LBP that contains chemically conserved residues, in close proximity to substrate and azole ligands (1, 22).
Structural features of the ligand binding pocket that may contribute to LDM function.
Two conserved groups of hydrogen bond acceptors/donors run along the SEC and into the active site (Fig. 2). In CaLDM, group 1 is comprised of the main chain carbonyls of Y505SSM508, P375, M306, and G307 plus the main chain and side chain carbonyls of Q142. Group 2 also involves the side chain carbonyl of Q142, the main chain carbonyl of S378, the two heme propionates, and the hydrogen bond-donating hydroxyl groups of Y64, Y118, and Y132. The main chain amide and the side chain imidazole of H377 separate the two tracks. These conserved features may be important for the entry and positioning in precatalytic and catalytic states for substrates such as lanosterol and eburicol during the three monooxygenase reaction cycles that sequentially replace the 14α-methyl group with an alcohol and an aldehyde and then its removal as formic acid, as well as during egress of the reaction product into the putative exit channel. We speculate that group 1 could allow the 3β-hydroxyl of lanosterol, its only polar group, to be guided through an otherwise hydrophobic landscape to engage in a precatalytic state that involves formation of a hydrogen bond with the main chain carbonyl of CaLDM G303, as suggested for ScLDM G310 in complex with lanosterol (PDB entry 4LXJ) (8). The catalytic state could then initiate via formation of a hydrogen bond between the hydroxyl group of the substrate and the side chain carbonyl of Q142. This would afford stabilizing interactions for the intermediate alcohol, aldehyde plus product water, and formic acid in the vicinity of the heme propionates and the hydroxyl groups of Y118 and Y132. The Y132 hydroxyl group forms a hydrogen bond network with waters, the heme propionates, and inhibitor tertiary alcohols, as shown for CaLDM in complex with VT-1161 (PDB entry 5TZ1) and for structurally aligned components of ScLDM in complex with VT-1161 (PDB entry 5UL0), FLC (PDB entry 4WMZ), and VCZ (PDB entry 5ZE1). Once the 14-methyl group of lanosterol has been removed, the more hydrophobic reaction product, 4,4-dimethylcholesta-8,14,24-trien-3β-ol, could reverse out of the active site and deep into the SEC, using group 1 residues to provide polar interactions for the 3β-hydroxyl group. The product would then migrate into the PPEC and project the hydroxyl head group and its pair of 4-methyl groups into the aqueous milieu for further reaction.
Only 4 amino acid residues within the LBP are absolutely conserved among the ascomycete and basidiomycete fungal pathogens, the human host, and the plant host wheat and soybean LDMs (Fig. 3A). In ScLDM these conserved residues are Y126, F134, Q150, and H317. Their locations within the active site, and their potential interactions with the substrate, are consistent with involvement in sterol demethylase catalytic function and with other aspects of the catalytic cycle, including access to hydronium ions and regulation of substrate entry and product egress.
FIG 3.
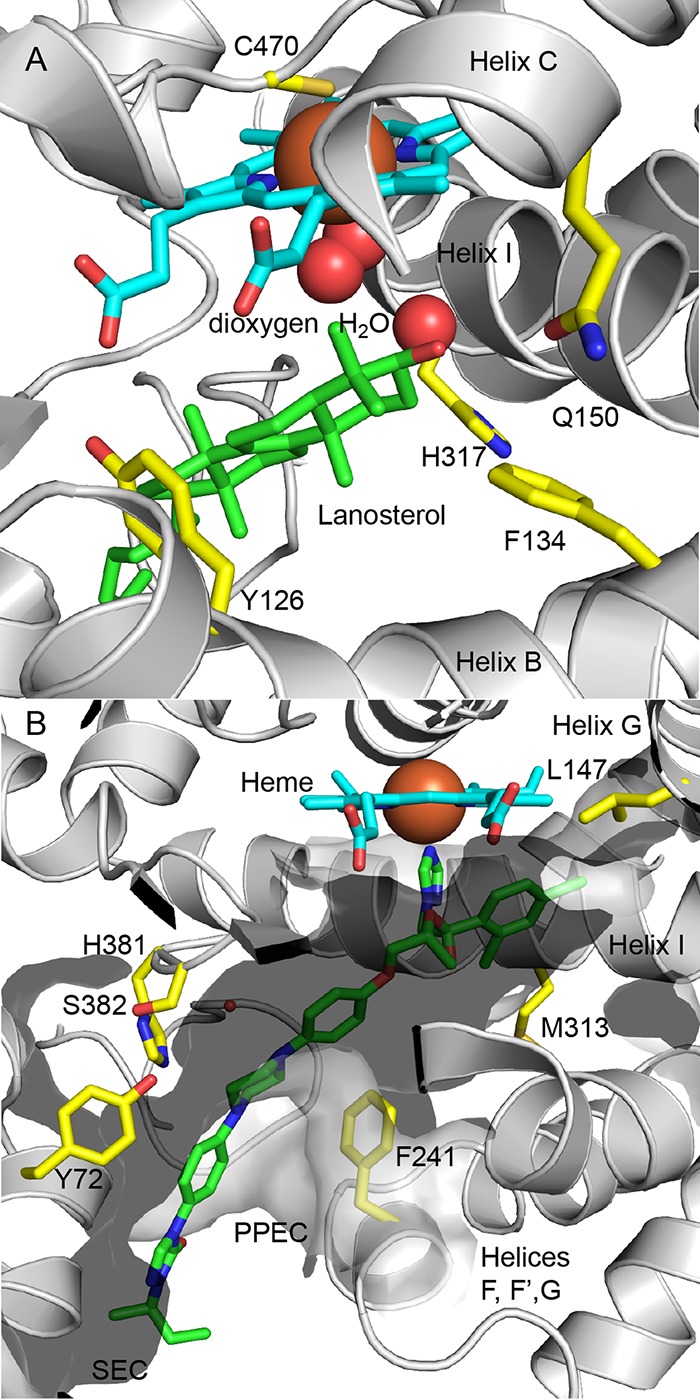
Key amino acid residues in the LBP of ScLDM. (A) Amino acid residues in the active site of ScLDM conserved among fungal pathogens and their human and plant hosts. The side chains of the conserved amino acid residues are shown in yellow. The carbons of lanosterol and the heme are shown in green and cyan, respectively, nitrogens are blue, oxygens are red, and iron is orange. (B) Amino acid residues in the LBP conserved among ascomycete and basidiomycete pathogens not found in human or plant hosts and not subject to known mutations. The surface of the LBP is shown. Heme carbons are shown in cyan, ITC carbons in green, and the side chain carbons of residues of interest in yellow. Nitrogens are shown in blue, oxygen in red, and iron in orange.
A water molecule is visualized in the wall of LBP of ScLDM in complex with lanosterol (PDB entry 4LXJ), where helix I is kinked (Fig. 3A). The water hydrogen bonds with the main chain carbonyls of helix I M313 and G314 and the main chain amides of H317 and T318 and is within 4.6 Å of the molecular oxygen. The location of this water is consistent with a role in proton delivery to the LBP via the S channel gated by H317 and D233 (14). Despite retaining the kink in helix I, this water is not detected in the structures of the S. cerevisiae and Candida enzymes in complex with VT-1161, ITC, or PCZ due to the proximity of the heme-binding azole group to helix I. Thus, the azole groups in these drugs occupy the active site and block the enzyme reaction by binding to the heme and inhibit access of protons to the active site.
Innate azole resistance and mutations conferring acquired azole resistance.
The ScLDM6×His, CgLDM6×His, and CaLDM6×His crystal structures and the functional expression of these enzymes provide tools to explore the impact of mutations on fungal LDMs (10). We have modeled residues that are mutated or occur as innate substitutions conferring azole resistance and assessed their direct or indirect effects on the LBP and the ligands it encapsulates. This information has enabled investigation of the mechanism in instances of innate or acquired resistance to individual, groups, or the entire class of azole drugs (Table 4).
TABLE 4.
Single-amino-acid substitutions in C. albicans LDM that confer azole resistance
| Mutationa | Present in LBP | Predicted effect |
|---|---|---|
| A61V | +/− | Mouth of SEC modified |
| F105L | − | Modification of environment beside heme ring D propionate |
| A107T | − | C-cap of surface helix containing F105 modified |
| A114S | − | Modification of environment near both heme propionates |
| Y118A | + | LBP beside heme propionate D enlarged; loss of water-mediated H-bond interactions with tertiary hydroxyl of FLC, VCZ, and VT-1161 and heme ring D propionate |
| F126S | + | Enlarged and more polar LBP in helix B beside helix I G303 |
| Y132F/Hb,c,d | + | Loss of H-bond with heme ring C propionate and water-mediated H-bonds with tertiary hydroxyl of FLC, VCZ, and VT-1161 |
| K143R/Q | + | Side chain involved in ionic bond with heme ring C propionate modified and conformation of β-bulge affected |
| T229A | − | H-bond between αF and αF′ modified near P230 and F234 |
| I253V | − | Modification of SRS2 helix G beside helix F |
| H283D | − | Modification of interaction with helix C F148 |
| G307S | + | Formation of helix I S307-OH H-bond to triazole group |
| F380S | + | Enlarged and more polar nexus of SEC and PPEC |
| S405Fd | − | Loss of H-bond with H377 in SEC |
| F449Vd/M/S | − | Modification of K143 heme ring C propionate environment |
| G464Sb,d | − | Water-mediated main chain H-bond network with heme ring D replaced with side chain interaction |
| R467Kb,d | + | Possible K467 side chain interaction with N136 may affect main chain H-bond with K143 side chain |
| I471Tb,c | + | Polarity increased in environment of heme beside helix I |
Single mutations in CaLDM found in azole-resistant clinical isolates are shown. Mutated residues within 4 Å of ITC are in italics. Mutations shown by expression in S. cerevisiae to confer azole resistance are shown in boldface. Mutations occurring outside the LBP are discussed in the supplemental material. References reporting the mutations that we considered included references 2–4 and 23–28.
Mutations shown to affect azole binding, substrate affinity, and/or enzyme activity.
Mutations investigated using the structurally aligned residue in ScLDM6×His.
Mutations investigated by expressing mutant LDM in drug-susceptible C. albicans host.
Despite an incidence of azole resistance of about 3.5% of C. albicans clinical isolates and about 30% for C. glabrata, only C. albicans clinical isolates thus far have been shown experimentally to confer azole resistance through mutations in LDM. Of >140 mutations identified in CaLDM (2, 3), most occur in combinations which give a sensitive enzyme, but some combinations give a functional enzyme in which azole resistance is enhanced additively or synergistically. Flowers et al. identified several single-site mutations in CaLDM that give at least 4-fold resistance to FLC and used the crystal structure of full-length ScLDM in complex with lanosterol (PDB entry 4LXJ) to show that these mutations were located in proximity to the heme, the substrate entry channel, and the FSL (4). Table 4 lists single-site mutations detected in the LDMs of azole-resistant clinical isolates of C. albicans. We have used the CaLDM6×His crystal structure to classify these mutations in two groups. The first group contributes to the LBP surface and may directly or indirectly affect the binding of azole drugs. The second group is located outside the LBP and may affect indirectly the binding of azole drugs. The mutations not in the LBP are discussed in the supplemental material.
Mutations in the LBP may affect the binding of azole drugs directly. CaLDM A61 contributes to the mouth of the SEC. Consistent with this location, the A61V mutation has been found to affect the binding of the long-tailed triazole PCZ but not FLC, VCZ, or ITC to LDM (26). The CaLDM structure shows the alanine methyl group is about 4.3 Å from N3 of the 1,2,4-triazol-3-one group in the tail of ITC, while a valine methyl group would clash with the tail group. The hydrophobic valine methyl group could interact more negatively with the bulkier and more polar tail of PCZ than with ITC while not affecting the binding of the shorter-tailed azoles FLC, VCZ, and VT-1161, provided their access to the active site is not significantly impeded.
The ScLDM6×His Y140F/H mutations in the LDM active site, analogous to CaLDM6×His Y132F/H, have been extensively characterized biochemically and structurally (10). They result in the loss of a hydrogen bond between Y132 and the heme ring C propionate plus the disruption of a water-mediated hydrogen bond network involving the tertiary alcohol of FLC, VCZ, and VT-1161 not found with ITC or PCZ. Furthermore, the Y140F mutation in ScLDM displaced VCZ 0.5 Å closer to helix I within the active site.
The conserved Y118 residue in the C. albicans LDM active site is within 4 Å of ITC and forms a hydrogen bond with heme ring D propionate. The CaLDM Y118A mutation should significantly increase the size of the active site adjacent to Y132, reducing affinity for azole drugs, particularly the short-tailed azole drugs, including FLC and VCZ, that bind entirely within the active site and are part of a water-mediated hydrogen bond network involving Y118, Y132, and both heme propionates. The role of the conserved F126 residue in the CaLDM active site is not understood. Its aromatic group is within 4 Å of ITC and projects parallel to helix I beside G303, the residue predicted to form a precatalytic hydrogen bond with lanosterol (see PDB entry 4LXJ) (8). The F126S mutation will increase the volume and polarity of the active site in proximity of the dihalogenated headgroup characteristic of most triazole drugs. This should reduce affinity for short- and long-tailed azole drugs but would do so most extensively for short-tailed azoles. Conversely, the F380S mutation at the nexus of the SEC and PPEC should affect affinity for long-tailed azoles but possibly not short-tailed azoles.
The side chain of CaLDM G307S in helix I is predicted to clash with the dihalogenated head group of FLC or ITC and disrupt triazole binding to the heme (4). This would be expected to modify the binding of all similar triazole drugs that target LDM.
The hydrophobic side chain of I471 in CaLDM projects beyond the hydrophobic edge of the heme into the LBP surface bordered by helix C and helix I. This contribution to the LBP is conserved in the C. glabrata and S. cerevisiae enzymes. The Y132H I471T mutations in the Darlington strain of C. albicans confer azole resistance in a synergistic manner that can be mimicked in S. cerevisiae. Modeling the mutant structure suggests increased polarity in the active site beside the hydrophobic edge of the heme and helix I. How this confers synergistic resistance to the short-tailed triazoles has yet to be elucidated.
New information for structure-directed drug discovery.
Our structure-based interpretation of the impact of mutations and substitutions establishes a platform for further biochemical and structural studies using overexpressed mutant enzymes. For example, the conformation of M509 in ScLDM in complex with VCZ (PDB entries 5HS1 and 5EAH) is different from that when other azole drugs, such as FLC and ITC, are bound (PDB entries 4WMZ and 5EQB). The binding of VCZ isolates the active site from the SEC and creates a new channel that reaches the cytosol. In ScLDM Y140F the VCZ is located 0.5 Å closer to helix H, and M509 adopts the conformation found in crystal structures with other azole drugs. The water-mediated hydrogen bond between VCZ and M509 seen in the wild-type enzyme structure is lost and the SEC has access to the active site.
We have used phylogenetic analysis to identify potential contact points for a pharmacophore that will help identify inhibitors specific for the LDMs of ascomycete and basidiomycete fungal pathogens. Only 6 of the ∼48 residues within the LDM LBP (Y72, L147, F241, M313, H381, and S382 in S. cerevisiae) are absolutely conserved among ascomycete and basidiomycete fungal pathogens of humans and at least two of the major fungal pathogens of plants (Table 3, Fig. 3B, and Fig. S4). In addition, these residues are not found in the sterol demethylases of their hosts, and they are not subject to known mutations that confer azole resistance. For example, residues equivalent to ScLDM H381 and S382 have been variously implicated in the binding of azole drugs, such as FLC, VCZ, PCZ, ITC, VT-1161, and VT-1598 (9, 10, 14, 18). The fungal phylum Zygomycota has LDMs more closely related to the animal and plant enzymes than the ascomycetes and basidiomycetes. It includes some common pathogens of horticultural crops and rare but emerging pathogens of humans. In the two zygomycete LDM paralogs F1 and F5, the residues aligned to ScLDM Y72, L147, M313, and H381 are replaced with F, while those aligned to S382 are replaced with an N or Q (29). Despite this limitation, which does not significantly affect the two groupings contributing polarity or charge to the LBP, the crystal structures obtained for the full-length C. glabrata, C. albicans, and S. cerevisiae LDM provide important tools for structure-directed antifungal discovery that can be extended to most known fungal pathogens of humans, animals, and plants.
Future prospects.
This report provides structure-function insight needed to inform the discovery of broad-spectrum fungicides that will more effectively target fungal LDMs, not be prone to LDM-mediated drug resistance, and avoid targeting of host drug-metabolizing enzymes.
In particular, the available crystal structures show the catalytic domains and, hence, LBPs of fungal LDMs to be relatively rigid, a valuable attribute for modeling and drug discovery. Our target database of LDMs can now be expanded by using the crystal structures as templates for homology models of the LDMs of other fungal pathogens of interest. The crystal structures and the models, which can include important details such as key water molecules in the LBPs, are being applied in virtual screens using ligand-based pharmacophores or in docking experiments designed to identify compounds and drug-like fragments likely to bind in the LBP of fungal but not host LDMs. An important focus is to identify highly potent, fungus-specific LDM inhibitors, preferably with low-nanomolar affinities, and especially compounds likely to avoid the impact of known mutations, such as those described in this report. Testing of the most highly ranked virtual hits in S. cerevisiae-based phenotypic and biochemical screens, followed by measurements of the susceptibility of clinical isolates, provide the tools to identify scaffolds for broad-spectrum azole and non-azole antifungals that specifically target the LDMs of fungal pathogens.
MATERIALS AND METHODS
Yeast strains and culture media.
The yeast strains used in the present study are shown in Table S2 in the supplemental material. Yeast strains were grown on YPD medium, containing 1% (wt/vol) Bacto-yeast extract (BD Difco Laboratories Inc., Franklin Lakes, NJ), 2% (wt/vol) Bacto-peptone (BD Difco), and 2% (wt/vol) glucose. Synthetic defined (SD) medium was used for selection of transformants expressing CgLDM6×His or CaLDM6×His. It contained 2% (wt/vol) glucose, 0.67% (wt/vol) yeast nitrogen base without amino acids (BD Difco), 1.8% (wt/vol) agar (Oxoid Ltd., Hampshire, UK), and either uracil dropout (Qbiogene, Irvine, CA) or histidine dropout (Formedium, Norfolk, UK) complete supplement mixture. Liquid SD media with complete supplement mixture (Formedium), containing 10 mM morpholineethanesulfonic acid and 20 mM HEPES buffered with Tris to pH 6.8, was used for MIC80 determinations.
Materials.
Desalted oligonucleotides (Table S3), FLC, ITC, VCZ, PCZ, amphotericin B, and lanosterol were purchased from Sigma-Aldrich, Ltd. (St. Louis, MO). VT-1161 was prepared by MicroCombiChem (Wiesbaden, Germany) using the methods described by Hoekstra et al. (30). Micafungin was supplied by Sapphire Bioscience (Redfern, NSW, Australia). Colony PCRs were carried out using TaKaRa DNA polymerase (TaKaRa Bio Inc., Shiga, Japan). All other PCRs were performed using KOD Hot Start DNA polymerase (Novagen, Madison, WI). PCR cleanup and DNA gel extraction were carried out using kits from Qiagen, Pty. Ltd. (Limburg, Netherlands). Genomic DNA from yeast was isolated using the Y-DER kit from Thermo Fisher (Waltham, MA). Yeast DNA transformation was carried out using an Alkali Cation yeast transformation kit from Qbiogene (Irvine, CA). DNA transformation cassettes and genes inserted at the S. cerevisiae PDR5 or ERG11 locus were confirmed by DNA sequence analysis, performed at the Genetic Analysis Services facility (University of Otago, Dunedin, New Zealand). The presence of recombinant protein was verified by mass spectrometry using an LTQ Orbitrap XL hybrid ion trap-Orbitrap mass spectrometer (Thermo Fischer Scientific, New Zealand) at the Centre for Protein Research (University of Otago, Dunedin, New Zealand).
Construction of recombinant strains.
The S. cerevisiae ADΔ and AD2Δ hosts (8, 10, 31) were used to create the strains used in this study. The host ADΔ has the URA3 ORF deleted, while AD2Δ also has the HIS1 ORF deleted. Both host strains are deleted of 7 pleiotropic drug resistance (PDR) ABC transporters and the PDR3 transcriptional regulator but include the mutant pdr1-3 transcriptional regulator that drives constitutive expression from the PDR5 locus (29). Strains of S. cerevisiae that express full-length recombinant LDM6×His (Table S2) were constructed using transformation cassettes that encode C-terminal hexahistidine (6×His)-tagged ORFs of C. albicans or C. glabrata ERG11 (alignment of primary sequences is shown in Fig. S1), together with a PGK transcription terminator and the URA3 selection marker downstream. The transformation cassettes were transferred by homologous recombination into the PDR5 locus of the S. cerevisiae host strains. Two open reading frames of C. albicans ERG11 were tested. Strain Y2372 had the native ERG11 ORF, while strain Y2473 had the triplet 787CTG, encoding serine in C. albicans but leucine in S. cerevisiae, modified to 787AGC to encode serine in S. cerevisiae. Both ORFs gave the same physiological responses when expressed in S. cerevisiae, and results obtained using the latter ORF only are reported in the present study. The ORFs inserted into the PDR5 locus or HIS1, used to disrupt the native ERG11 locus, together with flanking sequences, were confirmed by DNA sequence analysis. The genomic DNA sequence of each transformant used in the present study was confirmed from at least 720 nucleotides upstream to 4 nucleotides downstream of the recombinant ERG11 ORFs.
Enzyme purification.
CaLDM6×His from strain Y2473 or CgLDM6×His from strain Y2374 was purified by Ni-NTA affinity and size exclusion chromatography essentially as described by Monk et al. (8). Recombinant enzymes were extracted from crude membranes using 17 mM n-decyl-β-d-maltoside (DM; 10× critical micelle concentration) and ultracentrifugation. Samples for crystallography were partially purified by Ni-NTA chromatography using imidazole as the eluent and then purified to near homogeneity by size exclusion chromatography using a Superdex 200 10/300 column in the presence of 150 mM NaCl, 6.8 mM DM (4× cmc), and 50 mM HEPES, pH 8.0 (SEC buffer), at 8°C. The protein peak was pooled and concentrated to ≥20 mg/ml by centrifugal filtration in SEC buffer containing 40 μM ITC using 50-kDa Amicon Ultra-4 filters (Sigma).
X-ray crystallography.
A hanging-drop vapor-diffusion method was used to crystallize both CaLDM6×His and CgLDM6×His. The reservoir solution contained 20 to 50% polyethylene glycol 400 (PEG-400) in 500 mM glycine-NaOH and 30 to 40% PEG-400 in 500 mM glycine-NaOH, respectively, at a pH range of 8 to 9.5. Crystals were obtained using 4-μl drops comprised of 2 μl of the concentrated LDM6×His preparation (20 mg/ml) together with 2 μl of reservoir solution. After 3 to 4 weeks of incubation at 18°C, CgLDM formed shallow boat-shaped crystals in diffusion against 40% PEG-400 at pH 9.0, and CaLDM gave thin rhombohedron-shaped crystals in diffusion against 35% PEG-400 at pH 8.8. The crystals were flash-cooled in liquid nitrogen prior to data collection. Data sets were collected on the MX2 beamline (ADSC Quantum 315r detector) at the Australian Synchrotron (Melbourne, Australia), with crystals kept frozen under a cryostream at −180°C. Indexing and integration of data were done using iMosflm (32) and scaling with SCALA (33). Phaser-MR (34) from Phenix was used to carry out molecular replacement using ScLDM6×His in complex with lanosterol (PDB entry 4LXJ) as a template (8). Refinement and modeling were performed using phenix.refine (35) and Coot (36). Water molecules were added if at least one hydrogen bond was detected (2.5 to 3.3 Å), and the inhibitors were modeled into the appropriate density in the active site and substrate channel. The crystallographic information files (.cif) for triazole inhibitors were obtained from the Grade Global Phasing online tool (Global Phasing Ltd., Cambridge, UK). Structures were visualized in Coot (34) and PyMOL (Schrodinger LLC).
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by grants to B.C.M. from the Marsden Fund of the Royal Society of New Zealand and the Health Research Council of New Zealand. E. Lamping is acknowledged for creating S. cerevisiae strain Y2411, used in the present study, and Y1030, the parent strain for Y2374.
We have no conflicts of interest to declare.
M.V.K. carried out genetic manipulations, determined phenotypes of recombinant organisms, purified the enzymes, set up crystal trials, and edited the manuscript. M.S. and R.J.W. purified enzymes and contributed to crystal trials and structural resolution. M.A.W. carried out genetic manipulations. A.A.S. assisted with structure determination and edited the manuscript. J.D.A.T. assisted with structure determination and building and assessment of structural models and edited the manuscript. B.C.M. developed and directed the project, obtained funding, interpreted data, and wrote the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01134-18.
REFERENCES
- 1.Lepesheva GI, Hargrove TY, Kleshchenko Y, Nes WD, Villalta F, Waterman MR. 2008. CYP51: a major drug target in the cytochrome P450 superfamily. Lipids 43:1117–1125. doi: 10.1007/s11745-008-3225-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Becher R, Wirsel SG. 2012. Fungal cytochrome P450 sterol 14alpha-demethylase (CYP51) and azole resistance in plant and human pathogens. Appl Microbiol Biotechnol 95:825–840. doi: 10.1007/s00253-012-4195-9. [DOI] [PubMed] [Google Scholar]
- 3.Morio F, Loge C, Besse B, Hennequin C, Le Pape P. 2010. Screening for amino acid substitutions in the Candida albicans Erg11 protein of azole-susceptible and azole-resistant clinical isolates: new substitutions and a review of the literature. Diagn Microbiol Infect Dis 66:373–384. doi: 10.1016/j.diagmicrobio.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 4.Flowers SA, Colon B, Whaley SG, Schuler MA, Rogers PD. 2015. Contribution of clinically derived mutations in ERG11 to azole resistance in Candida albicans. Antimicrob Agents Chemother 59:450–460. doi: 10.1128/AAC.03470-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snelders E, Huis In 't Veld RA, Rijs AJ, Kema GH, Melchers WJ, Verweij PE. 2009. Possible environmental origin of resistance of Aspergillus fumigatus to medical triazoles. Appl Environ Microbiol 75:4053–4057. doi: 10.1128/AEM.00231-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snelders E, van der Lee HA, Kuijpers J, Rijs AJ, Varga J, Samson RA, Mellado E, Donders AR, Melchers WJ, Verweij PE. 2008. Emergence of azole resistance in Aspergillus fumigatus and spread of a single resistance mechanism. PLoS Med 5:e219. doi: 10.1371/journal.pmed.0050219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berger S, El Chazli Y, Babu AF, Coste AT. 2017. Azole resistance in Aspergillus fumigatus: a consequence of antifungal use in agriculture? Front Microbiol 8:1024. doi: 10.3389/fmicb.2017.01024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monk BC, Tomasiak TM, Keniya MV, Huschmann FU, Tyndall JD, O'Connell JD III, Cannon RD, McDonald JG, Rodriguez A, Finer-Moore JS, Stroud RM. 2014. Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc Natl Acad Sci U S A 111:3865–3870. doi: 10.1073/pnas.1324245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sagatova AA, Keniya MV, Wilson RK, Monk BC, Tyndall JD. 2015. Structural insights into binding of the antifungal drug fluconazole to Saccharomyces cerevisiae lanosterol 14alpha-demethylase. Antimicrob Agents Chemother 59:4982–4989. doi: 10.1128/AAC.00925-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sagatova AA, Keniya MV, Wilson RK, Sabherwal M, Tyndall JD, Monk BC. 2016. Triazole resistance mediated by mutations of a conserved active site tyrosine in fungal lanosterol 14alpha-demethylase. Sci Rep 6:26213. doi: 10.1038/srep26213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyndall JD, Sabherwal M, Sagatova AA, Keniya MV, Negroni J, Wilson RK, Woods MA, Tietjen K, Monk BC. 2016. Structural and functional elucidation of yeast lanosterol 14alpha-demethylase in complex with agrochemical antifungals. PLoS One 11:e0167485. doi: 10.1371/journal.pone.0167485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sagatova AA, Keniya MV, Tyndall JDA, Monk BC. 2017. The impact of homologous resistance mutations from pathogenic yeast on Saccharomyces cerevisiae lanosterol 14alpha-demethylase. Antimicrob Agents Chemother 62:e02242-17. doi: 10.1128/AAC.02242-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hargrove TY, Wawrzak Z, Lamb DC, Guengerich FP, Lepesheva GI. 2015. Structure-functional characterization of cytochrome P450 sterol 14alpha-demethylase (CYP51B) from Aspergillus fumigatus and molecular basis for the development of antifungal drugs. J Biol Chem 290:23916–23934. doi: 10.1074/jbc.M115.677310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hargrove TY, Friggeri L, Wawrzak Z, Qi A, Hoekstra WJ, Schotzinger RJ, York JD, Guengerich FP, Lepesheva GI. 2017. Structural analyses of Candida albicans sterol 14alpha-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J Biol Chem 292:6728–6743. doi: 10.1074/jbc.M117.778308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoekstra WJ, Hargrove TY, Wawrzak Z, da Gama Jaen Batista D, da Silva CF, Nefertiti AS, Rachakonda G, Schotzinger RJ, Villalta F, Soeiro Mde N, Lepesheva GI. 2016. Clinical candidate VT-1161's antiparasitic effect in vitro, activity in a murine model of Chagas disease, and structural characterization in complex with the target enzyme CYP51 from Trypanosoma cruzi. Antimicrob Agents Chemother 60:1058–1066. doi: 10.1128/AAC.02287-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warrilow AG, Hull CM, Parker JE, Garvey EP, Hoekstra WJ, Moore WR, Schotzinger RJ, Kelly DE, Kelly SL. 2014. The clinical candidate VT-1161 is a highly potent inhibitor of Candida albicans CYP51 but fails to bind the human enzyme. Antimicrob Agents Chemother 58:7121–7127. doi: 10.1128/AAC.03707-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warrilow AG, Parker JE, Price CL, Nes WD, Garvey EP, Hoekstra WJ, Schotzinger RJ, Kelly DE, Kelly SL. 2016. The investigational drug VT-1129 is a highly potent inhibitor of Cryptococcus species CYP51 but only weakly inhibits the human enzyme. Antimicrob Agents Chemother 60:4530–4538. doi: 10.1128/AAC.00349-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hargrove TY, Garvey EP, Hoekstra WJ, Yates CM, Wawrzak Z, Rachakonda G, Villalta F, Lepesheva GI. 2017. Crystal structure of the new investigational drug candidate VT-1598 in complex with Aspergillus fumigatus sterol 14alpha-demethylase provides insights into its broad-spectrum antifungal activity. Antimicrob Agents Chemother 61:e00570-17. doi: 10.1128/AAC.00570-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keniya MV, Ruma YN, Tyndall JDA, Monk BC. 2018. Heterologous expression of full-length lanosterol 14α-demethylases of prominent fungal pathogens Candida albicans and Candida glabrata provides tools for antifungal discovery. Antimicrob Agents Chemother 62:e01131-18. doi: 10.1128/AAC.01131-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warrilow AG, Melo N, Martel CM, Parker JE, Nes WD, Kelly SL, Kelly DE. 2010. Expression, purification, and characterization of Aspergillus fumigatus sterol 14-alpha demethylase (CYP51) isoenzymes A and B. Antimicrob Agents Chemother 54:4225–4234. doi: 10.1128/AAC.00316-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu X, Nandekar P, Mustafa G, Cojocaru V, Lepesheva GI, Wade RC. 2016. Ligand tunnels in T. brucei and human CYP51: insights for parasite-specific drug design. Biochim Biophys Acta 1860:67–78. doi: 10.1016/j.bbagen.2015.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lepesheva GI, Waterman MR. 2011. Structural basis for conservation in the CYP51 family. Biochim Biophys Acta 1814:88–93. doi: 10.1016/j.bbapap.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parker JE, Warrilow AG, Price CL, Mullins JG, Kelly DE, Kelly SL. 2014. Resistance to antifungals that target CYP51. J Chem Biol 7:143–161. doi: 10.1007/s12154-014-0121-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanglard D, Ischer F, Koymans L, Bille J. 1998. Amino acid substitutions in the cytochrome P-450 lanosterol 14alpha-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob Agents Chemother 42:241–253. doi: 10.1093/jac/42.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiang MJ, Liu JY, Ni PH, Wang S, Shi C, Wei B, Ni YX, Ge HL. 2013. Erg11 mutations associated with azole resistance in clinical isolates of Candida albicans. FEMS Yeast Res 13:386–393. doi: 10.1111/1567-1364.12042. [DOI] [PubMed] [Google Scholar]
- 26.Chau AS, Mendrick CA, Sabatelli FJ, Loebenberg D, McNicholas PM. 2004. Application of real-time quantitative PCR to molecular analysis of Candida albicans strains exhibiting reduced susceptibility to azoles. Antimicrob Agents Chemother 48:2124–2131. doi: 10.1128/AAC.48.6.2124-2131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morschhauser J. 2002. The genetic basis of fluconazole resistance development in Candida albicans. Biochim Biophys Acta 1587:240–248. doi: 10.1016/S0925-4439(02)00087-X. [DOI] [PubMed] [Google Scholar]
- 28.Goldman GH, da Silva Ferreira ME, dos Reis Marques E, Savoldi M, Perlin D, Park S, Godoy Martinez PC, Goldman MH, Colombo AL. 2004. Evaluation of fluconazole resistance mechanisms in Candida albicans clinical isolates from HIV-infected patients in Brazil. Diagn Microbiol Infect Dis 50:25–32. doi: 10.1016/j.diagmicrobio.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 29.Caramalho R, Tyndall JDA, Monk BC, Larentis T, Lass-Florl C, Lackner M. 2017. Intrinsic short-tailed azole resistance in mucormycetes is due to an evolutionary conserved amino acid substitution of the lanosterol 14alpha-demethylase. Sci Rep 7:15898. doi: 10.1038/s41598-017-16123-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoekstra WJ, Garvey EP, Moore WR, Rafferty SW, Yates CM, Schotzinger RJ. 2014. Design and optimization of highly-selective fungal CYP51 inhibitors. Bioorg Med Chem Lett 24:3455–3458. doi: 10.1016/j.bmcl.2014.05.068. [DOI] [PubMed] [Google Scholar]
- 31.Lamping E, Monk BC, Niimi K, Holmes AR, Tsao S, Tanabe K, Niimi M, Uehara Y, Cannon RD. 2007. Characterization of three classes of membrane proteins involved in fungal azole resistance by functional hyperexpression in Saccharomyces cerevisiae. Eukaryot Cell 6:1150–1165. doi: 10.1128/EC.00091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans P. 2006. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 34.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.