

Antiretroviral therapy (ART) does not cure HIV-1 infection due to the persistence of proviruses in long-lived resting T cells. Strategies targeting these latently infected cells will be necessary to eradicate HIV-1 in infected individuals.
KEYWORDS: HIV, HIV latency, HIV persistence, PKC agonist, ingenols
ABSTRACT
Antiretroviral therapy (ART) does not cure HIV-1 infection due to the persistence of proviruses in long-lived resting T cells. Strategies targeting these latently infected cells will be necessary to eradicate HIV-1 in infected individuals. Protein kinase C (PKC) activation is an effective mechanism to reactivate latent proviruses and allows for recognition and clearance of infected cells by the immune system. Several ingenol compounds, naturally occurring PKC agonists, have been described to have potent latency reversal activity. We sought to optimize this activity by synthesizing a library of novel ingenols via esterification of the C-3 hydroxyl group of the ingenol core, which itself is inactive for latency reversal. Newly synthesized ingenol derivatives were evaluated for latency reversal activity, cellular activation, and cytotoxicity alongside commercially available ingenols (ingenol-3,20-dibenzoate, ingenol 3-hexanoate, and ingenol-3-angelate) in HIV latency cell lines and resting CD4+ T cells from aviremic participants. Among the synthetic ingenols that we produced, we identified several compounds that demonstrate high efficacy and represent promising leads as latency reversal agents for HIV-1 eradication.
INTRODUCTION
Durable blockade of viral replication by combination antiretroviral therapy (ART) has transformed HIV-1 infection from a lethal condition characterized by progressive immune deficiency into a manageable illness (1–4). However, long-term ART does not result in HIV-1 eradication due the persistence of inducible proviruses in long-lived memory CD4+ T cells (5–7). Stochastic or antigen-driven activation of latently infected CD4+ T cells induces viral transcription and will rekindle active replication and disease progression if ART is stopped. Latently infected cells are not efficiently recognized by the immune system and do not decay significantly during the life span of a person living with HIV-1 (8–10). Targeting this reservoir for elimination is an essential component of HIV-1 cure strategies.
Early attempts at depleting the latent reservoir in vivo made use of a strategy of global T cell activation, which was ineffective and toxic (11–13). Subsequent strategies have utilized compounds identified in vitro as potential latency reversal agents (LRAs) that induce proviral transcription while avoiding T cell activation. Histone deacetylase (HDAC) inhibitors have been tested in several pilot eradication trials (14), as they appeared in vitro to offer an acceptable balance between proviral transcriptional activation and minimal cellular activation. The FDA-approved drug disulfiram, demonstrated to reactivate latent HIV-1 in in vitro latency models, has also been well tolerated in vivo (15, 16). However, none of these trials have demonstrated significant reservoir depletion in vivo with these LRAs (17–26).
Protein kinase C (PKC) agonists represent a promising alternative mechanism for latency reversal, as they have long been known for their ability to induce HIV-1 transcription in vitro (27–29). PKC enzymes are serine/threonine kinases that are activated by the second messenger diacylglycerol (DAG) (30). PKC agonists mimic DAG, binding to one or more intracellular PKC isoforms to initiate downstream signaling. Activated PKC isoforms phosphorylate (and destabilize) IκB, which then releases RelA, the p65 subunit of NF-κB. NF-κB can then enter the nucleus and bind to cognate binding sites in the proviral long terminal repeat (LTR), which initiates viral transcription. The role of targeting PKC-NF-κB signaling as a means to reactivate latent HIV-1 has been reviewed in detail (30, 31). Several groups have reported latency reversal using protein kinase C (PKC) agonists that far exceed what can be achieved with an HDAC inhibitor (HDACi) or disulfiram (32–35). PKC agonists can induce T cell activation, and the potential for adverse effects related to immune activation has limited their clinical development to date. The only clinical trial making use of a PKC agonist for HIV-1 eradication reported no adverse effects due to bryostatin-1 (36). However, the investigators used doses of bryostatin-1 that did not achieve detectable systemic concentrations in a majority of trial participants, and latency reversal was not observed.
Ingenols are naturally occurring diterpene compounds originally isolated from members of Euphorbia, a family of flowering plants. Euphorbia species have been integral components of traditional medicine practices across many cultures for millennia (37). Semisynthetic ingenols have been engineered in order to optimize their latency reversal activity (38, 39). Ingenol-3-mebutate (also known as ingenol-3-angelate) is FDA approved as a topical therapy for actinic keratosis (40) and has demonstrated efficacy in multiple HIV-1 latency systems (41, 42). Ingenol-3-hexanoate, also known as ingenol B, has recently been administered to nonhuman primates in combination with vorinostat (43). One of the two rhesus macaques exposed to ingenol B and vorinostat demonstrated increased simian immunodeficiency virus (SIV) loads in both the central nervous system (CNS) and the periphery in response to LRA treatment, and also developed markers of systemic and CNS inflammation.
We hypothesized that rational design of a library of novel ingenol derivatives would allow us to identify compounds able to maximize viral reactivation. Ingenols that have been described to reverse proviral latency contain lipophilic moieties at the C-3 alcohol on the ingenol core compound, which itself is inactive for latency reversal (34, 38, 39, 44). We therefore synthesized a library of novel ingenols via esterification of the C-3 alcohol on the ingenol core compound. Newly synthesized ingenol derivatives were then evaluated for latency reversal activity, cellular activation, and cytotoxicity alongside commercially available ingenols (ingenol-3,20-dibenzoate, ingenol-3-hexanoate, and ingenol-3-angelate) in cell lines and resting CD4+ T cells from aviremic participants ex vivo.
RESULTS
A focused library of C-3-modified ingenols was prepared in collaboration with the University of Utah Synthetic and Medicinal Chemistry Core facility. The unmodified ingenol core molecule does not induce latency reversal in cellular systems (34, 38) and therefore represents an ideal chemical scaffold devoid of background activity. Latency reversal efficacy of ingenol derivatives can therefore be attributed to chemical modifications made to the inactive ingenol core. C-3 modifications were selected to probe and expand the structure-activity relationship based upon commercially available ingenol compounds whose latency reversal potential has been previously described (34, 38, 39, 44). We hypothesized that the nature (saturated versus unsaturated, rigid versus flexible, and cyclic versus linear) along with the length of the hydrophobic side chains would confer various degrees of potency to the resulting compounds. We did not know a priori which configurations would be most optimal for conferring latency reactivation. For initial tests of activity of the novel ingenol derivatives, we chose the Jurkat T cell line (J-Lat 10.6) and observed a broad range of activity after 24 h of exposure.
Straight-chain esters were designed to evaluate the effect of both the length and saturation of the carbon chain (Fig. 1a), in comparison to ingenol B, also known as ingenol-3-hexanoate, which has been shown to reverse latency in multiple in vitro models (31, 34, 38), as well as in a nonhuman primate model of HIV latency (43). Among saturated linear esters, we observed improved latency reversal activity with increasing ester carbon length (Fig. 1b). Ingenol-3-hexanoate (Ing B) and ingenol-3-octanoate (Ing 3-O) reversed latency at lower concentrations than ingenol-3-acetate (Ing 3-A) and ingenol-3-butyrate (Ing 3-B). However, all four of the above-mentioned compounds were outperformed by ingenol-3-mono-2-octene (Ing 3-T), which contains an α,β-unsaturated ester. On the other hand, the presence of additional γ,δ-unsaturation (Ing 3-D) weakened latency reversal. The presence of a polar group or an amine within a straight ester chain also diminished activity (comparing Ing B with Ing 3-E and Ing 3-N).
FIG 1.

Latency reversal activity of straight-chain saturated and unsaturated ingenol-C-3-esters. Linear esters were added to the 3-carbon position of the ingenol core molecule (full and abbreviated nomenclature shown in panel A, along with ingenol core structure in black and esters in color), with the exception of ingenol-3-hexanoate, which was obtained from the UNC Care Collaboratory Pharmacology Core. Straight-chain ingenol-3-esters were tested for latency reversal efficacy using a latent HIV-1 cell line (J-Lat 10.6) containing a full-length integrated HIV-1 provirus that expresses green fluorescent protein (GFP) upon proviral transcription. Jurkat cells were exposed to compounds across a dose range from 1 nM to 3,000 nM. After 24-hour exposure, flow cytometry was performed to quantify GFP-positive cells, which was compared to positive control (PMA stimulation) to obtain percent maximal HIV-1 reactivation. (B) Fitted dose-response curves with median and 95% confidence intervals are based on five independent in vitro experiments.
α- or β-Branched esters were modeled on the structure of ingenol-3-angelate (Ing M), an FDA-approved compound for the topical treatment of actinic keratosis (40), which is highly efficacious in vitro in reversing HIV latency alone and in combination with other LRAs (41) (Fig. 2a). Ingenol-3-acrylate (Ing 3-R) resulted in latency reversal at significantly lower concentrations than Ing M (50% effective concentration [EC50], 58 nM for Ing 3-R compared to 211 nM for Ing M) despite their structural similarity. The efficacy of Ing 3-R led us to pursue acrylate diesterification at C-3 and C-20 (Ing D-R) and tri-esterification at C-3, C-5, and C-20 (Ing T-R). Interestingly, these additional ester groups resulted in abrogation of latency reversal activity (Fig. 2b).
FIG 2.

Latency reversal activity of branched ingenol-C-3-esters. Branched esters were added to the 3-carbon position of the ingenol core molecule (structures, full and abbreviated nomenclature shown in panel A), with the exception of ingenol-3-angelate (Ing M), which was commercially available. As in Fig. 1, branched ingenol-3-esters were tested for latency reversal efficacy using a latent HIV-1 cell line (J-Lat 10.6) containing a full-length integrated HIV-1 provirus that expresses GFP upon proviral transcription. Compounds were tested across a dose range from 1 nM to 3,000 nM. After 24 h of exposure, flow cytometry was performed to quantify GFP-positive cells, which was compared to positive control (PMA stimulation) to obtain percent maximal HIV-1 reactivation. (B) Fitted dose-response curves with median and 95% confidence intervals are based on five independent in vitro experiments. Based on the efficacy of Ing 3-R, ingenol di- and triacrylate compounds were synthesized (Ing D-R and T-R). The addition of these ester groups abolished all latency reactivation activity.
A variety of aromatic and carbocyclic ingenol-C-3-esters were evaluated alongside ingenol dibenzoate (Ing DB), which contains an aromatic ring at C-3 and has previously shown to be efficacious at reversing latency in resting CD4+ T cells from aviremic participants (44) (Fig. 3a). Surprisingly, the two monoesters, Ing 3-X and Ing 3-P, which contain a carbocycle, were significantly more efficacious than Ing DB (Fig. 3b). The five most efficacious ingenols (Ing 3-R, Ing 3-P, Ing 3-X, Ing 3-T, and Ing M) spanned the three structural classes. α,β-Unsaturation is a shared feature of four of these five top synthetic ingenols but is not requisite for activity, as demonstrated by the comparable potency of Ing 3-X.
FIG 3.

Latency reversal activity of aromatic and carbocyclic ingenol-C-3-esters. Aromatic and carbocyclic esters were added to the 3-carbon position of the ingenol core molecule (structures, full and abbreviated nomenclature shown in panel A), with the exception of ingenol-3,20-dibenzoate, which was commercially available. Aromatic and carbocyclic ingenol-3-esters were tested for latency reversal efficacy using a latent HIV-1 cell line (J-Lat 10.6) containing a full-length integrated HIV-1 provirus that expresses GFP upon proviral transcription. Compounds were tested across a dose range from 1 nM to 3,000 nM. After 24 h of exposure, flow cytometry was performed to quantify GFP-positive cells, which was compared to positive control (PMA stimulation) to obtain percent maximal HIV-1 reactivation. (B) Fitted dose-response curves with median and 95% confidence intervals are shown based on five independent in vitro experiments.
In order to ensure that PKC activation is the predominant mechanism of latency reversal of these ingenol compounds, we evaluated for latency reversal in the presence or absence of known PKC inhibitors. The pan-PKC inhibitor Gö6983 significantly decreased latency reversal induced by highly active ingenol derivatives Ing 3-R and Ing 3-X in resting CD4+ T cells from aviremic participants, establishing that these ingenol compounds are acting through PKC to induce latency reversal (Fig. S1a and b in the supplemental material). We next investigated the activities of unique PKC isoforms in the presence of ingenol derivatives using resting CD4+ T cells from HIV-1-uninfected participants to determine whether the efficacy of ingenol derivatives could be attributed to specific PKC isoforms (45) (Fig. 4 and S2). PKC phosphorylation was quantified by flow cytometry after 30 min of exposure to ingenols, bryostatin-1, 12-myristate 13-acetate (PMA), or no compound (negative control). Ingenols with minimal latency reversal activity (Ing core and Ing 3-A) showed no significant phosphorylation of any PKC isoform. The highly active ingenols Ing M, Ing 3-R, and Ing 3-X significantly increased the phosphorylation of PKC isoforms PKCβ, PKCδ, PKCθ, and protein kinase D (PKD) above baseline. Interestingly, this PKC isoform activation pattern did not significantly differ between structurally distinct ingenols, nor did the PKC isoform activation profile distinguish these ingenols from chemically distinct PKC agonists bryostatin-1 and PMA (Fig. 4 and S2). This suggests a common pathway of PKC activation resulting in latency reversal that is independent of the chemical structure of the PKC agonist.
FIG 4.
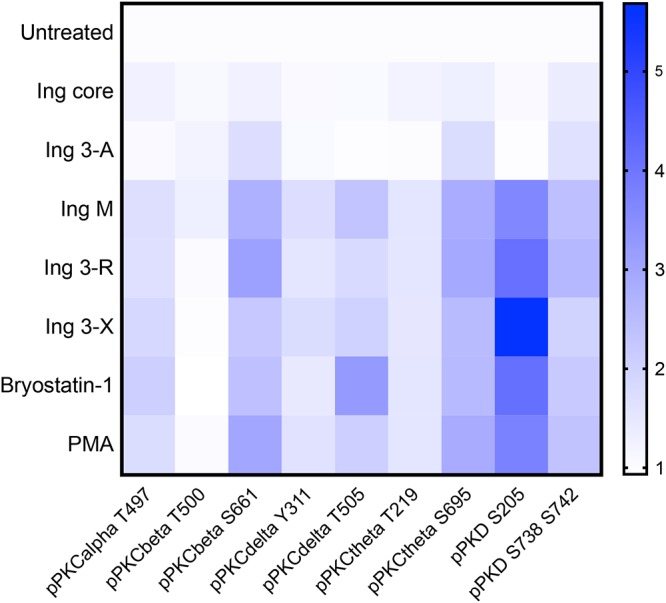
PKC isoform activation after ingenol exposure. Resting primary CD4 cells isolated from HIV-1-uninfected participants (n = 5) underwent a 30-min exposure to ingenol derivatives (100 nM), bryostatin-1 (100 nM), or PMA (10 ng/ml). Cells underwent intracellular staining with anti-pPKC antibodies and were analyzed by flow cytometry. Changes in PKC isoform phosphorylation were quantified by the fold change in mean fluorescence intensity compared to untreated (negative-control) cell cultures (increasing mean fluorescence intensity [MFI] fold change represented above as blue color scale). Ingenol core and Ing 3-A, which have little to no latency reversal activity, did not induce any PKC isoform phosphorylation in resting primary CD4 cells isolated from HIV-1-uninfected participants (n = 5). Highly active ingenol derivatives Ing M, Ing 3-R, and Ing 3-X significantly induced phosphorylation of PKC isoforms PKCβ, PKCδ, PKCθ, and PKD. This PKC isoform phosphorylation pattern did not significantly differ among these ingenols or structurally distinct PKC agonists PMA or bryostatin-1.
We next sought to test the PKC activity and cytotoxicity of synthetic ingenols in resting CD4+ T cells in vitro as a means to explore the therapeutic windows of these compounds. Cell surface expression of CD69, a biomarker of early T cell activation, is well known to directly correlate with PKC activity (46–48). Recent studies with bryostatin-1 and bryostatin analogs have demonstrated a direct correlation between CD69 expression and HIV-1 latency reversal in vitro (48, 49). We identified a similar positive correlation between the EC50 of synthetic ingenols for the induction of CD69 expression on primary resting CD4+ T cells and the EC50 for latency reversal in J-Lat 10.6 from experiments described above (Fig. 5). The cytotoxicity of synthetic ingenols was evaluated by quantifying changes in cell membrane integrity by flow cytometry. Resting CD4+ T cells from aviremic HIV-1-infected individuals (n = 3) were exposed to concentrations of synthetic ingenols ranging from 0.1 μM to 50 μM for 48 h in vitro. None of the ingenols induced significant declines in cell viability at concentrations between 0.1 μM and 10 μM. Three ingenol derivatives (3-T, 3-D, and T-R) induced significant decreases in cellular viability at 50 μM (Fig. S3a to c).
FIG 5.
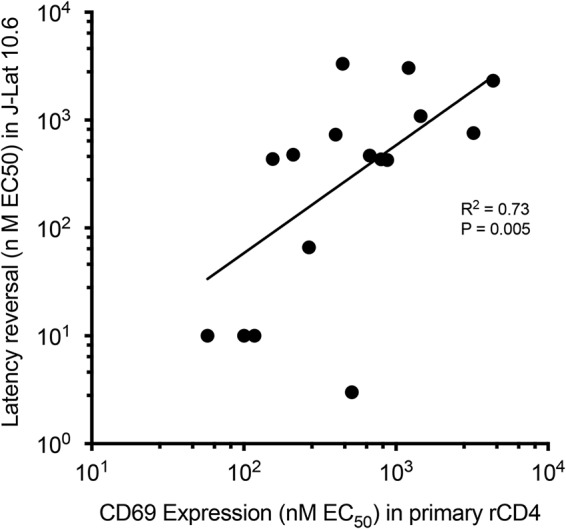
CD69 expression correlates with latency reversal activity. Using resting CD4+ T cells from aviremic HIV-1-positive participants, the percentage of cells expressing CD69 was measured after 48-h in vitro exposure to ingenol derivatives across a concentration range of 1 nM to 10,000 nM. Fitted dose-response curves from four independent experiments were used to calculate the EC50 of CD69 expression for each ingenol derivative. Each black circle represents a unique ingenol derivative whose location is determined by EC50 for CD69 expression in primary recombinant CD4 (rCD4) cells (x axis) and EC50 for latency reversal in J-Lat 10.6 (y axis; determined from experiments shown in Fig. 1 to 3). Linear regression analysis revealed a significant direct correlation between latency reversal activity and CD69 expression (R2 = 0.73, P = 0.005).
J-Lat 10.6 cells are a transformed T lymphocyte cell line and contain a monoclonal proviral integration site. In order to test ingenol in a biological system that would take into account polyclonal proviral integration sites and the genetic variability associated with donor-to-donor differences, we evaluated the relative latency reversal potency of eight active ingenol derivatives (selected based on latency reversal EC50 from J-Lat 10.6 experiments described above) using resting CD4+ T cells from aviremic individuals on ART (Fig. 6; see Table S1 for participant characteristics). Aliquots of between five and eight million resting CD4+ T cells were exposed to ingenol derivatives at 40 nM, antibodies to CD3 and CD28 (positive control), or medium alone (negative control). After 48 h in culture, cell-associated RNA was extracted and viral mRNA copies quantified via quantitative PCR (qPCR) for each culture condition (50). Small quantities of viral transcripts are often detected in medium-alone (negative) control cultures under these culture conditions. Therefore, ingenol efficacy was determined by comparing cell-associated viral transcripts from ingenol-exposed cells to negative-control cultures and expressed as a fold change over baseline.
FIG 6.

Ingenol derivatives reactivate latent HIV-1 in resting CD4+ T cells from aviremic HIV-1-positive participants ex vivo. The eight most efficacious ingenol derivatives based on calculated EC50s from J-Lat 10.6 experiments (Fig. 1 to 3) were evaluated for latency reversal using resting CD4+ T cells from aviremic HIV-1-positive participants (participant characteristics in Table S1). After a 48-h exposure (40 nM concentration for all ingenol derivatives), fold increases in cell-associated HIV-1 RNA compared to the medium-alone condition (negative control) were determined by qPCR. Using a nonparametric Mann-Whitney analysis, Ing M, Ing 3-X, and Ing 3-R all demonstrated statistically significant increases in cell-associated viral transcripts compared to medium alone (**, P < 0.005 for Ing M and Ing 3-X; ****, P < 0.0001 for Ing 3-R).
Ingenol M, 3-X, and 3-R significantly reversed proviral latency in cells from aviremic participants, determined by the increased fold change of cell-associated HIV-1 RNA compared to that under medium-alone conditions (P < 0.005 for Ing M and 3-X, P < 0.0001 for Ing 3-R). Ing 3-R, in keeping with the results of Jurkat cell line described above, demonstrated the highest latency reversal of any compound tested. Ing 3-R reversed latency more efficiently than ingenol 3-angelate (Ing M), despite the structural similarities of these two compounds (Fig. 7a). However, at 40 nM, latency reversal induced by Ing 3-R was indistinguishable from the positive control (T cell activation via CD3 and CD28 antibodies; Fig. 7b).
FIG 7.

Latency reversal of Ing M, Ing 3-R, and αCD3/CD28. (A) Despite structural similarities between Ing M and Ing 3-R, paired analysis of nine independent ex vivo experiments comparing fold change increases in cell-associated viral transcripts in resting CD4+ T cells from aviremic HIV-1-positive participants demonstrated a statistically significant difference favoring Ing 3-R (Wilcoxon matched-pairs signed-rank test, P < 0.005; n = 9). (B) The same paired analysis comparing fold change increases in cell-associated viral transcripts between Ing 3-R and positive control (αCD3/CD28) did not reveal any statistically significant difference between these conditions (Wilcoxon matched-pairs signed-rank test, P = 0.25; n = 9).
DISCUSSION
We created a small library of novel ingenol derivatives that act through the same pathway (protein kinase C activation) as previously described ingenol compounds, but they are significantly more potent with regard to viral reactivation in resting CD4+ T cells from aviremic participants on ART. While the ingenol core molecule is inactive with regard to latency reversal and CD69 activation, esterified ingenols demonstrated a wide range of activity in cell lines and participant cells ex vivo. The most potent of these compounds shared an α-β-unsaturated moiety adjacent to carbon-3, shedding light on the structure-function relationship of PKC activation. The most active of these ingenol analogs, ingenol-3-acrylate (Ing 3-R), is able to reactivate latent proviruses to the same degree as the positive control (T cell receptor stimulation with antibodies against CD3 and CD28), making this one of the most potent LRAs described to date. Given the modest results of clinical trials testing existing LRAs (51), these compounds represent exciting scaffolds as “next-generation” latency reversal agents.
PKC activation, while promising as a latency reversal strategy in vitro, presents a challenge with regard to the possibility of off-target effects that may narrow the in vivo therapeutic window of compounds acting through this mechanism. We and others have demonstrated that ingenols induce proinflammatory cytokine release from peripheral blood mononuclear cells (PBMCs) (50). Bryostatin-1 has advanced toward the clinic faster than any compound in this mechanistic class but has raised concerns in early stage clinical trials in cancer due to adverse effects linked to systemic inflammation (52, 53). While our PKC isoform experiments appear to demonstrate a common PKC activation profile among the ingenols tested, further exploration of PKC isoform specificity may allow for the development of PKC agonists that target isoforms responsible for latency reversal while minimally activating isoforms inducing proinflammatory cytokine release. Previous work using diacylglycerol (DAG) and phorbol as chemical scaffolds provides evidence that PKC-isoform-specific agonists can be engineered that uncouple proviral reactivation from in vitro production of tumor necrosis factor alpha (TNF-α) (54, 55). Future medicinal chemistry approaches in regard to our ingenol library will focus on PKC isoform specificity and will also take into account the instability of these compounds due to ester migration (56).
Previous comparisons of cell lines and primary cell models of HIV-1 latency have demonstrated discordance with regard to the efficacy of latency reversal agents (57). In this study, we found that the EC50 of ingenol derivatives in J-Lat 10.6 cells correlated well with latency reversal in primary resting CD4+ T cells from aviremic participants (Fig. S4). Four of the five ingenols with the lowest EC50s in J-Lat 10.6 cells were among the top five ingenols in primary cells. Similarly, ingenols with minimal activity in J-Lat experiments demonstrated no significant latency reversal in primary cells. Ultimately, the potential efficacy and toxicity of PKC activation as a latency reversal strategy cannot be fully addressed in vitro. Murine and nonhuman primate models are being employed to answer these critical translational questions (43, 48, 58). A clinical trial testing the safety and efficacy of Euphorbia kansui, a medicinal plant that contains 14 naturally occurring ingenols and has been shown to reactivate latent HIV-1 in vitro (47), is under way (ClinicalTrials.gov identifier NCT02531295). The concerns for a narrow therapeutic window and the potential for in vivo toxicity with existing PKC agonists, including bryostatin-1 and ingenol derivatives, have limited clinical trials of these agents (36). A natural product, such as E. kansui, that contains active ingenol derivatives (59) and has been part of traditional Chinese medicine for centuries may provide a novel strategy for safe in vivo administration of PKC agonists.
The most potent ingenol derivatives demonstrated latency reversal efficacy approaching that of the positive control (Fig. 6 and 7) after 48 h of exposure. Previous work has demonstrated that a single in vitro exposure to a strong stimulus, including antibodies to CD3 and CD28, does not efficiently reactivate all potentially inducible proviruses in culture (60–62). Repeated LRA exposures will likely be necessary to reactivate a sufficient fraction of inducible proviruses that in turn will allow for detectable perturbation of the latent reservoir in vivo. Given the potency of the ingenol derivatives described here, immediate next steps include exploration of the fractional reactivation of these compounds in comparison to other LRAs and αCD3/CD28. Several groups have recently described synergy between ingenols and the bromodomain inhibitor JQ1 (35, 41). We have also observed synergistic latency reversal between JQ1 and several ingenol derivatives in J-Lat 10.6 cells (data not shown) and plan to further evaluate this relationship using primary cells from aviremic participants ex vivo. A better understanding of the structure-activity relationship, PKC-isoform specificity, chemical stability, synergy with LRAs from complementary mechanistic classes, and in vivo toxicity of ingenols, including the derivatives we describe here, will help define a role for this promising class of compounds among viable strategies for HIV-1 eradication.
MATERIALS AND METHODS
Participants.
Aviremic HIV-1-infected participants on ART were recruited for phlebotomy according to an approved and active institutional review board (IRB) protocol at the University of Utah (IRB_0058246), as described previously (44). Inclusion criteria for this study required viral suppression (<50 HIV-1 RNA copies/ml) for a minimum of 6 months, ART initiation during chronic HIV-1 infection (greater than 6 months since seroconversion), and compliance with a stable ART regimen for a minimum of 12 months per participant and provider report. Informed consent and phlebotomy were performed in the Center for Clinical and Translational Science Clinical Services Core at the University of Utah Medical Center. Participant characteristics are provided in Table S1. Healthy donor peripheral blood mononuclear cells (PBMCs) were obtained via peripheral phlebotomy according to a separate approved and active IRB protocol at the University of Utah (IRB_0067637).
Reagents.
Ingenol core was purchased from StruChem (Suzhou, China). Ingenol 3,20-dibenzoate and ingenol 3-angelate were purchased from Santa Cruz Biotechnology. Ingenol 3-hexanoate was obtained from the Martin Delaney Collaboratory of AIDS Researchers for Eradication (CARE) Pharmacology Core, University of North Carolina, Chapel Hill, NC. PKC inhibitors were purchased from Cayman Chemical (Gö6976; CAS no. 136194-77-9) and Santa Cruz Biotechnology (Gö6983; CAS no. 133053-19-7). CD3/CD28 antibody-coated magnetic beads (Dynabeads human T-activator CD3/CD28 beads) were purchased from Thermo Fisher Technologies.
Chemical synthesis.
Ingenol was used as a scaffold to generate a focused library of synthetic derivatives in collaboration with the University of Utah Synthetic and Medicinal Chemistry Core Facility. Protection of the C-5 and C-20 alcohols as an acetonide permitted selective esterification of the C-3 alcohol, as previously reported (56). Subsequent hydrolysis of the acetonide yielded a library of ingenol-3-monoesters (shown in Fig. 1 to 3). Seventeen unnatural ingenol-C-3-esters were generated. Compound identities were established using 1H-nuclear magnetic resonance (1H-NMR) and 13C-NMR spectroscopy and purity via liquid chromatography-mass spectrometry (LC-MS). All compounds were >95% pure as judged by LC-MS prior to biological evaluation.
Cell isolation and culture.
The HIV-1-infected Jurkat T cell line (J-Lat 10.6) was obtained from the NIH AIDS Reagent Program (www.aidsreagent.org) and cultured in RPMI-based medium supplemented with 10% fetal calf serum. Human primary PBMCs were obtained via peripheral phlebotomy, as described above, and resting CD4+ T cells were isolated using a magnetic bead negative selection kit from StemCell Technologies (EasySep human CD4+ T cell isolation kit, catalog no. 17952), according to the manufacturer's instructions. Ex vivo cell culture assays with resting CD4+ T cells from HIV-1-infected aviremic participants were performed as previously described (44, 50). For PKC inhibition experiments, aliquots of five million resting CD4+ T cells were exposed to 300 nM PKC inhibitor Gö6976 or Gö6983 for 4 h, followed by exposure to 300 nM ingenol 3-R or 3-X for 48 h. Cell-associated RNA was isolated via TRIzol purification for quantitative PCR, as described below.
qPCR.
Quantitative PCR (qPCR) to detect HIV-1 mRNA transcripts was performed on cell-associated RNA (caRNA) isolated from cultured cells, as described previously (44, 50). Briefly, RNA isolation and purification were performed using TRIzol RNA isolation (Invitrogen) according to the manufacturer's instructions. First-step reverse transcriptase PCR was performed in a 20-μl reaction mixture containing 4× Quanta Biosciences qScript SuperMix (catalog no. 101414-106). The cycling parameters were 25°C for 5 min, followed by 42°C for 30 min and then 85°C for 5 min. Using cDNA obtained from the first-step PCR, second-step qPCR to detect HIV-1 mRNA transcripts was performed in triplicate 20-μl reactions using a TaqMan polymerase enzyme (Applied Biosystems TaqMan Universal master mix II with UNG, catalog no. 4440045; Thermo Fisher). The sequence of forward primer VQA Fwd P9501 is 5′-CAGATGCTGCATATAAGCAGCTG-3′, and that of the reverse primer VQA Rev 7T24 is 5′-TTTTTTTTTTTTTTTTTTTTTTTTGAAGCAC-3′; the 6-carboxyfluorescein (FAM) probe sequence is FAMCCTGTACTGGGTCTCTCTGGTBHQ1 (BHQ1, black hole quencher 1). The cycling parameters were one cycle at 50°C for 2 min, followed by one cycle at 95°C for 10 min, followed by 55 cycles of 95°C for 15 s and 60°C for 1 min.
Flow cytometry for GFP detection and cell viability.
After in vitro culture, J-Lat cells and primary CD4+ T cells were washed with 1× phosphate-buffered saline (PBS) prior to staining with 0.1 μl fixable viability dye eFluor 450 (eBioscience catalog no. 65-0863-14; Thermo Fisher) per 105 cells for 30 min at 4°C. Primary CD4+ T cells were stained with antibodies against CD69 conjugated to an antigen-presenting cell (APC) fluorophore (APC anti-human CD69 antibody; BioLegend). Cells were then washed and resuspended in 1× PBS prior to flow cytometry acquisition evaluating for cellular viability, green florescent protein (GFP) expression (J-Lat 10.6), or CD69 expression (primary CD4+ T cells). Flow cytometry was performed with a BD FACSCelesta flow cytometer with FACSDiva acquisition software (Becton Dickinson, Mountain View, CA) prior to analysis with FlowJo (TreeStar, Inc., Ashland, OR).
Flow cytometry for PKC isoform phosphorylation.
A total of 0.5 × 106 to 1.0 × 106 primary CD4 cells were treated with ingenols, PMA, or bryostatin-1 or left untreated and incubated for 30 min at 37°C. Cells were washed with 1× PBS prior to staining with 0.1 μl of fixable viability dye eFluor450 (catalog no. 65-0863; Thermo Fisher Scientific) for 10 min at 4°C. Primary CD4 cells were washed with PBS and fixed and permeabilized by incubation in 100 μl of BD Cytofix/Cytoperm (catalog no. 554722; BD Biosciences) solution for 30 min. The cells were washed in BD Perm/Wash buffer (catalog no. 554723; BD Biosciences), separated into individual aliquots for phospho-PKC isoform analysis, and stained with 2 μl of the following primary antibodies in BD Perm/Wash buffer: pPKCα T497 (catalog no. AB-PK763; Kinexus), pPKCβ T500 (catalog no. AB-PK766; Kinexus), pPKCβ S661 (catalog no. AB-PK766; Kinexus), pPKCδ Y311 (catalog no. sc-377560; Santa Cruz), pPKCδ T505 (catalog no. 9374P; Cell Signaling), pPKCθ S965 (catalog no. AB-PK772; Kinexus), pPKCθ T219 (catalog no. GTX109691; GeneTex), pPKD S205 (catalog no. AB-PK770; Kinexus), and pPKD S738-S742 (catalog no. AB-PK771; Kinexus) for 30 min at 4°C. Two untreated samples were incubated in BD Perm/Wash buffer without primary antibody for use as controls. Samples were washed with BD Perm/Wash buffer and then stained with 0.1 μl APC conjugated anti-rabbit (catalog no. A-21244; Thermo Fisher Scientific) or anti-mouse antibody (catalog no. A-21235; Thermo Fisher Scientific) for 30 min at 4°C. Cells were then washed and resuspended in PBS supplemented with 3% fetal bovine serum (FBS) prior to flow cytometry data acquisition. Flow cytometry was performed with a BD FACSCanto flow cytometer with FACSDiva acquisition software (Becton Dickinson, Mountain View, CA) and analyzed using FlowJo software (TreeStar, Inc., Ashland, OR).
Statistical analysis.
Statistical significance was analyzed using software from GraphPad Prism version 7.0c (GraphPad Software, San Diego, CA).
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to our study participants for their willingness to take part in ongoing translational research, including the experiments described in this paper. We greatly appreciate the ongoing support of the University of Utah Clinical Trials Office, Clinic 1A and the CCTS Clinical Services Core.
This work was supported by NIH grants R33AI122377 (to V.P.), R01AI124843 (principal investigator [PI], Sumit Chanda) and Doris Duke Charitable Foundation Clinical Scientist Development Award DDCF2016102 (to A.M.S.). V.P. also receives support from NIH grants and UM1AI126620 (BEAT-HIV Delaney Collaborative to Cure HIV-1 Infection by Combination Immunotherapy), which is cofunded by the NIAID, NIMH, NINDS, and NIDA.
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01361-18.
REFERENCES
- 1.Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, Richman DD, Valentine FT, Jonas L, Meibohm A, Emini EA, Chodakewitz JA. 1997. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med 337:734–739. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 2.Egger M, May M, Chene G, Phillips AN, Ledergerber B, Dabis F, Costagliola D, D'Arminio Monforte A, de Wolf F, Reiss P, Lundgren JD, Justice AC, Staszewski S, Leport C, Hogg RS, Sabin CA, Gill MJ, Salzberger B, Sterne JA, ART Cohort Collaboration . 2002. Prognosis of HIV-1-infected patients starting highly active antiretroviral therapy: a collaborative analysis of prospective studies. Lancet 360:119–129. doi: 10.1016/S0140-6736(02)09411-4. [DOI] [PubMed] [Google Scholar]
- 3.Mocroft A, Vella S, Benfield TL, Chiesi A, Miller V, Gargalianos P, d'Arminio Monforte A, Yust I, Bruun JN, Phillips AN, Lundgren JD. 1998. Changing patterns of mortality across Europe in patients infected with HIV-1. EuroSIDA Study Group. Lancet 352:1725–1730. [DOI] [PubMed] [Google Scholar]
- 4.Palella FJ Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. 1998. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med 338:853–860. [DOI] [PubMed] [Google Scholar]
- 5.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 6.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A 94:13193–13197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD. 1997. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 8.Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. 2003. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med 9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 9.Crooks AM, Bateson R, Cope AB, Dahl NP, Griggs MK, Kuruc JD, Gay CL, Eron JJ, Margolis DM, Bosch RJ, Archin NM. 2015. Precise quantitation of the latent HIV-1 reservoir: implications for eradication strategies. J Infect Dis 212:1361–1365. doi: 10.1093/infdis/jiv218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siliciano JM, Siliciano RF. 2015. The remarkable stability of the latent reservoir for HIV-1 in resting memory CD4+ T cells. J Infect Dis 212:1345–1347. doi: 10.1093/infdis/jiv219. [DOI] [PubMed] [Google Scholar]
- 11.Chun TW, Engel D, Mizell SB, Hallahan CW, Fischette M, Park S, Davey RT Jr, Dybul M, Kovacs JA, Metcalf JA, Mican JM, Berrey MM, Corey L, Lane HC, Fauci AS. 1999. Effect of interleukin-2 on the pool of latently infected, resting CD4+ T cells in HIV-1-infected patients receiving highly active anti-retroviral therapy. Nat Med 5:651–655. doi: 10.1038/9498. [DOI] [PubMed] [Google Scholar]
- 12.Kulkosky J, Nunnari G, Otero M, Calarota S, Dornadula G, Zhang H, Malin A, Sullivan J, Xu Y, DeSimone J, Babinchak T, Stern J, Cavert W, Haase A, Pomerantz RJ. 2002. Intensification and stimulation therapy for human immunodeficiency virus type 1 reservoirs in infected persons receiving virally suppressive highly active antiretroviral therapy. J Infect Dis 186:1403–1411. doi: 10.1086/344357. [DOI] [PubMed] [Google Scholar]
- 13.Prins JM, Jurriaans S, van Praag RM, Blaak H, van Rij R, Schellekens PT, ten Berge IJ, Yong SL, Fox CH, Roos MT, de Wolf F, Goudsmit J, Schuitemaker H, Lange JM. 1999. Immuno-activation with anti-CD3 and recombinant human IL-2 in HIV-1-infected patients on potent antiretroviral therapy. AIDS 13:2405–2410. doi: 10.1097/00002030-199912030-00012. [DOI] [PubMed] [Google Scholar]
- 14.Rasmussen TA, Schmeltz Sogaard O, Brinkmann C, Wightman F, Lewin SR, Melchjorsen J, Dinarello C, Ostergaard L, Tolstrup M. 2013. Comparison of HDAC inhibitors in clinical development: effect on HIV production in latently infected cells and T-cell activation. Hum Vaccin Immunother 9:993–1001. doi: 10.4161/hv.23800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spivak AM, Andrade A, Eisele E, Hoh R, Bacchetti P, Bumpus NN, Emad F, Buckheit R III, McCance-Katz EF, Lai J, Kennedy M, Chander G, Siliciano RF, Siliciano JD, Deeks SG. 2014. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1-infected adults on antiretroviral therapy. Clin Infect Dis 58:883–890. doi: 10.1093/cid/cit813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elliott JH, McMahon JH, Chang CC, Lee SA, Hartogensis W, Bumpus N, Savic R, Roney J, Hoh R, Solomon A, Piatak M, Gorelick RJ, Lifson J, Bacchetti P, Deeks SG, Lewin SR. 2015. Short-term administration of disulfiram for reversal of latent HIV infection: a phase 2 dose-escalation study. Lancet HIV 2:e520–e529. doi: 10.1016/S2352-3018(15)00226-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Archin NM, Bateson R, Tripathy MK, Crooks AM, Yang KH, Dahl NP, Kearney MF, Anderson EM, Coffin JM, Strain MC, Richman DD, Robertson KR, Kashuba AD, Bosch RJ, Hazuda DJ, Kuruc JD, Eron JJ, Margolis DM. 2014. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. J Infect Dis 210:728–735. doi: 10.1093/infdis/jiu155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Archin NM, Cheema M, Parker D, Wiegand A, Bosch RJ, Coffin JM, Eron J, Cohen M, Margolis DM. 2010. Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infection. PLoS One 5:e9390. doi: 10.1371/journal.pone.0009390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Archin NM, Eron JJ, Palmer S, Hartmann-Duff A, Martinson JA, Wiegand A, Bandarenko N, Schmitz JL, Bosch RJ, Landay AL, Coffin JM, Margolis DM. 2008. Valproic acid without intensified antiviral therapy has limited impact on persistent HIV infection of resting CD4+ T cells. AIDS 22:1131–1135. doi: 10.1097/QAD.0b013e3282fd6df4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM. 2012. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, Smith MZ, Spelman T, McMahon J, Velayudham P, Brown G, Roney J, Watson J, Prince MH, Hoy JF, Chomont N, Fromentin R, Procopio FA, Zeidan J, Palmer S, Odevall L, Johnstone RW, Martin BP, Sinclair E, Deeks SG, Hazuda DJ, Cameron PU, Sekaly RP, Lewin SR. 2014. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog 10:e1004473. doi: 10.1371/journal.ppat.1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, Winckelmann A, Palmer S, Dinarello C, Buzon M, Lichterfeld M, Lewin SR, Ostergaard L, Sogaard OS. 2014. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV 1:e13–e21. doi: 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- 23.Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, Wiegand A, Landay AL, Coombs RW, Richman DD, Mellors JW, Coffin JM, Bosch RJ, Margolis DM. 2005. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet 366:549–555. doi: 10.1016/S0140-6736(05)67098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Søgaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, Kjaer AS, Schleimann MH, Denton PW, Hey-Cunningham WJ, Koelsch KK, Pantaleo G, Krogsgaard K, Sommerfelt M, Fromentin R, Chomont N, Rasmussen TA, Ostergaard L, Tolstrup M. 2015. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog 11:e1005142. doi: 10.1371/journal.ppat.1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leth S, Schleimann MH, Nissen SK, Hojen JF, Olesen R, Graversen ME, Jorgensen S, Kjaer AS, Denton PW, Mork A, Sommerfelt MA, Krogsgaard K, Ostergaard L, Rasmussen TA, Tolstrup M, Sogaard OS. 2016. Combined effect of Vacc-4x, recombinant human granulocyte macrophage colony-stimulating factor vaccination, and romidepsin on the HIV-1 reservoir (REDUC): a single-arm, phase 1B/2A trial. Lancet HIV 3:e463–e472. doi: 10.1016/S2352-3018(16)30055-8. [DOI] [PubMed] [Google Scholar]
- 26.Vibholm L, Schleimann MH, Hojen JF, Benfield T, Offersen R, Rasmussen K, Olesen R, Dige A, Agnholt J, Grau J, Buzon M, Wittig B, Lichterfeld M, Petersen AM, Deng X, Abdel-Mohsen M, Pillai SK, Rutsaert S, Trypsteen W, De Spiegelaere W, Vandekerchove L, Ostergaard L, Rasmussen TA, Denton PW, Tolstrup M, Sogaard OS. 2017. Short-course Toll-like receptor 9 agonist treatment impacts innate immunity and plasma viremia in individuals with human immunodeficiency virus infection. Clin Infect Dis 64:1686–1695. doi: 10.1093/cid/cix201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bocklandt S, Blumberg PM, Hamer DH. 2003. Activation of latent HIV-1 expression by the potent anti-tumor promoter 12-deoxyphorbol 13-phenylacetate. Antiviral Res 59:89–98. doi: 10.1016/S0166-3542(03)00034-2. [DOI] [PubMed] [Google Scholar]
- 28.Kulkosky J, Culnan DM, Roman J, Dornadula G, Schnell M, Boyd MR, Pomerantz RJ. 2001. Prostratin: activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood 98:3006–3015. doi: 10.1182/blood.V98.10.3006. [DOI] [PubMed] [Google Scholar]
- 29.Williams SA, Chen LF, Kwon H, Fenard D, Bisgrove D, Verdin E, Greene WC. 2004. Prostratin antagonizes HIV latency by activating NF-kappaB. J Biol Chem 279:42008–42017. doi: 10.1074/jbc.M402124200. [DOI] [PubMed] [Google Scholar]
- 30.McKernan LN, Momjian D, Kulkosky J. 2012. Protein kinase C. One pathway towards the eradication of latent HIV-1 reservoirs. Adv Virol 2012:805347. doi: 10.1155/2012/805347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang G, Dandekar S. 2015. Targeting NF-kappaB signaling with protein kinase C agonists as an emerging strategy for combating HIV latency. AIDS Res Hum Retroviruses 31:4–12. doi: 10.1089/aid.2014.0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bullen CK, Laird GM, Durand CM, Siliciano JD, Siliciano RF. 2014. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat Med 20:425–429. doi: 10.1038/nm.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laird GM, Bullen CK, Rosenbloom DI, Martin AR, Hill AL, Durand CM, Siliciano JD, Siliciano RF. 2015. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J Clin Invest 125:1901–1912. doi: 10.1172/JCI80142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang G, Mendes EA, Kaiser P, Sankaran-Walters S, Tang Y, Weber MG, Melcher GP, Thompson GR III, Tanuri A, Pianowski LF, Wong JK, Dandekar S. 2014. Reactivation of HIV latency by a newly modified Ingenol derivative via protein kinase Cdelta-NF-kappaB signaling. AIDS 28:1555–1566. doi: 10.1097/QAD.0000000000000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Darcis G, Kula A, Bouchat S, Fujinaga K, Corazza F, Ait-Ammar A, Delacourt N, Melard A, Kabeya K, Vanhulle C, Van Driessche B, Gatot JS, Cherrier T, Pianowski LF, Gama L, Schwartz C, Vila J, Burny A, Clumeck N, Moutschen M, De Wit S, Peterlin BM, Rouzioux C, Rohr O, Van Lint C. 2015. An in-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo HIV-1 latency models identified bryostatin-1+JQ1 and ingenol-B+JQ1 to potently reactivate viral gene expression. PLoS Pathog 11:e1005063. doi: 10.1371/journal.ppat.1005063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gutiérrez C, Serrano-Villar S, Madrid-Elena N, Perez-Elias MJ, Martin ME, Barbas C, Ruiperez J, Munoz E, Munoz-Fernandez MA, Castor T, Moreno S. 2016. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 30:1385–1392. doi: 10.1097/QAD.0000000000001064. [DOI] [PubMed] [Google Scholar]
- 37.Ernst M, Grace OM, Saslis-Lagoudakis CH, Nilsson N, Simonsen HT, Ronsted N. 2015. Global medicinal uses of Euphorbia L. (Euphorbiaceae). J Ethnopharmacol 176:90–101. doi: 10.1016/j.jep.2015.10.025. [DOI] [PubMed] [Google Scholar]
- 38.Pandeló José D, Bartholomeeusen K, da Cunha RD, Abreu CM, Glinski J, da Costa TB, Bacchi Rabay AF, Pianowski Filho LF, Dudycz LW, Ranga U, Peterlin BM, Pianowski LF, Tanuri A, Aguiar RS. 2014. Reactivation of latent HIV-1 by new semi-synthetic ingenol esters. Virology 462–463:328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abreu CM, Price SL, Shirk EN, Cunha RD, Pianowski LF, Clements JE, Tanuri A, Gama L. 2014. Dual role of novel ingenol derivatives from Euphorbia tirucalli in HIV replication: inhibition of de novo infection and activation of viral LTR. PLoS One 9:e97257. doi: 10.1371/journal.pone.0097257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alchin DR. 2014. Ingenol mebutate: a succinct review of a succinct therapy. Dermatol Ther (Heidelb) 4:157–164. doi: 10.1007/s13555-014-0061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang G, Mendes EA, Kaiser P, Wong DP, Tang Y, Cai I, Fenton A, Melcher GP, Hildreth JE, Thompson GR, Wong JK, Dandekar S. 2015. Synergistic reactivation of latent HIV expression by ingenol-3-angelate, PEP005, targeted NF-kB signaling in combination with JQ1 induced p-TEFb activation. PLoS Pathog 11:e1005066. doi: 10.1371/journal.ppat.1005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warrilow D, Gardner J, Darnell GA, Suhrbier A, Harrich D. 2006. HIV type 1 inhibition by protein kinase C modulatory compounds. AIDS Res Hum Retroviruses 22:854–864. doi: 10.1089/aid.2006.22.854. [DOI] [PubMed] [Google Scholar]
- 43.Gama L, Abreu CM, Shirk EN, Price SL, Li M, Laird GM, Pate KA, Wietgrefe SW, O'Connor SL, Pianowski L, Haase AT, Van Lint C, Siliciano RF, Clements JE, LRA-SIV Study Group . 2017. Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS 31:5–14. doi: 10.1097/QAD.0000000000001267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spivak AM, Bosque A, Balch AH, Smyth D, Martins L, Planelles V. 2015. Ex vivo bioactivity and HIV-1 latency reversal by ingenol dibenzoate and panobinostat in resting CD4+ T cells from aviremic patients. Antimicrob Agents Chemother 59:5984–5991. doi: 10.1128/AAC.01077-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freeley M, Kelleher D, Long A. 2011. Regulation of protein kinase C function by phosphorylation on conserved and non-conserved sites. Cell Signal 23:753–762. doi: 10.1016/j.cellsig.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 46.López-Cabrera M, Santis AG, Fernandez-Ruiz E, Blacher R, Esch F, Sanchez-Mateos P, Sanchez-Madrid F. 1993. Molecular cloning, expression, and chromosomal localization of the human earliest lymphocyte activation antigen AIM/CD69, a new member of the C-type animal lectin superfamily of signal-transmitting receptors. J Exp Med 178:537–547. doi: 10.1084/jem.178.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cary DC, Fujinaga K, Peterlin BM. 2016. Euphorbia kansui reactivates latent HIV. PLoS One 11:e0168027. doi: 10.1371/journal.pone.0168027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marsden MD, Loy BA, Wu X, Ramirez CM, Schrier AJ, Murray D, Shimizu A, Ryckbosch SM, Near KE, Chun TW, Wender PA, Zack JA. 2017. In vivo activation of latent HIV with a synthetic bryostatin analog effects both latent cell “kick” and “kill” in strategy for virus eradication. PLoS Pathog 13:e1006575. doi: 10.1371/journal.ppat.1006575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marsden MD, Wu X, Navab SM, Loy BA, Schrier AJ, DeChristopher BA, Shimizu AJ, Hardman CT, Ho S, Ramirez CM, Wender PA, Zack JA. 2018. Characterization of designed, synthetically accessible bryostatin analog HIV latency reversing agents. Virology 520:83–93. doi: 10.1016/j.virol.2018.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spivak AM, Larragoite ET, Coletti ML, Macedo AB, Martins LJ, Bosque A, Planelles V. 2016. Janus kinase inhibition suppresses PKC-induced cytokine release without affecting HIV-1 latency reversal ex vivo. Retrovirology 13:88. doi: 10.1186/s12977-016-0319-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spivak AM, Planelles V. 2016. HIV-1 eradication: early trials (and tribulations). Trends Mol Med 22:10–27. doi: 10.1016/j.molmed.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith BD, Jones RJ, Cho E, Kowalski J, Karp JE, Gore SD, Vala M, Meade B, Baker SD, Zhao M, Piantadosi S, Zhang Z, Blumenthal G, Warlick ED, Brodsky RA, Murgo A, Rudek MA, Matsui WH. 2011. Differentiation therapy in poor risk myeloid malignancies: results of a dose finding study of the combination bryostatin-1 and GM-CSF. Leuk Res 35:87–94. doi: 10.1016/j.leukres.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Philip PA, Rea D, Thavasu P, Carmichael J, Stuart NS, Rockett H, Talbot DC, Ganesan T, Pettit GR, Balkwill F, Harris AL. 1993. Phase I study of bryostatin 1: assessment of interleukin 6 and tumor necrosis factor alpha induction in vivo. the Cancer Research Campaign Phase I Committee. J Natl Cancer Inst 85:1812–1818. doi: 10.1093/jnci/85.22.1812. [DOI] [PubMed] [Google Scholar]
- 54.Hamer DH, Bocklandt S, McHugh L, Chun TW, Blumberg PM, Sigano DM, Marquez VE. 2003. Rational design of drugs that induce human immunodeficiency virus replication. J Virol 77:10227–10236. doi: 10.1128/JVI.77.19.10227-10236.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Márquez N, Calzado MA, Sanchez-Duffhues G, Perez M, Minassi A, Pagani A, Appendino G, Diaz L, Munoz-Fernandez MA, Munoz E. 2008. Differential effects of phorbol-13-monoesters on human immunodeficiency virus reactivation. Biochem Pharmacol 75:1370–1380. doi: 10.1016/j.bcp.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 56.Liang X, Grue-Sorensen G, Mansson K, Vedso P, Soor A, Stahlhut M, Bertelsen M, Engell KM, Hogberg T. 2013. Syntheses, biological evaluation and SAR of ingenol mebutate analogues for treatment of actinic keratosis and non-melanoma skin cancer. Bioorg Med Chem Lett 23:5624–5629. doi: 10.1016/j.bmcl.2013.08.038. [DOI] [PubMed] [Google Scholar]
- 57.Spina CA, Anderson J, Archin NM, Bosque A, Chan J, Famiglietti M, Greene WC, Kashuba A, Lewin SR, Margolis DM, Mau M, Ruelas D, Saleh S, Shirakawa K, Siliciano RF, Singhania A, Soto PC, Terry VH, Verdin E, Woelk C, Wooden S, Xing S, Planelles V. 2013. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog 9:e1003834. doi: 10.1371/journal.ppat.1003834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marsden MD, Zack JA. 2015. Studies of retroviral infection in humanized mice. Virology 479–480:297–309. doi: 10.1016/j.virol.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang P, Lu P, Qu X, Shen Y, Zeng H, Zhu X, Zhu Y, Li X, Wu H, Xu J, Lu H, Ma Z, Zhu H. 2017. Reactivation of HIV-1 from latency by an ingenol derivative from Euphorbia kansui. Sci Rep 7:9451. doi: 10.1038/s41598-017-07157-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, Siliciano RF. 2013. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cillo AR, Sobolewski MD, Bosch RJ, Fyne E, Piatak M Jr, Coffin JM, Mellors JW. 2014. Quantification of HIV-1 latency reversal in resting CD4+ T cells from patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A 111:7078–7083. doi: 10.1073/pnas.1402873111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Z, Gurule EE, Brennan TP, Gerold JM, Kwon KJ, Hosmane NN, Kumar MR, Beg SA, Capoferri AA, Ray SC, Ho YC, Hill AL, Siliciano JD, Siliciano RF. 2018. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc Natl Acad Sci U S A 115:E2575–E2584. doi: 10.1073/pnas.1720665115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.