

Abstract
Since its original discovery over a century ago, the water-gas shift reaction (WGSR) has played a crucial role in industrial chemistry, providing a source of H2 to feed fundamental industrial transformations such as the Haber–Bosch synthesis of ammonia. Although the production of hydrogen remains nowadays the major application of the WGSR, the advent of homogeneous catalysis in the 1970s marked the beginning of a synergy between WGSR and organic chemistry. Thus, the reducing power provided by the CO/H2O couple has been exploited in the synthesis of fine chemicals; not only hydrogenation-type reactions, but also catalytic processes that require a reductive step for the turnover of the catalytic cycle. Despite the potential and unique features of the WGSR, its applications in organic synthesis remain largely underdeveloped. The topic will be critically reviewed herein, with the expectation that an increased awareness may stimulate new, creative work in the area.
Keywords: carbon monoxide, heterogeneous catalysis, homogeneous catalysis, reduction, transition metals
Graphical Abstract

1. Introduction
The water-gas shift reaction (WGSR) is a transformation of long-standing industrial relevance that converts a mixture of carbon monoxide and water into hydrogen and carbon dioxide [Eq. (1)].
| (1) |
Many industrial manufacturing processes rely on synthesis gas (CO + H2) as the raw material.[1] This mixture can be obtained either by coal gasification or, more commonly, from steam reforming of natural gas. Depending upon the method of production, synthesis gas will constitute different ratios of CO and H2. The WGSR is employed to adjust this ratio to the optimal value, or to remove CO altogether by converting it to CO2. Thus, the WGSR finds application in the Haber–Bosch process for ammonia synthesis,[2] in which high purity H2 is obtained from synthesis gas by removal of CO (which would poison the Fe-based ammonia synthesis catalyst). Also, it is used to tune the H2/CO ratio in the production of methanol[3] and in the Fischer–Tropsch process for the synthesis of hydrocarbons.[4]
The WGSR is not limited to industrial applications, but has also been used sporadically in organic synthesis as a method to carry out reductive processes. In these instances, H2 is not a direct product of the reaction, but rather, the reductive potential of the WGSR is channeled toward the reduction of organic substrates, or exploited to insure turnover of catalytic cycles. The hydroformylation reaction is an excellent example to illustrate this concept. In its original incarnation, the hydroformylation reaction converts terminal alkenes into homologous aldehydes utilizing a mixture of CO and H2 over a Co or Rh catalyst [Eq. (2)].[5] In 1953, Reppe and Vetter introduced a modified protocol that used H2O in lieu of H2, and Fe(CO)5 as the catalyst in basic media [Eq. (3)].[6] Under these conditions, the WGSR generates intermediate metal-hydride complexes that account for the overall reductive process. Formally, this can be seen as formation of H2 in situ.
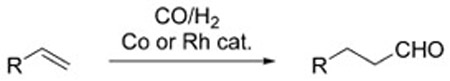 |
(2) |
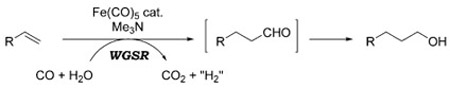 |
(3) |
In addition to hydroformylation, the operation of the WGSR has also been documented in several other reductive processes, including nitro reduction, reductive amination, hydrogenation of carbonyls and alkenes.[7]
Our group became interested in the WGSR in 2008, after the serendipitous discovery that the WGSR was operative in a transformation being studied in our laboratories.[8] At that time, the goal was to develop an allylation reaction of aldehydes that was truly catalytic in metal (i.e. without stoichiometric amounts of metal reducing agents). Two decades earlier, Watanabe and co-workers had reported the formation of homoallylic alcohols from allyl acetate and aldehydes under ruthenium catalysis using triethylamine as the stoichiometric reducing agent (Scheme 1a).[9] The reaction required harsh conditions and high pressures of CO to stabilize the low-valent ruthenium carbonyl catalyst, but nevertheless suited our requirement of being catalytic in metal. In an attempt to identify a more practical protocol, reaction conditions were surveyed in our laboratories using a six-well autoclave. To our surprise, it was found that the reaction proceeded much faster after the autoclave had been cooled and opened in air for sampling and monitoring, a fact that was attributed to the presence of oxygen or water. Control experiments showed indeed that addition of water significantly accelerated the reaction and allowed for milder conditions to be employed (Scheme 1b). Moreover, it was found that the conversion improved using a substoichiometric amount of triethylamine (0.1 equiv), whereas the use of excess amine was detrimental. Evidently, in our case the base was not the stoichiometric reducing agent, as initially shown by Watanabe et al.[10] Thus, it was concluded that the turnover of the catalytic cycle was originating instead from the combined action of CO and H2O in the WGSR.
Scheme 1.

Catalytic allylation of aldehydes.
The topic of the WGSR in its developments and industrial facets has been reviewed extensively.[11] However, only one review, dating back to 1985, describes the application of the WGSR to organic synthesis.[7] The purpose of this review is to fill this gap and provide a compendium of examples wherein, knowingly or not, the WGSR has been used as a way of promoting organic reactions. To ensure a better understanding of the context, the discussion of the WGSR in organic synthesis will be preceded by a historical and mechanistic prelude.
2. The Water-Gas Shift Reaction
2.1. Historical Overview
The WGSR was first reported by Ludwig Mond in 1888,[12] but its importance was not recognized until the development of the Haber–Bosch process for the synthesis of ammonia.[2,13] The water gas (a mixture of CO and H2 obtained from steam and incandescent coal) was employed as the source of hydrogen. However, removal of excess CO by liquefaction proved to be challenging on a large scale. The WGSR provided an efficient method for the removal of CO by conversion to CO2 (more easily removed by dissolution in water), with the benefit of producing additional H2 for ammonia synthesis. In an attempt to optimize the process, researchers at BASF initiated an intensive research program in the early 1910s and eventually settled on a Fe2O3-Cr2O3 heterogeneous catalyst. This system, named the high temperature (HT) shift catalyst, operated above 400 °C and could achieve CO levels of 2–4 % at the exit of the reactor.[14] In the early 1960s, the CO conversion was further improved by the discovery of a series of Cu-ZnO catalysts, called low temperature (LT) shift catalysts, which operated at lower temperature (200 °C) and afforded an exit CO concentration of 0.1–0.3 %.[15] The higher efficiency observed at lower temperature is a direct consequence of the thermodynamics of the WGSR (see below). Consequently, further research efforts focused on the identification of more active catalysts that could operate at still lower temperatures.
In 1932 Hieber et al. reported that metal carbonyl complexes reacted with aqueous bases to form metal carbonyl hydrides. Following acidification, H2 and CO2 were released [Eq. (4)].[16]
| (4) |
In the 1970s, the analogy between these observations and the steps in the WGSR was eventually recognized. Thus, metal carbonyls were investigated as potential low-temperature, homogeneous WGSR catalysts, leading to a series of patents[17] and four independent publications in 1977. Ford et al.[18] and Pettit et al.[19] reported that several transition metal carbonyl complexes are active WGSR catalysts when combined with organic or inorganic bases at temperatures as low as 100 °C. In contrast, Eisenberg et al.[20] and Zudin et al.[21] were able to show that the WGSR can also take place in acidic media. These seminal reports launched decades of intensive research aimed at identifying more efficient homogeneous catalysts. Hundreds of soluble transition metal complexes, representing over 20 d-block elements, have been examined for their ability to catalyze the WGSR and the results have been thoroughly chronicled.[22]
More recently, a renewed interest in heterogeneous WGSR catalysis led to the discovery of extremely active, supported gold catalysts. The initial investigations by Andreeva et al. showed that Fe2O3-supported Au catalyzes the WGSR in the 160–200 °C range, whereas Fe2O3 alone is poorly reactive.[23] Titania (TiO2)-,[24] ceria (CeO2)-,[25] and zirconia (ZrO2)-supported[26] gold catalysts are equally reliable. The activity of supported gold catalysts is highly dependent on the method of deposition, the structure and size of the nanoparticles. Investigation in the area is still ongoing, especially with regard to the reaction mechanism, the nature of active sites and their relationship to catalytic activity.[27]
Remarkably mild conditions for the WGSR have been achieved through the use of photocatalysis.[28] In 1980, Sato and White reported that UV-irradiated Pt/TiO2 catalyzed the WGSR at room temperature and below![29] Following this initial disclosure, a number of soluble transition metal complexes were found to be active photocatalysts for the WGSR at ambient pressure and temperature.[30] Ir-bipyridine complexes, which are widely utilized for their photocatalytic properties, have been investigated by Ziessel in their capacity to promote the WGSR.[31]
Despite the advances in the low temperature WGSR brought about by the use of homogeneous and photocatalysis, these systems have yet to match the levels of performance and robustness of heterogeneous catalysts. Thus, the HT and LT shift catalysts, originally intended for the synthesis of ammonia and improved over the course of the years, still represent today the basis for the catalysis of the WGSR on an industrial scale.
2.2. Thermodynamics and Mechanism
The WGSR is a reversible, moderately exothermic reaction (ΔH0 = −9.8 kcalmol−1). As such, according to the Van ’tHoff equation, the equilibrium constant decreases with increasing temperature. This aspect, demonstrated experimentally by Haber as early as 1909,[32] explains the higher efficiency of the LT shift catalysts, although at the expense of the reaction rate. In accordance with Le Châtelier’s principle, the equilibrium constant is not affected by the pressure since the number of moles of reactant and products is independent of the position of the equilibrium.
The kinetics and mechanism of the WGSR under heterogeneous catalysis have been widely investigated and have been the subject of debate over decades.[33] Two limiting mechanisms have been proposed: 1) the associative mechanism, which involves the adsorption of H2O and CO, the formation of an intermediate on the surface of the catalyst, and its decomposition into CO2 and H2, and 2) the regenerative or redox mechanism, which entails reduction of H2O to H2 with concomitant oxidation of the catalytic surface, followed by oxidation of CO to CO2. Although the consensus mechanism for the HT catalysts is the regenerative, redox proposal, the mechanistic scenario for LT catalysts appears much less well understood and is highly dependent upon the metal and the experimental conditions.[34]
The discovery of homogeneous catalysts has opened up the possibility for the detection and characterization of reactive intermediates, leading to a more refined and relatively well understood mechanistic picture.[22b] The catalytic cycle shown in Scheme 2 represents the generally accepted mechanism for the WGSR catalyzed by a metal–carbonyl complex (i) under basic conditions. The cycle consists of five steps: A) nucleophilic activation of coordinated CO to form hydroxycarbonyl complex ii, B) decarboxylation to form metal hydride iii, C) protonation, D) reductive elimination of H2 from the dihydrido complex iv, and E) binding of CO to the coordinatively unsaturated intermediate v.
Scheme 2.

General catalytic cycle of the WGSR under basic conditions.
In the Fe(CO)5 system investigated by King et al., the mechanism follows the general catalytic cycle with step A being turnover limiting.[35] DFT studies also support this catalytic cycle, although alternative proposals have been formulated.[36] The mechanism appears to be more complex with Ru, Os, Rh and Ir carbonyl complexes due to the formation of metal clusters.[37] A different pathway has been suggested for Group 6 (Cr, Mo, W) metal–carbonyl complexes. Activation of free CO forms anionic formate (F), which coordinates to the metal (G). Decarboxylation then takes place from a formate–metal complex vi rather than a hydroxycarbonyl–metal complex ii.[38]
The occurrence of the WGSR under acidic conditions is a much less general phenomenon. For instance, whereas Ir4(CO)12, Fe(CO)5 and Ru3(CO)12 are all active catalysts in alkaline solution, only the latter is active in the presence of H2SO4.[22a,39] A plausible mechanism was proposed by Ford et al. using Ru3(CO)12, which generates the active Ru2(CO)9 catalyst vii in situ (Scheme 3).[40]
Scheme 3.

Catalytic cycle of the WGSR under acidic conditions using Ru3(CO)12/H2SO4.
Under acidic conditions, coordinated CO must be sufficiently activated toward nucleophilic attack by water rather than hydroxide (step J, as opposed to A). This activation can be achieved if protonation of the metal center (H) occurs prior to nucleophilic attack of CO. Protonation raises the oxidation state by two, decreases the extent of CO back-bonding and therefore renders CO more susceptible to nucleophilic attack. Similar mechanistic proposals have been formulated for related acid-catalyzed processes.[41]
The mechanism of the WGSR under photocatalysis has been described for [Ir(bipy)Cp*Cl]+Cl− (xii, R = COOH) in a landmark study by Ziessel (Scheme 4).[31c] Following the initial CO coordination and activation steps (M and N), turnover-limiting decarboxylation yields the neutral, coordinatively unsaturated complex xv, the intermediacy of which has been established by direct isolation. Protonation of the metal center furnishes the hydrido intermediate xvi, which is prone to photoexcitation and protonation of the excited state xvii, and reductive elimination of H2 thereby (R). The requirement for light in the final protonation step was demonstrated by the lack of reactivity of complex xvi in the dark.
Scheme 4.

Catalytic cycle of the WGSR under photocatalysis using xii.
3. The Water-Gas Shift Reaction in Organic Synthesis
The vast amount of research done since the 1970s on homogeneous catalysis has demonstrated the possibility of harnessing the reducing potential of the WGSR to drive reductive organic transformations. The obvious advantages of this synergy include low impact of waste, scalability and availability of precursors. However, despite all of the research into improving the efficiency of the WGSR, the process nowadays is mainly utilized for the generation of hydrogen using heterogeneous catalysis, while its applications to the synthesis of fine chemicals remain underdeveloped. The following sections will illustrate the potential for application of the WGSR to a myriad of organic transformations and identify areas of future development.
The reduction potential of the WGSR can be exploited in organic synthesis in two different ways. In the first approach, the WGSR provides reducing equivalents that can be directly incorporated into the final product (Figure 1a). Essentially this corresponds to the delivery of H2 as would be achieved through a hydrogenation reaction, although free H2 is not directly involved. In the second approach, the reducing potential of the WGSR is applied to a metal catalyst that has to be reduced to its active form to reenter the catalytic cycle (Figure 1b). In the absence of the WGSR, transformations of this type would require a stoichiometric amount of a reducing agent, usually a low-valent metal (Mn, Zn, SmI2, SnCl2, etc.). It should be highlighted that, whereas in the former case the WGSR offers modest to no practical advantage over a standard hydrogenation, the latter case eliminates the need for a stoichiometric amount of metal, thus reducing the waste stream and enhancing atom economy.
Figure 1.

WGSR in organic transformations.
Another important distinction involves the type of transformation that is accomplished. The vast majority of reactions employing the WGSR entail the reduction of a functional group without a carbon–carbon bond forming event. These transformations usually fall into the category of incorporation of H2 (as in the reduction of nitro groups, imines, alkenes and carbonyls), although there are exceptions (deoxygenation of epoxides). Only a limited number of WGSR-promoted reactions accomplish the formation of a carbon–carbon bond. Among those, some rely on CO as both the reducing agent and the source of carbon (hydroformylation and other carbonylation reactions), whereas others utilize an external building block to forge the new carbon-carbon bond (allylation, alkylation). The following sections are arranged on the basis of this classification.
This review covers only transformations that employ the CO/H2O couple as the reducing agent in the presence of a metal catalyst. Reductions carried out with CO alone (without water and/or hydroxide) will not be covered. Likewise, reactions that employ a metal carbonyl complex in stoichiometric amounts are not included in this review.
3.1. Non-C–C Bond Forming Reactions
3.1.1. Nitro Reduction
The reduction of nitroarenes to anilines represents the most common application of the WGSR to organic synthesis. Traditionally, this transformation is carried out using stoichiometric amounts of reducing agents (such as Fe or Zn powder, Na2S, SnCl2), or catalytic hydrogenation over Pd, Pt or Ni.[42] The use of the WGSR as the source of reducing equivalents allows for a mild and chemoselective process. The nitro-to-amine transformation is a 6e−-reduction, therefore three equivalents of CO are required per mole of nitro compound [Eq. (5)]
| (5) |
The reduction of nitrobenzene to aniline with CO/H2O was first reported by Iqbal in 1971 using Rh catalysts and organic bases (Scheme 5a).[43] More systematic studies were undertaken by Pettit et al. in 1978 using Fe, Rh, Ru, Os, Ir carbonyl complexes and aqueous solutions of trialkylamines (Scheme 5b).[44]
Scheme 5.

Initial reports on nitroarene reduction using WGSR conditions.
The metal complexes under examination are active catalysts for the WGSR and do generate H2 when exposed to CO/H2O. However, Pettit et al. suggested the involvement of a metal-hydride intermediate [HM(CO)n]− as the reducing agent (formed via CO activation/decarboxylation, as iii in Scheme 2),[45] as opposed to in situ generation of H2. This point was demonstrated by the incorporation of deuterium in the product when the reaction was carried out in the presence of CO/D2O/H2 [Eq. (6)]. From these results, it appears
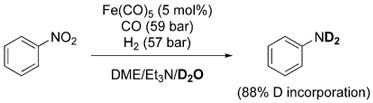 |
(6) |
evident that H2 is not causing the reduction of nitrobenzene, even though it may be generated under the reaction conditions.
In 1980, Alper and Amaratunga described a system capable of operating under mild conditions (ambient pressure and temperature).[46] The system is comprised of Ru3(CO)12 and BnEt3N+Cl− as the phase transfer catalyst in a mixture of 5m aqueous NaOH/benzene/2-methoxyethanol [Eq. (7)]. The increased reactivity is attributed to the solubility of the putative ammonium–ruthenate intermediate [BnEt3N]+ [HRu3(CO)11]− in the organic phase.
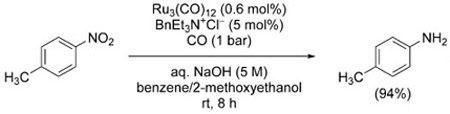 |
(7) |
These initial reports stimulated dozens of further elaborations,[47] and the topic is still an active field of research. A summary of catalysts and reaction conditions that have been reported is provided in Table 1. Most of the protocols employ basic or neutral conditions, although there are examples of acidic media (entries 12 and 14). Generally, the reaction conditions have been optimized for nitrobenzene as a benchmark and extended to a limited number of nitroarenes, making it impossible to come to conclusions regarding their generality. A few methods, however, possess remarkable generality and functional group tolerance, and will be discussed separately (entries 6, 17, 22, 35, 39). The most user-friendly protocols appear to be those that employ phase-transfer catalysis (entries 4 and 11) or additives such as amines and phosphines (entries 15 and 16), as they operate under mild conditions (room temperature and ambient pressure) with readily available reagents. The use of selenium as the catalyst, which generates H2Se under WGSR conditions, is also well established (entries 5, 26, 32).
Table 1:
Reaction conditions for the reduction of aromatic nitro groups.
| Entry | Metal catalyst | Additives | T [°C] | Pco [bar] | Ref. |
|---|---|---|---|---|---|
| 1 | Rh2O3, RhCl3·3H2O, Rh carbonyl complexes | pyridine, N-methylpyrrolidine | 50–150 | 50–121 | [43] |
| 2 | Mn, Re, Fe, Ru, Os, Rh, Ir, Pt carbonyl complexes | Me3N, Et3N | 25–187 | 34–117 | [44, 48] |
| 3 | Rh6(CO)16 | N,N-dimethylbenzylamine (DMBA), polymer-bound DMBA | 80–120 | 7–55 | [49] |
| 4 | Ru3 (CO)12 | NaOH/BnEt3N+Cl− | 25 | 1 | [46] |
| 5 | Se | Et3N | 80 | 30 | [50] |
| 6 | Rh carbonyl complexes | aliphatic amines and diamines, pyridine, substituted pyridines, polymer-bound amines | 80 | 1–8 | [51–53] |
| 7 | [Ru(cod)py4](BPh4)2 | – | 80–145 | 15–61 | [54] |
| 8 | PtCl2(PPh3)2 | Et3N/SnCl4 | 20–100 | 15–59 | [55, 56] |
| 9 | Fe2O3/Al2O3 | alkali metal carbonates | 300–350 | 1 | [57] |
| 10 | Rh6(CO)16, Ru3(CO)12 | phenanthroline, substituted phenanthrolines, bipyridine, TMEDA | 165 | 30 | [58, 59] |
| 11 | Co2(CO)8/[Rh(1,5-hexadiene)Cl]2 | NaOH/(C12H25)Me3N+Cl− | 25 | 1 | [60] |
| 12 | Ru and Os carbonyl complexes | KOH, HCI, HBr, HBF4, CH3COOH, CF3COOH | 120 | 69 | [61] |
| 13 | Co carbonyl complexes | NaOH/(C16H33)Me3N+Br− | 25 | 1 | [62] |
| 14 | (η4-Ph4C4C=O)Ru(CO)3 | Na2CO3, NaHCO2, CH3COOH, CF3COOH | 105 | 34 | [63] |
| 15 | Rh carbonyl complexes/amines and diamines | NaOH | 25 | 1 | [64] |
| 16 | Rh and Ru carbonyl complexes/phosphines and bis-phosphines | NaOH | 25–50 | 1 | [65] |
| 17 | Ru3(CO)12 | alkyl amines | 150–180 | 20 | [66–68] |
| 18 | Ru3(CO)12/N,N′-diaryl-diiminoacenaphthene ligands | – | 150–180 | 30 | [69] |
| 19 | S, CS2, H2S, COS/ V2O5, NH4VO3 | NaOH, NH4OH, NaOCH3, Et3N | 70–150 | 120–140 | [70] |
| 20 | RhCl3·3 H2O on PVP[a] | – | 100 | 1 | [71] |
| 21 | Ru3(CO)12/α,α-dimethylbis(2-benzothiazolyl)methane | – | 150–200 | 10–50 | [72] |
| 22 | PdCl2 | NaOH/(3-C6H4SO3−Na+)3P (TPPTS), BINAS (see Figure 2) |
100 | 120 | [73] |
| 23 | Pd, PdCl2/Fe, Fe2O3/I2 | pyridine | 150–180 | 25–90 | [74] |
| 24 | K+, Cs+, (PPh3)2N+ [Rh(CO)4]− | – | 200 | 40–81 | [75] |
| 25 | [Rh(nbd)(ppy)]ClO4[a] | KOH | 130 | 1 | [76] |
| 26 | Se | pyridine, Et3N | 180–200 | 4 | [77] |
| 27 | [Rh(CO)2(amine)2]PF6, [Ir(cod)(amine)2]PF6 | pyridine, substituted pyridines | 60–150 | 0.5–30 | [78,79] |
| 28 | [Rh(CO)2(amine)2]PF6, [Ir(cod)(amine)2]PF6, [Rh(cod)(amine)2]PF6, Rh2(CO)4Cl2 on PVP | 70–130 | 0.5–2 | [80–83] | |
| 29 | Rh6(CO)16/dimethylaminoethylated hydroxypropyl-chitosan |
80 | 10 | [84] | |
| 30 | Ru3(CO)9(PEO-DPPSA)3[a] | – | 100–140 | 20–50 | [85] |
| 31 | RhCl3·3 H2O | alkyl amines, 2-picoline, TMEDA | 100 | 1 | [86] |
| 32 | Se | NaOAc, Na2CO3, NaOH, Et3N, DBU | 85–160 | 1–10 | [87] |
| 33 | CuCl2·2 H2O on PVP | – | 100–150 | 7–27 | [88] |
| 34 | Au/Fe(OH)x | – | 100–120 | 5–15 | [89] |
| 35 | Au/TiO2 | – | 25 | 5 | [90] |
| 36 | Ag, Au, Pd, Rh, Pt on hydrotalcite | – | 150 | 9 | [91] |
| 37 | Ru/MgF2 | – | 175 | 20 | [92] |
| 38 | dendrimer-encapsulated [Rh5(CO)15]− | – | 80 | 10 | [93] |
| 39 | Co3O4/N-doped graphene | – | 125 | 30 | [94] |
| 40 | PdCl2(py)2/Fe/l2 | pyridine, substituted pyridines | 100–180 | 40 | [95] |
PVP=poly(4-vinylpyridine); ppy=polypyridine chelating ligand; PEO-DPPSA=poly(ethylene oxide)-substituted 4-(diphenylphosphino) benzenesulfonamide.
One of the remarkable features of the nitro reduction under WGSR conditions is the high chemoselectivity, which often cannot be guaranteed using other reduction protocols.[96] For example, all three reduction-susceptible functional groups of 4-chloro-3-nitroacetophenone are reduced unselectively by a standard hydrogenation over Pd/C [Eq. (8)].[97] Conversely, under WGSR conditions, reduction of the nitro group occurs selectively even in the presence of other
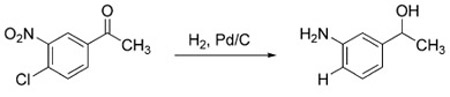 |
(8) |
functional groups prone to reduction (alkenes, alkynes, halides, nitriles, ketones, aldehydes, esters, etc., Figure 2).[53,68,73,90,94]
Figure 2.

Chemoselective reduction of nitroarenes.
A noteworthy sequence of complementary reduction methods allows for the preparation of 5, a tritiated photoaffinity ligand for the dopamine reuptake transporter protein (Scheme 6).[98] Reduction of the nitro group in 1 under Alper’s phase-transfer conditions affords amine 2 without reduction of the unsaturated amide. Lithium aluminum hydride was used to carry out the deoxygenation of the amide, followed by the introduction of the tritium label by hydrogenation of 3 (T2, Pd/C).
Scheme 6.

Sequential reduction reactions for the synthesis of 5.
WGSR conditions display a marked preference for the reduction of aromatic as compared to aliphatic nitro groups; aliphatic nitro groups are normally unreactive (Figure 3a).[49,55] However, conditions have been developed that can effect the reduction of aliphatic nitro groups to amines (Figure 3b)[46,90] or the partial reduction to nitriles (Figure 3c).[52,99]
Figure 3.

Reduction of aliphatic nitro groups.
Mechanistic investigations of the reduction of nitro groups by CO/H2O are abundant. From a qualitative standpoint, the participation of H2 is clearly excluded on the basis of important facts: 1) the reduction is often less efficient if H2 is used instead of CO/H2O ;[61] 2) some of the systems used for the nitro reduction are not active WGSR catalysts and do not produce CO2 and H2 in the absence of a nitro compound;[68,86,95] 3) even for those systems that are capable of producing H2, competition/labelling experiments demonstrate that the nitro group is not directly reduced by H2;[44] 4) functional groups that are susceptible to reduction by H2 do not react (Figure 2).
The generally accepted catalytic cycle, supported by the isolation of some reactive intermediates,[100] involves two sequential deoxygenation steps (Scheme 7). Following the formation of cycloadduct xviii between the nitro group and ligated CO decarboxylation produces the η2-nitrosoarene complex xix. Insertion of CO leads to the four-membered cycloadduct xx which decarboxylates to form the nitrenoid complex xxi. The nitrenoid affords the aniline product after a further 2e−-reduction, either WGSR-driven (via metal-hydride xxii),[101] or via formation of an isocyanate and its subsequent hydrolysis/decarboxylation.[56,68,102] An exception to this general catalytic cycle is the reduction using the Se/amine system. The reaction of selenium with CO/aqueous amine produces H2Se,[103] which is believed to be the actual reducing agent.[50,87b]
Scheme 7.

Catalytic cycle for the reduction of nitroarenes.
3.1.2. Downstream Reactions From Nitro Reduction
The reduction of nitro groups to amines represents only a small portion of the vast amount of reductive processes that can be accomplished from nitro compounds using CO as the reducing agent. As illustrated in Scheme 7, the reaction of a nitro group with CO generates a nitrenoid intermediate xxi which, in the absence of water, may undergo different reaction pathways.[97,104,105] Thus, depending on the reaction conditions, nitro compounds can be converted into isocyanates, azo compounds, imines, ureas, or carbamates (Figure 4a). The nitrenoid can also react intramolecularly with a tethered functional group (olefinic or aromatic C–H, alcohol, amine, etc.), lending itself to the synthesis of heterocycles (Figure 4b).
Figure 4.

Possible reaction pathways following the CO-mediated reduction of nitroarenes.
These sets of reductive transformations, which do not rely on the CO/H2O couple, are outside the scope of this review. However, similar approaches have been used to combine the WGSR-driven nitro reduction with a tandem transformation.
The reductive amination reaction from nitroarenes and carbonyl compounds falls into this category, and proceeds through the in situ formation of an imine, which is reduced under WGSR conditions. The first example was reported by Watanabe et al. in 1980.[106] In the presence of [Rh(cod)Cl]2 or [Rh(nbd)Cl]2, nitroarenes and linear, aliphatic aldehydes furnish reductive amination products in moderate yield [Eq. (9)]. Under these conditions, N,N-dialkylated anilines result.
An improved variant possessing increased synthetic utility was recently reported by Park and Chung.[107] The use of
 |
(9) |
a Co2Rh2/C heterogeneous catalyst enabled the synthesis of a number of secondary amines from nitroarenes, aromatic aldehydes and ketones (Figure 5). These conditions avoid double alkylation, but the substrate scope remains limited to aromatic nitro compounds and benzaldehydes.
Figure 5.

Reductive amination from nitroarenes and carbonyls.
It might be tempting to assume that the nitro compound is reduced to the aniline first and condensation with the aldehyde follows, but this appears not to be the case. Indeed, imines form readily from nitroarenes and aldehydes by a CO-mediated reduction without the need for H2O.[108] In addition, control experiments wherein the aldehyde is combined directly with the aniline yield no product, showing that the aniline is not a competent intermediate.[106b] Therefore, it was suggested that the imine is formed directly from the nitrenoid intermediate xxiii (Scheme 8) via oxaziridine xxiv and nitrone xxv, which is reduced by CO to the imine.[109] The reductive amination product is then obtained via WGSR reduction (Section 3.1.3).
Scheme 8.

Mechanism for the formation of an imine by CO reduction of a nitro compound.
Over the course of their studies on reductive amination from nitroarenes, Watanabe et al. also found that, under modified conditions (RhCl(PPh3)3 and PdCl2), the formation of dialkylated anilines was suppressed and quinolines were obtained instead [Eq. (10)].[106] Although the transformation is overall redox-neutral, the aniline formed via WGSR reduction of the nitroarene is along the reaction pathway. Quinoline formation occurs by self-condensation of the aldehyde, conjugate addition of the aniline, and oxidative cyclization (Skraup quinoline synthesis).
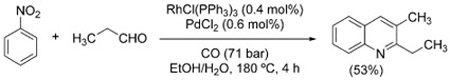 |
(10) |
Similarly, the syntheses of phenanthrolines [Eq. (11)][110] and quinolines from 2-nitrochalcones [Eq. (12)][111,112] have been reported. In the latter case, the intermediacy of the
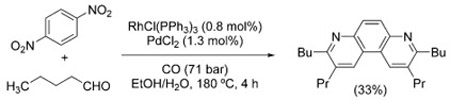 |
(11) |
 |
(12) |
aniline obtained via WGSR reduction was suggested by a set of clever competition experiments.[113]
Lastly, although ureas are generally synthesized from nitroarenes and amines under anhydrous conditions (Figure 4), symmetrical arylureas can be obtained directly from the corresponding nitroarenes via CO/H2O reduction/carbonylation, using selenium as the catalyst [Eq. (13)].[114]
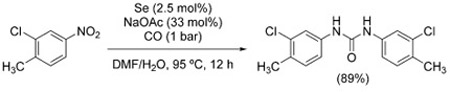 |
(13) |
3.1.3. Reduction of Imines and Reductive Amination
The reductive amination employing nitroarenes and carbonyl compounds was discussed in Section 3.1.2. In addition to this approach, the synthesis of substituted amines by reductive amination can be achieved directly from an amine precursor and a carbonyl compound, thus bypassing the first nitro-to-amine reduction step. The obvious advantage is the wider range of products that can be produced. Whereas reductive amination from nitro compounds can lead only to secondary anilines (or tertiary anilines with two identical substituents), no such limitations exist when a primary or secondary amine is used as the starting material.
As is the case with the reductive amination from nitro compounds, the reaction relies on the in situ generation of an imine (or iminium ion) that is reduced under WGSR conditions. This crucial step has been documented in the reduction of N-benzylideneaniline with catalytic amounts of Fe(CO)5 [Eq. (14)].[115]
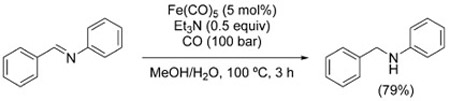 |
(14) |
The first example of reductive amination under WGSR conditions was reported by Watanabe et al. in 1978.[116] A number of amines combined with formaldehyde or benzaldeyde to produce the corresponding methylated or benzylated amines in variable yield using Rh catalysts [Eq. (15)]. The same process can be carried out in the presence of Co2(CO)8/dppe with comparable results.[117] Ketones can participate in the reaction, as well [Eq. (16)].[52]
The last two years have witnessed a renewed interest in the WGSR-driven reductive amination. Several improved
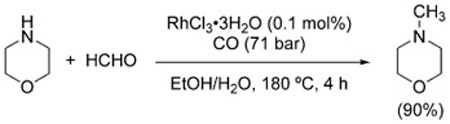 |
(15) |
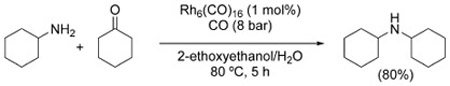 |
(16) |
variants have been developed that allow for the alkylation of primary and secondary amines with aldehydes and ketones, using homogeneous[118,119] or heterogeneous catalysts.[107,120] Quite surprisingly, none of the recent reports acknowledge the pioneering cases mentioned above. Reaction conditions and illustrative examples are provided in Figure 6.
Figure 6.

Reductive amination under WGSR conditions.
Since the reaction protocols employ little to no water, the role of water deserves a comment. The condensation between a carbonyl compound and an amine liberates one equivalent of water, which in principle should suffice to carry out the subsequent reduction of the imine bond. The formation of reduction products even in the absence of added water is therefore justified (Figure 6c). In the presence of molecular sieves, which sequester the water originating from imine condensation, the reaction stops at the imine stage.[107] On the other hand, addition of a superstoichiometric amount of water is detrimental.[118]
By means of labeling and competition experiments, Chusov and List ruled out the involvement of free H2. The reaction proceeded at a much lower rate when CO was replaced by H2. Only 11% D incorporation was measured when D2 (1 bar) was added to the reaction mixture, whereas D incorporation reached 74% when N,N-deuterated aniline was used. In addition, a Rh-hydride complex was detected by 1HNMR reaction monitoring. On the basis of these observations, the authors proposed a catalytic cycle involving oxidative addition of a Rh carbonyl complex into the C-OH bond of the intermediate hemiaminal.[118] This step seems rather unlikely, given the tendency of hemiaminals to lose water and the fact that oxidative addition of Rh into C–OH bonds is unknown. It seems more reasonable that the intermediate imine would undergo reduction by a Rh—H complex (observed in situ!) generated via the WGSR.
3.1.4. Reduction of Heterocycles
The reduction of nitrogen-containing heterocycles can be driven by the WGSR under forcing conditions. Murahashi et al. reported that quinolines and isoquinolines are reduced to the corresponding 1,2,3,4-tetrahydroquinolines and 1,2,3,4-tetrahydroisoquinolines (with concomitant N-formylation in the latter case) in the presence of Rh6(CO)16 at 150 °C and 55 bar of CO (Figure 7).[121] Only the N-containing ring undergoes reduction. This feature offers complementarity to direct hydrogenation methods, which provide 5,6,7,8-tetrahydroquinolines selectively [Eq. (17)].
Figure 7.

Reduction of quinolines and isoquinolines.
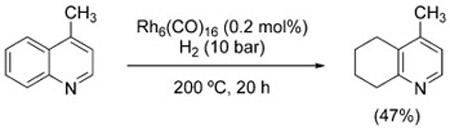 |
(17) |
Other nitrogen-containing heterocycles undergo reduction under WGSR conditions as well, but the reported methods have little synthetic utility due to the low turnover numbers or complexity of the reaction mixtures.[122–124] Oxygen- and sulfur-containing heterocycles, as well as polynuclear aromatic hydrocarbons, are unreactive or poorly reactive under WGSR conditions.[123,124]
3.1.5. Reduction of Carbonyls
The reduction of carbonyls to primary and secondary alcohols is a relatively facile transformation under WGSR conditions and several catalytic systems have been investigated in this capacity (Table 2). The early reports described harsh conditions with limited applicability (entries 1–4). While aromatic and branched aliphatic aldehydes were reduced to the corresponding alcohols, linear aldehydes underwent aldol condensation/1,2-reduction (entry 1). Improved results were obtained with the Rh6(CO)16/TMPDA system described by Kaneda et al., which allowed for a milder process compatible with several classes of aldehydes, but not as widely applicable in the case of ketones (entry 5 and Figure 8a). Recently, Beller et al. reported the efficient and scalable reduction of aldehydes using Knölker’s catalyst (entry 7 and Figure 8b). Ketones are not reactive under these conditions.
Table 2:
Reaction conditions for the reduction of carbonyls.
| Entry | Metal catalyst | Additives | T[°C] | Pco [bar] | Substrates | Ref. |
|---|---|---|---|---|---|---|
| 1 | RhCl3·3H2O | Et3N | 200 | 51 | aliphatic aldehydes | [126] |
| 2 | Fe, Ru, Rh carbonyl complexes | KOH | 125 | 55 | aromatic aldehydes, acetophenone | [37a] |
| 3 | Fe3(CO)12 | Et3N | 100 | 100 | acetone | [127] |
| 4 | RhH2(O2COH)[P(iPr)3]2 | – | 150 | 15 | benzaldehyde, acetophenone | [128] |
| 5 | Rh6(CO)16 | TMPDA[a] | 30–80 | 5–20 | aliphatic and aromatic aldehydes, ketones | [129] |
| 6 | Rh6(CO)16/dimethylaminoethylated hydroxypropyl-chitosan | – | 80 | 10 | benzaldehyde | [84] |
| 7 | Knölker’s catalyst | K2CO3 | 100 | 10 | aliphatic and aromatic aldehydes | [130] |
| 8 | Knölker’s catalyst | Na2CO3 | 120 | in situ release | aromatic aldehydes | [131] |
N,N,N′,N′-tetramethyl-1,3-propanediamine.
Figure 8.

Reduction of aldehydes and ketones.
The reduction of α,β-unsaturated aldehydes is more challenging due to the competition between the 1,2- and 1,4-reduction modes (see Section 3.1.6). For example, the reduction of α,β-unsaturated aldehydes using Knölker’s catalyst afforded mixtures of saturated and allylic alcohols [Eq. (18)].[130]
Kaneda et al. were able to obtain selective carbonyl reduction by fine tuning of the reaction conditions. In the case
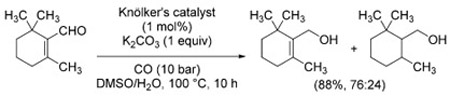 |
(18) |
of Rh6(CO)16, it was shown that the site-selectivity is highly dependent on the base used in the reaction. The use of TMPDA is crucial for the achievement of 1,2-reduction (even the similar TMEDA gives poorer results), whereas 4-DMAP reverses the selectivity in favor of 1,4-reduction [Eq. (19)].[129b] Good 1,2-selectivity is also displayed by ceria-supported gold nanoparticle catalysts [Eq. (20)].[125] In contrast, direct hydrogenation using the same catalyst is unselective.
 |
(19) |
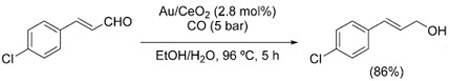 |
(20) |
3.1.6. Reduction of Alkenes and Alkynes
The hydrogenation of unactivated olefins under WGSR conditions is rather unexplored and only two cases have been reported. Ethylene is hydrogenated to ethane using K2PtCl4/SnCl2 in acidic medium (HCl/acetic acid) under CO (0.4 bar) at 88 °C.[132] However, the same conditions failed to provide clean hydrogenation of propylene. Hydrogen added to the reaction mixture remained unconsumed, thus lending support to the intermediacy of a Pt–H intermediate formed from H2O. Styrene is reduced to ethylbenzene in good yields using Fe3(CO)12 and basic conditions, though with competing hydrohydroxymethylation (Section 3.2.1) depending upon the solvent mixture [Eq. (21)].[133]
Conversely, the hydrogenation of activated olefins (α,β-unsaturated aldehydes, ketones, esters, amides, and nitriles) has been investigated more thoroughly. Already in 1973, Joh
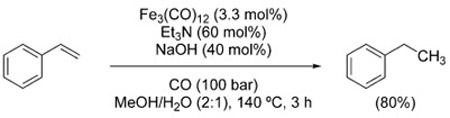 |
(21) |
et al. had shown that the C–C double bond of a variety of unsaturated carbonyl compounds underwent selective 1,4-reduction, whereas unactivated olefins were unreactive.[134]
The initial conditions were quite harsh (98 bar, 130 °C, Figure 9a), but the addition of Et3N allowed for a milder process to be developed (1 bar, 30 °C, Figure 9b).[135] Selenium, often used as an alternative to transition metals in WGSR-driven reductions, is also effective in the selective reduction of activated olefins (Figure 9c).[136] Other Rh-based catalytic systems were studied in the 1,4-reduction of conjugated olefins, however with limited exploration of the substrate scope. [58b, 128, 129b, 137, 138]
Figure 9.

Selective 1,4-reduction of conjugated olefins.
The only reported example of alkyne hydrogenation under WGSR conditions involves heterogeneous catalysis. Using TiO2-supported gold nanoparticle catalysts, Cao et al. were able to achieve the chemo- and stereoselective reduction of a wide range of alkynes to (Z)-alkenes in high yields and without overreduction. (Figure 10).[139]
Figure 10.

Selective reduction of alkynes to alkenes.
3.1.7. Deoxygenation of Epoxides
The deoxygenation of epoxides to the corresponding alkenes under WGSR conditions has been known since 1968, when Watanabe et al. reported the reduction of styrene oxide with K2Fe(CO)4.[140] However, the development of synthetically useful reduction protocols occurred only in the last decade with the advent of nanoparticle catalysts. Hydrotalcite-supported silver[141] and gold,[142] as well as titania-supported gold nanoparticles,[143] are efficient promoters of the reduction of a variety of epoxides (Figure 11). Thanks to these contributions, the use of WGSR conditions represents one of the mildest and most efficient catalytic methods to date for this transformation.
Figure 11.

Deoxygenation of epoxides with nanoparticle catalysts.
From a mechanistic standpoint, it should be emphasized that the deoxygenation of epoxides is conceptually different from the reactions discussed above because it does not lead to the incorporation of reducing equivalents (H) into the substrate. The reducing power provided by the CO/H2O couple ensures turnover of the catalytic cycle by reducing the putative metal–oxo intermediate xxvii to the active form of the metal xxvi (Scheme 9). Metal–oxo intermediates are believed to be involved in the deoxygenation of epoxides.[144] However, their intermediacy in the deoxygenation reaction under WGSR conditions has not been explicitly documented and, therefore, alternative mechanisms cannot be discounted at this time.
Scheme 9.

WGSR in the deoxygenation of epoxides.
Regardless of the mechanistic pathway, an important aspect concerns the role of water. Although water does not appear in the balanced equation, its presence is of course required for the occurrence of the WGSR. Control experiments demonstrated that the reaction did not proceed in the absence of water (Scheme 10a). However, a catalytic amount of water is needed to initiate the reaction (Scheme 10b).[143]
Scheme 10.

Control experiments with water.
3.1.8. Other Deoxygenation Reactions
Several other deoxygenation reactions have been carried out using CO/H2O as the reducing agent. A summary is provided in Table 3.
Table 3:
Deoxygenation reactions using the CO/H2O couple.
3.1.9. Desulfurization
The desulfurization of thiophenols and benzylic mercaptans was described by Alper et al. in 1985 using Co2(CO)8 under rather forcing conditions [Eq. (22)].[147] Interestingly, the proposed mechanistic pathway involves generation of carbonyl sulfide, rather than carbon dioxide, and is supported by its direct observation.
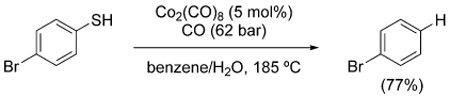 |
(22) |
3.1.10. Dehalogenation
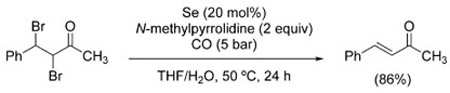
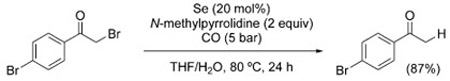
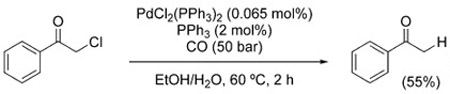
The examples shown in Figure 2 and thereafter illustrate the reluctance of carbon-halogen bonds to undergo reduction under WGSR conditions. Indeed, only a handful of cases of hydrodehalogenation have been reported. In 1990, Sonoda et al. described the use of Se/Et3N for the debromination/elimination of vicinal dibromides [Eq. (23)], as well as the dehalogenation of α-halo ketones [Eq. (24)].[148] Similarly, α-chloroacetophenone undergoes dechlorination in the presence of PdCl2(PPh3)2 [Eq. (25)].[149]
 |
(23) |
 |
(24) |
 |
(25) |
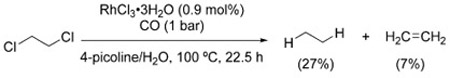
Ford et al. showed that 1,2-dichloroethane can be dechlorinated to a mixture of ethane and ethylene with RhCl3/4-picoline [Eq. (26)].[150] From the observation that the ethylene/ethane ratio decreases as the reaction progresses, it was suggested that ethylene forms first via a reduction/elimination pathway and is then further reduced to ethane by the same catalytic system.
 |
(26) |
Harsh conditions are needed for the dechlorination of chlorobenzene, which has been achieved, albeit in low yield, using CuCl2 supported on poly(4-vinylpyridine) (Scheme 11a).[151] In contrast, deiodination of aryl iodides occurs readily at 60 °C and 1 bar of CO (Scheme 11b).[152]
Scheme 11.

Hydrodehalogenation of aryl halides.
3.2. C–C Bond Forming Reactions
3.2.1. Hydroformylation and Hydrohydroxymethylation
The hydroformylation reaction is a powerful C–C bond forming process with extensive industrial applications. The traditional Co- and Rh-based catalysts for this transformation are capable of converting alkenes into homologous aldehydes using syngas.[5] The possibility of using the CO/H2O couple was investigated by Reppe and Vetter as early as 1953.[6] Besides being one of the earliest applications of the WGSR to organic synthesis, the Reppe-type hydroformylation reaction represents an exception in the landscape of WGSR-driven organic reactions, because CO acts both as carbon source and reducing agent (Scheme 12).
Scheme 12.

Reducing equivalents in hydroformylation reactions.
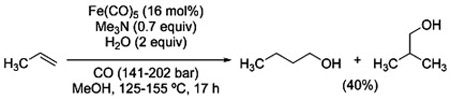
The seminal report by Reppe, part of a thorough study on the chemistry of hydrido-metal-carbonyl complexes, illustrates the conversion of terminal alkenes into homologated alcohols (via the corresponding aldehydes) using Fe(CO)5 in combination with trimethylamine [Eq. (27)]. Strictly speaking, therefore, the term hydrohydroxymethylation would be more appropriate for this transformation.
These results established the basis for further elaborations that took place beginning in the 1970s, contemporaneous with the development of homogeneous catalysis for the WGSR. Thus, Pettit et al. investigated the use of other metal–carbonyl complexes in the Reppe-type hydroformylation of propylene
 |
(27) |
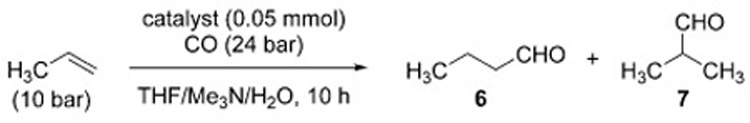
and obtained higher efficiency and selectivity for the linear aldehyde 6 relative to the Fe(CO)5 system (Table 4).[19]
Table 4:
Performance of metal carbonyls in the Reppe-type hydroformylation reaction.
 | ||||
|---|---|---|---|---|
| Entry | Metal catalyst | T [°C] | TON[a] | 6:7 ratio |
| 1 | Fe(CO)5 | 110 | 5.2 | 1:1 |
| 2 | Ru3(CO)12 | 100 | 47 | 11.5:1 |
| 3 | H4Ru4(CO)12 | 100 | 79 | 11:1 |
| 4 | Os3(CO)12 | 180 | 13 | 1.9:1 |
| 5 | Rh6(CO)16 | 125 | 300 | 1.4:1 |
| 6 | Ir4(CO)12 | 125 | 250 | 1.8:1 |
| 7 | (Bu4N)[Pt3(CO)6]5 | 125 | 0.5 | 1.9:1 |
Turnover number (TON)=[mmol(6)+mmol(7)]/mmol(catalyst)
Similarly, Laine compared the performance of Ru and Rh carbonyls in alkaline solution and found that, whereas Ru carbonyls display a higher selectivity for the linear aldehyde, Rh carbonyls are less selective, and capable of reducing the aldehyde product to the alcohol.[153] Numerous other variations of the Reppe-type hydroformylation reaction have been reported, encompassing a wide array of metal catalysts and using either basic, neutral, or acidic conditions (Table 5.). However, unlike some other WGSR-driven reactions, the hydroformylation under WGSR conditions has not risen to the status of a well-established synthetic method. In most cases, harsh conditions are needed and the desired aldehyde is obtained in low yield even from simple, linear olefins. The process is fraught with multiple challenges, ranging from the control of the site selectivity, to the occurrence of hydrogenation of the alkene, overreduction of the aldehyde to the alcohol, or aldol condensation. In contrast, traditional hydro-formylation approaches (with CO/H2) appear much more robust and synthetically useful.[154]
Table 5:
Reaction conditions for the hydroformylation reaction.
| Entry | Metal catalyst | Additives | T [°C] | Pco [bar] | Ref. |
|---|---|---|---|---|---|
| 1 | Rh2O3/Fe(CO)5 | N-methylpyrrolidine | 100–200 | 203 | [158] |
| 2 | Fe, Ru, Os, Rh, Ir, Pt carbonyl complexes | Me3N | 100–180 | 24 | [19] |
| 3 | Ru3(CO)12, H4Ru4(CO)12, Rh6(CO)16 | KOH | 135–150 | 55 | [153] |
| 4 | Co2(CO)8/dppe, other phosphines | none, Et3N | 135–195 | 12–150 | [159] |
| 5 | RhH2(O2COH)[P(iPr)3]2 | – | 115 | 15 | [128] |
| 6 | Rh6(CO)16 | aliphatic amines and diamines, pyridine, substituted pyridines |
80 | 5 | [137] |
| 7 | Rh2(CO)3(dppm)2 | LiCl, p-TsOH | 90 | 0.4 | [160] |
| 8 | Fe(CO)5 | NaOH, Et3N | 110–140 | 10–27 | [161] |
| 9 | Rh2(StBu)2(CO)2(TPPTS)2, [Rh(cod)(TPPTS)2](ClO4), RhH(CO)(TPPTS)3 [a] |
– | 80 | 8 | [162] |
| 10 | K[Ru(EDTA-H)Cl]·2H2O | – | 110–130 | 10–40 | [163] |
| 11 | RhCl3·3 H2O on PVP[a] | – | 85–120 | 0.3–1.7 | [164] |
| 12 | [Rh(cod)(amine)2]PF6, [Rh(CO)2(amine)2]PF6 on PVP |
– | 100–140 | 10–56 | [165] |
| 13 | [Ru(OCOEt)(CO)2]n | EtCOOH/EtCOONa | 140 | 18–88 | [166] |
| 14 | [Rh2(μ-S2CBn2)(cod)2] | – | 80 | 1.3–22 | [167] |
TPPTS=(3-C6H4SO3−Na+)3P, PVP=poly(4-vinylpyridine).
The boundary between hydroformylation and hydrohydroxymethylation is not clear-cut. The hydroformylation reaction is often accompanied by overreduction of the aldehyde to the alcohol. The ease of carbonyl reduction under the forcing conditions required for hydroformylation has been exploited in the formulation of protocols that yield hydrohydroxymethylation products predominantly. This is true for the original report by Reppe, as well as a number of other cases.[133,155,156] For example, allyl alcohol can be converted to 1,4-butanediol under WGSR conditions in the presence of Rh6(CO)16 and TMPDA [Eq. (28)].[155a] Again, however, these methods are of little synthetic utility due to the limited substrate scope and poor efficiency.
 |
(28) |
The mechanism of Reppe-type hydroformylation has been examined by means of identification of reactive intermediates and kinetic studies. The proposed mechanism parallels the earlier proposal by Heck and Breslow for the hydroformylation with Co2(CO)8 and CO/H2,[157] the only difference being that the reactive metal hydride iii is generated via WGSR rather than by oxidative addition of H2 (Scheme 13).[19] Besides being involved in the coordination/migratory insertion of the olefin to form xxx, intermediate iii is accountable for the reduction of the aldehyde to the alcohol.[153] Intermediate iii can also react along the traditional WGSR manifold to produce H2, which is detected in the reaction mixtures.[164,165]
Scheme 13.

Catalytic cycle for the Reppe-type hydroformylation reaction.
One of the key observations is that, in the case of Fe(CO)5, the reaction displays second order kinetic behavior with respect to Fe(CO)5.[161] This fact is consistent with an alternative mechanistic proposal whereby the acyl iron intermediate xxxiii liberates the product not by means of protonation/reductive elimination (as in Scheme 13), but by attack of iron hydride xxxiv (Scheme 14).[168]
Scheme 14.

Alternative binuclear mechanism for hydroformylation.
3.2.2. Hydroaminomethylation
If the aldehyde produced by hydroformylation is intercepted by an amine, the imine (or iminium ion) formed in situ can undergo reduction, resulting in an overall hydroaminomethylation process [Eq. (29)].
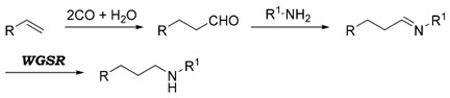 |
(29) |
Interestingly, the hydroaminomethylation sequence was first reported by Reppe and Vetter in the context of their seminal work on the CO/H2O-mediated hydroformylation reaction,[6] and only later revisited (and vastly elaborated) using syngas.[169] The initial conditions used by Reppe were quite harsh (Fe(CO)5, 105–145 °C, 100–200 bar) and the process was inefficient, but further studies identified more effective catalysts and reaction conditions. Iqbal obtained improved results with a mixed Fe(CO)5/Rh2O3 system [Eq. (30)].[170]
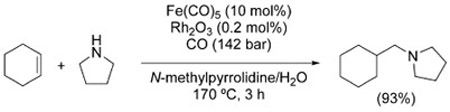 |
(30) |
Other catalysts (metal carbonyls[171,172] and cationic Rh complexes)[173] behave similarly. However, the control of the site-selectivity (linerar vs. branched) and the forcing conditions remained the major issues which hampered further synthetic developments for decades.
Significant advances have been achieved in the last two years thanks to the contributions of Beller and co-workers. Using a combination of Ru3(CO)12 and K2CO3, the hydroaminomethylation reaction proceeds cleanly with a wide range of olefins and primary/secondary amines (Figure 12a).[174] Remarkably, the site-selectivity for the linear product is high (typically > 95:5) even in the absence of ligands. Under these conditions, the catalyst displays selectivity for terminal olefins, whereas internal olefins are unreactive. However, in a modified approach, which employs a phosphinoimidazole ligand, internal olefins become reactive and undergo isomerization to furnish the linear amine product when possible (Figure 12b).[175]
Figure 12.

Hydroaminomethylation reaction under WGSR conditions.
3.2.3. Other Carbonylation Reactions
A large number of carbonylation reactions are carried out using mixtures of CO and H2O, namely hydroxycarbonylation of alkenes,[176] alkynes,[177] and halides[47b] (including the Monsanto process).[178] However, a careful distinction must be made as to the role of CO and H2O. The aforementioned transformations are redox-neutral and employ CO and H2O as reactants, not as a reducing couple. Since the WGSR is not involved, those transformations will not be covered here. Besides hydroformylation and related processes, only a limited number of carbonylation reactions employing CO/H2O are reductive and rely on the WGSR. These include cyclocarbonylation reactions and the carbonylation of alkenes to symmetrical ketones.
The reductive cyclocarbonylation under WGSR conditions was pioneered and considerably developed by Takahashi and co-workers. In an initial report, the reaction of internal alkynes with CO and Rh4(CO)12 under basic conditions afforded furanones (Figure 13a).[179] The reaction was subsequently extended to terminal alkynes under modified conditions (Figure 13b).[180] This pathway is a prerogative of the CO/H2O system, as the use of CO/H2 caused hydrogenation of the alkyne and hydrohydroxymethylation.[181] The site-selectivity under these conditions is poor and mainly substrate-induced, but it was later shown that the selectivity could be significantly improved using more polar solvents, although at the expenses of the conversion.[182]
Figure 13.
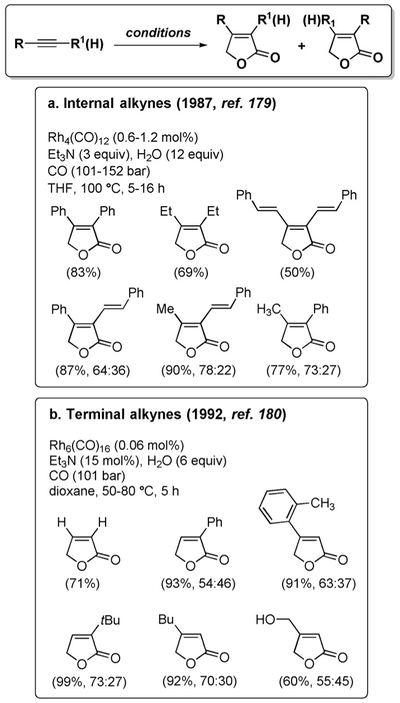
Synthesis of furanones by reductive cyclocarbonylation.
The involvement of the WGSR as the source of reducing equivalents was initially established by carrying out the reaction with D2O, which resulted in full incorporation of D at the 5-position of the furanone.[179] A mechanistic proposal was then put forward which proceeds by double carbonylation of the alkyne and rearrangement to form xxxviii, generation of a Rh-hydride intermediate xl via WGSR, and subsequent reductive elimination to the product (Scheme 15).[182]
Scheme 15.

Catalytic cycle for the formation of furanones.
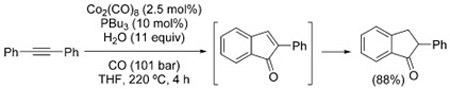
Similar approaches to the synthesis of furanones were also described by other research groups, using PdI2,[183] Co2Rh2/C,[184] and Ru3(CO)12.[185] Interestingly, Takahashi et al. found that, while Co2(CO)8 is inactive in the formation of furanones, at higher temperatures it promotes the formation of indanones from aryl-substituted alkynes, through a C–H insertion process [Eq. (31)].[186] The reaction proceeds via the corresponding indenone, which undergoes reduction under WGSR conditions and was shown to be a competent intermediate.[187]
The propensity of alkynes toward reductive cyclocarbonylation has been pivotal to achieve the synthesis of various other cyclic structures.[188] In particular, by introducing a reactive, tethered functional group, fused cycles such as
 |
(31) |
indolones,[189] tricyclic lactones,[190] isocoumarins,[191] benzofuranones,[192] and isochromanones[193] could be synthesized (Scheme 16).
Scheme 16.

Synthesis of other fused cycles by cyclocarbonylation.
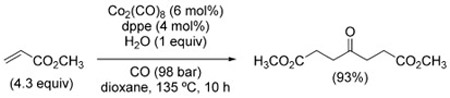
The synthesis of symmetrical ketones from alkenes is another instance of reductive carbonylation reaction that can be carried out under WGSR conditions. The reaction represents an alternative pathway to hydroformylation that becomes predominant when operating in an excess of the olefin. Being related to hydroformylation, it suffers from limitations such as need for harsh conditions and poor control over site-selectivity. Indeed, the method is largely underdeveloped and only a few cases have been reported.[194] For example, dimethyl-4-oxopimelate has been synthesized from methyl acrylate using Co2(CO)8/dppe in 93% yield with respect to H2O (the limiting reagent) [Eq. (32)].[194d]
 |
(32) |
3.2.4. Reductive Alkylation
As was illustrated for reductive amination reactions, stabilized carbanions can be alkylated with aldehydes under WGSR conditions. The reaction proceeds via formation of an
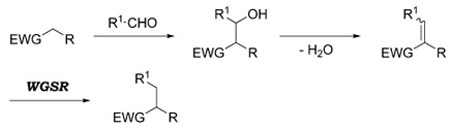 |
(33) |
aldol adduct, elimination of water, and reduction of the activated double bond [Eq. (33)].
The first example of this transformation was reported by Watanabe et al. in 1978.[195] Several ketones were α-methylated with aqueous formaldehyde in the presence of RhCl3·3H2O. Yields were low and mixtures of mono- and dimethylated products were obtained (Scheme 17).
Scheme 17.

Reductive alkylation of ketones.
In a similar fashion, picolines can be homologated with formaldehyde to afford ethylpyridines [Eq. (34)],[196] and arylacetonitriles are α-methylated to arylpropionitriles [Eq. (35)].[197]
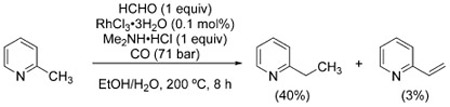 |
(34) |
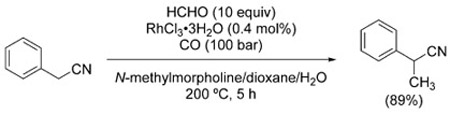 |
(35) |
In an attempt to streamline the synthesis of methyl isobutyl ketone from acetone, Rossi et al. discovered that WGSR conditions can be employed to this end using a Cu/Al2O3 catalyst [Eq. (36)].[198]
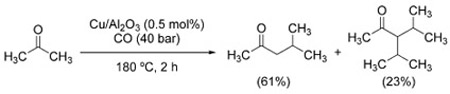 |
(36) |
A more useful synthetic protocol involves the reaction of active methylene compounds with carbonyls in what is formally a reductive Knoevenagel condensation. Chusov et al. reported that methyl cyanoacetate can be alkylated in good yield with aldehydes and ketones using homogeneous (Figure 14a)[199] or heterogeneous (Figure 14b)[120] Rh catalysts.
Figure 14.

Reductive Knoevenagel condensation.
As is the case in the reductive amination, addition of water is not necessary because one equivalent of water is liberated from the condensation reaction. Indeed, it was shown that decarboxylation of the ester moiety in the product takes place when water is added to the reaction mixture (Figure 14c). The authors again propose a catalytic cycle which involves oxidative addition of Rh into the C–OH bond of the aldol adduct,[199] but this proposal is arbitrary and lacks experimental evidence, as discussed in Section 3.1.3. More likely, the Knoevenagel condensation adduct undergoes reduction by a Rh–H species formed via the WGSR.
3.2.5. Carbonyl Allylation
The allylation of carbonyl compounds is a fundamental reaction in organic synthesis owing to its versatility and capacity to build adjacent stereogenic centers with high levels of stereocontrol.[200] The reaction is generally conducted with a pre-formed allylmetal species, typically an allylic borane, silane, or stannane. Catalytic variants have also been developed. Some of the catalytic protocols employ an allylic halide or alcohol derivative in combination with a metal catalyst and a sacrificial metal reducing agent in stoichiometric amount. An example is represented by the allylation under Nozaki–Hiyama–Kishi/Fürstner conditions, which combines catalytic amounts of CrCl2 (or CrCl3) and superstoichiometric amounts of Mn.[201] Although catalytic in the metal that carries out the allylation reaction itself, protocols of this kind are de facto not catalytic, because a stoichiometric amount of metal reducing agent is still required. Truly catalytic methods include the transfer hydrogenation approach described by Krische.[202] Given the prominence and abundant use of allylation reactions in total synthesis endeavors, the discovery that catalysis is possible by means of the WGSR represents an important advance, especially from the point of view of atom economy and environmental concerns. In fact, under WGSR conditions, the only byproducts are CO2 and the conjugate base of the allyl nucleofuge!
The first report from these laboratories, in 2008, identified optimal reaction conditions for the allylation of aldehydes with allyl acetate, using RuCl3 hydrate.[8] Under mild conditions (70–80 °C and 2 bar of CO), a variety of aromatic, aliphatic and α,β-unsaturated aldehydes could be allylated with excellent functional group compatibility (Figure 15).
Figure 15.

Allylation of aldehydes.
One of the key findings is that only 10 mol% of Et3N is needed. The use of a stoichiometric or superstoichiometric amount of base is detrimental. On the other hand, the reaction does not proceed in the absence of base. These results clearly rule out the participation of triethylamine as the stoichiometric reducing agent. Rather, it seems that the base may be involved in the reduction of RuCl3 to the catalytically active Ru0, and/or act as a buffer for the acid liberated as the byproduct. Another important observation concerns the role of halide. The reaction proceeds at a much reduced rate when Ru3(CO)12 is employed instead of RuCl3·n H2O, but the rate is restored upon addition Bu4N+Cl−. It seems that the neutral Ru0–carbonyl complex is not effective in the formation of a π-allylruthenium intermediate, but it can be converted into a more reactive anionic Ru0–Cl complex by the addition of a chloride source. On the basis of these observations, the catalytic cycle shown in Scheme 18 was formulated.
Scheme 18.

Catalytic cycle for the allylation reaction.
The suggested catalytically active species xli is produced either by chloride displacement of CO in Ru3(CO)12, or reduction of RuCl3 to Ru0 by WGSR or perhaps Et3N.[203] Oxidative addition of allyl acetate into Ru0 forms π-allylruthenium(II) complex xlii, which adds to the aldehyde (xliii) and generates the product after protonolysis. This process leaves behind a RuII–hydroxide complex xliv, which is prone to CO migratory insertion to form the Ru-hydroxycarbonyl intermediate xlv. From xlv, the usual WGSR steps (decarboxylation, reductive elimination) regenerate the active Ru0 catalyst along with carbon dioxide and acetic acid as the byproducts.
Recently, the scope of the allylation reaction has been expanded to include substituted allylic acetates. Under similar reaction conditions, allylic acetates bearing a variety of substituents at the 2-position (methyl, phenyl, ester, ketone, acetal) react with aldehydes in good yield (Figure 16).[204] Ketones are not suitable electrophilic partners under these conditions.
Figure 16.

Extension to 2-substituted allylic acetates.
In addition, a recent report by Vasylyev and Alper describes the use of [Rh(TMEDA)(CO)2]+[RhCl2(CO)2]− as an allylation catalyst in the presence of Cs2CO3·n H2O serving both as the base and the water source [Eq. (37)].[205]
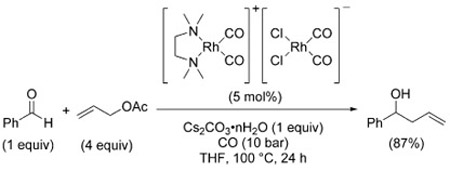 |
(37) |
3.3. Future Opportunities
The water-gas shift reaction has found application in a diverse set of organic reactions. One of the most intriguing, yet underdeveloped aspects is the capacity of the CO/H2O couple to act as a terminal reductant, therefore enabling turnover of catalytic cycles. The carbonyl allylation reaction embodies this approach, resulting in an atom-economic process which is essentially waste-free. In this capacity, the WGSR is aligned with the first of the “grand challenges” identified by the NRC report “Sustainability in the Chemical Industry”, which calls for the reduction of waste and the identification of environmentally benign strategies.[206] Given the number of reductive transformations currently carried out with pre-formed organometallic species or (super)stoichiometric amounts of reducing agents, the impact of the WGSR could be broad, especially in process chemistry.
In the design of new methods that capitalize on the WGSR, one has to keep in mind the electrochemical limits posed by the CO/H2O couple. With a half-cell reduction potential E°= −0.53 V, the WGSR has approximately the same reducing power as gallium(0). Therefore, it is not likely to be successful in regenerating highly electropositive metals (E° < 0 V). On the other hand, metals with positive reduction potentials are good candidates, as they generate an overall positive (hence thermodynamically favorable) electromotive force ΔE° (Scheme 19).
Scheme 19.

Electrochemical limitations for WGSR-driven reductions.
This range corresponds roughly to the right half of the d-transition metal block, and explains why the allylation reaction worked efficiently with Ru (E°Ru(II)/Ru(0) = 0.45 V, ΔE° = 0.98 V), but failed with Fe (E°Fe(II)/Fe(0) = −0.45 V, ΔE° = 0.08 V) or Mn (E°Mn(II)/Mn(0) = −1.19 V, ΔE° = −0.66 V).[207] In view of such electrochemical requirements, opportunities for expanding the range of applicability of the WGSR to many other reductive processes can be envisioned, including additions to carbonyls and imines, conjugate addition, opening of epoxides, pinacol coupling, reductive homocoupling and heterocoupling of halides, etc.
4. Summary and Outlook
Aside from its industrial use in the production of hydrogen, the potential of the WGSR is still underappreciated. This review has highlighted the broad applicability of the WGSR in organic synthesis, ranging from bulk reductions to the synthesis of fine chemicals. Nevertheless, the methods that involve the WGSR are often underdeveloped and lack robustness. The scarcity of interest into developing improved methods with CO/H2O is motivated by the fact that their hydrogenative (or stoichiometric) counterparts are so reliable and well established that there is no need to seek opportunities elsewhere. In addition, the conditions required to initiate the WGSR are often harsh and incompatible with sensitive substrates, although the fine tuning of reaction conditions and the revival of heterogeneous catalysis (in the form of nanoparticle catalysts) have demonstrated the achievability of mild, practical and operationally simple protocols.
Nevertheless, several desirable features are unique to the WGSR. First, is the ability to act as an atom-economic, non-wasteful terminal reductant, which could be leveraged in a variety of reductive transformations. Second, is the high degree of chemoselectivity, as illustrated in the reduction of nitroarenes, conjugated carbonyls, and alkynes. Third, is the access to reaction pathways that are not available using traditional methods, as in cyclocarbonylation reactions.
In our view, opportunities abound for creative applications of the WGSR in organic synthesis. We hope that this overview achieves its intended goal of capturing the imagination of researchers and stimulating new avenues of investigation that leverage the potential of the WGSR.
Acknowledgements
We are grateful to the National Science Foundation (NSF CHE-1151566) for generous financial support. A.A. thanks the University of Illinois and Eli Lilly and Co. for graduate fellowships.
Biographies

Andrea Ambrosi obtained a Laurea Triennale (B.S.) from the University of Torino (Italy) and a Laurea Magistrale (M.S.) from the University of Milano in 2010. While in Milano, he worked under the direction of Prof. Cesare Gennari on the total synthesis of dictyostatin. He is currently a member of the Denmark group at the University of Illinois, where his research interests include the cross-coupling of organosilicon compounds as well as other organometallic reactions.

Scott E. Denmark obtained an S.B. degree from MIT in 1975 and his D.Sc.Tech. (under the direction of Albert Eschenmoser) from the ETH Zürich in 1980. That same year he began his career at the University of Illinois and since 1991 he has been the Reynold C. Fuson Professor of Chemistry. His research interests include the invention of new synthetic reactions, exploratory organoelement chemistry and the origin of stereocontrol in fundamental carbon–carbon bond forming processes.
References
- [1].Häring HW in Industrial Gases Processing, Wiley-VCH, Weinheim, 2008, pp. 135–184. [Google Scholar]
- [2].Tamaru K in Catalytic Ammonia Synthesis (Ed.: Jennings JR), Springer, New York, 1991, pp. 1–18. [Google Scholar]
- [3].“Methanol”: Ott J, Gronemann V, Pontzen F, Fiedler E, Grossmann G, Kersebohm DB, Weiss G, Witte C in Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000. [Google Scholar]
- [4].“Coal Liquefaction”: Kaneko T, Derbyshire F, Makino E, Gray D, Tamura M, Li K in Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000. [Google Scholar]
- [5].Ojima I, Tsai C-Y, Tzamarioudaki M, Bonafoux D, Org. React 2000, 56, 1–354. [Google Scholar]
- [6].Reppe W, Vetter H, Liebigs Ann. Chem 1953, 582, 133–161. [Google Scholar]
- [7].Escaffre P, Thorez A, Kalck P, J. Mol. Catal 1985, 33, 87–118. [Google Scholar]
- [8].Denmark SE, Nguyen ST, Org. Lett 2009, 11, 781–784. [DOI] [PubMed] [Google Scholar]
- [9].Tsuji Y, Mukai T, Kondo T, Watanabe Y, J. Organomet. Chem 1989, 369, C51–C53. [Google Scholar]
- [10].Kondo T, Ono H, Satake N, Mitsudo T, Watanabe Y, Organometallics 1995,14, 1945–1953. [Google Scholar]
- [11].a) Newsome DS, Catal. Rev 1980, 21, 275–318 [Google Scholar]; b) Lloyd L, Ridler DE, Twigg MV in Catalyst Handbook (Ed.: Twigg MV), Manson, London, 1996, pp. 283–339; [Google Scholar]; c) Hinrichsen K-O, Kochloefl K, Muhler M in Handbook of Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2008; [Google Scholar]; d) Ratnasamy C, Wagner JP, Catal Rev 2009, 51, 325–440 [Google Scholar]; e) Mendes D, Mendes A, Madeira LM, Iulianelli A, Sousa JM, Basile A, Asia-Pac. J. Chem. Eng 2010, 5, 111–137; [Google Scholar]; f) Smith RJB, Loganathan M, Murthy SS, Int J. Chem. React. Eng 2010,8, R4; [Google Scholar]; g) Water Gas Shift Reaction. Research, Developments and Applications (Ed.: Smirniotis GKRG), Elsevier, Amsterdam, 2015. [Google Scholar]
- [12].Mond L, Langer C, British Patent 12608, 1888.
- [13].Reddy GK, Smirniotis PG in Water Gas Shift Reaction (Ed.: Smirniotis GKRG), Elsevier, Amsterdam, 2015, pp. 1–20. [Google Scholar]
- [14].Reddy GK, Smirniotis PG in Water Gas Shift Reaction (Ed.: Smirniotis GKRG), Elsevier, Amsterdam, 2015, pp. 21–45. [Google Scholar]
- [15].Reddy GK, Smirniotis PG in Water Gas Shift Reaction (Ed.: Smirniotis GKRG), Elsevier, Amsterdam, 2015, pp. 47–100. [Google Scholar]
- [16].Hieber W, Leutert F, Z. Anorg. Allg. Chem 1932, 204, 145–164. [Google Scholar]
- [17].a) Fenton DM, US Patent 3490872,1970;; b) Fenton DM, US Patent 3539298, 1970;; c) Fenton DM, US Patent 3781364, 1973.
- [18].Laine RM, Rinker RG, Ford PC, J. Am. Chem. Soc 1977, 99, 252–253. [Google Scholar]
- [19].Kang H, Mauldin CH, Cole T, Slegeir W, Cann K, Pettit R, J. Am. Chem. Soc 1977, 99, 8323–8325. [Google Scholar]
- [20].Cheng C-H, Hendriksen DE, Eisenberg R, J. Am. Chem. Soc 1977, 99, 2791–2792. [Google Scholar]
- [21].Zudin VN, Likholobov VA, Yermakov YI, Yeremenko NK, Kinet. Katal 1977,18, 524. [Google Scholar]
- [22].a) Ford PC, Acc. Chem. Res 1981, 14, 31–37; [Google Scholar]; b) Laine RM, Crawford EJ, J. Mol. Catal 1988, 44, 357–387; [Google Scholar]; c) Ford PC, Rokicki A, Adv. Organomet. Chem 1988, 28, 139–217; [Google Scholar]; d) King RB, J. Organomet. Chem 1999, 586, 2–17; [Google Scholar]; e) Jacobs G, Davis BH, Catalysis 2007, 20, 122–285; [Google Scholar]; f) Reddy GK, Smirniotis PG in Water Gas Shift Reaction (Ed.: Smirniotis GKRG), Elsevier, Amsterdam, 2015, pp. 169–205. [Google Scholar]
- [23].Andreeva D, Idakiev V, Tabakova T, Andreev A, Giovanoli R, Appl. Catal A 1996,134, 275–283. [Google Scholar]
- [24].Sakurai H, Ueda A, Kobayashi T, Haruta M, Chem. Commun 1997, 271–272. [Google Scholar]
- [25].Fu Q, Weber A, Flytzani-Stephanopoulos M, Catal. Lett 2001, 77, 87–95. [Google Scholar]
- [26].Idakiev V, Tabakova T, Naydenov A, Yuan ZY, Su BL, Appl. Catal. B 2006, 63, 178–186. [Google Scholar]
- [27].a) Tao F, Ma Z, Phys. Chem. Chem. Phys 2013, 15, 15260–15270; [DOI] [PubMed] [Google Scholar]; b) Rodriguez JA, Senanayake SD, Stacchiola D, Liu P, Hrbek J, Acc. Chem. Res 2014, 47, 773–782; [DOI] [PubMed] [Google Scholar]; c) Flytzani-Stephanopoulos M, Acc. Chem. Res 2014, 47, 783–792; [DOI] [PubMed] [Google Scholar]; d) Liu X, He L, Liu Y-M, Cao Y, Acc. Chem. Res 2014, 47, 793–804. [DOI] [PubMed] [Google Scholar]
- [28].Reddy GK, Smirniotis PG in Water Gas Shift Reaction (Ed.: Smirniotis GKRG), Elsevier, Amsterdam, 2015, pp. 207–223. [Google Scholar]
- [29].Sato S, White JM, J. Am. Chem. Soc 1980, 102, 7206–7210. [Google Scholar]
- [30].a) Cole-Hamilton DJ, J. Chem. Soc. Chem. Commun 1980, 1213–1215; [Google Scholar]; b) King AD, King RB Jr., Sailers EL III, J. Am. Chem. Soc 1981, 103, 1867–1868; [Google Scholar]; c) Choudhury D, Cole-Hamilton DJ, J. Chem. Soc. Dalton Trans 1982, 1885–1893; [Google Scholar]; d) Nagorski H, Mirbach MJ, Mirbach MF, J. Organomet. Chem 1985, 297,171–176; [Google Scholar]; e) Pac C, Miyake K, Matsuo T, Yanagida S, Sakurai H, J. Chem. Soc. Chem. Commun 1986, 1115–1116. [Google Scholar]
- [31].a) Ziessel R, J. Chem. Soc. Chem. Commun 1988,16–17; [Google Scholar]; b) Ziessel R, Angew. Chem. Int. Ed. Engl 1991, 30, 844–847 [Google Scholar]; Angew. Chem 1991, 103, 863–866; [Google Scholar]; c) Ziessel R, J. Am. Chem. Soc 1993, 115, 118–127. [Google Scholar]
- [32].Haber F, Z. Phys. Chem 1909, 68, 726–752. [Google Scholar]
- [33].Rhodes C, Hutchings GJ, Ward AM, Catal Today 1995, 23, 43–58. [Google Scholar]
- [34].a) Burch R, Phys. Chem. Chem. Phys 2006, 8, 5483–5500; [DOI] [PubMed] [Google Scholar]; b) Reddy GK, Smirniotis PG in Water Gas Shift Reaction (Ed.: Smirniotis GKRG), Elsevier, Amsterdam, 2015, pp. 225–261. [Google Scholar]
- [35].a) King AD, King RB Jr., Yang DB, J. Am. Chem. Soc 1980, 102, 1028–1032; [Google Scholar]; b) Sunderlin LS, Squires RR, J. Am. Chem. Soc 1993, 115, 337–343. [Google Scholar]
- [36].a) Torrent M, Solà M, Frenking G, Organometallics 1999, 18, 2801–2812; [Google Scholar]; b) Barrows SE, Inorg. Chem 2004, 43, 8236–8238; [DOI] [PubMed] [Google Scholar]; c) Rozanska X, Vuilleumier R, Inorg. Chem 2008, 47, 8635–8640; [DOI] [PubMed] [Google Scholar]; d) Chen Y, Zhang F, Xu C, Gao J, Zhai D, Zhao Z, J. Phys. Chem. A 2012, 116, 2529–2535; [DOI] [PubMed] [Google Scholar]; e) Schulz H, Görling A, Hieringer W, Inorg. Chem 2013, 52, 4786–4794. [DOI] [PubMed] [Google Scholar]
- [37].a) Thomson WJ, Laine RM, ACS Symp. Ser 1981, 152,133–145; [Google Scholar]; b) Bricker JC, Nagel CC, Shore SG, J. Am. Chem. Soc 1982, 104,1444–1445; [Google Scholar]; c) Vandenberg DM, Suzuki TM, Ford PC, J. Organomet. Chem 1984, 272, 309–320; [Google Scholar]; d) Gross DC, Ford PC, J. Am. Chem. Soc 1985, 107, 585–593; [Google Scholar]; e) Trautman RJ, Gross DC, Ford PC, J. Am. Chem. Soc 1985, 107, 2355–2362; [Google Scholar]; f) Ishida H, Tanaka K, Morimoto M, Tanaka T, Organometallics 1986, 5, 724–730. [Google Scholar]
- [38].a) King AD, King RB, Yang DB, J. Am. Chem. Soc 1981, 103, 2699–2704; [Google Scholar]; b) King RB, King AD Jr., Yang DB, ACS Symp. Ser 1981, 152, 123–132; [Google Scholar]; c) Weiller BH, Liu J-P, Grant ER, J. Am. Chem. Soc 1985, 107, 1595–1604. [Google Scholar]
- [39].Ford PC, Rinker RG, Ungermann C, Laine RM, Landis V, Moya SA, J. Am. Chem. Soc 1978, 100, 4595–4597. [Google Scholar]
- [40].a) Ford PC, Yarrow P, Cohen H, ACS Symp. Ser 1981, 152, 95–105; [Google Scholar]; b) Yarrow P, Cohen H, Ungermann C, Vandenberg D, Ford PC, Rinker RG, J. Mol. Catal 1983, 22, 239–256. [Google Scholar]
- [41].a) Baker EC, Hendriksen DE, Eisenberg R, J. Am. Chem. Soc 1980, 102,1020–1027; [Google Scholar]; b) Mahajan D, Creutz C, Sutin N, Inorg. Chem 1985, 24, 2063–2067. [Google Scholar]
- [42].“Synthesis of anilines”: Hartwig JF, Shekhar S, Shen Q, Barrios-Landeros F in Patai’s Chemistry of Functional Groups, Wiley, New York, 2009. [Google Scholar]
- [43].Iqbal AFM, Tetrahedron Lett. 1971, 12, 3385–3388. [Google Scholar]
- [44].Cann K, Cole T, Slegeir W, Pettit R, J. Am. Chem. Soc 1978, 100, 3969–3971. [Google Scholar]
- [45].a) Landesberg JM, Katz L, Olsen C, J. Org. Chem 1972, 37, 930–936; [Google Scholar]; b) Watanabe Y, Mitsudo T-A, Yamashita M, Takegami Y, Bull. Chem. Soc. Jpn 1975, 48, 1478–1479; [Google Scholar]; c) Des Abbayes H, Alper H, J. Am. Chem. Soc 1977, 99, 98–101. [Google Scholar]
- [46].Alper H, Amaratunga S, Tetrahedron Lett. 1980, 21, 2603–2604. [Google Scholar]
- [47].a) Nomura K, J. Mol. Catal. A 1998, 130,1–28; [Google Scholar]; b) Nomura K, Catal Surv. Jpn 1998, 2, 59–69. [Google Scholar]
- [48].Cole T, Ramage R, Cann K, Pettit R, J. Am. Chem. Soc 1980, 102, 6182–6184. [Google Scholar]
- [49].Ryan RC, Wilemon GM, Dalsanto MP, Pittman CU Jr., J. Mol. Catal 1979, 5, 319–330. [Google Scholar]
- [50].Miyata T, Kondo K, Murai S, Hirashima T, Sonoda N, Angew. Chem. Int. Ed. Engl 1980, 19, 1008 [Google Scholar]; Angew. Chem 1980, 92, 1040–1041. [Google Scholar]
- [51].Kaneda K, Hiraki M, Imanaka T, Teranishi S, J. Mol. Catal 1981, 12, 385–387. [Google Scholar]
- [52].Kaneda K, Fujita K, Takemoto T, Imanaka T, Bull. Chem. Soc. Jpn 1991, 64, 602–612. [Google Scholar]
- [53].Kaneda K, Kuwahara H, Imanaka T, J. Mol. Catal 1994, 88, L267–L270. [Google Scholar]
- [54].Okano T, Fujiwara K, Konishi H, Kiji J, Chem. Lett 1981, 10, 1083–1086. [Google Scholar]
- [55].Watanabe Y, Tsuji Y, Ohsumi T, Takeuchi R, Tetrahedron Lett. 1983, 24,4121–4122. [Google Scholar]
- [56].Watanabe Y, Tsuji Y, Ohsumi T, Takeuchi R, Bull. Chem. Soc. Jpn 1984, 57, 2867–2870. [Google Scholar]
- [57].Takemura Y, Onodera K, Ouchi K, Ind. Eng. Chem. Prod. Res. Dev 1983, 22, 539–542. [Google Scholar]
- [58].a) Alessio E, Zassinovich G, Mestroni G, J. Mol. Catal 1983, 18,113–116; [Google Scholar]; b) Alessio E, Vinzi F, Mestroni G, J. Mol Catal 1984, 22, 327–339. [Google Scholar]
- [59].Alessio E, Clauti G, Mestroni G, J. Mol Catal 1985, 29, 77–98. [Google Scholar]
- [60].a) Hashem KE, Petrignani J-F, Alper H, J. Mol Catal 1984, 26, 285–287; [Google Scholar]; b) Joó F, Alper H, Can. J. Chem 1985, 63,1157–1160. [Google Scholar]
- [61].Sanchez-Delgado RA, Oramas BA, J. Mol Catal 1986, 36, 283–291. [Google Scholar]
- [62].Miura M, Shinohara M, Nomura M, J. Mol. Catal 1988, 45, 151–153. [Google Scholar]
- [63].Shvo Y, Czarkie D, J. Organomet. Chem 1989, 368, 357–365. [Google Scholar]
- [64].a) Nomura K, Ishino M, Hazama M, J. Mol. Catal 1991, 66, L1–L3; [Google Scholar]; b) Nomura K, Ishino M, Hazama M, J. Mol. Catal 1991, 66, L11–L13; [Google Scholar]; c) Nomura K, Ishino M, Hazama M, Bull. Chem. Soc. Jpn 1991, 64, 2624–2628. [Google Scholar]
- [65].a) Nomura K, Ishino M, Hazama M, J. Mol. Catal 1991, 65, L5–L7; [Google Scholar]; b) Nomura K, Ishino M, Hazama M, J. Mol. Catal 1991, 66, L19–L21; [Google Scholar]; c) Nomura K, Ishino M, Hazama M, J. Mol. Catal 1993, 78, 273–282. [Google Scholar]
- [66].Nomura K, Chem. Lett 1991, 20, 1679–1682. [Google Scholar]
- [67].Nomura K, J. Mol. Catal 1992, 73, L1–L4. [Google Scholar]
- [68].Nomura KK, J. Mol. Catal. A 1995, 95, 203–210. [Google Scholar]
- [69].a) Ragaini F, Cenini S, Tollari S, J. Mol. Catal 1993, 85, L1–L5; [Google Scholar]; b) Ragaini F, Cenini S, Gasperini M, J. Mol. Catal. A 2001, 174, 51–57; [Google Scholar]; c) Viganò M, Ragaini F, Buonomenna MG, Lariccia R, Caselli A, Gallo E, Cenini S, Jansen JC, Drioli E, ChemCatChem 2010, 2, 1150–1164. [Google Scholar]
- [70].Macho V, Vojček L, Schmidtova M, Haruštiak M, J. Mol. Catal 1994, 88, 177–184. [Google Scholar]
- [71].Mdleleni MM, Rinker RG, Ford PC, J. Mol. Catal 1994, 89, 283–294. [Google Scholar]
- [72].Ragaini F, Pizzotti M, Cenini S, Abbotto A, Pagani GA, Demartin F, J. Organomet. Chem 1995, 489, 107–112. [Google Scholar]
- [73].Tafesh AM, Beller M, Tetrahedron Lett. 1995, 36,9305–9308. [Google Scholar]
- [74].a) Skupińska J, Smółka G, Każmierowicz W, Ilmużyńska J, React. Kinet. Catal Lett 1995, 54, 59–64; [Google Scholar]; b) Skupińska J, Zukowska A, Chajewski A, React. Kinet. Catal. Lett 2001, 72, 21–27. [Google Scholar]
- [75].Ragaini F, Cenini S, J. Mol. Catal. A 1996, 105, 145–148. [Google Scholar]
- [76].Moya SA, Pastene R, Sariego R, Sartori R, Aguirre P, Le Bozec H, Polyhedron 1996, 15, 1823–1827. [Google Scholar]
- [77].Skupińska J, Smółka G, React Kinet. Catal. Lett 1998, 63, 313–316. [Google Scholar]
- [78].a) Linares C, Mediavilla M, Pardey AJ, Baricelli P, Pardey CL, Moya SA, Catal. Lett 1998, 50, 183–185; [Google Scholar]; b) Longo C, Alvarez J, Fernández M, Pardey AJ, Moya SA, Baricelli PJ, Mdleleni MM, Polyhedron 2000, 19, 487–493; [Google Scholar]; c) Fernández C, Lujano E, Macias U, Marcano J, Baricelli PJ, Longo C, Moya SA, Solórzano MG, Ortega MC, Pardey AJ, Catal Lett 2004, 95, 143–150; [Google Scholar]; d) Bartolini M, Molina J, Ortega MC, Sojo P, Pardey AJ, Longo C, Moya SA, Feazell RP, J. Chil. Chem. Soc 2007, 52, 1254–1256. [Google Scholar]
- [79].Linares C, Mediavilla M, Pardey AJ, Longo C, Baricelli P, Moya SA, Bol. Soc. Chil Quim 1998, 43, 55–59. [Google Scholar]
- [80].Mediavilla M, Fernández M, Pardey AJ, Baricelli P, Longo C, Sartori R, Moya SA, Bol. Soc. Chil Quim 1998, 43, 359–362. [Google Scholar]
- [81].Pardey AJ, Fernández M, Longo C, Lujano E, Baricelli P, Guerrero J, Moya SA, React Kinet. Catal Lett 1998, 65, 315–320. [Google Scholar]
- [82].a) Pardey AJ, Fernández M, Alvarez J, Urbina C, Moronta D, Leon V, Longo C, Baricelli PJ, Moya SA, J. Mol. Catal. A 2000, 164, 225–234; [Google Scholar]; b) Mayora J, Fernandez M, Alvarez J, Ortega M, Pardey AJ, Longo C, Baricelli PJ, Lujano E, Moya SA, Bol Soc. Chil Quim 2001, 46, 121–129; [Google Scholar]; c) Farkas ME, Rodríguez E, Longo C, Monasterios M, Ortega MC, Rivas AB, Pardey AJ, Lòpez R, Moya SA, J. Chil. Chem. Soc 2006, 51, 829–835. [Google Scholar]
- [83].Pardey AJ, Fernández M, Rivas AB, Ortega MC, Urbina C, Moronta D, Longo C, Mediavilla M, Baricelli PJ, Moya SA, Inorg. Chim. Acta 2002, 329, 22–30. [Google Scholar]
- [84].Zhu H, Mizugaki T, Ebitani K, Kaneda K, J. Appl. Polym. Sci 2001, 80, 447–453. [Google Scholar]
- [85].Jiang J, Mei J, Wang Y, Wen F, Jin Z, Appl Catal A 2002, 224, 21–25. [Google Scholar]
- [86].Mdleleni MM, Rinker RG, Ford PC, J. Mol. Catal. A 2003, 204, 125–131. [Google Scholar]
- [87].a) Liu X. – z., Lu S.-w., Chem. Lett 2003, 32,1142–1143 [Google Scholar]; b) Liu X, Lu S, J. Mol. Catal. A 2004, 212, 127–130; [Google Scholar]; c) Liu X. – z., Lu S.-w., J. Mol. Catal. A 2009, 300, 36–40. [Google Scholar]
- [88].a) Pardey AJ, Rojas AD, Yánez JE, Betancourt P, Scott C, Chinea C, Urbina C, Moronta D, Longo C, Polyhedron 2005, 24,511–519; [Google Scholar]; b) Yánez JE, Rivas AB, Alvarez J, Ortega MC, Pardey AJ, Longo C, Feazell RP, J. Coord. Chem 2006, 59, 1719–1728. [Google Scholar]
- [89].Liu L, Qiao B, Chen Z, Zhang J, Deng Y, Chem. Commun 2009, 653–655. [DOI] [PubMed] [Google Scholar]
- [90].He L, Wang L-C, Sun H, Ni J, Cao Y, He H-Y, Fan K-N, Angew. Chem. Int. Ed 2009, 48, 9538–9541 [DOI] [PubMed] [Google Scholar]; Angew. Chem 2009, 121, 9702–9705. [Google Scholar]
- [91].Mikami Y, Noujima A, Mitsudome T, Mizugaki T, Jitsukawa K, Kaneda K, Chem. Lett 2010, 39, 223–225. [DOI] [PubMed] [Google Scholar]
- [92].Pietrowski M, Green Chem. 2011, 13, 1633–1635. [Google Scholar]
- [93].Maeno Z, Mitsudome T, Mizugaki T, Jitsukawa K, Kaneda K, Chem. Commun 2014, 50, 6526–6529. [DOI] [PubMed] [Google Scholar]
- [94].Westerhaus FA, Sorribes I, Wienhöfer G, Junge K, Beller M, Synlett 2015, 26, 313–317. [Google Scholar]
- [95].Krogul A, Litwinienko G, Org. Process Res. Dev 2015, 19, 2017–2021. [Google Scholar]
- [96].a) Blaser H-U, Steiner H, Studer M, ChemCatChem 2009, 1, 210–221; [Google Scholar]; b) Pietrowski M, Curr. Org. Synth 2012, 9, 470–487. [Google Scholar]
- [97].Tafesh AM, Weiguny J, Chem. Rev 1996, 96, 2035–2052. [DOI] [PubMed] [Google Scholar]
- [98].Thurkauf A, de Costa B, Berger P, Paul S, Rice KC, Labelled Compd J. Radiopharm. 1991, 29, 125–129. [Google Scholar]
- [99].a) Kaneda K, Mori T, Kobayashi M, Imanaka T, Teranishi S, Chem. Lett 1985, 14, 1339–1342; [Google Scholar]; b) Kaneda K, Takemoto T, Imanaka T, Chem. Lett 1988, 17,1759–1762 [Google Scholar]; c) Kaneda K, Takemoto T, Kitaoka K, Imanaka T, Organometallics 1991, 10, 846–850. [Google Scholar]
- [100].a) Pizzotti M, Porta F, Cenini S, Demartin F, Masciocchi N, J. Organomet. Chem 1987, 330, 265–278; [Google Scholar]; b) Han SH, Geoffroy GL, Polyhedron 1988, 7, 2331–2339; [Google Scholar]; c) Leconte P, Metz F, Mortreux A, Osborn JA, Paul F, Petit F, Pillot A, J. Chem. Soc. Chem. Commun 1990,1616–1617; [Google Scholar]; d) Glueck DS, Wu J, Hollander FJ, Bergman RG, J. Am. Chem. Soc 1991, 113, 2041–2054; [Google Scholar]; e) Gargulak JD, Berry AJ, Noirot MD, Gladfelter WL, J. Am. Chem. Soc 1992, 114, 8933–8945; [Google Scholar]; f) Ragaini F, Cenini S, Demartin F, Organometallics 1994, 13, 1178–1189; [Google Scholar]; g) Skoog SJ, Campbell JP, Gladfelter WL, Organometallics 1994, 13, 4137–4139; [Google Scholar]; h) Skoog SJ, Gladfelter WL, J. Am. Chem. Soc 1997, 119, 11049–11060. [Google Scholar]
- [101].Bhaduri S, Gopalkrishnan KS, Clegg W, Jones PG, Sheldrick GM, Stalke D, J. Chem. Soc. Dalton Trans 1984, 1765–1767. [Google Scholar]
- [102].Paul F, Fischer J, Ochsenbein P, Osborn JA, Organometallics 1998, 17, 2199–2206. [Google Scholar]
- [103].Sonoda N, Kondo K, Nagano K, Kambe N, Morimoto F, Angew. Chem. Int. Ed. Engl 1980, 19,308–309 [Google Scholar]; Angew. Chem 1980, 92, 317–318. [Google Scholar]
- [104].Ragaini F, Cenini S, Gallo E, Caselli A, Fantauzzi S, Curr. Org. Chem 2006, 10, 1479–1510. [Google Scholar]
- [105].Cenini S, Ragaini F, Catalytic Reductive Carbonylation of Organic Nitro Compounds, Springer, Dordrecht, 1997. [Google Scholar]
- [106].a) Watanabe Y, Suzuki N, Shim SC, Yamamoto M, Mitsudo T.-a, Takegami Y, Chem. Lett 1980, 9, 429–430; [Google Scholar]; b) Watanabe Y, Suzuki N, Tsuji Y, Shim SC, Mitsudo T.-a., Bull. Chem. Soc. Jpn 1982, 55, 1116–1120. [Google Scholar]
- [107].Park JW, Chung YK, ACS Catal. 2015, 5, 4846–4850. [Google Scholar]
- [108].Cenini S, Ragaini F in Catalytic Reductive Carbonylation of Organic Nitro Compounds, Springer, Dordrecht, 1997, pp. 132–176. [Google Scholar]
- [109].Iqbal AFM, J. Org. Chem 1972, 37, 2791–2793. [Google Scholar]
- [110].Watanabe Y, Suzuki N, Tsuji Y, Bull Chem. Soc. Jpn 1982, 55, 2445–2449. [Google Scholar]
- [111].Cenini S, Bettettini E, Fedele M, Tollari S, J. Mol. Catal. A 1996, 111,37–41. [Google Scholar]
- [112].Umeda R, Kouno H, Kitagawa T, Okamoto T, Kawashima K, Mashino T, Nishiyama Y, Heteroat. Chem 2014, 25, 698–703. [Google Scholar]
- [113].Ragaini F, Sportiello P, Cenini S, J. Organomet. Chem 1999, 577, 283–291. [Google Scholar]
- [114].a) Wang X, Lu S, Yu Z, Adv. Synth. Catal 2004, 346,929–932; [Google Scholar]; b) Wang X, Li P, Yuan X, Lu S, J. Mol. Catal. A 2006, 253, 261–264. [Google Scholar]
- [115].Radhi MA, Pályi G, Markó L, J. Mol. Catal 1983, 22, 195–203. [Google Scholar]
- [116].Watanabe Y, Yamamoto M, Mitsudo T.-a., Takegami Y, Tetrahedron Lett. 1978, 19, 1289–1290. [Google Scholar]
- [117].Sugi Y, Matsuda A, Bando K.-i., Murata K, Chem. Lett 1979, 8, 363–364. [Google Scholar]
- [118].Chusov D, List B, Angew. Chem. Int. Ed 2014, 53, 5199–5201 [DOI] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 5299–5302. [Google Scholar]
- [119].Kolesnikov PN, Yagafarov NZ, Usanov DL, Maleev VI, Chusov D, Org. Lett 2015, 17, 173–175. [DOI] [PubMed] [Google Scholar]
- [120].Yagafarov NZ, Usanov DL, Moskovets AP, Kagramanov ND, Maleev VI, Chusov D, ChemCatChem 2015, 7, 2590–2593. [Google Scholar]
- [121].a) Murahashi SI, Imada Y, Hirai Y, Tetrahedron Lett 1987, 28, 77–80; [Google Scholar]; b) Murahashi S-I, Imada Y, Hirai Y, Bull. Chem. Soc. Jpn 1989, 62, 2968–2976. [Google Scholar]
- [122].Laine RM, Thomas DW, Cary LW, J. Org. Chem 1979, 44, 4964–4966. [Google Scholar]
- [123].Fish RH, Thormodsen AD, Cremer GA, J. Am. Chem. Soc 1982, 104, 5234–5237. [Google Scholar]
- [124].a) Lynch TJ, Banah M, McDougall M, Kaesz HD, Porter CR, J. Mol. Catal 1982, 17, 109–113; [Google Scholar]; b) Lynch TJ, Banah M, Kaesz HD, Porter CR, J. Org. Chem 1984, 49,1266–1270. [Google Scholar]
- [125].He L, Yu F-J, Lou X-B, Cao Y, He H-Y, Fan K-N, Chem. Commun 2010, 46, 1553–1555. [DOI] [PubMed] [Google Scholar]
- [126].Watanabe Y, Takatsuki K, Takegami Y, Tetrahedron Lett. 1978, 19, 3369–3370. [Google Scholar]
- [127].Markó L, Radhi MA, Ötvös I, J. Organomet. Chem 1981, 218, 369–376. [Google Scholar]
- [128].Okano T, Kobayashi T, Konishi H, Kiji J, Bull. Chem. Soc. Jpn 1981, 54, 3799–3805. [Google Scholar]
- [129].a) Kaneda K, Yasumura M, Imanaka T, Teranishi S, J. Chem. Soc. Chem. Commun 1982, 935–936; [Google Scholar]; b) Kaneda K, Kobayashi M, Imanaka T, Teranishi S, Nippon Kagaku Kaishi 1985, 494–502. [Google Scholar]
- [130].Tlili A, Schranck J, Neumann H, Beller M, Chem. Eur. J 2012, 18,15935–15939. [DOI] [PubMed] [Google Scholar]
- [131].Natte K, Li W, Zhou S, Neumann H, Wu X-F, Tetrahedron Lett. 2015, 56, 1118–1121. [Google Scholar]
- [132].Cheng C-H, Kuritzkes L, Eisenberg R, J. Organomet. Chem 1980, 190, C21–C24. [Google Scholar]
- [133].Palágyi J, Markó L, J. Organomet. Chem 1982, 236, 343–347. [Google Scholar]
- [134].a) Kitamura T, Sakamoto N, Joh T, Chem. Lett 1973, 379–382; [Google Scholar]; b) Kitamura T, Joh T, Hagihara N, Chem. Lett 1975, 203–206. [Google Scholar]
- [135].Joh T, Fujiwara K, Takahashi S, Bull. Chem. Soc. Jpn 1993, 66, 978–980. [Google Scholar]
- [136].Tian F, Lu S, Synlett 2004, 1953–1956. [Google Scholar]
- [137].Kaneda K, Yasumura M, Hiraki M, Imanaka T, Teranishi S, Chem. Lett 1981, 1763–1766. [Google Scholar]
- [138].Kaspar J, Spogliarich R, Cernogoraz A, Graziani M, J. Organomet. Chem 1983, 255, 371–376. [Google Scholar]
- [139].Li S-S, Liu X, Liu Y-M, He H-Y, Fan K-N, Cao Y, Chem. Commun 2014, 50, 5626–5628. [DOI] [PubMed] [Google Scholar]
- [140].Takegami Y, Watanabe Y, Mitsudo T, Kanaya I, Masada H, Bull. Chem. Soc. Jpn 1968, 41, 158–161. [Google Scholar]
- [141].Mikami Y, Noujima A, Mitsudome T, Mizugaki T, Jitsukawa K, Kaneda K, Tetrahedron Lett. 2010, 51, 5466–5468. [Google Scholar]
- [142].a) Mitsudome T, Noujima A, Mikami Y, Mizugaki T, Jitsukawa K, Kaneda K, Chem. Eur. J 2010, 16, 11818–11821; [DOI] [PubMed] [Google Scholar]; b) Noujima A, Mitsudome T, Mizugaki T, Jitsukawa K, Kaneda K, Molecules 2011, 16, 8209–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Ni J, He L, Liu Y-M, Cao Y, He H-Y, Fan K-N, Chem. Commun 2011, 47, 812–814. [DOI] [PubMed] [Google Scholar]
- [144].a) Baeckvall JE, Boekman F, Blomberg MRA, J. Am. Chem. Soc 1992, 114, 534–538; [Google Scholar]; b) Bi S, Wang J, Liu L, Li P, Lin Z, Organometallics 2012, 31, 6139–6147. [Google Scholar]
- [145].Okano T, Fujiwara K, Konishi H, Kiji J, Bull. Chem. Soc. Jpn 1982, 55, 1975–1976. [Google Scholar]
- [146].a) Cavinato G, Ronchin L, Toniolo L, J. Mol. Catal. A 1995, 102,129–134; [Google Scholar]; b) Cavinato G, Toniolo L, J. Mol. Catal. A 1996, 105, 9–15. [Google Scholar]
- [147].Shim SC, Antebi S, Alper H, Tetrahedron Lett. 1985, 26, 1935–1938. [Google Scholar]
- [148].Nishiyama Y, Katsuen S, Jounen H, Hamanaka S, Ogawa A, Sonoda N, Heteroat. Chem 1990, 1, 467–474. [Google Scholar]
- [149].Cavinato G, Pasqualetto M, Ronchin L, Toniolo L, J. Mol. Catal.. A 1997, 125, 15–22. [Google Scholar]
- [150].Trabuco E, Ford PC, J. Mol. Catal. A 1999, 148, 1–7. [Google Scholar]
- [151].Pardey AJ, Morillo B, Alvarez J, Yanez JE, Ortega M, Longo C, Catal. Lett 2005, 104, 141–150. [Google Scholar]
- [152].Brunet J-J, Taillefer M, J. Organomet. Chem 1988, 348, C5–C8. [Google Scholar]
- [153].Laine RM, J. Am. Chem. Soc 1978, 100, 6451–6454. [Google Scholar]
- [154].a) Franke R, Selent D, Börner A, Chem. Rev 2012, 112,5675–5732; [DOI] [PubMed] [Google Scholar]; b) Pospech J, Fleischer I, Franke R, Buchholz S, Beller M, Angew. Chem. Int. Ed 2013, 52, 2852–2872; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 2922–2944. [Google Scholar]
- [155].a) Kaneda K, Imanaka T, Teranishi S, Chem. Lett 1983, 12, 1465–1466; [Google Scholar]; b) Kaneda K, Kuwahara H, Imanaka T, J. Mol. Catal 1992, 72, L27–L30. [Google Scholar]
- [156].Hirva P, Venalainen T, Pakkanen TA, J. Catal 1994, 148, 722–728. [Google Scholar]
- [157].Heck RF, Breslow DS, J. Am. Chem. Soc 1961, 83, 4023–4027. [Google Scholar]
- [158].Henrici-Olivé G, Olivé S, Transition Met. Chem 1976, 1, 77–93. [Google Scholar]
- [159].a) Murata K, Matsuda A, Bando K-I, Sugi Y, J. Chem. Soc. Chem. Commun 1979, 785–786; [Google Scholar]; b) Murata K, Matsuda A, Bull. Chem. Soc. Jpn 1981, 54, 245–248. [Google Scholar]
- [160].Kubiak CP, Woodcock C, Eisenberg R, Inorg. Chem 1982, 21, 2119–2129. [Google Scholar]
- [161].Massoudi R, Kim JH, King RB, King AD, J. Am. Chem. Soc 1987, 109, 7428–7433. [Google Scholar]
- [162].Escaffre P, Thorez A, Kalck P, J. Chem. Soc. Chem. Commun 1987, 146–147. [Google Scholar]
- [163].Khan MMT, Halligudi SB, Abdi SHR, J. Mol. Catal 1988, 48, 7–9. [Google Scholar]
- [164].Mdleleni MM, Rinker RG, Ford PC, Inorg. Chim. Acta 1998, 270, 345–352. [Google Scholar]
- [165].a) Hung-Low F, Uzcátegui GC, Ortega MC, Rivas AB, Yanez JE, Alvarez J, Pardey AJ, Longo C, Catal. Today 2005, 107–108, 273–281; [Google Scholar]; b) Pardey AJ, Uzcátegui GC, Hung-Low F, Rivas AB, Yánez JE, Ortega MC, Longo C, Aguirre P, Moya SA, J. Mol. Catal. A 2005, 239, 205–214. [Google Scholar]
- [166].Fachinetti G, Funaioli T, Marchetti F, Chem. Commun 2005, 2912–2914. [DOI] [PubMed] [Google Scholar]
- [167].Vargas R, Rivas AB, Suárez JD, Chaparros I, Ortega MC, Pardey AJ, Longo C, Pérez-Torrente JJ, Oro LA, Catal. Lett 2009, 130, 470–475. [Google Scholar]
- [168].Barborak JC, Cann K, Organometallics 1982, 1, 1726–1728. [Google Scholar]
- [169].a) Eilbracht P, Bärfacker L, Buss C, Hollmann C, Kitsos-Rzychon BE, Kranemann CL, Rische T, Roggenbuck R, Schmidt A, Chem. Rev 1999, 99, 3329–3366; [DOI] [PubMed] [Google Scholar]; b) Crozet D, Urrutigoity M, Kalck P, ChemCatChem 2011, 3, 1102–1118. [Google Scholar]
- [170].Iqbal AFM, Helv. Chim. Acta 1971, 54, 1440–1445. [DOI] [PubMed] [Google Scholar]
- [171].Laine RM, J. Org. Chem 1980, 45, 3370–3372. [Google Scholar]
- [172].Murata K, Matsuda A, Masuda T, J. Mol. Catal 1984, 23, 121–132. [Google Scholar]
- [173].Jachimowicz F, Raksis JW, J. Org. Chem 1982, 47, 445–447. [Google Scholar]
- [174].Gülak S, Wu L, Liu Q, Franke R, Jackstell R, Beller M, Angew. Chem. Int. Ed 2014, 53, 7320–7323 [DOI] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 7448–7451. [Google Scholar]
- [175].Liu J, Kubis C, Franke R, Jackstell R, Beller M, ACS Catal 2016, 907–912. [Google Scholar]
- [176].Kiss G, Chem. Rev 2001, 101, 3435–3456. [DOI] [PubMed] [Google Scholar]
- [177].Quintero-Duque S, Dyballa KM, Fleischer I, Tetrahedron Lett. 2015, 56, 2634–2650. [Google Scholar]
- [178].“Acetic Acid”: Le Berre C, Serp P, Kalck P, Torrence GP in Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000. [Google Scholar]
- [179].Doyama K, Joh T, Onitsuka K, Shiohara T, Takahashi S, J. Chem. Soc. Chem. Commun 1987, 649–650. [Google Scholar]
- [180].Joh T, Nagata H, Takahashi S, Chem. Lett 1992, 21, 1305–1308. [Google Scholar]
- [181].Joh T, Doyama K, Onitsuka K, Shiohara T, Takahashi S, Organometallics 1991, 10, 2493–2498. [Google Scholar]
- [182].Joh T, Nagata H, Takahashi S, Inorg. Chim. Acta 1994, 220, 45–53. [Google Scholar]
- [183].a) Gabriele B, Salerno G, Costa M, Chiusoli GP, Tetrahedron Lett. 1999, 40, 989–990; [Google Scholar]; b) Chiusoli GP, Costa M, Cucchia L, Gabriele B, Salerno G, Veltri L, J. Mol. Catal. A 2003, 204–205, 133–142. [Google Scholar]
- [184].Park KH, Kim SY, Chung YK, Org. Biomol. Chem 2005, 3, 395–398. [DOI] [PubMed] [Google Scholar]
- [185].Huang Q, Hua R, Catal. Commun 2007, 8, 1031–1035. [Google Scholar]
- [186].Doyama K, Fujiwara K, Joh T, Maeshima K, Takahashi S, Chem. Lett 1988, 17, 901–904. [Google Scholar]
- [187].Joh T, Boyama K, Fujiwara K, Maeshima K, Takahashi S, Organometallics 1991, 10, 508–513. [Google Scholar]
- [188].a) Zhang S-W, Kaneko T, Yoneda E, Sugioka T, Takahashi S, Inorg. Chim. Acta 1999, 296, 195–203; [Google Scholar]; b) Zhang S-W, Sugioka T, Takahashi S, J. Mol. Catal. A 1999, 143, 211–228; [Google Scholar]; c) Huang Q, Hua R, Chem. Eur. J 2009, 15, 3817–3822. [DOI] [PubMed] [Google Scholar]
- [189].Hirao K, Morii N, Joh T, Takahashi S, Tetrahedron Lett. 1995, 36, 6243–6246. [Google Scholar]
- [190].Sugioka T, Zhang S-W, Morii N, Joh T, Takahashi S, Chem. Lett 1996, 25, 249–250. [Google Scholar]
- [191].Sugioka T, Yoneda E, Onitsuka K, Zhang S-W, Takahashi S, Tetrahedron Lett. 1997, 38, 4989–4992. [Google Scholar]
- [192].Yoneda E, Sugioka T, Hirao K, Zhang S-W, Takahashi S, J. Chem. Soc. Perkin Trans. 1 1998, 477–483. [Google Scholar]
- [193].Yoneda E, Kaneko T, Zhang S-W, Takahashi S, Tetrahedron Lett. 1998, 39, 5061–5064. [Google Scholar]
- [194].a) Murata K, Matsuda A, Chem. Lett 1980, 9, 11–12; [Google Scholar]; b) Murata K, Matsuda A, Bull. Chem. Soc. Jpn 1981, 54, 249–252; [Google Scholar]; c) Murata K, Matsuda A, Bull. Chem. Soc. Jpn 1981, 54, 2089–2092; [Google Scholar]; d) Murata K, Matsuda A, Bull. Chem. Soc. Jpn 1982, 55, 2195–2199. [Google Scholar]
- [195].Watanabe Y, Shimizu Y, Takatsuki K, Takegami Y, Chem. Lett 1978, 7, 215–216. [Google Scholar]
- [196].Watanabe Y, Takatsuki K, Yamamoto M, Sakamoto F, Tsuji Y, Nippon Kagaku Kaishi 1985, 507–511. [Google Scholar]
- [197].Takeuchi K, Sugi Y, Matsuzaki T, Arakawa H, Bando K-I, C1 Mol. Chem 1986, 1, 407–410. [Google Scholar]
- [198].Pina CD, Falletta E, Rossi M, Gargano M, Giannoccaro P, Ciriminna R, Pagliaro M, Appl. Catal. A 2007, 321, 35–39. [Google Scholar]
- [199].Kolesnikov PN, Usanov DL, Barablina EA, Maleev VI, Chusov D, Org. Lett 2014, 16, 5068–5071. [DOI] [PubMed] [Google Scholar]
- [200].a) Denmark SE, Almstead NG in Modern Carbonyl Chemistry (Ed.: Otera J), Wiley-VCH, Weinheim, 2000, pp. 299–401; [Google Scholar]; b) Denmark SE, Fu J, Chem. Rev 2003, 103, 2763–2794; [DOI] [PubMed] [Google Scholar]; c) Yus M, González-Gómez JC, Foubelo F, Chem. Rev 2011, 111, 7774–7854; [DOI] [PubMed] [Google Scholar]; d) Yus M, González-Gómez JC, Foubelo F, Chem. Rev 2013, 113, 5595–5698. [DOI] [PubMed] [Google Scholar]
- [201].a) Fürstner A, Chem. Rev 1999, 99, 991–1045; [DOI] [PubMed] [Google Scholar]; b) Takai K, Org. React 2004, 64, 253–597. [Google Scholar]
- [202].a) Patman RL, Bower JF, Kim IS, Krische MJ, Aldrichimica Acta 2008, 41, 95–104; [PMC free article] [PubMed] [Google Scholar]; b) Bower JF, Kim IS, Patman RL, Krische MJ, Angew. Chem. Int. Ed 2009, 48, 34–46; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2009, 121, 36–48; [Google Scholar]; c) Bower J, Krische MJ, Top. Organomet. Chem 2011, 34, 107–138; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Moran J, Krische MJ, Pure Appl Chem 2012, 84, 1729–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Saito M, Nakagawa H, Shigetomi T, Goto K, US patent 0344510, 2015.
- [204].Denmark SE, Matesich ZD, J. Org. Chem 2014, 79, 5970–5986. [DOI] [PubMed] [Google Scholar]
- [205].Vasylyev M, Alper H, J. Org. Chem 2010, 75, 2710–2713. [DOI] [PubMed] [Google Scholar]
- [206].Sustainability in the Chemical Industry: Grand Challenges and Research Needs. A Workshop Report; The National Academies Press, Washington, DC, 2005. [Google Scholar]
- [207].Matesich Z, Ambrosi A, unpublished results from these laboratories. [Google Scholar]