

Abstract
An electrochemical protocol has been developed for α-oxygenation of cyclic carbamates by using a bicyclic aminoxyl as a mediator and water as the nucleophile. The mediated electrochemical method enables substrate oxygenation to proceed approximately 1 V lower than the redox potential of carbamate substrate. This feature allows for functional-group compatibility that is inaccessilble with conventional Shono oxidations that proceed via direct electrochemical substrate oxidation. This chemistry also represents the first α-functionalization of non-activated cyclic carbamates with oxoammonium as the oxidant.
Keywords: Shono oxidation, Mediated electrolysis, Oxygenation, Carbamate, Hydride transfer, Aminoxyls
Reaching your low potential:
Use of aminoxyls as hydride-transfer mediators enables electrochemical aoxygenation of unactivated cyclic carbamates at low electrode potentials. This method greatly expands the substrate scope beyond conventional Shono oxidations.
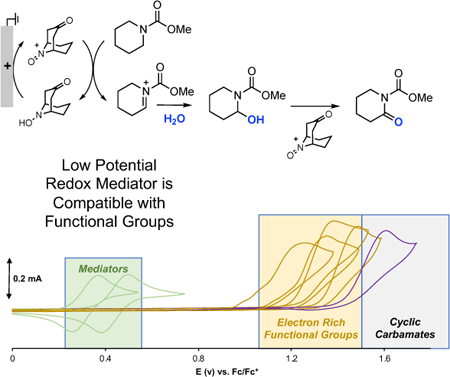
Electrolysis is a versatile approach to achieve redox transformations in organic synthesis that provides a compelling alternative to traditional reagent-based methods.[1,2,3] Shono oxidations (Scheme 1a)[4] are among the most widely employed electrochemical methods for the functionalization of C-H bonds adjacent to nitrogen atoms, and they have been featured in the synthesis of natural products,[5] synthesis and late-stage functionalization of pharmaceuticals,[6] and preparation of drug metabolites.[7] The reactions proceed via direct electron transfer to the electrode, and a sequence of electron-proton-electron transfer (ET-PT-ET) steps affords an iminium species that is trapped by a nucleophile, such as MeOH or H2O. The functional group compatibility of this method for both substrates and nucleophiles is often limited, however, by the high electrode potentials required to initiate electron transfer from the substrate. Various strategies, such as “cation pool” and flow-based methods, have been employed to expand the nucleophile compatibility, but functional groups on the amine-derived substrate remain limited.[8,9] Indirect (i.e., mediated) electrolysis methods provide a means to circumvent this issue.[3] The ET-PT-ET sequence involved in traditional Shono oxidations is formally equivalent to a hydride transfer. A mediator that reacts with substrate via concerted hydride transfer could alleviate the requirement for a high-potential electron transfer step, thereby permitting operation at lower electrode potentials and increasing the functional group compatibility. Herein, we demonstrate this concept through the use of derivatives of the bicyclic aminoxyl ABNO (9-azabicyclo[3.3.1]nonane N-oxyl) as the mediator. The resulting methods show considerably improved scope and functional group compatibility relative to conventional Shono conditions, as well as previously reported indirect electrolysis methods.[10,11,12]
Scheme 1.

Alpha-functionalization of cyclic carbamates.
The functional group limitations of Shono oxidations are illustrated by the attempted α-oxygenation of the 4-(4-methoxyphenyl)-piperidine carbamate 1a. When 1a was subjected to conventional Shono oxidation conditions with water as the nucleophile, the substrate was completely consumed without forming the desired oxygenated product. In contrast, when the unsubstituted piperidine carbamate 1b was subjected to identical conditions, the α-hydroxylated product was obtained in 89% yield (Scheme 2, top). The basis for these observations is evident from cyclic voltammetry (CV) data. Substrate 1a and anisole exhibit oxidation peaks at 1.28 and 1.38 V, respectively vs Fc/Fc+. A nalysis of 1b, however, reveals a peak potential at 1.61 V. These results show that the electron-rich aromatic ring is more readily oxidized than the carbamate fragment in 1a, thereby accounting for the observed decomposition under Shono conditions.
Scheme 2.

Limitations in conventional Shono oxidation..
Use of a hydride-transfer mediator could provide a means to alleviate the functional-group incompatibility evident in Scheme 2. To our knowledge, the only precedent for mediated Shono-type oxidation of unactivated substrates, such as 1a, was reported by Torii and coworkers, using a RuO2/Cl− dual-mediator system.[10] Preliminary tests of this system, however, led to chlorination of the methoxyarene ring rather than oxygenation of the nitrogen heterocycle (see Supporting Information for details). Oxoammonium reagents, derived from one-electron oxidation of aminoxyls, have been used as oxidants for the α -functionalization of activated (benzylic and allylic) carbamates and amides.[11,13] These observations, together with recent studies of aminoxyl-based mediators in electroorganic chemistry by us[14] and others,[15,16,17] prompted us to consider whether aminoxyls could mediate Shono-type α -oxidation of unactivated substrates with water as the nucleophile (cf. Scheme 1b).[18] To test this hypothesis, we initiated experiments with 1a as substrate and evaluated a series of aminoxyls with different steric profiles and redox potentials (Figure 1; redox potentials correspond to the one-electron aminoxyl/oxoammonium redox couple). Constant current conditions (3 mA) were employed, using a reticulated vitreous carbon (RVC) anode and Pt wire counter electrode in an undivided cell (see Supporting Information for full optimization data). The results showed that TEMPO (2,2,6,6-tetramethylpiperidine N-oxyl) and its derivatives were insufficiently reactive to promote substrate oxidation, irrespective of the aminoxyl redox potential. In each case, the reactivity reported previously for α-functionalization of non-activated carbamates with stoichiometric oxoammonium-based substrate was recovered unreacted, even with 4-oxo-TEMPO as mediator. These observations are consistent with the lack of reactivity with oxidants such as TEMPO+ and Bobbitt’s salt.[13] The desired reactivity was observed, however, with the less sterically hindered bicyclic aminoxyls ABNO, PhCO2ABNO, and ketoABNO. Only low yields were observed with the commercially available derivatives, ABNO and ketoABNO. The majority of substrate (81%) was recovered unreacted with ABNO. This low reactivity is attributed to the lower redox potential and hydride acceptor ability of ABNO. Somewhat improved yield was observed with ketoABNO (36%); however, the reaction solution of ketoABNO and 1a was darkened relative to solutions ketoABNO and other substrates (see below). The origin of this observation is not known, but it may reflect formation of a non-productive adduct between the electrophilic ketoABNO+ species and the electron-rich aromatic ring (i.e., a donor-acceptor interaction), which could hinder the desired reactivity and/or lead to side reactions (only 32% of 1a was recovered). The modified ABNO derivative, PhCO2ABNO, which has a redox potential intermediate between ABNO and ketoABNO, was then synthesized and tested as a mediator.[19] This aminoxyl led to considerable improvement in the yield of 2a (62%, with 23% recovery of 1a). The reaction proceeded smoothly at room temperature in a simple undivided cell.
Figure 1.

Reactions with different mediators (1H NMR yields with mesitylene as internal standard).
These bicyclic aminoxyl derivatives were then tested in mediated electrolysis conditions with substrates bearing different functional groups, and the results w ere compared with conventional Shono oxidation conditions (Scheme 3). The Shono conditions led to poor results in each case. For substrates 1c and 1d, the best yield was observed with PhCO2ABNO as mediator (67 and 64%), while the best yield for 2e was obtained with ketoABNO as the mediator (60%).
Scheme 3. Comparison of different α -functionalization conditions for a series of different substrates.

[a] Yields of hydroxylated products. [b] 40 equiv of H2O @ 3 mA, 40 mol% ABNO. [c] 30 equiv of H2O @ 5 mA, 60 mol% PhCO2ABNO. [d] 50 equiv of H2O @ 5 mA, 60 mol% ketoABNO.1H NMR yield was shown with mesitylene as internal standard.
These results prompted us to examine the compatibility of the mediated system toward diverse functional groups, and a robustness screen[20] was conducted by testing the oxidation of 1b in the presence of various additives with PhCO2ABNO as mediator (Table 1, see Supporting Information for an analogous screen with ketoABNO). The data reveal that the reactions proceed well in the presence of m any different functional groups, including several that are oxidatively sensitive. For example, thiophene, benzothiophene, and indole derivatives and anisole were tolerated under the mediated conditions, with >92% of the additive recovered after bulk electrolysis. CV analysis of the individual additives (see below Table 1) shows that none of these functional groups is compatible with conventional Shono oxidation conditions; each of the additives undergoes oxidation at potentials below the potential of 1b. The results in Table 1 also draw attention to the chemoselectivity of the reaction. For example, substrate 1b undergoes selective conversion to 2b in the presence of N-methyl-2-piperidone (11) and 1-(phenylsulfonyl)piperidine (12), both of which could potentially undergo α-oxidation.
Table 1.
Robustness screen for mediated electrolysis with PhCO2ABNO.[a]
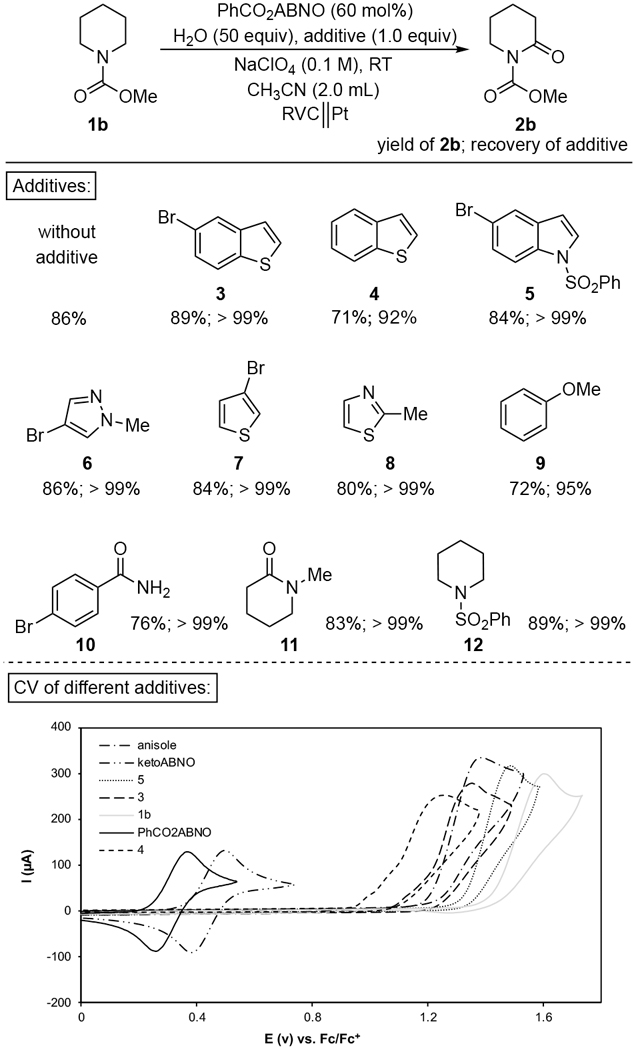 |
Constantcurrent @ 3 mA with PhCO2ABNO as mediator.
Under the optimized conditions, a range of substituted piperidine carbamate derivatives were tested for their ability to undergo α-oxygenation with PhCO2ABNO or ketoABNO as the mediator (Table 2). Good yields were obtained with substrates bearing diverse functional groups, such as substituted arenes, esters, ethers, ketones, halides and sulfonyl groups. Substituted pyrrolidines also proved to be good candidates for the α- oxygenation reaction, furnishing the desired products in improved yields relative to those observed with the piperidine carbamate substrates. Collectively, these results demonstrate the broadest functional group compatibility reported to date for Shono-type oxidations.
Table 2.
 |
These reactions were conducted with 0.2 mmol scale in 2 mL CH3CN (see Supporting Information for conditions).
Isolated yield was shown.
Analysis of the reaction time course provided valuable, and unexpected, insights into the reaction pathway. Formation of the α-oxygenation products corresponds to net four-electron oxidation of the carbamate substrates. The reaction is initiated by one-electron oxidation of the aminoxyl mediator to generate an oxoammonium species that can promote two-electron hydride- transfer oxidation of the substrate, resulting in formation of the hydroxylamine form of the mediator. Oxidation of this species at the electrode will regenerate the oxoammonium species to allow for further substrate oxidation. We anticipated that substrate- derived iminium ion resulting from hydride transfer would be trapped by water to generate a hemiaminal that could undergo the second two-electron oxidation by the mediator to furnish the desired product (Figure 2b, path A). The reaction time course with 1b as the substrate and ketoABNO as the mediator was analyzed, and the data revealed build-up and decay of an intermediate during the reaction (Figure 2a). Subsequent isolation and characterization of the intermediate by1H and13C NMR spectroscopy and high-resolution mass spectrometry revealed this interm ediate to be an adduct of the substrate and ketoABNO (2b’), rather than the expected hemiaminal (Figure 2b). This outcome is rationalized by the nucleophilicity of hydroxylamines (and their tautomeric amine oxides),[21] and by the generation of ketoABNO-H in direct proximity to the iminium ion in the hydride transfer reaction. This species and the time course in Figure 2a also accounts for the high mediator loading in these reactions, as nearly all of the mediator is present in the form of 2b’ at the maximum concentration of this species. Control experiments suggest that this species undergoes negligible hydrolysis under the reaction conditions. Therefore, we propose that formation of 2b forms via hydride transfer from the intermediate 2b’ to generate the iminium species 2b”, which then undergoes hydrolysis. A control experiment confirmed that 2b’ undergoes direct conversion to 2b in the presence of ketoABNO as a mediator.
Figure 2.

Reaction time course (a) and proposed mechanism (b).
In summary, the results described herein represent the first electrochemical Shono-type oxidation of unactivated cyclic amine derivatives that show broad scope and functional group tolerance. This advance has been achieved by use of an aminoxyl mediator that undergoes electrochemical generation at a potential approximately 1.0 V lower than is needed for direct one-electron oxidation of the substrate. The potential needed for oxoammonium generation is also much lower than the potentials associated with outer-sphere ET oxidation of heterocycles and other common substituents and functional groups. The concepts illustrated in this study, specifically the redox-potential-lowering ability of hydride transfer mediators, has significant implications for the development of electrochemical synthetic methods that can achieve broader functional-group tolerance. The mechanistic insights gain from this study provide a foundation for ongoing studies directed toward the development of improved mediated electrolysis methods that operate with lower catalyst loading.
Supplementary Material
Acknowledgements
The authors thank Dr. A. J. J. Lennox and J. E. Nutting for helpful discussions during the course of this project. Financial support was provided from an SIOC fellow ship (for FW), AbbVie, and the NIH (R01 G M 100143 for SSS). Spectroscopic instrumentation was partially supported by the NIH (S10 O D020022) and the NSF (C H E-1048642).
Footnotes
Supporting Information for this article is given via a link at the end of the document.
Conflict of interest:
The authors declare no conflict of interest.
References
- [1].a) Shono T, Electroorganic Synthesis, Academic Press, London, 1990; [Google Scholar]; b) Little RD, Weinberg NL, Electroorganic Synthesis, Marcel Dekker, New York, 1991; [Google Scholar]; c) Torii S, Novel Trends in Electroorganic Synthesis, Springer-Verlag, Tokyo, 1998; [Google Scholar]; d) Lund H, Hammerich O, Organic Electrochemistry, 5th ed, CRC Press, Boca Raton, 2015. [Google Scholar]
- [2].For recent reviews, see:Sperry JB, Wright DL, Chem. Soc. Rev 2006, 35, 605;Yoshida J.-i., Kataoka K, Horcajada R, Nagaki A, Chem. Rev 2008, 108, 2265;Francke R, Beilstein J. Org. Chem 2014, 10, 2858;Yan M, Kawamata Y, Baran PS, Chem. Rev 2017, 117, 13230;Jiang Y, Xu K, Zeng C, Chem. Rev 2018, DOI: 10.1021/acs.chemrev.7b00271;Pletcher D, Green RA, Brown RCD, Chem. Rev 2018, DOI: 10.1021/acs.chemrev.7b00360;Yoshida J.-i., Shimizu A, Hayashi R, Chem. Rev 2017. DOI: 10.1021/acs.chemrev.7b00475;Tang S, Liu Y, Lei A, Chem 2018, 4, 27;Wiebe A, Gieshoff T, Mohle S, Rodrigo E, Zirbes M, Waldvogel SR, Angew. Chem. Int. Ed 2018, DOI: 10.1002/anie.201711060;Möhle S, Zirbes M, Rodrigo E, Gieshoff T, Wiebe A, Waldvogel SR, Angew. Chem. Int. Ed 2018, DOI: 10.1002/anie.201712732;Yang Q-L, Fang P, Mei T-S, Chin. J. Chem 2018, 36, 338.
- [3].For reviews on mediated electrolysis, see:Steckhan E, Angew. Chem. Int. Ed 1986, 28, 683;Ogibin YN, Elinson MN, Nikishin GI, Russ. Chem. Rev 2009, 78, 89;Francke R, Little RD, Chem. Soc. Rev 2014, 43, 2492.
- [4].a) Shono T, Hamaguchi H, Matsumura Y J. Am. Chem. Soc 1975, 97, 4264; [Google Scholar]; b) Shono T, Matsumura Y, Tsubata K, J. Am. Chem. Soc 1981, 103, 1172. [Google Scholar]
- [5].a) Shono T, Matsumura Y, Uchida K, Tsubata K, Makino A, J. Org. Chem 1984, 49, 300; [Google Scholar]; b) Myers EL, de Vries JG, Aggarwal VK, Angew. Chem., Int. Ed 2007, 46, 1893; [DOI] [PubMed] [Google Scholar]; d) Kabeshov MA, Musio B, Murray PRD, Browne DL, Ley SV, Org. Lett 2014, 16, 4618. [DOI] [PubMed] [Google Scholar]
- [6].a) Moeller KD, Wong PL, Bioorg. Med. Chem. Lett 1992, 2, 739; [Google Scholar]; b) Frankowski KJ, Liu R, Milligan GL, Moeller KD, Aubé J, Angew. Chem. Int. Ed 2015, 54, 10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Shono T, Toda T, Oshino N, Drug Metab. Dispos 1981, 9, 481. [PubMed] [Google Scholar]; b) Shono T, Toda T, Oshino N, J. Am. Chem. Soc 1982, 104, 2639. [Google Scholar]
- [8].For leading references, see:Suga S, Okajima M, Fujiwara K, Yoshida J.-i., J. Am. Chem. Soc 2001, 123, 7941;Yoshida J.-i., Suga S, Chem. Eur. J 2002, 8, 2650.
- [9].Modification of the substrate with an “electroauxiliary” provides a strategy to enhance the substrate scope. For leading references, see ref. 2b and the following:Yoshida J.-i., Isoe S, Tetrahedron Lett 1987, 28, 6621;Suga S, Watanabe M, Yoshida J.-i., J. Am. Chem. Soc 2002, 124, 14824;Suga S, Watanabe M, Song C-H, Yoshida J.-i., Electrochemistry 2006, 74, 672.
- [10].Torii S, Inokuchi T, Yukawa T, Chem. Lett 1984, 1063. [Google Scholar]
- [11].Li C, Zeng C-C, Hu L-M, Yang F-L, Yoo SJ, Little RD, Electrochim. Acta 2013, 114, 560. [Google Scholar]
- [12].Zeng, Little and coworkers (ref. 11) reported a dual-mediator Br-/TEMPO system for two-phase electrochemical α-oxygenation, but the method was limited to activated 1,2,3,4-tetrahydroisoquinoline derivatives. As shown in Figure 1, TEMPO is ineffective with the substrates described in the present study [Google Scholar]
- [13].For examples, see:Richter H, Fröhlich R, Daniliuc C-G, Mañcheno OG, Angew. Chem. Int. Ed 2012, 51, 8656;Sun S, Li C, Floreancig PE, Lou H, Liu L, Org. Lett 2015, 17, 1684;Yan C, Liu Y, Wang Q, Org. Lett 2015, 17, 5714;Wang G, Mao Y, Liu L, Org. Lett 2016, 18, 6476;Long H, Wang G, Lu R, Xu M, Zhang K, Qi S, He Y, Bu Y, Liu L, Org. Lett 2017, 19, 2146.
- [14].a) Rafiee M, Miles KC, Stahl SS, J. Am. Chem. Soc 2015, 137, 14751; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Badalyan A, Stahl SS, Nature 2016, 535, 406; [DOI] [PubMed] [Google Scholar]; c) Das A, Stahl SS, Angew. Chem. Int. Ed 2017, 56, 8892; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rafiee M, Wang F, Hruszkewycz DP, Stahl SS J. Am. Chem. Soc 2018, 140, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].For representative leading references, see:Hickey DP, McCammant MS, Giroud F, Sigman MS, Minteer SD, J. Am. Chem. Soc 2014, 136, 15917;Rafiee M, Karimi B, Alizadeh S, ChemElectroChem 2014, 1, 455;Hickey DP, Schiedler DA, Matanovic I, Doan PV, Atanassov P, Minteer SD, Sigman MS, J. Am. Chem. Soc 2015, 137, 16179;Horn EJ, Rosen BR, Chen Y, Tang J, Chen K, Eastgate MD, Baran PS, Nature 2016, 533, 77;Qian X-Y, Li S-Q, Song J, Xu H-C, ACS Catal 2017, 7, 2730;Wu Y, Yi H, Lei A, ACS Catal 2018, 8, 1192;Schille B, Giltzau NO, Francke R, Angew. Chem., Int. Ed 2018, 57, 422.
- [16].For early precedents, see:Semmelhack MF, Chou CS, Cortes DA, J. Am. Chem. Soc 1983, 105, 4492;Inokuchi T, Matsumoto S, Torii S, J. Org. Chem 1991, 56, 2416.
- [17].For a comprehensive review, see:Nutting JE, Rafiee M, Stahl SS Chem. Rev 2018, submitted for publication.
- [18].For leading references on Shono oxidation with water as nucleophile, see, ref. 7 and the following:Okita M, Wakamatsu T, Ban Y, J. Chem. Soc., Chem. Commun 1979, 749.
- [19].For the synthesis of N-oxyl mediators, see:Lauber MB, Stahl SS, ACS Catal 2013, 3, 2612 and ref. 15c.
- [20].Collins KD, Glorius F, Nat. Chem 2013, 5, 597. [DOI] [PubMed] [Google Scholar]
- [21].For relevant discussion, see:Jencks WP, Carriuolo J, J. Am. Chem. Soc 1960, 82, 1778;Kirby AJ, Davies JE, Brandão TAS, da Silva PF, Rocha WR, Nome F, J. Am. Chem. Soc 2006, 128, 12374.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.