

Abstract
Late Na+ current (INaL) significantly contributes to shaping cardiac action potentials (AP) and increased INaL is associated with cardiac arrhythmias. β-adrenergic receptor (βAR) stimulation and its downstream signaling via protein kinase A (PKA) and Ca2+/calmodulin-dependent protein kinase II (CaMKII) pathways are known to regulate INaL. However, it remains unclear how each of these pathways regulates INaL during the AP under physiological conditions. Here we performed AP-clamp experiments in rabbit ventricular myocytes to delineate the impact of each signaling pathway on INaL at different AP phases to understand the arrhythmogenic potential. During the physiological AP (2 Hz, 37°C) we found that INaL had a basal level current independent of PKA, but partially dependent on CaMKII. PAR activation (10 nM isoproterenol, ISO) further enhanced INaL via both PKA and CaMKII pathways. However, PKA predominantly increased INaL early during the AP plateau, whereas CaMKII mainly increased INaL later in the plateau and during rapid repolarization. We also tested the role of key signaling pathways through exchange protein activated by cAMP (Epac), nitric oxide synthase (NOS) and reactive oxygen species (ROS). Direct Epac stimulation enhanced INaL similar to the PAR-induced CaMKII effect, while NOS inhibition prevented the βAR-induced CaMKII-dependent INaL enhancement. ROS generated by NADPH oxidase 2 (NOX2) also contributed to the ISO-induced INaL activation early in the AP. Taken together, our data reveal differential modulations of INaL by PKA and CaMKII signaling pathways at different AP phases. This nuanced and comprehensive view on the changes in INaL during AP deepens our understanding of the important role of INaL in reshaping the cardiac AP and arrhythmogenic potential under elevated sympathetic stimulation, which is relevant for designing therapeutic treatment of arrhythmias under pathological conditions.
Keywords: Late sodium current, action potential, beta-adrenergic stimulation, protein kinase A, Ca2+/calmodulin-dependent kinase II, nitric oxide synthase
Graphical abstract:
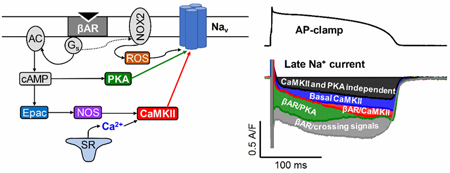
1. INTRODUCTION
Activation of Na+ channel upon excitation leads to a fast, transient Na+ current (INaT) generating the upstroke of action potential (AP). However, under a sustained depolarization such as the plateau phase of AP in ventricular cardiomyocytes, a tiny fraction of Na+ channels may remain open/reopen generating a non-inactivating or persistent Na+ current referred as late Na+ current (INaL) [1]. INaL significantly contributes to shaping cardiac AP and pathological augmentation of INaL is associated with increased risk for cardiac arrhythmias [2]. Several gating modalities of Na+ channels with different voltage-and time- dependent properties have been identified that contribute to increased INaL in pathological states, including early channel bursting [3], late scattered opening [4], window current [5] and non-equilibrium gating [6]. Mutations of Na+ channels linked to long QT syndrome 3 (LQT3) in patients cause increased INaL, and may preferentially affect one of these gating modalities resulting in distinct molecular determinants [7]. Beside inherited genetic defects, several signaling pathways modulate INaL, and are associated with heart diseases like ischemia, cardiomyopathies and heart failure [2]. One such regulatory mechanism is Ca2+/calmodulin-dependent protein kinase II (CaMKII), which causes complex INa gating changes including elevated INaL [8, 9]. CaMKII is upregulated in pathologic states and is associated with enhanced INaL contributing to arrhythmias and cardiac dysfunction [10–12]. β-adrenergic receptor (βAR) stimulation has also been shown to regulate Na+ channels through both cAMP-dependent and independent pathways[13]. βAR stimulation increases whole-cell Na+ channel conductance by protein kinase A (PKA) phosphorylation [14], predominantly via enhanced Na+ channel trafficking to the sarcolemma [15]. In addition, a PKA-independent Gs protein mediated effect on Na+ channel gating has also been suggested [13, 16]. However, most previous studies of cardiac Na+ channel regulation focused on INaT rather than INaL [17–19], and most were conducted under non-physiological conditions. Typically, INaL was recorded under a rectangular voltage command to −20/−30 mV at very low pulse frequency and with strong intracellular Ca2+ buffering. So, no direct data is available as to βAR regulation of INaL during physiological APs, and how different pathways may contribute to those INaL changes. Therefore, the goal of our study was to determine how PAR stimulation modulates INaL during the physiological AP with physiological ionic conditions in rabbit ventricular cardiomyocytes.
βAR stimulation induces complex signaling, including crosstalk between PKA and CaMKII pathways in cardiac myocytes [20]. Much prior work on PKA-CaMKII crosstalk during cardiac βAR stimulation targeted Ca2+ handling [21–24]. However, because INaL is known to be regulated by both PKA[25]and CaMKII [8, 9], we focused here on dissecting PKA- versus CaMKII-dependent mechanisms of INaL modulation during the cardiac AP. Moreover, both PKA and CaMKII can be regulated by posttranslational modifications causing autonomous activity. CaMKIIô autophosphorylation (Thr287), oxidation (Met280 281) and S-nitrosylation (Cys273/290) are well-established modulators of CaMKII activity [26, 27]. The exchange protein directly activated by cAMP (Epac) can also induce CaMKII activation upon PAR stimulation [28, 29]. Similarly, oxidation [30] and S-nitrosylation [31] of PKA have been shown to activate type I PKA independent of cAMP via interprotein disulfide bound formation between the two regulatory subunits. However, it is unknown whether such autonomous PKA activation occurs upon acute βAR stimulation physiologically in adult myocytes. It has also been shown that stimulation of Gq proteins by angiotensin II results in PKA-dependent enhancement of INaT, but CaMKII-dependent enhancement of INaL [32]. However, it remains unknown whether similar mechanism occur upon βAR stimulation and how different posttranslational modifications might affect INaL via PKA and CaMKII autonomous activation. Hence, we aimed to study how PKA and CaMKII mediate INaL under physiological conditions and whether Epac, reactive oxygen species (ROS) and nitric oxide signaling influence these PAR-induced INaL changes during the cardiac AP. Our results indicate that physiological pacing alone causes CaMKII-dependent augmentation of INaL, whereas PAR activation induces additional INaL that is mediated by both PKA and CaMKII (early and late in the AP, respectively).
2. METHODS
All animal handling and laboratory procedures were in accordance with the approved protocols of the Institutional Animal Care and Use Committee at University of California, Davis confirming to the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (8th edition, 2011).
2.1. Animal model, cell isolation
New Zealand White rabbits (male, 3–4 months old, 2.5–3 kg) were purchased from Charles River Laboratories (Wilmington, MA, USA). Rabbits were first injected with heparin (1000 U/kg) and then anesthetized with isoflurane inhalation (3–5%). After achieving deep anesthesia, a standard enzymatic technique was used to isolate ventricular myocytes at 37°C as previously described [33, 34]. Briefly, hearts were mounted on a Langendorff system and retrogradely perfused for 5 min with an oxygenated solution containing (in mmol/L): NaCl 138, KCl 5.4, CaCl2 0.05, MgCl2 1, NaH2PO4 0.33, NaHCO3 10, HEPES 10, glucose 6, pyruvic acid 2.5; at pH=7.4. When blood was removed from the coronary circulation, we added 1 mg/mL type II collagenase (305 U/mg; Worthington Biochemical Co., Lakewood, NJ, USA), 0.05 mg/ml protease type XIV (Sigma-Aldrich Co., St. Louis, MO, USA) and 1 mg/ml bovine serum albumin, which was perfused for ~30 min to enzymatically dissociate cells. The left ventricle minced and Ca2+ concentration [Ca2+]o was gradually restored to 1.2 mmol/L.
2.2. Electrophysiology
Isolated myocytes were placed in a temperature-controlled Plexiglas chamber (Cell Microsystems Inc., Research Triangle Park, NC, USA) and continuously perfused with a bicarbonate-containing Tyrode (BTY) solution with the following composition (in mmol/L): NaCl 124, NaHCO3 25, KCl 4, CaCl2 1.2, MgCl2 1, HEPES 10, and glucose 10; pH=7.4. Electrodes were fabricated from borosilicate glass (World Precision Instruments Inc., Sarasota, FL, USA) having tip resistances of 2–2.5 MQ when filled with internal solution. In experiments aimed to preserve the physiological Ca2+ homeostasis during AP, the internal solution contained (in mmol/L): K-Aspartate 110, KCl 25, NaCl 5, Mg-ATP 3, HEPES 10, cAMP 0.002, phosphocreatine dipotassium salt 10, and EGTA 0.01; pH was set to 7.2 with KOH. In another set of experiments intracellular Ca2+ concentration, [Ca2+]i was buffered to nominally zero by using an internal solution containing (in mmol/L): K-Aspartate 100, KCl 25, NaCl 5, Mg-ATP 3, HEPES 10, cAMP 0.002, phosphocreatine dipotassium salt 10, and BAPTA 10; pH=7.2.
AP-clamp experiments were conducted as previously described [33, 35, 36]. Briefly, the steps are: (1) Recording the cell’s steady-state AP under I-clamp at given pacing frequency. (2) Applying this AP to the same cell as the V-clamp command pulse at the same pacing frequency. The net current (reference current) during the AP at steady-state should be zero (Fig. 1A). (3) Isolation of the current of interest (compensation current; Fig. 1A, third panel) uses specific blockers to remove only that from the net current. (4) The current of interest is obtained by subtraction, (i.e. reference current - compensation current). In some experiments, a pre-recorded “typical” AP waveform was used as the V-clamp command (canonical AP-clamp) and delivered at 2 Hz steady-state frequency to measure INaL using tetrodotoxin (TTX, 10 μM) or the specific INaL blocker GS-458967 (GS, 1 μM) [37]. Drug-sensitive current (IGS or ITTX) was calculated by subtracting the average compensation current when TTX or GS achieved steady- state inhibition (3 min of perfusion) from the reference current right before TTX or GS application (average of 60 consecutive traces in each case). In all cases INaL was taken as the IGS measured in this way, beginning after steady state had been achieved for pretreatment with various inhibitors (next section). This INaL density was calculated after normalizing to cell capacitance, determined in each cell using 10 ms hyperpolarizing pulses from −10 mV to −20 mV. Average cell capacitance was 144.98±0.99 pA/pF in the measured 233 cells from 46 animals.
Figure 1. Action potential-clamp measurement of INaL using tetrodotoxin and GS-458967.
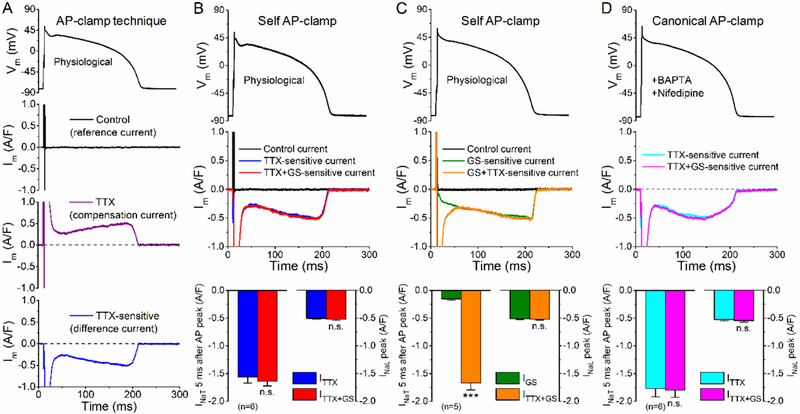
Overview of the action potential-clamp technique. First, using an AP as voltage command a pre-drug control or reference current is recorded (above). Next, when a drug is applied, a compensation current is recorded specific to the drug action (middle). The drug-sensitive current is obtained as the difference current (i.e. subtracting the compensation current from the reference current) (below). (B) Effect of TTX (10 μM), then GS-458967 (GS, 1 μM) under self AP-clamp in rabbit ventricular myocytes. The cell’s own steady-state AP was applied as voltage command (upper panel). Application of TTX inhibited both the transient Na+ current (INaT) and the late Na+ current (INaL). GS in the presence of TTX did not cause any additional current inhibition. Peak INaT was out of scale, thus INaT amplitude was reported 5 ms after the peak of AP when the Na+ channels are already largely inactivated. (C) Effect of GS, then TTX under self AP-clamp in rabbit ventricular myocytes. GS also partially inhibited INaT, but it mostly inhibits INaL. TTX in the presence of GS further inhibited INaT, but it did not have any additional effect on INaL. (D) Canonical AP-clamp (using a prerecorded, typical rabbit AP) to measure INaL. To ensure that the recorded currents are not contaminated with L-type Ca2+ current and the Na+/Ca2+ exchanger, 10 μM nifedipine in the extracellular solution and 10 mM BAPTA in the pipette solutions were applied, respectively. Just as in panel B, no additional effect of GS was observed when applied after TTX in a cumulative manner. Columns and bars represent mean±SEM. Asterisks denote significant difference using paired, two-tailed Student’s t test. ***p<0.001.
To avoid contamination with INaT, GS-458967-sensitive currents were analyzed starting from 10 ms after the AP peak, except for when we compared the effect of TTX and GS-458967 on early INa, where inhibited currents are reported starting 5 ms after AP peak. Note that INaT peak density cannot be reliably measured under AP-clamp with physiological conditions despite rigorous series resistance compensation, because the capacitive transient (stimulation spike) still overlaps with the AP upstroke. Axopatch 200B amplifier (Axon Instruments Inc., Union City, CA, USA) was used for AP and INaL measurements and the signals were digitized at 50 kHz by a Digidata 1440A A/D converter (Molecular Devices, Sunnyvale, CA, USA) under software control (pClamp 10, Molecular Devices). Signal amplification was set to achieve high resolution in the range of INaL magnitude. The series resistance was typically 3–5 MQ and it was compensated by 90% to achieve good voltage control. Experiments were discarded if the series resistance was higher or increased by >10%. Reported AP voltages are already corrected for liquid junction potentials. All experiments were conducted at 36±0.1°C.
2.3. Chemicals and cell treatments
Chemicals and reagents were purchased from Sigma-Aldrich if not specified otherwise. GS-458967 was obtained from Gilead Sciences, Inc. (Foster City, CA, USA). Gp91ds-tat peptide was from AnaSpec (Fremont, CA, USA).
Cell pretreatments with different drugs occurred for ~2 hours prior the seal formation and the given drug was also continuously present in both the perfusing and pipette solutions. To inhibit CaMKII, KN-93 (1 pM) and the more specific autocamtide-2-related inhibitory peptide (AIP, 1 μM, myristoylated) were used and compared with KN-92 (1 μM). To inhibit PKA, H-89 (1 μM) and the more specific protein kinase inhibitor-(14–22)-amide (PKI, 1 pM, myristoylated) were used. cAMP-dependent pathways were also examined using the Rp-isomer of adenosine-3’,5’-cyclic monophosphorothioate (Rp-cAMPS, 100 μM) as a competitive inhibitor of cAMP binding. To investigate the influence of ROS pathway on INaL modulation, a reductant and ROS scavenger “cocktail” was applied containing reduced glutathione (GSH, 10 mM) and N-acetyl cysteine (NAC, 10 mM). The involvement of NADPH oxidase 2 (NOX2) pathway was further tested using its specific inhibitor gp91ds-tat (1 μM). As positive control, H2O2 (100 μM) was applied. To examine the effect of nitric oxide signalling, non-specific nitric oxide synthase (NOS) inhibitor Nω-nitro-L-arginine methyl ester (L-NAME, 1 mM) pretreatment was used. Epac was directly activated using 8-(4-Chlorophenylthio)-2’-O-methyladenosine 3’,5’-cyclic monophosphate (8- pCPT, 3 μM). To examine the [Ca2+]i-dependence of pathways mediating βAR response, the pipette solution was supplemented with 10 mM BAPTA (with no added Ca2+), and we waited for 10 min after cell break-in to allow the agent to diffuse sufficiently into the cell, meanwhile this effect was monitored using a voltage step pulse to +5 mV arising at every 1 s to follow the loss of Ca2+-dependent inactivation of L-type Ca2+ current and myocyte contraction. In all other AP-clamp experiments the recordings were started 5 min after membrane rupture.
βAR responses were evoked adding 10 nM isoproterenol (ISO) to the appropriate perfusion solution. ISO response reached a steady-state in 2 min which was maintained usually for ~5 min before some desensitization was observed. GS was applied 2 min after ISO application and the GS-sensitive current traces were analyzed following 3 min of perfusion. If any sign of Ca2+ current rundown was observed in periodic tests or either ISO or GS effect did not reach steady-state, those experiments were excluded from the analysis.
2.4. Statistical analysis
Data are expressed as Mean±SEM. The number of cells in each experimental group is reported in the figures, and the cells in each group came from three to eight individual animals. Given the biological variability among cells, each cell was treated as independent in the statistical tests, although multiple cells may come from one animal. Statistical significance of differences was evaluated using paired Student’s t test to compare two groups and one-way or two-way ANOVA to compare multiple groups, followed by a Bonferroni posttest for pairwise comparisons. Differences were deemed significant if p<0.05 and denoted *p<0.05, **p<0.01, and ***p<0.001.
3. RESULTS
3.1. Profile of INaL under AP-clamp using TTX and GS-458967
INaL was measured as specific blocker-sensitive current under AP-clamp (Fig. 1A) using physiological conditions (internal and external solutions mimicking physiological ionic composition, preserved intracellular Ca2+ homeostasis, 2 Hz steady-state pacing frequency, and at 36°C).
First, we compared the effect of the selective INaL inhibitor, GS-458967 (GS, 1 pM) with tetrodotoxin (TTX, 10 μM). Accordingly, TTX-sensitive (ITTX) and GS-sensitive currents (IGS) were measured under AP-clamp using the cell’s own steady-state AP. TTX inhibited both the transient and late Na+ current (INaT and INaL) under AP-clamp (Fig. 1B). Because of overlap with the capacitive transient during AP upstroke INaT peak cannot be reliably measured using AP-clamp with physiological solutions at 36°C (despite rigorous series resistance compensation). However, the huge INaT (hundreds of A/F) inactivates rapidly, but the TTX-sensitive early decaying peak in Fig. 1A-B may include terminal decay of Inst (i.e. the last 1–2%). On the other hand, the much smaller sustained INaL was present throughout the entire AP, and achieved a peak density of −0.50±0.02 A/F during AP phase 3, as driving force (ENa - Em) increases (Fig. 1B). Cumulative application of GS in the presence of TTX did not inhibit additional current (Fig. 1B). This indicates that under our conditions, GS does not influence other ionic currents not already blocked by TTX. When the order was reversed (Fig. 1C) GS only slightly inhibited INaT (9.0% of that for ITTX at 5 ms after AP peak), whereas INaL (as either IGS or IGS+ITTX) late in the AP was identical. Thus, IGS provides a useful measure of INaL during the cardiac AP under our physiological ionic conditions.
While this self AP-clamp technique is the most physiological technique to determine current profile during the cell’s own AP, each myocyte has a slightly different AP duration (APD) which can also alter INaL density [33]. Thus, we used a canonical rabbit AP waveform (prerecorded) in subsequent AP- clamp experiments, to obtain more controlled mechanistic insight into INaL modulation (Fig. 1D; in this panel only, Ca2+ current and transients were suppressed by nifedipine and with 10 mM BAPTA in the pipette). Again, 1 μM GS had no further effect after prior TTX application.
3.2. Tetrodotoxin-sensitivity of INaL under AP-clamp
The TTX-sensitivity of INaL measured under AP-clamp can suggest whether it is primarily due to cardiac Na+ channel isoforms that are relatively TTX-resistant (μM range as for the predominant cardiac isoform NaV1.5) or TTX-sensitive (nM range as for neuronal Na+ channel isoforms). To prevent complications of TTX partial inhibition of L-type Ca2+ current at high concentrations (IC50=55 μM) [38], these experiments included nifedipine. Fig. 2A shows that TTX dose-dependently inhibited INaL. The observed IC50 values were 1.18±0.20 μM (for INaL net charge) and 1.08±0.07 μM (for INaL peak density) under AP-clamp (Fig. 2B). Importantly, ≤5% of total INaL was inhibited by 100 nM TTX suggesting that INaL is predominantly mediated by TTX-resistant Na+ channel isoforms in healthy rabbit ventricular myocytes.
Figure 2. Tetrodotoxin-sensitivity ofINaL under AP-clamp.
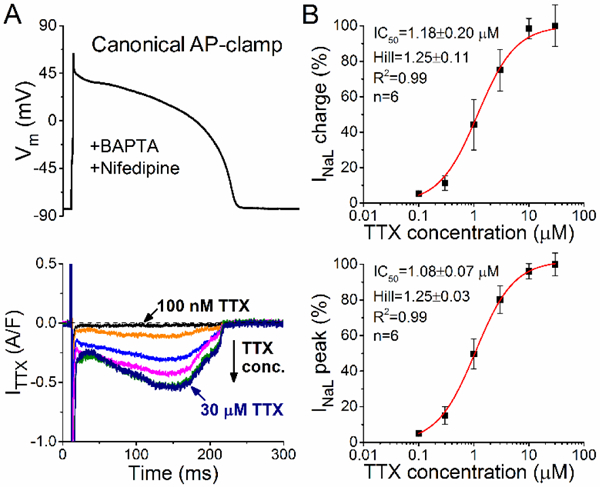
Representative tetrodotoxin (TTX)-sensitive current traces under AP-clamp at 2 Hz steady-state pacing. Increasing TTX concentrations (0.1, 0.3, 1, 3, 10, 30 μM) were applied in a cumulative manner. Pipette solution contained 10 mM BAPTA and extracellular Tyrode solution was supplemented with 10 ^M nifedipine. (B) TTX dose-response effect on INaL net charge and INaL peak. Inhibition of INaL was normalized to that obtained with 30 μM TTX in each cell. IC50 values and Hill coefficients were determined by fitting data to the Hill equation, indicated by solid lines.
3.3. CaMKII, but not PKA regulates basal INaL under AP-clamp
Next, we studied the modulation of INaL by basal PKA or CaMKII activity under AP-clamp with physiological intracellular Ca2+ transients at 2 Hz and 36°C. Inhibition of CaMKII by extracellular application of KN-93 (1 μM) significantly decreased IGS magnitude throughout the AP (Fig. 3A), most prominently in the phase 3 at −60 mV (−0.45±0.02 A/F in control vs. −0.24±0.04 A/F in KN-93, Fig. 3F). The same reduction in IGS was observed when KN-93 was applied intracellularly through the patch pipette. Moreover, the more specific autocamtide-2-related inhibitory peptide, AIP (1 pM) caused similar decrease of IGS (Fig. 3A,C-F). In contrast, the inactive KN-93analogue, KN-92 (1 μM) had no effect on IGS (Fig. 3C-F). These data indicate that roughly half of basal INaL during the physiological AP is secondary to CaMKII activity.
Figure 3. CaMKII-dependent and PKA-dependent regulation of basal INaL under AP-clamp.
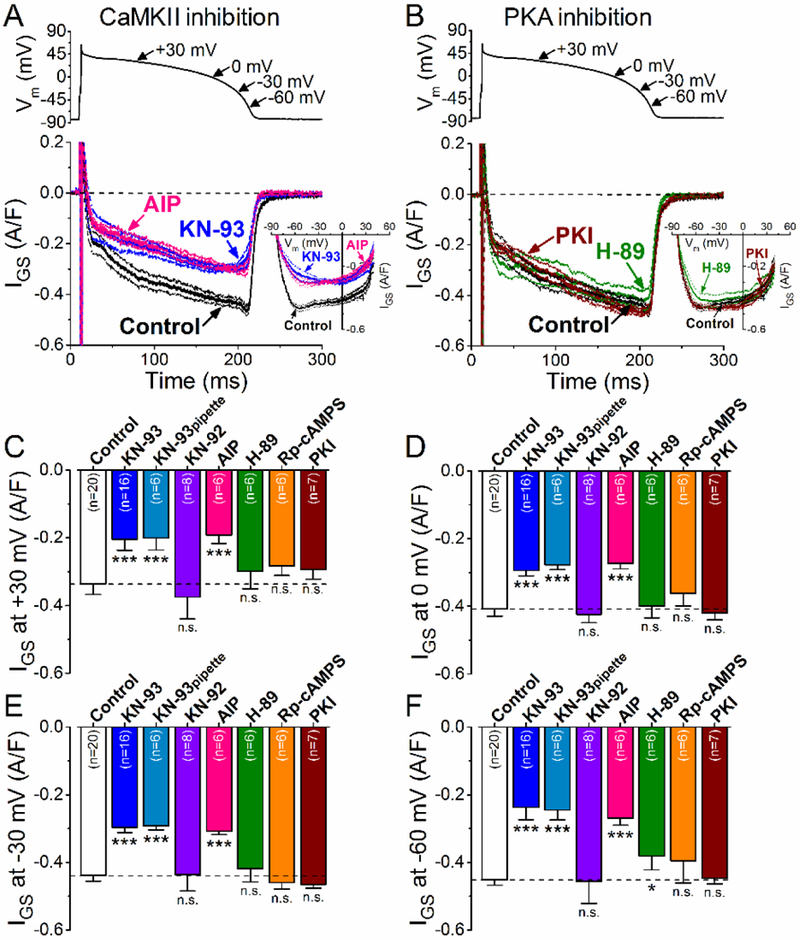
(A) GS-458967-sensitive current (IGs) traces (mean±SEM) under AP-clamp in control and after CaMKII inhibition using KN-93 (1 pM) or AIP (1 μM). The physiological intracellular Ca2+ cycling was preserved, and 2 Hz steady-state pacing was applied. I-V trajectories of IGS under AP-clamp are shown in inset. (B ) IGS traces (mean±SEM) under AP-clamp in control and after PKA inhibition using H-89 (1 μM) or PKI (1 μM). I-V relationships are shown in the inset. (C-E) IGS density at different voltages during AP repolarization. CaMKII inhibition with KN-93 or AIP significantly decreased INaL at all voltages, whereas KN-92 (1 μM) had no effect on IGS. The decrease in INaL was most prominent at −60 mV which may reflect the window component of INaL. On the contrary, PKA inhibition using PKI or Rp-cAMPS (100 μM) had no effect on IGS, whereas H-89 may exhibit some off-target effect at −60 mV. Columns and bars represent mean±SEM. Asterisks denote significant difference using two-way ANOVA with Bonferroni posttest. *p<0.05, **p<0.01, ***p<0.001.
We also tested the effect of basal PKA activity on INaL under AP-clamp (Fig. 3B). Inhibiting PKA using H-89 had no effect on IGS except for a slight inhibition in the late phase 3 of the AP (~15% at - 60 mV) (Fig. 3C-F). However, more specific inhibitors of PKA, Rp-cAMPS (100 μM) and protein kinase inhibitor peptide (PKI, 1 μM) had no effect on IGS (Fig. 3B-F). Thus, we infer that INaL is not modulated by basal PKA activity under baseline conditions.
3.4. βAR stimulation upregulates INaL under AP-clamp via both CaMKII and PKA
βAR stimulation with 10 nM ISO led to a substantial increase of IGS under AP-clamp (Fig. 4A). Surprisingly, this increase was most prominent during the early AP plateau phase at relatively positive Vm, where driving force for Na+ entry is low (Fig. 4A), The IGS-Vm relationship below the IGS time course for Control is fairly linear, indicating little change in conductance between −60 and +25 mV. ISO more than doubled IGS density at +30 mV (from −0.34±0.03 A/F in control to −0.78±0.02 A/F in ISO, Fig. 4E,) and also at 0 and −30 mV (Fig. 4F,G). However, ISO did not change IGS density at −60 mV (Fig. 4H), such that ISO shifted maximal IGS from −60 mV in control to ~0 mV after βAR stimulation (Fig. 4A, IGS-Vm).
Figure 4. βAR stimulation upregulates INaL under AP-clamp via both CaMKII and PKA signaling.
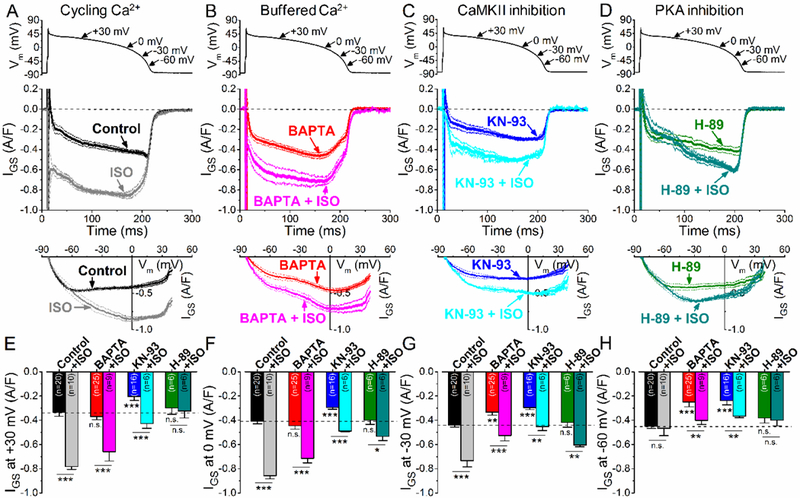
(A-D) GS-458967-sensitive current (IGs) traces (mean±SEM) under AP-clamp with and without isoproterenol stimulation (ISO, 10 nM) in control (A, cycling Ca2+), in the presence of 10 mM BAPTA in the pipette solution (B, buffered Ca2+), in the presence of CaMKII inhibitor KN-93 (C, 1 μM) and in the presence of the PKA inhibitor H-89 (D, 1 μM). Lower panels show the corresponding I-V trajectories of IGS under AP-clamp. (E-H) IGS density at different voltages during AP repolarization. ISO significantly upregulated IGS throughout the AP, except for −60 mV in control and in H-89. Ca2+-buffering using BAPTA decreased basal IGS at phase 3 of the AP, but not during the plateau phase. However, ISO increased IGS in BAPTA under the AP plateau similarly as is control. ISO also increased IGS in KN-93, but the increase in IGS during the plateau phase of the AP was significantly reduced compared to control. Interestingly, ISO increased IGS also at −60 mV both in BAPTA and in KN-93. H-89 pretreatment abolished the ISO-induced increase in IGS at the early plateau phase, and significantly diminished the effect of ISO on IGS at phase 3 of the AP. Columns and bars represent mean±SEM. Asterisks denote significant difference using two-way ANOVA with Bonferroni posttest. *p<0.05, **p<0.01, ***p<0.001.
Next, we examined how buffering [Ca2+]i with 10 mM BAPTA affects INaL density and profile under AP-clamp (Fig. 4B). BAPTA did not change IGS significantly at positive Vm (Red vs. Black in Fig. 4E,F), but reduced IGS significantly at −30 mV and even more at −60 mV (Fig. 4G,H), similar to the effects of CaMKII inhibition (Fig. 3A vs. BAPTA in Figs. 1D and3B). This is consistent with the baseline CaMKII-dependence of INaL being due to regular 2 Hz Ca2+ transients at baseline (Fig 3A; absent with 10 mM BAPTA). ISO still enhanced IGS with BAPTA, but mainly at +30 and 0 mV and much less at negative Vm (−30 and −60 mV) where CaMKII effects were largest (Black/gray vs Red/pink in Fig. 4E- H).
To dissect the contributions of PKA and CaMKII to the PAR-induced INaL enhancement, we pretreated cells with KN-93 (1 μM) to inhibit CaMKII or H-89 (1 μM) to inhibit PKA. In KN-93 pretreated cells, ISO significantly increased IGS, but the magnitude of ISO effect was significantly reduced compared to control (Fig. 4C). Qualitatively, the ISO-induced IGS in KN-93 resembled that in BAPTA, with larger increases at positive Vm (vs. near −60 mV; Fig. 4B vs. Fig. 4C). In contrast, the ISO-induced IGS enhancement in the early plateau (at +30 mV) was completely abolished by PKA inhibition by H-89 pretreatment (Fig. 4D, E). Nonetheless, ISO still increased IGS later during the AP (more negative Vm), with the largest difference near Vm= −30 mV (consistent with CaMKII predominance after PKA inhibition).
3.5. CaMKII and PKA differentially modulates INaL in different AP phases
So far, we infer that ISO increases INaL via PKA early in the AP plateau, but via CaMKII later in the AP plateau. However, both H-89 and KN-93 might have off-target effects [35]. So we repeated the above experiments with additional PKA and CaMKII inhibitors that differ molecularly and may be more selective. First, we pretreated myocytes with the selective CaMKII inhibitory peptide, AIP (1 μM) that inhibits CaMKII both upon Ca2+/CaM-activation and also when CaMKII is autonomously activated (via autophosphorylation, oxidation or S-nitrosylation). ISO still increased IGS in AIP-treated cells at the early plateau phase (Fig. 5A, C), but smaller increases were observed at −30 mV and no change at −60 mV (Fig. 5E,F). These results agree well qualitatively with the KN-93 and BAPTA results from Fig. 4, and with CaMKII having most prominent effects on INaL late in the AP plateau.
Figure 5. βAR stimulation differently modulates INaL under AP-clamp via CaMKII and PKA.
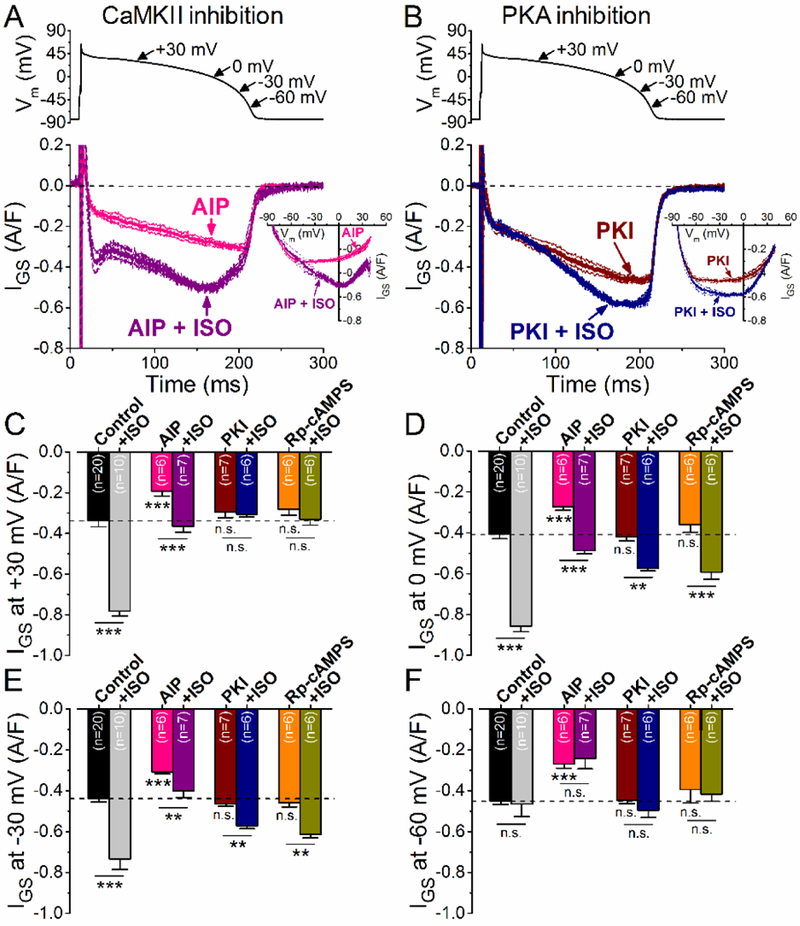
(A-B ) GS-458967-sensitive current (IGs) traces (mean±SEM) under AP-clamp with and without isoproterenol stimulation (ISO, 10 nM) in the presence of highly selective peptide inhibitors of CaMKII (A, AIP, 1 μM) and PKA (B, PKI, 1 μM). Corresponding I-V trajectories are shown in the inset. (C-E) Effect of ISO on IGS density at different voltages during AP repolarization in the presence of AIP, PKI and Rp-cAMPS (100 μM). Similar results were obtained using the selective peptide inhibitors as using KN-93 and H-89 (shown in Fig. 3), confirming the different modulation of INaL by CaMKII and PKI upon PAR stimulation. CaMKII inhibitor AIP significantly reduced the amplitude of ISO stimulation on IGS. Importantly, no IGS increase at −60 mV was observed following ISO application in AIP pretreated cells in contrast to cells pretreated with KN-93 (compare with Fig. 3H). PKA inhibitor PKI and Rp-cAMPS completely abolished the ISO effect on IGS at +30 mV, and significantly diminished the IGS upregulation both at 0 and −30 mV. Results in control cells are shown for comparison. The physiological intracellular Ca2+ cycling was preserved, and 2 Hz steady-state pacing was applied. Columns and bars represent mean±SEM. Asterisks denote significant difference using two-way ANOVA with Bonferroni posttest. **p<0.01, ***p<0.001.
We also blocked PKA using the specific inhibitory peptide PKI (1 μM) and cAMP analog Rp- cAMPS (100 μM, Fig. 5B-F). PKI or Rp-cAMPS completely abolished the ISO-induced enhancement in early IGS, but did not prevent the enhanced IGS late in the plateau (Fig. 5B,D,E). These findings agree with the H-89 results shown in Fig. 4D, and with PKA effects being most prominent in the early plateau.
These data suggest distinctive effects of PKA and CaMKII on mediating the ISO-induced enhancement of INaL. PKA predominantly mediates the ISO effect on increasing INaL during the early plateau phase of the AP, whereas CaMKII contributes in the late plateau and the phase 3 of the AP.
3.6. Effect on Epac activation and ROS signaling on INaL under AP-clamp
Physiologically, Epac2 (a parallel cAMP target to PKA) has been shown to mediate βAR-induced activation of CaMKII and arrhythmogenic SR Ca leak [29]. Here we tested whether the Epac-selective agonist 8-pCPT-2’-O-Me-cAMP (8-pCPT, 3 μM, Fig. 6A,B) could mimic PAR effects on INaL. Indeed, 8- pCPT increased IGS during the late plateau (at 0 mV) and phase 3 of AP (at 0 and −30 mV, Fig. 6F,G), but not during the early AP plateau (at +30 mV, Fig. 6E). Importantly, BAPTA completely prevented the 8- pCPT-induced IGS enhancement (Fig. 6C). The 8-pCPT data implicates Epac as a potential mediator of the ISO-induced increase in IGS late in the AP, likely via activating CaMKII (rather than PKA).
Figure 6. Effect of Epac activation and ROS on INaL under AP-clamp.
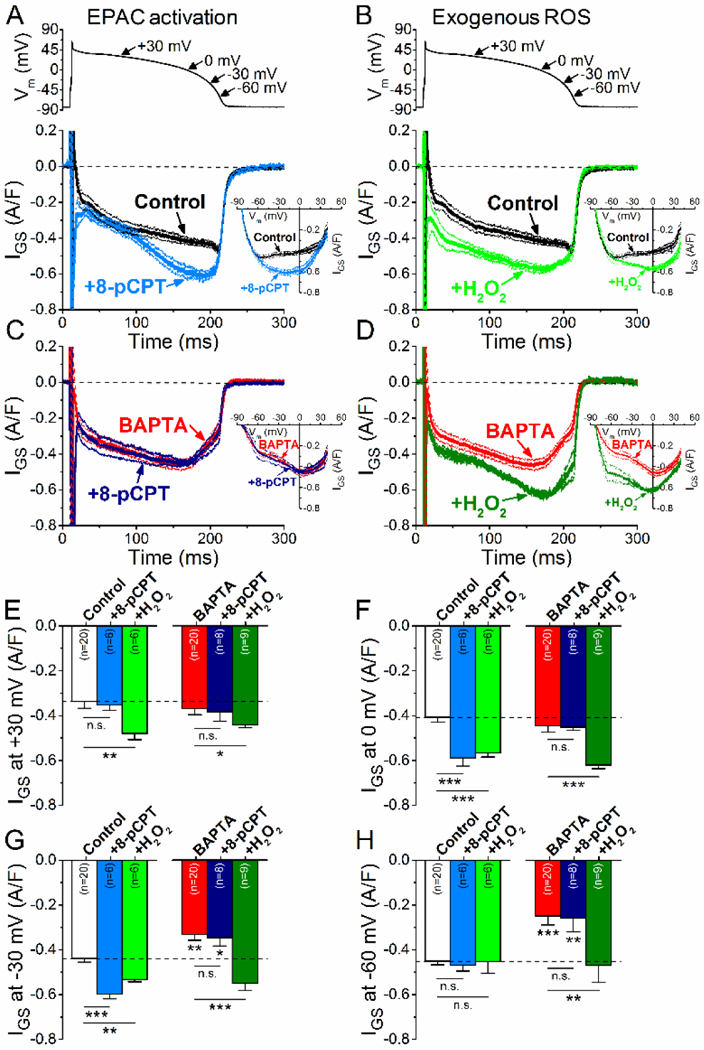
(A) GS-458967-sensitive current (IGs) traces (mean±SEM) selective Epac activator 8-pCPT-2’-O-Me- cAMP (8-pCPT, 3 μM) treatment under AP-clamp with preserved Ca2+ cycling. I-V relationships of IGS after Epac activation are shown in inset. (B) IGS traces (mean±SEM) following pretreatment with H2O2 (100 μM) under preserved Ca2+ cycling. I-V relationships of IGS after H2O2 application are shown in inset. (C) IGS traces (mean±SEM) following 8-pCPT pretreatment measured with 10 mM BAPTA in the pipette solution. I-V trajectories are shown in the inset. (D) IGS traces (mean±SEM) following H2O2 pretreatment measured with buffered [Ca2+]i. I-V trajectories are shown in the inset. (E-H) IGS density at different voltages during AP repolarization. H2O2, but not Epac, increased IGS at +30 mV (E). H2O2 increased IGS at 0 mV and −30 mV regardless of [Ca2+]i. 8-pCPT increased IGS during phase 3 of AP, but only with preserved Ca2+ cycling. (F-G) H2O2 increased IGS at −60 mV in the presence of 10 mM BAPTA (H). Results in control cells are shown for comparison. Columns and bars represent mean±SEM. Asterisks denote significant difference using two-way ANOVA with Bonferroni posttest. *p<0.05, **p<0.01, ***p<0.001.
Increased production of reactive oxygen species (ROS) may also promote autonomous activation of CaMKII and pKa. We used H2O2 (100 μM) to examine the effect of increased ROS on INaL (Fig. 6B). H2O2 significantly increased IGS both at early and late AP plateau phases (at +30 mV and 0 mV, Fig. 6E-F); however, only a slight increase in IGS was observed at −30 mV (Fig. 6G) and none at −60 mV under AP-clamp when measured with preserved Ca2+ cycling (Fig. 6B). H2O2 caused similar effect at −30 and 0 mV under AP-clamp with BAPTA in the pipette solution (Fig. 6D,E,F). However, in this case, significant increase in IGS was observed also at −30 mV and −60 mV (Fig. 6G-H). These results suggest that ROS might contribute to early INaL enhancement during the AP, but that it may also contribute to CaMKII-dependent effects, especially late component of INaL at −60 mV.
3.7. Effect of ROS and NOS inhibition on INaL modulation during PAR stimulation
To test the effect of endogenous physiological ROS production on INaL, we pretreated the cells with ROS scavengers (reduced glutathione, GSH, 10 mM; and N-acetyl cysteine, NAC, 10 mM). This treatment did not change basal IGS under AP-clamp, except for a slight decrease at −60 mV (Fig. 7A,F), consistent with most of the basal CaMKII-dependent INaL being independent of ROS. However, ROS scavengers limited the ISO-induced IGS enhancement. The effect was most prominent at the early plateau phase (at +30 mV, Fig. 7C), but smaller effects were observed at −30 mV (Fig. 7E). We also tested the involvement of NADPH oxidase 2 (NOX2) in mediating the ISO-induced INaL enhancement. The NOX2 specific inhibitor gp91ds-tat (1 pM) partially inhibited the early IGS enhancement at +30 mV (Fig. 7A,C), but no significant limitation was found later in the AP at more negative membrane voltages (Fig. 7D-F). Thus, we conclude that endogenous ROS production, partially via NOX2, is involved in the IGS enhancement in the early phase of the AP (the range where PKA-dependent effects were largest). However, oxidation of CaMKII may also occur upon PAR stimulation, contributing to the ISO-induced INaL.
Figure 7. Effect of ROS and NOS inhibition on INaL during PAR stimulation.
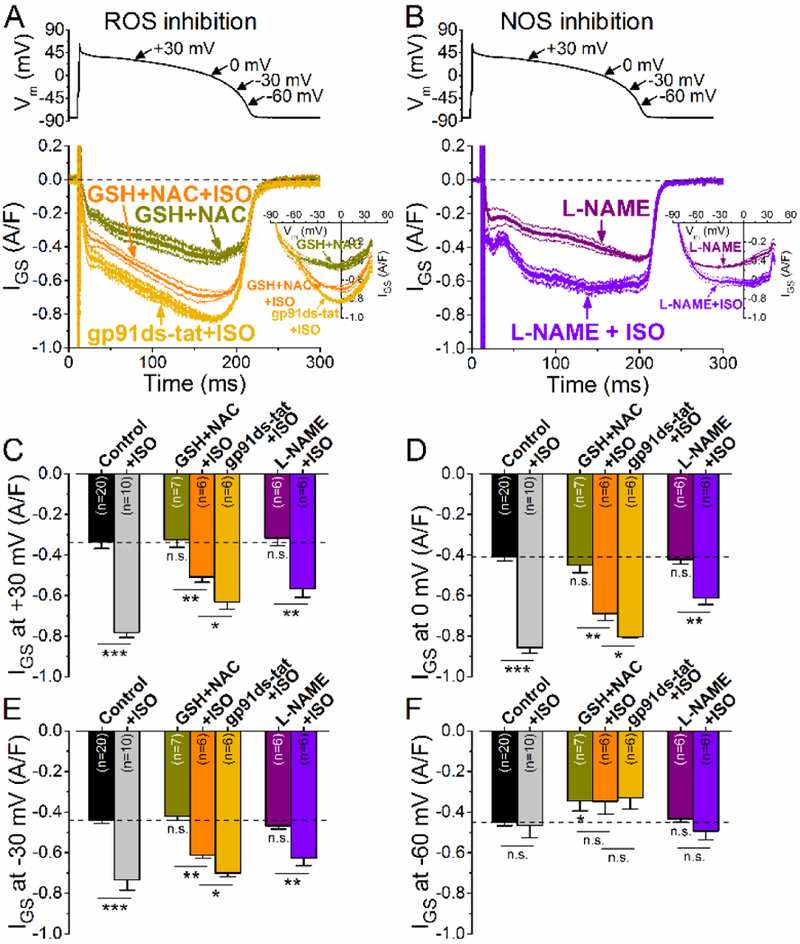
GS-458967-sensitive current (IGs) traces (mean±SEM) under AP-clamp with and without isoproterenol stimulation (ISO, 10 nM) in the presence of reduced glutathione (GSH, 10 mM) + N-acetyl cysteine (NAC, 10 mM), as well as NOX2 inhibitor peptide gp91ds-tat (1 μM). The corresponding I-V trajectories are shown in inset. (B) IGS traces (mean±SEM) under AP-clamp in L-NAME (1 mM) pretreated cells with and without ISO stimulation. (C-F) IGS density at different voltages during AP repolarization. Both NOX2 inhibition and NOS inhibition diminished the effect of ISO on IGS at +30 mV. GSH+NAC further decreased the effect of ISO on IGS. Results in control cells are shown for comparison. Columns and bars represent mean±SEM. Asterisks denote significant difference using two-way ANOVA with Bonferroni posttest. *p<0.05, **p<0.01, ***p<0.001.
Recent studies have also implicated myocyte nitric oxide signaling in βAR-induced CaMKII activation in myocytes [27, 39–42], so we tested whether inhibition of nitric oxide synthase (NOS) would alter ISO-induced INaL enhancement. Pretreatment of myocytes with non-specific NOS inhibitor L-NAME (1 mM) did not alter basal IGS (Fig. 7B), suggesting that nitrosylation is not involved in the basal CaMKII-induced INaL. However, L-NAME significantly reduced the ISO-induced enhancement of IGS during the AP plateau (Fig. 7B-D), similar to that seen with KN-93, AIP or BAPTA. These data are consistent with a significant role of NOS in mediating the ISO-induced enhancement of INaL, especially the CaMKII-dependent component during the plateau phase of the AP. This may be similar to the Epac2- NOS1-CaMKII5 pathway implicated in βAR-induced increase in RyR2-mediated SR Ca2+ leak [43].
4. DISCUSSION
Our study shows that βAR stimulation increases cardiac INaL during the physiological AP via both PKA and CaMKII signaling pathways, preferentially at more positive (early) vs. more negative (late) Vm, respectively. The CaMKII-mediated effect on INaL were already partially active during normal APs at 2 Hz, 36°C and normal Ca2+ transients, while PKA inhibition had no effect under these baseline physiological conditions. The CaMKII- and PKA-mediated effects on INaL appear to be additive and may synergize. This upregulation of INaL upon PAR stimulation may significantly alter the balance between the depolarizing and repolarizing currents during the plateau phase of the AP, where overall conductance is low [44]. Moreover, INaL during physiological APs can peak during AP repolarization, where early afterdepolarizations (EADs) arise under pathologic conditions [33, 45]. Therefore, under pathological conditions like heart failure - where both INaL, and CaMKII activity are elevated, and repolarization reserve is reduced [46] - PAR stimulation may increase INaL and lead to AP prolongation and increased arrhythmia risk.
4.1. INaL gating differs from INaT and is TTX-sensitive and GS-sensitive
Na+ channels exhibit transient openings that generate INaT, but during sustained depolarization can also exhibit additional openings (early bursting mode, late scattered mode) that contribute to INaL [1, 4]. During AP repolarization, an apparent window component of INaL has also been reported in well- controlled biophysical studies [5, 8, 9, 17], and a similar Na+ current was implicated in neuronal pacemaking [47, 48], despite the tiny overlap of steady state activation and availability Vm-dependence. Additionally, the slowly decreasing Vm during the cardiac AP plateau can enhance INaL, via a unique nonequilibrium gating scheme [6]. So, the time- and Vm-dependence of Na+ channel gating is complex, may differ widely between INaT and INaL, and only a small subset of Na+ channels may exhibit gating modes that mediate INaL. Furthermore, INaL has mostly been studied in conditions far from the physiological AP (e.g. square pulses), intracellular Ca2+ buffering and low stimulation frequency, with limited INaL data during physiological AP [33, 44].
Single-channel INaL records exhibit burst and late scattered modes of Na+ channel opening. In square Vm steps Maltsev and Undrovinas [4] reported an early larger burst mode, which declines in 50100 ms, and a smaller late scattered opening mode that declines only slightly during 200 ms. Burst mode open probability declines faster with membrane depolarization, but late scattered openings seemed less voltage-dependent. Slow Vm ramps indicated a noninactivating INaL (at 0 mV) which at more negative potentials resembled a window current [49, 50]. To account for the complex INaL Vm-dependence, we analyzed Igs at 4 different AP voltages: (1) Early plateau (+30 mV; ~60 ms after AP peak) which likely includes INaL burst mode, (2) Late plateau (0 mV) where a transition to more late scattered openings are expected, (3) Early phase 3 (−30 mV) as repolarization accelerates, and (4) Rapid repolarization (- 60 mV) where driving force is rapidly increasing as Vm-dependent deactivation may be progressing. These latter two phases may reflect the non-inactivating current and window type INaL. These helped classify INaL Vm ranges that were preferentially influenced by PKA or CaMKII activity.
INaL measured under AP-clamp was significant throughout the AP plateau, but increased during late the plateau phase as driving force increases (Fig. 1). Using TTX and GS-458967 in AP-clamp experiments produced identical INaL traces, as expected if both block INaL (and not other currents). In contrast, fast INaT was significantly less affected by GS-458967 vs. TTX, in agreement with higher INaL selectivity reported for GS-458967 [37]. Measure of the much larger INaT was impractical here during physiological AP-clamp, so we used only GS-458967 to study INaL regulation by PKA and CaMKII.
The TTX titrations in Fig. 2 were used to identify the main Na+ channel isoforms that likely mediate INaL as measured here. TTX inhibited >95% of INaL with an IC50 value of ≈1 μM, suggesting that predominantly TTX-resistant Na+ channel isoforms mediate INaL, including the predominant “cardiac” Nav1.5 (but not excluding Nav1.8 or Nav1.9). However, TTX-sensitive Na+ channels (Nav1.1–1.4, Nav1.6– 1.7) are less likely to contribute to INaL here. This agrees with the reported IC50 values of 1–2 ^M for TTX INaL inhibition in rabbit [51], guinea-pig [33, 52] and human [1] ventricular myocytes, although TTX- sensitive Na+ channel isoforms have been reported to contribute to INaL in heart failure [53]. Moreover, TTX-resistant Nav1.8 channels have also been suggested to be expressed and contribute to INaL in mouse, rabbit [54] and failing human [55] ventricular myocytes. Further studies would be needed to resolve the exact contribution of different Na+ channel isoforms, splice variants and their regulation by CaMKII and PKA to INaL in health and disease.
4.2. Nearly half of the basal physiological INaL is CaMKII-dependent
We found that [Ca2+]i and CaMKII affect the magnitude of Igs under AP-clamp already in control (Fig. 3). Buffering [Ca2+]i and CaMKII inhibition (either by KN-93 or AIP) strongly decreased Igs during AP phase 3, especially between −30 and −60 mV. CaMKII inhibition also decreased Igs earlier during the AP plateau (Figs. 3–4). Calmodulin (CaM) and CaMKII have complex effects on Na+ channel gating, with CaMKII shifting steady-state inactivation to more negative Vm, slowing inactivation, promoting intermediate inactivation and slowing recovery from inactivation, but not altering maximal conductance or activation Vm-dependence [8, 9, 56–58]. In addition to those loss of function effects CaMKII also increases myocyte INaL. Moreover, Ca2+/CaM alone can alter steady-state INa inactivation [9, 58, 59], but does not alter INaL [9], in agreement with our study. Importantly, we demonstrated that basal CaMKII- activity at 2 Hz pacing under physiological conditions (with endogenous Ca2+ levels) nearly doubles INaL in rabbit ventricular myocytes vs. what is seen without Ca2+ transients or when CaMKII is inhibited (Fig 3). This agrees with recent data in guinea pig [52]. The gradual repolarization during the AP also promotes INaL that is attributable to non-equilibrium gating. These factors may account for prior underestimates of physiological INaL when studies are done under non-physiological conditions (buffered [Ca2+] and square pulses).
AIP and KN-93 exerted the same effect on basal IGS (Fig. 3), despite having different mechanisms of inhibition (KN-93 competes with CaM binding [60], while AIP mimics autoinhibition of basal and autonomous CaMKII [61]). Inhibition of ROS and NOS did not alter basal IGS appreciably (Fig. 7) suggesting that oxidation and S-nitrosylation that are known to promote autonomous CaMKII [26, 27] are not required for the basal CaMKII effect on INaL. In marked contrast, basal PKA activity did not contribute to IGS under AP-clamp (Fig. 3) in agreement with previous square pulse studies [9].
4.3. βAR-induced INaL is dependent on both PKA and CaMKII signaling
Importantly, βAR stimulation upregulated IGS under AP-clamp and both PKA and CaMKII are required for the full effect (Figs. 4–5). Notably, PKA and CaMKII affected IGS predominantly in different phases of the AP. INaL enhancement early in the AP plateau (+30 mV) upon βAR stimulation was exclusively dependent upon PKA, suggesting that PKA may particularly enhance the early burst mode INaL openings. This is consistent with effects of a PKA-dependent long QT3-associated mutation, D1790G, that promotes early burst opening of the Na+ channel [62]. Conversely, PAR activation had no effect on INaL measured during rapid repolarization (−60 mV). While BAPTA and KN-93 uncovered a potential CaMKII-independent effect of ISO (Fig. 4H), this was not seen with more selective CaMKII block via AIP (Fig. 5F).
At intermediate Vm during repolarization (0 and −30 mV) the βAR-induced IGS was progressively less PKA-dependent and more CaMKII-dependent. For some data this is hard to appreciate because KN-93, AIP and BAPTA all reduce basal INaL prior to ISO activation. First, we consider the AIP and PKI data in Fig. 5 and assume the ISO effect with AIP is all due to PKA and that with PKI is all due to CaMKII. The IGS vs. Vm curves for PKA effect are superimposable from −90 to −45 mV and then split progressively despite a decrease in driving force. This indicates that PKA influences opening preferentially at more positive Vm. Conversely, these IGS - Vm curves for CaMKII effect (with PKI) diverge already below −60 mV but start converging at 0 mV and are identical at +25–40 mV. This indicates that CaMKII promotes INaL most strongly at negative Vm and later in the AP plateau. Using Fig. 5C-F we can also infer that the PAR-induced INaL increase is entirely PKA-dependent at +30 mV, and declines to 65% and 57% during the plateau (0 and −30 mV) and becomes entirely CaMKII- dependent between −30 and −60 mV. We speculate that PKA promotes preferentially the burst mode, while CaMKII promotes the late scattered openings seen at the single channel level. Further study will be required to test this speculation and also to identify specific amino acids phosphorylated by PKA and CaMKII in this process (and dozens of candidate sites exist [63–66]). Key candidates on NaV1.5 could include Ser525 and Ser528 for PKA vs. Ser516 and Ser571 for CaMKII [64–66].
The CaMKII-dependent activation of INaL with ISO was partially dependent on NO production and was mimicked by direct Epac activation (Figs. 6A and7B). This is reminiscent of the recently elucidated pathway by which βAR activates RyR2 and SR Ca leak, mediated by cAMP-dependent Epac2 activation which causes NOS1-dependent S-nitrosylation/activation of CaMKIIS to phosphorylate RyR2 [24, 27, 29, 39, 43]. So that same pathway may impact Na+ channels as well. The INaL enhancement with ISO was also partially dependent on ROS and NOX2, especially in the early AP phase where PKA- dependent effects were strongest (Fig. 7A). This might reflect some ROS-dependent modulation of the PAR-PKA-INaL pathway, but our data do not resolve a molecular mechanism for such an effect. Angiotensin II was reported to induce PKA-dependent enhancement of INaT via NOX2-mediated ROS production [32]. In that study, the angiotensin-II and ROS induced INaL was attributed to CaMKII vs. PKA, but INaL was measured only late in square pulses which might have favored detection of CaMKII as the mediator. Since oxidation may lead to autonomous activation of both CaMKII and PKA, further studies are needed to better clarify the upstream signaling whereby ROS leads to INaL enhancement during βAR stimulation.
4.4. Physiological magnitude of INaL in rabbit ventricular myocytes
The magnitude of INaL and the contributions of multiple Na+ channel gating components have become a greater focus in cardiac research, because altered Na+ channel gating has been linked to abnormal AP activities and disturbed cellular Na+ homeostasis [2, 67]. Here we examine for the first time the detailed time course of INaL under physiological conditions in response to βAR stimulation. Comparing INaL amplitude with other studies is complicated by different conditions, which have often been done at sub- physiological temperature, with square pulses at single Vm (vs. AP-clamp), measured only at end of pulse, and non-physiological intracellular solutions (including Ca2+ buffering).
We measured peak INaL at −30 mV under AP-clamp as −0.44±0.02 A/F (~65 pA) with preserved Ca2+ cycling, −0.31±0.01 with CaMKII inhibition and −0.33±0.02 A/F (~50 pA) with 10 mM BAPTA (Fig. 4). These values agree well with prior studies in rabbit ventricular myocytes (between 0.25 and 0.4 A/F at −20/−30 mV with strong Ca2+ buffering [10, 68–70]. Clamping [Ca2+]i at 600–1,000 nM in rabbit myocytes also increased INaL to 0.45 A/F and 0.55 A/F, respectively [70], and similar effects of [Ca2+]i-dependence of INaL were seen in canine cardiomyocytes [58]. Human ventricular myocytes from healthy donors had similar INaL magnitude [1, 12], but significantly larger INaL was reported in guinea-pig [33, 71] and rat [50, 72] ventricular myocytes. Other studies compared INaL magnitude to INaT peak density and INaL was reported to be 0.15–0.3% of INaT [8, 9]. We could not directly measure peak INaT in our physiological AP-clamp, but prior estimates in rabbit ventricular myocytes at 36°C [73] gave peak INaT in the range of −395 to −438±27 A/F. Thus, our INaL peak density is ≈0.13% of peak INaT during physiological AP in rabbits.
4.5. Study Limitations
We used pharmacological agents in freshly isolated adult rabbit ventricular myocytes, because we sought to use an animal with AP plateau phase resembling the human cardiac AP. This made it impractical to use genetically modified mice (or rabbits) to knockout CaMKII or PKA, NOS, Epac, NOX2. Even gene silencing in culture for 2–3 days can significantly alter ion channels and signaling pathways [74]. To minimize the inherent limitations of small molecule inhibitors, we used multiple agents with different structure and mechanism of action wherever practical to confirm target effects. For example, we used TTX to verify the utility of using 1 GS-458967 to measure INaL, AIP and KN-93 to inhibit CaMKII and H-89, PKI and Rp-cAMPS to inhibit PKA in experiments leading to major conclusions. Another limitation arises from the complexity of PAR signaling and its crosstalk with other signaling pathways. Because of these potential complications, we were cautious not to combine two or more inhibitors to further isolate signaling pathway.
4.6. Conclusions
In summary, our data reveal that basal INaL during the physiological AP and Ca2+ transients at 2 Hz is already boosted significantly by the basal level CaMKII activity. PAR stimulation further increases INaL and is mediated in concert by both PKA-dependent and CaMKII-dependent pathways. The PKA- dependent increase in INaL is predominantly in the early AP plateau (at more positive Vm), whereas CaMKII mainly increases INaL during the late AP plateau and rapid repolarization phase. The CaMKII- dependent effects appear to involve Epac and NOS signaling. Both CaMKII- and PKA-dependent effects might also include ROS signaling. Furthermore, there is a synergistic crosstalk between PKA and CaMKII signaling that further promotes ßAR-induced INaL. However, the precise identification of ROS and crosstalk require further studies. Taken together, our data reveal differential modulations of INaL at different AP phases by PKA and CaMKII pathways following β-adrenergic stimulation. This comprehensive and nuanced view on the fine-tuning of INaL during different AP phases deepens our understanding of the role of INaL in shaping cardiac AP and arrhythmogenic potentials, which will inform therapeutic development for treating arrhythmias in heart diseases whereby increased sympathetic tone, increased CaMKII-activity and oxidative stress are present under pathological conditions [11, 75, 76].
Highlights:
Measure the dynamic profile of INaL under cardiac AP during β-adrenergic stimulation
Reveal the differential contributions of PKA and CaMKII pathways in modulating INaL
Pacing-induced CaMKII activation upregulates basal INaL under AP-clamp
Both PKA and CaMKII upregulate INaL during β-adrenergic stimulation
PKA increases INaL in phase 2 and CaMKII increases INaL in phase 3 of the AP
Reactive oxygen species and nitric oxide may contribute to PKA and CaMKII effects
ACKNOWLEDGEMENTS
We thank Rafael Shimkunas, Zhong Jian, Mark Jaradeh, Logan R. J. Bailey, Austen J. Lucena and Johanna M. Borst for their help in animal care and cell isolation.
SOURCES OF FUNDING
This work was supported by the National Institute of Health R01-HL123526 (YCI), R01-HL90880 (LTI and YCI), R01-HL30077 (DMB); the American Heart Association 14GRNT20510041 (YCI); and the Hungarian Scientific Research Fund 0TKA101196 (TB).
Abbreviations:
- AIP
Autocamtide-2-related inhibitory peptide
- AP
Action potential
- βAR
β-adrenergic receptor
- CaM
Calmodulin
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- cAMP
Adenosine-3’,5’-cyclic monophosphate
- dV/dtmax
Maximal upstroke velocity of AP
- GS
GS-458967, late Na+ current inhibitor
- GSH
Reduced glutathione
- Epac
Exchange protein directly activated by cAMP
- IGS
GS-458967-sensitive current
- INaL
Late Na+ current
- INaT
Transient Na+ current
- ITTX
Tetrodotoxin-sensitive current
- ISO
Isoproterenol
- L-NAME
Nω-nitro-L-arginine methyl ester
- NAC
N-acetyl cysteine
- NOS
Nitric oxide synthase
- NOX2
NADPH oxidase 2
- PKA
Protein kinase A
- PKI
Protein kinase inhibitor peptide
- ROS
Reactive oxygen species
- Rp-cAMPS
Rp-adenosine-3’,5’-cyclic monophosphorothioate
- TTX
Tetrodotoxin
Footnotes
DISCLOSURES
Dr. Luiz Belardinelli is a former employee of Gilead Sciences, Inc, which is the patent holder of GS- 458967. Current affiliation of Dr. Belardinelli is InCarda Therapeutics, Inc. (Brisbane, CA, USA).
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–52. [DOI] [PubMed] [Google Scholar]
- [2].Belardinelli L, Giles WR, Rajamani S, Karagueuzian HS, Shryock JC. Cardiac late Na(+) current: proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Heart Rhythm. 2015;12:440–8. [DOI] [PubMed] [Google Scholar]
- [3].Undrovinas AI, Maltsev VA, Kyle JW, Silverman N, Sabbah HN. Gating of the late Na+ channel in normal and failing human myocardium. J. Mol. Cell. Cardiol. 2002;34:1477–89. [DOI] [PubMed] [Google Scholar]
- [4].Maltsev VA, Undrovinas AI. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc. Res. 2006;69:116–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Attwell D, Cohen I, Eisner D, Ohba M, Ojeda C. Steady-State Ttx-Sensitive (Window) Sodium Current in Cardiac Purkinje-Fibers. Pflugers Arch. 1979;379:137–42. [DOI] [PubMed] [Google Scholar]
- [6].Clancy CE, Tateyama M, Liu H, Wehrens XH, Kass RS. Non-equilibrium gating in cardiac Na+ channels: an original mechanism of arrhythmia. Circulation. 2003;107:2233–7. [DOI] [PubMed] [Google Scholar]
- [7].Bohnen MS, Peng G, Robey SH, Terrenoire C, Iyer V, Sampson KJ, et al. Molecular Pathophysiology of Congenital Long QT Syndrome. Physiol. Rev. 2017;97:89–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, et al. Ca2+/calmodulin- dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 2006;116:3127–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aiba T, Hesketh GG, Liu T, Carlisle R, Villa-Abrille MC, O’Rourke B, et al. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc. Res. 2010;85:454–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sossalla S, Wagner S, Rasenack EC, Ruff H, Weber SL, Schondube FA, et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts--role of late sodium current and intracellular ion accumulation. J. Mol. Cell. Cardiol. 2008;45:32–43. [DOI] [PubMed] [Google Scholar]
- [11].Toischer K, Hartmann N, Wagner S, Fischer TH, Herting J, Danner BC, et al. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J. Mol. Cell. Cardiol. 2013;61:111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, et al. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. 2013;127:575–84. [DOI] [PubMed] [Google Scholar]
- [13].Matsuda JJ, Lee H, Shibata EF. Enhancement of rabbit cardiac sodium channels by beta-adrenergic stimulation. Circ. Res. 1992;70:199–207. [DOI] [PubMed] [Google Scholar]
- [14].Frohnwieser B, Chen LQ, Schreibmayer W, Kallen RG. Modulation of the human cardiac sodium channel alpha-subunit by cAMP-dependent protein kinase and the responsible sequence domain. J. Physiol. 1997;498 ( Pt 2):309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhou J, Yi J, Hu N, George AL Jr., Murray KT. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ. Res. 2000;87:33–8. [DOI] [PubMed] [Google Scholar]
- [16].Lu T, Lee HC, Kabat JA, Shibata EF. Modulation of rat cardiac sodium channel by the stimulatory G protein alpha subunit. J. Physiol. 1999;518 ( Pt 2):371–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dybkova N, Wagner S, Backs J, Hund TJ, Mohler PJ, Sowa T, et al. Tubulin polymerization disrupts cardiac beta-adrenergic regulation of late INa. Cardiovasc. Res. 2014;103:168–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, et al. A beta(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest. 2010;120:3508–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Song Y, El-Bizri N, Rajamani S, Belardinelli L. Abstract 17193: Inhibiting Late Sodium Current Attenuates Isoproterenol-Induced Transient Inward Current and Delayed Afterdepolarizations in Ventricular Myocytes. Circulation. 2015;132:A17193-A. [Google Scholar]
- [20].Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: role of CaMKII. J. Mol. Cell. Cardiol. 2010;48:322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mani SK, Egan EA, Addy BK, Grimm M, Kasiganesan H, Thiyagarajan T, et al. beta-Adrenergic receptor stimulated Ncx1 upregulation is mediated via a CaMKII/AP-1 signaling pathway in adult cardiomyocytes. J. Mol. Cell. Cardiol. 2010;48:342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Grimm M, Ling H, Willeford A, Pereira L, Gray CB, Erickson JR, et al. CaMKIIdelta mediates beta- adrenergic effects on RyR2 phosphorylation and SR Ca(2+) leak and the pathophysiological response to chronic beta-adrenergic stimulation. J. Mol. Cell. Cardiol. 2015;85:282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Polakova E, Illaste A, Niggli E, Sobie EA. Maximal acceleration of Ca2+ release refractoriness by beta-adrenergic stimulation requires dual activation of kinases PKA and CaMKII in mouse ventricular myocytes. J. Physiol. 2015;593:1495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dries E, Santiago DJ, Johnson DM, Gilbert G, Holemans P, Korte SM, et al. Calcium/calmodulin- dependent kinase II and nitric oxide synthase 1-dependent modulation of ryanodine receptors during beta- adrenergic stimulation is restricted to the dyadic cleft. J. Physiol. 2016;594:5923–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tsurugi T, Nagatomo T, Abe H, Oginosawa Y, Takemasa H, Kohno R, et al. Differential modulation of late sodium current by protein kinase A in R1623Q mutant of LQT3. Life Scis. 2009;84:380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Erickson JR, Patel R, Ferguson A, Bossuyt J, Bers DM. Fluorescence resonance energy transfer- based sensor Camui provides new insight into mechanisms of calcium/calmodulin-dependent protein kinase II activation in intact cardiomyocytes. Circ. Res. 2011;109:729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Erickson JR, Nichols CB, Uchinoumi H, Stein ML, Bossuyt J, Bers DM. S-Nitrosylation Induces Both Autonomous Activation and Inhibition of Calcium/Calmodulin-dependent Protein Kinase II delta. J. Biol. Chem 2015;290:25646–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mangmool S, Shukla AK, Rockman HA. beta-Arrestin-dependent activation of Ca(2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J. Cell Biol. 2010;189:573–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, et al. Epac2 mediates cardiac beta1- adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation. 2013;127:913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Brennan JP, Bardswell SC, Burgoyne JR, Fuller W, Schroder E, Wait R, et al. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem. 2006;281:21827–36. [DOI] [PubMed] [Google Scholar]
- [31].Burgoyne JR, Eaton P. Transnitrosylating nitric oxide species directly activate type I protein kinase A, providing a novel adenylate cyclase-independent cross-talk to beta-adrenergic-like signaling. J. Biol. Chem. 2009;284:29260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wagner S, Dantz C, Flebbe H, Azizian A, Sag CM, Engels S, et al. NADPH oxidase 2 mediates angiotensin II-dependent cellular arrhythmias via PKA and CaMKII. J. Mol. Cell. Cardiol. 2014;75:206–15. [DOI] [PubMed] [Google Scholar]
- [33].Horvath B, Banyasz T, Jian Z, Hegyi B, Kistamas K, Nanasi PP, et al. Dynamics of the late Na(+) current during cardiac action potential and its contribution to afterdepolarizations. J. Mol. Cell. Cardiol. 2013;64:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hegyi B, Horvath B, Vaczi K, Gonczi M, Kistamas K, Ruzsnavszky F, et al. Ca(2+)-activated Cl(−) current is antiarrhythmic by reducing both spatial and temporal heterogeneity of cardiac repolarization. J. Mol. Cell. Cardiol. 2017;109:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hegyi B, Chen-Izu Y, Jian Z, Shimkunas R, Izu LT, Banyasz T. KN-93 inhibits IKr in mammalian cardiomyocytes. J. Mol. Cell. Cardiol. 2015;89:173–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen-Izu Y, Izu LT, Hegyi B, Banyasz T. Recording of Ionic Currents Under Physiological Conditions: Action Potential-Clamp and ‘Onion-Peeling’ Techniques In: Jue T, editor. Modern Tools of Biophysics. New York, NY: Springer; New York; 2017. p. 31–48. [Google Scholar]
- [37].Belardinelli L, Liu G, Smith-Maxwell C, Wang WQ, El-Bizri N, Hirakawa R, et al. A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J. Pharmacol. Exp. Ther. 2013;344:23–32. [DOI] [PubMed] [Google Scholar]
- [38].Hegyi B, Barandi L, Komaromi I, Papp F, Horvath B, Magyar J, et al. Tetrodotoxin blocks L-type Ca2+ channels in canine ventricular cardiomyocytes. Pflugers Arch. 2012;464:167–74. [DOI] [PubMed] [Google Scholar]
- [39].Curran J, Tang L, Roof SR, Velmurugan S, Millard A, Shonts S, et al. Nitric oxide-dependent activation of CaMKII increases diastolic sarcoplasmic reticulum calcium release in cardiac myocytes in response to adrenergic stimulation. PloS one. 2014;9:e87495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gutierrez DA, Fernandez-Tenorio M, Ogrodnik J, Niggli E. NO-dependent CaMKII activation during beta-adrenergic stimulation of cardiac muscle. Cardiovasc. Res. 2013;100:392–401. [DOI] [PubMed] [Google Scholar]
- [41].Coultrap SJ, Bayer KU. Nitric oxide induces Ca2+-independent activity of the Ca2+/calmodulin- dependent protein kinase II (CaMKII). J. Biol. Chem. 2014;289:19458–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang DM, Chai Y, Erickson JR, Brown JH, Bers DM, Lin YF. Intracellular signalling mechanism responsible for modulation of sarcolemmal ATP-sensitive potassium channels by nitric oxide in ventricular cardiomyocytes. J. Physiol. 2014;592:971–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pereira L, Bare DJ, Galice S, Shannon TR, Bers DM. beta-Adrenergic induced SR Ca2+ leak is mediated by an Epac-NOS pathway. J. Mol. Cell. Cardiol. 2017;108:8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hegyi B, Bossuyt J, Griffiths LG, Shimkunas R, Coulibaly Z, Jian Z, et al. Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc. Natl. Acad. Sci. U. S. A. 2018;115:E3036–E44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Horvath B, Hegyi B, Kistamas K, Vaczi K, Banyasz T, Magyar J, et al. Cytosolic calcium changes affect the incidence of early afterdepolarizations in canine ventricular myocytes. Can. J. Physiol. Pharmacol. 2015;93:527–34. [DOI] [PubMed] [Google Scholar]
- [46].Hegyi B, Bossuyt J, Ginsburg KS, Mendoza LM, Talken L, Ferrier WT, et al. Altered Repolarization Reserve in Failing Rabbit Ventricular Myocytes: Calcium and beta-Adrenergic Effects on Delayed- and Inward-Rectifier Potassium Currents. Circ. Arrhythm. Electrophysiol. 2018;11:e005852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Taddese A, Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. [DOI] [PubMed] [Google Scholar]
- [48].Yamada-Hanff J, Bean BP. Activation of Ih and TTX-sensitive sodium current at subthreshold voltages during CA1 pyramidal neuron firing. J. Neurophysiol. 2015;114:2376–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chandra R, Chauhan VS, Starmer CF, Grant AO. beta-adrenergic action on wild-type and KPQ mutant human cardiac Na+ channels: shift in gating but no change in Ca2+ : Na+ selectivity. Cardiovasc. Res. 1999;42:490–502. [DOI] [PubMed] [Google Scholar]
- [50].Rocchetti M, Sala L, Rizzetto R, Staszewsky LI, Alemanni M, Zambelli V, et al. Ranolazine prevents INaL enhancement and blunts myocardial remodelling in a model of pulmonary hypertension. Cardiovasc. Res. 2014;104:37–48. [DOI] [PubMed] [Google Scholar]
- [51].El-Bizri N, Li CH, Liu GX, Rajamani S, Belardinelli L. Selective inhibition of physiological late Na(+) current stabilizes ventricular repolarization. Am. J. Physiol. Heart Circ. Physiol. 2018;314:H236–H45. [DOI] [PubMed] [Google Scholar]
- [52].Song Y, Belardinelli L. Basal late sodium current is a significant contributor to the duration of action potential of guinea pig ventricular myocytes. Physiol. Rep. 2017;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mishra S, Reznikov V, Maltsev VA, Undrovinas NA, Sabbah HN, Undrovinas A. Contribution of sodium channel neuronal isoform Na(v)1.1 to late sodium current in ventricular myocytes from failing hearts. J. Physiol. 2015;593:1409–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yang T, Atack TC, Stroud DM, Zhang W, Hall L, Roden DM. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ. Res. 2012;111:322–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dybkova N, Ahmad S, Pabel S, Tirilomis P, Hartmann N, Fischer TH, et al. Differential regulation of sodium channels as a novel proarrhythmic mechanism in the human failing heart. Cardiovasc. Res. 2018. [DOI] [PubMed] [Google Scholar]
- [56].Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, et al. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 2012;287:19856–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, et al. Ca2+/calmodulin- dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation. 2012;126:2084–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H1597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Tan HL, Kupershmidt S, Zhang R, Stepanovic S, Roden DM, Wilde AA, et al. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415:442–7. [DOI] [PubMed] [Google Scholar]
- [60].Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, et al. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem. Biophys. Res. Commun. 1991;181:968–75. [DOI] [PubMed] [Google Scholar]
- [61].Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem. Biophys. Res. Commun. 1995;212:806–12. [DOI] [PubMed] [Google Scholar]
- [62].Tateyama M, Rivolta I, Clancy CE, Kass RS. Modulation of cardiac sodium channel gating by protein kinase A can be altered by disease-linked mutation. J. Biol. Chem. 2003;278:46718–26. [DOI] [PubMed] [Google Scholar]
- [63].Herren AW, Weber DM, Rigor RR, Margulies KB, Phinney BS, Bers DM. CaMKII Phosphorylation of Na(V)1.5: Novel in Vitro Sites Identified by Mass Spectrometry and Reduced S516 Phosphorylation in Human Heart Failure. J. Proteome Res. 2015;14:2298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, et al. Voltage-Gated Sodium Channel Phosphorylation at Ser571 Regulates Late Current, Arrhythmia, and Cardiac Function In Vivo. Circulation. 2015;132:567–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Herren AW, Bers DM, Grandi E. Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 2013;305:H431–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Murphy BJ, Rogers J, Perdichizzi AP, Colvin AA, Catterall WA. cAMP-dependent phosphorylation of two sites in the alpha subunit of the cardiac sodium channel. J. Biol. Chem. 1996;271:28837–43. [DOI] [PubMed] [Google Scholar]
- [67].Clancy CE, Chen-Izu Y, Bers DM, Belardinelli L, Boyden PA, Csernoch L, et al. Deranged sodium to sudden death. J. Physiol. 2015;593:1331–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Qian C, Ma J, Zhang P, Luo A, Wang C, Ren Z, et al. Resveratrol attenuates the Na(+)-dependent intracellular Ca(2+) overload by inhibiting H(2)O(2)-induced increase in late sodium current in ventricular myocytes. PloS one. 2012;7:e51358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Fu C, Hao J, Zeng M, Song Y, Jiang W, Zhang P, et al. Modulation of late sodium current by Ca(2+) -calmodulin-dependent protein kinase II, protein kinase C and Ca(2+) during hypoxia in rabbit ventricular myocytes. Exp. Physiol. 2017;102:818–34. [DOI] [PubMed] [Google Scholar]
- [70].Ma J, Luo A, Wu L, Wan W, Zhang P, Ren Z, et al. Calmodulin kinase II and protein kinase C mediate the effect of increased intracellular calcium to augment late sodium current in rabbit ventricular myocytes. Am. J. Physiol. Cell Physiol. 2012;302:C1141–51. [DOI] [PubMed] [Google Scholar]
- [71].Sakmann BF, Spindler AJ, Bryant SM, Linz KW, Noble D. Distribution of a persistent sodium current across the ventricular wall in guinea pigs. Circ. Res. 2000;87:910–4. [DOI] [PubMed] [Google Scholar]
- [72].Ahern GP, Hsu SF, Klyachko VA, Jackson MB. Induction of persistent sodium current by exogenous and endogenous nitric oxide. J. Biol. Chem. 2000;275:28810–5. [DOI] [PubMed] [Google Scholar]
- [73].Berecki G, Wilders R, de Jonge B, van Ginneken AC, Verkerk AO. Re-evaluation of the action potential upstroke velocity as a measure of the Na+ current in cardiac myocytes at physiological conditions. PloS one. 2010;5:e15772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Banyasz T, Lozinskiy I, Payne CE, Edelmann S, Norton B, Chen B, et al. Transformation of adult rat cardiac myocytes in primary culture. Exp. Physiol. 2008;93:370–82. [DOI] [PubMed] [Google Scholar]
- [75].Sag CM, Mallwitz A, Wagner S, Hartmann N, Schotola H, Fischer TH, et al. Enhanced late INa induces proarrhythmogenic SR Ca leak in a CaMKII-dependent manner. J. Mol. Cell. Cardiol. 2014;76:94–105. [DOI] [PubMed] [Google Scholar]
- [76].Bacic D, Carneiro JS, Bento AA, Nearing BD, Rajamani S, Belardinelli L, et al. Eleclazine, an inhibitor of the cardiac late sodium current, is superior to flecainide in suppressing catecholamine-induced ventricular tachycardia and T-wave alternans in an intact porcine model. Heart Rhythm. 2017;14:448–54. [DOI] [PubMed] [Google Scholar]