

Abstract
Escherichia coli UvrD is a superfamily 1 helicase/translocase involved in multiple DNA metabolic processes including methyl-directed mismatch DNA repair. Although a UvrD monomer can translocate along single-stranded DNA, a UvrD dimer is needed for processive helicase activity in vitro. E. coli MutL, a regulatory protein involved in methyl- directed mismatch repair, stimulates UvrD helicase activity, however, the mechanism is not well understood. Using single molecule fluorescence and ensemble approaches, we find that a single MutL dimer can activate latent UvrD monomer helicase activity. However, we also find that MutL stimulates UvrD dimer helicase activity. We further find that MutL enhances the DNA unwinding processivity of UvrD. Hence MutL acts as a processivity factor by binding to and presumably moving along with UvrD to facilitate DNA unwinding.
Keywords: DNA Mismatch repair, single molecule fluorescence, ATPase, processivity, allostery, protein assembly, SF1A helicase
Introduction
The DNA unwinding and translocase activities of superfamily 1 (SF1) and SF2 helicases can be regulated by the assembly state of the enzyme as well as through interactions with accessory proteins[1]. As isolated enzymes they display helicase and translocase activities in vitro, however, these enzymes rarely function in isolation in vivo, but often function as components of larger molecular complexes that catalyze a wide range of processes in DNA metabolism. Therefore, investigation of the interactions between helicases and their accessory proteins is needed to fully understand how they operate and are regulated in vivo.
E. coli UvrD is a prototypical SF1A DNA helicase/translocase involved in DNA repair[2, 3], replication[4-7] and recombination[8-10]. UvrD protein can self-associate into dimers and tetramers[11] and its assembly state regulates its properties. A UvrD monomer can processively and rapidly translocate in a 3’ to 5’ direction along single-stranded (ss) DNA but has little to no helicase activity in vitro[12-15] unless a force is applied to the DNA[16]. In the absence of accessory proteins, formation of at least a UvrD dimer is required to processively unwind duplex DNA in vitro[12-19]. The monomeric forms of the structurally homologous SF1 helicases, E. coli Rep and B. stearothermophilus PcrA, are also processive ssDNA translocases[20-23], and also display little helicase activity in vitro[22-24]. Activation of Rep and PcrA helicase activities also requires either self-assembly[22, 24, 25] or interaction with accessory proteins[26].
E. coli MutL protein plays a key role in methyl-directed mismatch repair (MMR) of DNA[27-30]. MutL interacts with UvrD[31] and stimulates its helicase activity in multiple- turnover[29, 32] DNA unwinding reactions. Although MutL was shown to stimulate UvrD-catalyzed unwinding of both 20 base pair (bp) and 92 bp partial duplex DNA under single round conditions such that additional UvrD could not re-initiate on the DNA, it was proposed that MutL functions by continually loading multiple UvrD molecules onto a DNA substrate without affecting its unwinding processivity[32]. Furthermore, the potential effects of UvrD assembly state were not considered in previous studies[32].
Using single molecule fluorescence and ensemble experiments, we show here that a single MutL dimer can activate the latent helicase activity of a UvrD monomer as well as stimulate the activity of a UvrD dimer. We further show that MutL does increase the DNA unwinding processivity of UvrD monomers and dimers indicating that MutL remains in complex with UvrD as it moves along the DNA.
Results
MutL activates the helicase activity of a UvrD monomer
To determine whether MutL can activate DNA unwinding by a UvrD monomer, we performed single molecule fluorescence resonance energy transfer (smFRET) DNA unwinding experiments[33] using total internal reflection fluorescence (TIRF) microscopy. A UvrD variant containing a biotin-avidity tag on its N-terminus was used to attach it to a polyethylene glycol (PEG) surface via a biotin-neutravidin linkage (Fig. 1a). The attachment of UvrD to the surface was performed at low UvrD concentrations (250 pM) to ensure that only UvrD monomers were immobilized on the surface. At this concentration the fraction of UvrD dimers in solution is less than 0.1 %[11]. As a further check, when immobilization was performed with Cy3-labeled UvrD only single photobleaching events were observed indicating the absence of dimers. To examine DNA unwinding we used a DNA substrate (DNA I) with an 18 bp duplex and a 3’-(dT)20 tail labeled with a fluorescent donor (Cy3) and acceptor (Cy5) at opposite ends of the duplex (Fig. 1a). Binding of a single DNA molecule to a UvrD monomer on the surface can be observed as the sudden appearance of a fluorescence signal. DNA unwinding is expected to be accompanied by an increase in Cy5 fluorescence and an anti-correlated decrease in Cy3 fluorescence[34], followed by acceptor strand release (Cy5 fluorescence disappearance and Cy3 fluorescence increase) and then donor strand release (loss of Cy3 fluorescence) upon complete unwinding (Fig. 1a).
Fig. 1. Activation of UvrD monomer helicase by MutL.
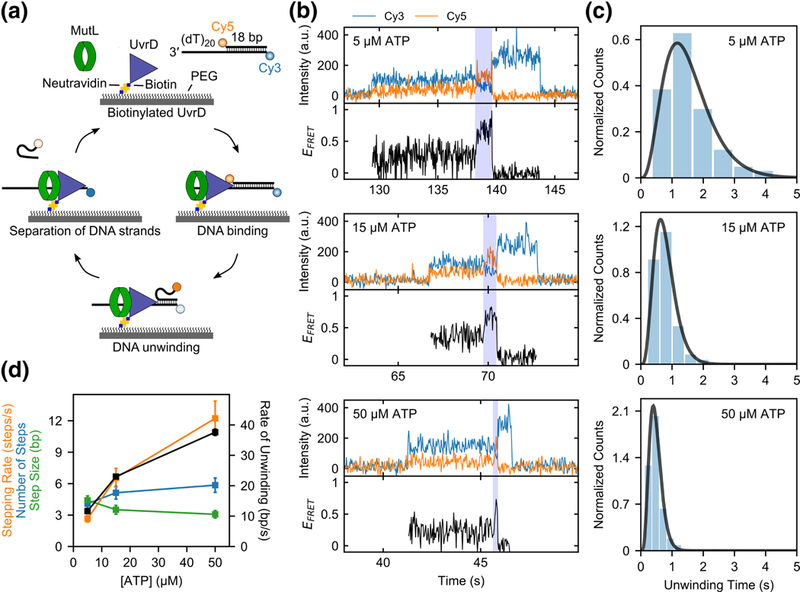
a, Cartoon showing the steps in a single molecule DNA unwinding event. b, Examples of single molecule time traces for UvrD monomer unwinding of 1 nM DNA in the presence of 50 nM MutL dimer at varying ATP concentrations (5 μM, 15 μM, and 50 μM) in imaging buffer at 25 °C. c, Histograms of the DNA unwinding time durations and the best fits (black lines) to a gamma distribution (see Eq. 1) at three ATP concentrations (5 μM, 15 μM, and 50 μM). d, Best fit values of the DNA unwinding parameters with standard errors shown as error bars.
Upon addition of DNA (1 nM) and ATP (5 μM) to the surface immobilized UvrD monomers in imaging buffer at 25 °C we observe DNA binding, but little DNA unwinding (<2%) (Supplementary Fig. 1a). This is consistent with previous studies indicating that UvrD monomers are unable to processively unwind even an 18 bp duplex DNA[12-15, 18, 19]. A rate constant for dissociation of DNA from a UvrD monomer of kd = 0.054 ± 0.002 s−1 was determined from analysis of the dwell times of DNA bound to UvrD (Supplementary Fig. 1b), which is consistent with a previous upper limit estimate of 0.12 s−1 determined from ensemble kinetic studies[12].
However, when an excess of MutL (50 nM dimer) was added along with DNA (1 nM) and ATP in imaging buffer, a significant number of DNA unwinding events were observed (Fig. 1b). The percentage of DNA molecules unwound was 33% (251/767) at 5 μM ATP, 42% (333/789) at 15 μM ATP, and 36% (310/854) at 50 μM ATP. The time duration of each DNA unwinding event (highlighted in blue in Fig. 1b) was measured as the time interval from the start of the increase in EFRET to the loss of Cy5 signal (acceptor strand release). The mean unwinding duration time, ṫ, decreases with increasing ATP concentration (Fig. 1c and Table 1), as expected for DNA unwinding. We analyzed the DNA unwinding time durations using the n-step sequential unwinding model in Supplementary Scheme 1 (Eq. 1) to determine the stepping rate, k, and the number of steps, n, from which the unwinding rate, r (bp/sec) and step size, m, can be calculated. The best fit kinetic parameters were determined as described by Neuman et al.[35] using a maximum likelihood estimation (MLE) analysis (Table 1 and Fig. 1d). As expected, the stepping rates, k, and unwinding rates, r, increase with increasing ATP concentration, while the number of steps, n, and step-sizes, m, are relatively constant. The unwinding rates measured for UvrD monomer in the presence of MutL are similar to the unwinding rates determined for UvrD dimer previously[13, 15, 19, 36]. These results demonstrate that the latent helicase activity of a UvrD monomer can be activated through interaction with the MutL protein.
Table 1.
| Kinetic parameters for MutL activated DNA unwinding by UvrD monomers
| [ATP] | 𝑡̅ (s) |
n (steps) |
k (s−1) |
m = L/n
(bp/step) |
r = L/t
(bp/s) |
|---|---|---|---|---|---|
| 5 μM | 1.54 ± 0.05 | 4.1 ± 0.4 | 2.6 ± 0.3 | 4.4 ± 0.4 | 11.7 ± 0.4 |
| 15 μM | 0.78 ± 0.02 | 5.1 ± 0.6 | 6.7 ± 0.9 | 3.5 ± 0.4 | 23.0 ± 0.7 |
| 50 μM | 0.48 ± 0.01 | 5.9 ± 0.7 | 12.2 ± 1.7 | 3.1 ± 0.4 | 37.6 ± 1.1 |
Best fit parameters (best fit ± s.e.) determined from maximum-likelihood estimation analysis (see Methods) of the data in Fig. 1c to Supplementary Scheme 1 (Eq. 1)
MutL relieves DNA substrate inhibition of UvrD-catalyzed DNA unwinding
The specific activity of UvrD-catalyzed DNA unwinding in vitro shows inhibition when the [DNA] exceeds the [UvrD][13]. This results from the fact that as the total [DNA] exceeds the total [UvrD], the population of UvrD monomers bound to DNA increases while the UvrD dimer-DNA population decreases. Since UvrD monomers show no helicase activity in vitro, the specific helicase activity decreases with increasing [DNA][11, 13]. Based on the observation that MutL can activate UvrD monomer helicase activity, we examined whether MutL can eliminate this DNA substrate inhibition. DNA unwinding was monitored under single round conditions using an “all or none” fluorescence stopped-flow DNA unwinding assay[36, 37] in buffer T at 25 °C (Fig. 2a). For this we used an 18 bp duplex DNA (DNA II) containing a 3’-(dT)20 tail with a Cy5 fluorophore attached to the long strand and Black Hole Quencher 2 (BHQ2) attached to the short strand. When in close proximity, BHQ2 quenches Cy5 fluorescence[38], hence, DNA strand separation is accompanied by an increase in Cy5 fluorescence. DNA unwinding was initiated by mixing pre-formed UvrD-DNA or MutL-UvrD-DNA complexes with buffer T containing 1 mM ATP, 2 mM MgCl2 and 2 μM protein trap. The protein trap, a 10 bp DNA hairpin (DNA X) possessing a 3’-(dT)40 ssDNA tail, binds to any free UvrD or UvrD that dissociates during unwinding thus preventing any reinitiation of DNA unwinding (see Supplementary Fig. 2) and ensuring that any unwinding is due to UvrD that is pre-bound to the DNA (single round conditions).
Fig. 2. DNA substrate inhibition of UvrD-catalyzed DNA unwinding is relieved by MutL.
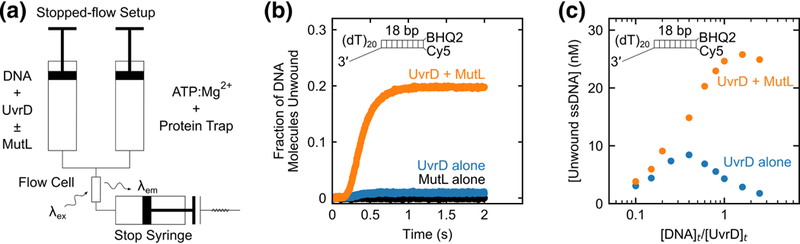
a, Schematic of the stopped-flow DNA unwinding experiment. In one syringe UvrD is pre-incubated with DNA labeled with Cy5 and BHQ2, with or without MutL in buffer T at 25 °C. UvrD-DNA-(±MutL) complex is rapidly mixed with ATP, MgCl2, and a protein trap to initiate DNA unwinding. DNA strand separation is accompanied by an enhancement in Cy5 fluorescence signal. b, DNA unwinding time courses from experiments performed at 125 nM 3’-(dT)20-ds18-BHQ2/Cy5 with 50 nM UvrD alone (blue), 625 nM MutL dimer alone (black) or 50 nM UvrD plus 625 nM MutL dimer (orange). c, Multiple DNA unwinding experiments were performed at constant 50 nM UvrD over a range of DNA concentrations in the presence (orange circles) or in the absence (blue circles) of saturating MutL (5-fold molar excess of MutL dimer over DNA) and the total unwinding amplitudes were plotted as a function of [DNA]t/[UvrD]t.
We first performed single round DNA unwinding experiments in the presence of a 2.5-fold excess of DNA (125 nM) over UvrD (50 nM) with and without MutL. The resulting DNA unwinding time courses (Fig. 2b) display the expected lag phase due to the fact that UvrD-catalyzed DNA unwinding occurs in multiple steps with similar rate constants, resulting in the formation of partially unwound intermediates prior to complete unwinding of double-stranded (ds) DNA[13, 36, 37]. We note that MutL alone does not support DNA unwinding (Fig. 2b). In the absence of MutL, <2% of the DNA substrate was unwound by UvrD (Fig. 2b), likely due to the small percentage of UvrD dimers present. However, at saturating MutL concentrations (625 nM dimer) (see Supplementary Fig. 3a,b) the same concentration of UvrD unwound ~20% of the DNA substrate (Fig. 2b). We note that in the Tris buffer that we used for the DNA unwinding studies performed above MutL exists as a mixture of dimers and higher oligomeric species[39].
Next, we performed a series of single round experiments at constant total UvrD concentration (50 nM) as a function of increasing DNA substrate concentration in the presence and absence of saturating MutL (Fig. 2c). With UvrD alone, we observe the expected DNA substrate inhibition. However, this DNA substrate inhibition is eliminated when excess MutL is included with UvrD. The amount of unwound ssDNA increases with increasing DNA substrate concentration until a plateau is reached at [DNA]t/[UvrD]t ≅ 0.8. These results support the conclusion that MutL stably activates the helicase activity of a UvrD monomer.
3’ ssDNA tail requirements for formation of a productive MutL-UvrD complex
We next investigated the effect of 3’ ssDNA tail length on MutL stimulation of UvrD monomer helicase activity. Single round stopped-flow DNA unwinding experiments were performed using DNA substrates (DNA II) with an 18 bp duplex and varying 3’-(dT)N tail lengths from N = 6 to 40 nt. By performing experiments in the presence of saturating MutL (625 nM dimer) and in DNA excess (125 nM) over UvrD (50 nM) we ensure that DNA molecules bind only one UvrD monomer. The time courses of DNA unwinding and the dependence of the total unwinding amplitudes on 3’ ssDNA tail length, N, are shown in Fig. 3a and 3b, respectively. No unwinding is detected for DNA substrates with N ≤ 8, whereas a sharp increase in DNA unwinding amplitude is observed for 3’ ssDNA tail lengths from 10 to 14 nt, with no further increase in amplitude for N > 14 nt.
Fig. 3. Effect of 3’ ssDNA tail length on the stimulation of UvrD monomer helicase by MutL.
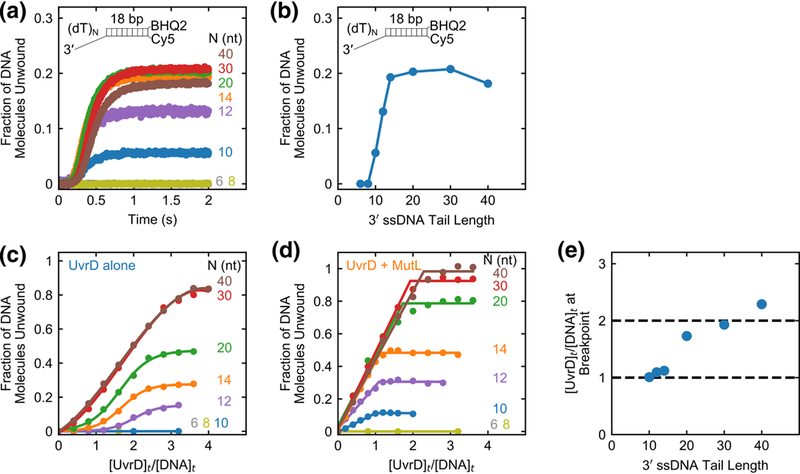
a, Stopped-flow DNA unwinding time courses monitoring Cy5 fluorescence performed at 50 nM UvrD, 625 nM MutL dimer, and 125 nM 3’-(dT)N-ds18-BHQ2/Cy5, with the indicated 3’-ssDNA tail lengths, N in nucleotides. b, The total unwinding amplitude plotted as a function of 3’ ssDNA tail length (N). c,d, The fraction of DNA molecules unwound, obtained under stoichiometric-binding conditions, is plotted as a function of the ratio of [UvrD]t/[DNA]t for a series of stopped-flow experiments with DNA of the indicated 3’-(dT)N. Experiments were performed by varying the [UvrD] for a constant DNA concentration (50 nM) with (c) UvrD alone or (d) UvrD plus 250 nM MutL dimer. e, Breakpoints from the plots in panel (d) are plotted as a function of 3’ ssDNA tail length (N).
Crystal structures of UvrD monomer in complex with partial duplex DNA suggest that UvrD contacts ~5 nt on the 3’ ssDNA tail[40]. Unwinding competition experiments indicate that UvrD shows some specificity for a 3’ ssDNA/dsDNA junction with 3’ tail lengths as short as N = 4 nt[13]. Thus, if MutL does not require any contact with the ssDNA tail, we would expect to detect unwinding of DNA substrates with 3’ ssDNA tail lengths as short as N = 4-8 nt. Hence the results above suggest that MutL interacts with at least some portion of the 3’ ssDNA tail in order to activate a UvrD monomer.
MutL stimulates the helicase activity of a UvrD dimer
We next examined the effect of MutL over a range of UvrD and DNA concentration ratios that will populate both UvrD monomers and dimers on the DNA[11- 13]. Single round stopped-flow DNA unwinding experiments were performed at constant DNA concentration (50 nM) as a function of UvrD concentration with and without MutL. The fraction of DNA molecules unwound is plotted as a function of UvrD to DNA ratio for UvrD alone (Fig. 3c) or UvrD plus saturating MutL (250 nM dimer) (Fig. 3d). For UvrD alone, no unwinding is observed for DNA substrates with N < 12 nt since a 3’ ssDNA tail with N ≥ 12 nt is needed to form a stable UvrD dimer-DNA complex[13]. In contrast, in the presence of MutL, UvrD can unwind DNA substrates with 3’ tail lengths as short as 10 nt (Fig. 3d) since a 10 nt tail is sufficient to stabilize a MutL-UvrD monomer-DNA complex consistent with the single molecule and stopped-flow experiments discussed above.
In the absence of MutL, the amplitudes of DNA unwinding display a sigmoidal dependence on [UvrD]t/[DNA]t with a breakpoint at [UvrD]t/[DNA]t ≅ 2-3 depending on the 3’-(dT)N length (Fig. 3c). This reflects the fact that DNA-unwinding activity requires at least a UvrD dimer bound per DNA substrate[13]. However, in the presence of MutL, the DNA unwinding amplitudes increase linearly with [UvrD]t/[DNA]t for each DNA substrate (Fig. 3d) with a breakpoint that increases from one UvrD per DNA for N = 10, 12 and 14 nt tails to two UvrD per DNA for N = 20, 30 and 40 nt tails (Fig. 3e). This indicates that on DNA substrates possessing 3’-(dT)N tails with N = 10, 12, or 14 nt, MutL activates a UvrD monomer. However, for DNA substrates possessing longer 3’- (dT)N tails of N ≥ 20 nt, MutL can stimulate the helicase activity of a UvrD dimer. We note that MutL eliminates the DNA substrate inhibition for the DNA substrates that can accommodate a UvrD dimer (N = 30, 40 nt).
MutL increases UvrD DNA unwinding processivity
DNA unwinding catalyzed by UvrD dimers has limited processivity[16, 36]. Since we have shown that MutL stimulates the helicase activity of UvrD monomers and dimers under single round conditions, this suggests the formation of a stable UvrD-MutL complex that can move along the DNA, i.e., that MutL might function as a processivity factor, keeping UvrD tethered to the DNA.
To test this hypothesis, we performed a series of single round stopped-flow experiments on 50 nM DNA substrates (DNA II-VIII) with varying duplex lengths (L = 18, 21, 25, 32, 40, 50, 80 bp) to measure the unwinding processivity of UvrD alone (160 nM) on 3’-(dT)20 tailed DNA substrates (Fig. 4a), UvrD (160 nM) plus MutL dimer (375 nM) on 3’-(dT)20 tailed DNA substrates (Fig. 4b), and UvrD (160 nM) plus MutL dimer (375 nM) on 3’-(dT)14 tailed DNA substrates (Fig. 4c). Note that a shorter 3’-(dT)14 tail is sufficient to bind only one UvrD monomer in the presence of MutL (see Fig. 3e). These time courses all display lag phases that increase with duplex length as expected for an “all or none” unwinding assay[36, 37]. We analyzed the time courses (Fig. 4a-c) using the n-step sequential unwinding model in Supplementary Scheme 2 (Eq. 3). The best fit parameters obtained from the non-linear least-squares (NLLS) analysis are given in Table 2.
Fig. 4. MutL increases the processivity of UvrD-catalyzed DNA unwinding.
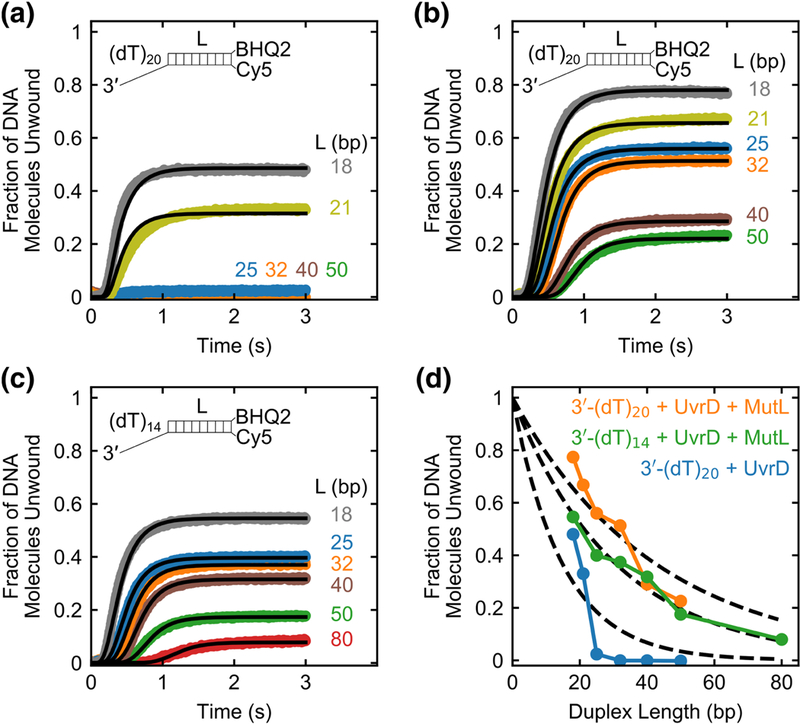
Stopped-flow DNA unwinding time courses monitoring Cy5 fluorescence are shown for a series of DNA substrates with the indicated duplex lengths, L (base pairs). (a) Experiments performed with 50 nM 3’-(dT)20 tailed DNA substrates with 160 nM UvrD alone. (b) Experiments performed with 50 nM 3’-(dT)2o tailed DNA substrates at 160 nM UvrD plus 375 nM MutL dimer. (c) Experiments performed with 50 nM 3’-(dT)14 tailed DNA substrates at 160 nM UvrD plus 375 nM MutL dimer. Solid lines are simulations using Eq. 3 and best-fit parameters obtained from global NLLS analysis of the unwinding time courses (Table 2). d, Fraction of DNA molecules unwound plotted as a function of duplex length (L) for 50 nM 3’-(dT)20 tailed DNA with 160 nM UvrD alone (blue); 50 nM 3’-(dT)20 tailed DNA with 160 nM UvrD plus 375 nM MutL dimer (orange); 50 nM 3’-(dT)14 tailed DNA with 160 nM UvrD plus 375 nM MutL dimer (green). The dashed lines show the best fit of each data set to Eq. 4 with the values of processivity P in Table 2.
Table 2.
| Kinetic parameters for DNA unwinding by UvrD alone and UvrD-MutL complexes
| Sample |
m (bp/step) |
kobs (s−1) |
kc (s−1) |
mkobs (bp/s) |
P | ⟨Nbp⟩ (bp) |
|---|---|---|---|---|---|---|
| 3’-(dT)20 + UvrD | 1.0 ± 0.2 | 78 ± 15 | 4.6 ± 0.1 | 80 ± 30 | 0.935 ± 0.012 | 15 ± 3 |
| 3’-(dT)20 + UvrD + MutL | 2.32 ± 0.05 | 29.5 ± 0.6 | 3.95 ± 0.02 | 68 ± 3 | 0.977 ± 0.002 | 42 ± 4 |
| 3’-(dT)14 + UvrD + MutL | 2.90 ± 0.05 | 26.9 ± 0.5 | 4.54 ± 0.02 | 78 ± 3 | 0.968 ± 0.001 | 31 ± 1 |
Best fit parameters (best fit ± s.e.) determined from non-linear least-squares analysis (see Methods) of the data in Fig. 4a-c to Supplementary Scheme 2 (Eq. 3)
The total DNA unwinding amplitude decreases with increasing duplex length (Fig. 4d). We analyzed these data using Eq. 4 which assumes that DNA unwinding is catalyzed by a single monodisperse population of helicases to obtain estimates of the unwinding processivity, P, and the average number of DNA base pairs unwound per protein binding event, ⟨Nbp⟩, for UvrD alone and UvrD in complex with MutL (Table 2). This analysis indicates that UvrD in complex with MutL on the DNA substrates with either a 3’-(dT)14 tail or 3’-(dT)20 tail on average unwinds 2-3 fold more DNA base pairs compared to UvrD alone on 3’-(dT)20 tailed DNA substrates (Table 2). The data for unwinding of DNA with a 3’-(dT)14 tail shows a good fit to Eq. 4 with P = 0.910 ± 0.003. However, the fit for the DNA with a 3’-(dT)20 tail is not as good. This may be due to the possible presence of a mixture of active UvrD species (UvrD dimers and UvrD dimer- MutL complexes), each of which may have different processivities. Since these are single round experiments that prevent re-binding of free UvrD, these results rule out a model in which MutL continuously loads free UvrD onto the DNA, but indicate that MutL serves to increase the DNA unwinding processivity of UvrD.
A single MutL dimer activates a UvrD monomer
We next sought to determine how many MutL dimers are required to activate a UvrD monomer. For these experiments, we used analytical ultracentrifugation and stopped-flow fluorescence techniques to determine the minimum stoichiometry of a MutL-UvrD-DNA complex that shows DNA unwinding activity. We performed these experiments in buffer M20/20, which is different from buffer T used in the experiments described above. We have previously shown that MutL exists as a mixture of dimers and higher oligomeric species in buffer T[39]. However, we have also shown that a MutL dimer is stable in buffer M20/20 that contains PO43- and this buffer inhibits formation of the higher oligomeric forms of MutL[39] (hydrodynamic properties of MutL2 are shown in Supplementary Table 1). In buffer M20/20 one MutL dimer binds to one 3’- (dT)20-ds18 DNA molecule with an apparent binding constant[39] K = (3.4 ± 0.4)x105 M” 1. However, the DNA unwinding activity of UvrD is lower in buffer M20/20 than in buffer T, which is why we used buffer T in the DNA unwinding experiments described above.
Sedimentation velocity and equilibrium approaches were used to examine the stoichiometry of MutL-UvrD-DNA complexes in buffer M20/20 at 25 °C. We used an 18 bp duplex DNA (DNA IX) possessing a 3’-(dT)i0 tail which is short enough to allow binding of only one UvrD monomer to the DNA[13] (see also Fig. 3d). A partial specific volume of vDNA = 0.563 ± 0.002 ml/g was measured for 3’-(dT)i0-ds18 DNA (DNA IX) by conducting sedimentation equilibrium experiments at 25 °C in buffer M20/20 (see Methods and Supplementary Fig. 4a). We used a UvrD variant, possessing a single Cys (UvrDACys-A100C)[41] that was labeled with Cy3. This UvrD-Cy3 variant retains both ssDNA translocase and DNA helicase activities[15, 16]. Sedimentation velocity experiments of UvrD monitoring 555 nm (Supplementary Fig. 4b) showed a single symmetrical peak at 2.15 ± 0.02 S, the position of which was independent of concentration indicating that UvrD was homogeneous and in a single assembly state. Sedimentation equilibrium experiments of UvrD-Cy3 (1 μM) at four rotor speeds (12, 15, 18, and 22k rpm) showed absorbance profiles that were well described by single exponentials (Fig. 5a). Initial analysis of the absorbance profiles using a partial specific volume for UvrD (vUvrDcaic = 0.7308 ml/g) calculated from its amino acid composition (see Methods) indicated molecular mass of 82.3 ± 1.2 kg/mol for UvrD-Cy3 consistent with monomeric UvrD-Cy3 (84.672 kg/mol). Using these sedimentation equilibrium data and constraining the molecular mass to the known value of 84.672 kg/mol for UvrD-Cy3 monomer, we estimated a more accurate partial specific volume (vUvrD-Cy3,exp= 0.737 ± 0.003 ml/g) for the UvrD-Cy3 monomer in buffer M20/20 at 25 °C. Values of s20,w and f/f0 for UvrD-Cy3 are given in Supplementary Table 1.
Fig. 5. A single MutL dimer can activate a UvrD monomer.
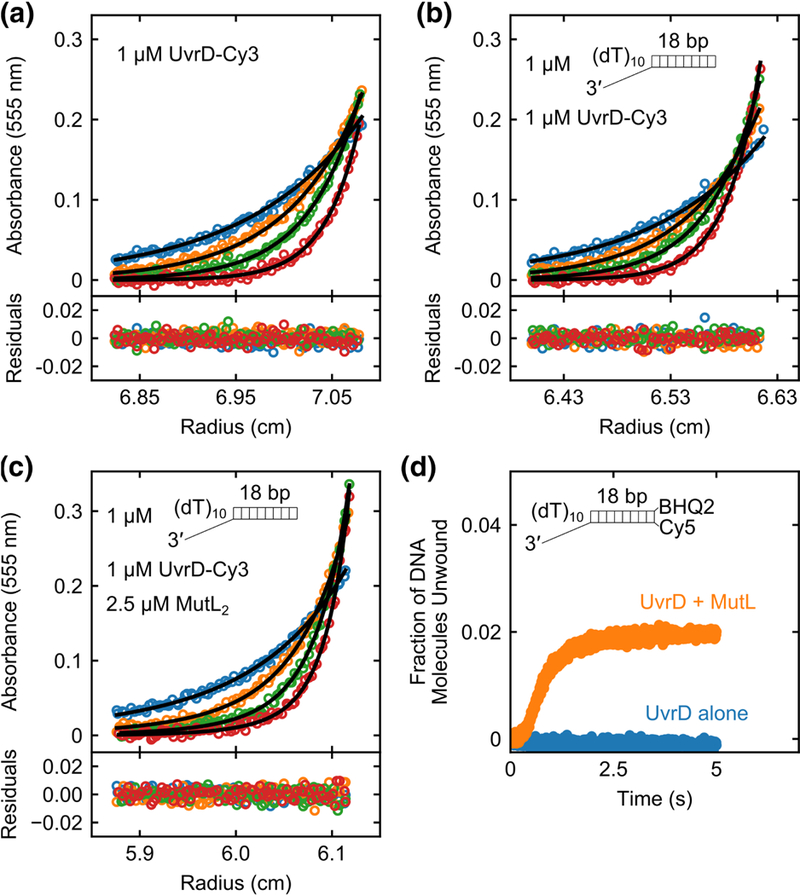
Sedimentation equilibrium experiments monitoring the Cy3 absorbance of UvrD-Cy3 at 555 nm were performed in buffer M20/20 at 25 °C. (a) UvrD-Cy3 alone (1 μM) shows a single species corresponding to a monomer at four rotor speeds (12k rpm (blue); 15k rpm (orange); 18k rpm (green); 22k rpm (red)). (b) UvrD-Cy3 (1 μM) plus 3’-(dT)10-ds18 (1 μM) shows a single species corresponding to a UvrD-Cy3 monomer-DNA complex at four rotor speeds (12k rpm (blue); 15k rpm (orange); 18k rpm (green); 22k rpm (red)). (c) UvrD- Cy3 (1 μM), 3’-(dT)10-ds18 (1 μM) and MutL (2.5 μM dimer) mixture shows two species containing UvrD-Cy3 at four rotor speeds (9k rpm (blue); 12k rpm (orange); 15k rpm (green); 18k rpm (red)). The two species correspond to a (UvrD-Cy3 monomer-DNA) complex and a (MutL dimer-UvrD-Cy3 monomer-DNA) complex. Smooth curves are simulations using best fit parameters with residuals shown below the plots as described in the text. d, Stopped-flow DNA unwinding time courses with 100 nM 3’-(dT)10-ds18- BHQ2/Cy5 DNA in buffer M20/20 at 25 °C were performed with 100 nM UvrD alone (blue) or 100 nM UvrD plus 250 nM MutL dimer (orange).
All remaining sedimentation experiments were performed in buffer M20/20 at 25 °C monitoring absorbance at 555 nm. At this wavelength, only the complexes that contain UvrD-Cy3 are detectable. Sedimentation velocity experiments were performed at four UvrD-Cy3 concentrations (0.6, 0.75, 0.85 and 1 μM) and a constant DNA concentration (1 μM). The resulting c(s) distributions show a single symmetrical peak at 2.63 ± 0.02 S (Supplementary Fig. 4c), the position of which did not change with UvrD concentration, indicating a distinct UvrD-DNA species. A peak corresponding to free UvrD monomer (2.15 S) was absent indicating that UvrD-Cy3 binds to the DNA stoichiometrically in buffer M20/20. To determine the molecular mass of the UvrD-DNA complex, we performed sedimentation equilibrium experiments with an equimolar mixture of UvrD-Cy3 (1 μM) and 3’-(dT)10-ds18 (1 μM) at four rotor speeds (12, 15, 18, and 22k rpm). The absorbance profiles fit well to a single exponential (Fig. 5b) giving a best fit molecular mass of 92.4 ± 1.4 kg/mol using a partial specific volume for the UvrD- DNA complex of 0.712 ml/g calculated as a weight average of the experimentally determined partial specific volumes of UvrD-Cy3 and 3’-(dT)10-ds18 in buffer M20/20 at 25 °C. This molecular mass is consistent with, although slightly lower than, that expected for a UvrD monomer-DNA complex (98.717 kg/mol). By constraining the molecular mass of the complex to its known value we calculated a partial specific volume, vUvrD-DNAexp = 0.727 ± 0.003 ml/g. Values of s20w and f/f0 for UvrD-Cy3-DNA complex are given in Supplementary Table 1.
We next performed sedimentation equilibrium experiments of a MutL-UvrD-Cy3- DNA complex using a 2.5-fold molar excess of MutL dimer (2.5 μM) over UvrD-Cy3 (1 μM) and 3’-(dT)10-ds18 (1 μM) at four rotor speeds (9, 12, 15, and 18k rpm). The absorbance profiles were well described by a two-exponential fit (Fig. 5c) indicating the presence of two species containing UvrD-Cy3, with one species corresponding to UvrD- Cy3-DNA and the other to MutL-UvrD-Cy3-DNA. No improvement in the quality of the fit was obtained by including a third exponential. By constraining the molecular mass of the lower molecular weight species to that of the UvrD-Cy3 monomer-DNA complex (98.717 kg/mol), we obtained a best fit value of 235 ± 6 kg/mol for the molecular mass of the MutL-UvrD-Cy3-DNA species using a weight average partial specific volume of 0.734 ml/g for (MutL)2-UvrD-DNA complex calculated from the partial specific volumes of UvrD-Cy3, MutL2, and 3’-(dT)10-ds18 in buffer M20/20 at 25 °C (see Methods and Supplementary Table 1). This molecular mass agrees well with the expected molecular mass for a MutL dimer-UvrD-Cy3 monomer-DNA complex (M = 234.540 kg/mol). Therefore, we conclude that the higher molecular mass species in buffer M20/20 contains only one MutL dimer and one UvrD-Cy3 monomer-bound to the DNA.
We next examined the DNA unwinding activity associated with the MutL2-UvrD- DNA complex at the same molar ratios (2.5:1:1) used in the sedimentation experiments under the identical solution conditions (buffer M20/20) at 25 °C. Based on the sedimentation experiments, this MutL-UvrD-DNA solution contains a mixture of DNA bound with a single UvrD monomer and DNA bound with a single UvrD monomer and a single MutL dimer. In single round DNA unwinding experiments performed at 100 nM 3’- (dT)10-ds18-BHQ2/Cy5 (DNA II) premixed with 100 nM UvrD plus 250 nM MutL dimer we observe significant DNA unwinding (Fig. 5d). No DNA unwinding was detected in the absence of MutL (Fig. 5d) consistent with there being no unwinding activity for the UvrD monomer-DNA complexes that are also present in the mixture. These results indicate that a single MutL dimer is sufficient to activate the helicase activity of a UvrD monomer.
MutL activation of UvrD helicase is specific
E. coli Rep is an SF1A helicase/translocase that shares ~40% sequence similarity with UvrD. Similar to UvrD, Rep monomers can translocate on ssDNA in an ATP-dependent manner (Supplementary Fig. 5a) but must form at least a dimer in order to activate its helicase activity[23-25, 42] (Supplementary Fig. 5b). We therefore examined whether MutL is able to activate the helicase activity of a Rep monomer. These experiments were performed with an excess of DNA substrate over Rep so that no more than one Rep monomer is bound per DNA molecule[23]. Single round unwinding experiments were performed using 50 nM 3’-(dT)20-ds18-BHQ2/Cy5 (DNA II) and 25 nM Rep alone or 25 nM Rep plus 250 nM MutL dimer in buffer T at 25 °C (Supplementary Fig. 5c). No stimulation of Rep-catalyzed DNA unwinding by MutL was observed under these conditions indicating that MutL stimulation is specific for UvrD reflecting a specific protein-protein interaction.
We next tested whether particular regions of UvrD are important for activation by MutL. The SF1A helicases, UvrD, Rep and PcrA all possess a relatively disordered C- terminal tail that is the least conserved region among these helicases and the C- terminal tail of UvrD has been reported to interact with MutL[31] as well as with RNA polymerase[43]. Hence, we examined UvrDΔ73 mutant, in which the last 73 C-terminal amino acids of UvrD are deleted, to see whether the C-terminal region of UvrD is important for activation by MutL. UvrDΔ73 retains helicase and ssDNA translocase activities[41], but monomeric UvrDΔ73 shows no helicase activity[11, 13]. Surprisingly, MutL is able to activate the helicase activity of monomeric UvrDA73 (Supplementary Fig. 5d) indicating that the C-terminal tail of UvrD is not essential for activation by MutL.
Discussion
UvrD possesses both ssDNA translocase and DNA helicase activities and functions in multiple DNA metabolic processes in vivo. These include methyl-directed mismatch DNA repair[2], nucleotide excision repair[3], replication restart[4, 5, 7, 44], recombination to remove RecA filaments from DNA[9, 10], and transcriptional control through interactions with RNA polymerase[45-47]. Although it is often assumed that the DNA helicase activity of UvrD is required for all of these functions this is not necessarily true. For some functions it may be that ssDNA translocase activity alone is sufficient as in its role to displace RecA filaments from ssDNA[1, 10, 48, 49]. Hence, it is of interest to understand how helicase activity vs. ssDNA translocase might be regulated. In this regard, it has been shown that functioning by itself in vitro, UvrD monomers are capable of rapid and processive ssDNA translocation, but are ineffective as DNA helicases, requiring dimerization for helicase activation[11-14, 19]. Hence, UvrD self-assembly is one way to separate and thus regulate its helicase and translocase activities. Such regulation is likely important in vivo since an unregulated helicase would likely be detrimental to the cell. In bacteria, UvrD-like helicases generally function as components of larger molecular machines[1, 26, 30, 50, 51]. Therefore, it is important to understand how these cellular partners regulate the various activities of UvrD.
MutL activates the UvrD monomer helicase and stimulates the UvrD dimer helicase
We show here that the latent UvrD monomer helicase activity can be activated through specific interactions with the accessory protein MutL. Even though activation of UvrD helicase activity in vitro requires dimerization[1, 11-13, 16, 17, 19], monomers of UvrD-like helicases contain all that is needed for helicase activity, but are auto-inhibited and require activation. Evidence for that comes from the studies of Rep and PcrA helicases. Activation of Rep monomer helicase activity can be accomplished through deletion of its 2B sub-domain[23, 42] or covalent crosslinking of the 2B sub-domain in a closed conformation[34]. Monomeric PcrA can be activated through interaction with an accessory protein, RepD[26]. In addition, limited helicase activity (<20 bp) has been observed for UvrD monomers when the DNA is under tension[16]. Interestingly, we find that MutL also stimulates the activity of UvrD dimers beyond that observed for UvrD dimers on their own[12, 13, 17]. Hence, the possibility remains that both UvrD monomers and dimers might both have functional roles in vivo.
MutL stimulation of UvrD is specific since we show here that MutL does not activate a Rep monomer although Rep is similar in structure to UvrD. This is consistent with previous multiple turnover unwinding studies[29]. The main regions that differ between Rep and UvrD are the C-terminal disordered tails and the 2B sub-domain. Based on yeast two-hybrid system experiments it was suggested that C-terminal domain of MutL and both N- and C-termini of UvrD are important for the interaction[31]. However, in our test experiment truncated UvrDΔ73 mutant lacking C-terminal tail was activated by MutL indicating that unstructured C-terminal tail of UvrD is dispensable for this activity. Interestingly in an analogous way, it was reported that C-terminal extension of UvrD interacts with UvrB, but truncated UvrDΔ73 version is still functional in nucleotide excision repair[52].
MutL functions as a processivity factor
Our single round DNA unwinding studies demonstrate that MutL increases the DNA unwinding processivity of UvrD, although this increase is moderate (2-3 fold increase, see Table 2). Such an increase can be explained by an increase in the rate of unwinding, mkobs, and/or a decrease in the dissociation rate, kd, of UvrD from DNA (Eq. 5). Our results indicate that activation of UvrD monomer by MutL does not significantly affect the rate of unwinding compared to UvrD dimers[13, 15, 19, 36] (Tables 1 and 2) suggesting that MutL acts to decrease the dissociation rate of UvrD from DNA. We infer from these results that MutL moves along the DNA in a complex with UvrD during unwinding although we lack direct evidence for this. MutL is known to bind DNA with a preference for ssDNA[32, 39, 53-55]. Based on the effects of 3’ ssDNA tail length, it appears that MutL requires contacts with the 3’ ssDNA tail of the DNA in order to form a productive complex with a UvrD monomer on the DNA. We also show that a single MutL dimer is sufficient to activate UvrD monomer helicase activity. MutL is comprised of C- terminal dimerization domain[54] and N-terminal ATPase domain which dimerizes upon ATP binding[53, 56]. Dimerization of the N-terminal domain leads to formation of a cavity in MutL dimer which can allow it to encircle the DNA substrate. Based on these observations, we propose a model where MutL clamps around the DNA and moves with UvrD during DNA unwinding.
In previous studies, Mechanic et al.[32] concluded that MutL functions to continually load multiple UvrD molecules onto a DNA substrate rather than to increase the DNA unwinding processivity of UvrD. A key experiment in their study involved the addition of a protein trap 30 seconds after initiation of multiple turnover unwinding of 148 bp blunt duplex DNA. This trap caused a cessation of further DNA unwinding. The authors argued that if MutL increased processivity then DNA unwinding would continue due to any UvrD that was already bound to the DNA. However, this would occur only if the processivity of unwinding were much greater than the moderate increase that we observe in our experiments. We also note that the rate limiting step in such multiple turnover unwinding of fully duplex DNA is the rate of initiation and not the rate of unwinding[57]. The actual rate of unwinding is relatively fast (on average it takes ~ 2 s to unwind a 148 bp duplex DNA). Therefore, even if MutL increased UvrD processivity, addition of a trap would still inhibit unwinding re-initiation and thus any detectable unwinding. Hence our observations and those of Mechanic et al.[32] appear to be consistent.
The limited DNA unwinding processivity of a MutL-UvrD complex might be explained by a molecular switch model proposed for MutL where the DNA binding affinity of MutL is modulated by ATP binding and ATP hydrolysis[53, 55, 58]. The processivity of this complex would be limited by the slow ATP hydrolysis rate of MutL. Notably, a MutL-E29A mutant, that binds but does not hydrolyze ATP, was shown to have greater stimulatory effect on UvrD compared to wild-type MutL[58] consistent with this interpretation.
In MMR, UvrD initiates DNA unwinding from a nick and proceeds past the mismatch, which can be as far removed as 1-2 kb in length[27]. The moderate increase in the DNA unwinding processivity of UvrD that we observe in the presence of MutL is inadequate to fully account for the unwinding of such long stretches of DNA. Hence the processivity of UvrD may be further enhanced through interactions with other proteins, such as MutS. It was shown that MutS and MutL enhance unwinding of a nicked DNA by UvrD in the presence of a mismatch[29]. Furthermore, it was shown that MutS, MutL, and mismatch-dependent unwinding by UvrD starts at the nick and unwinding is biased toward the shorter path between the nick and the mismatch[27] suggesting that helicase activity of UvrD can be influenced by multiple proteins.
It has been shown that homologous recombination is inhibited by DNA mismatch repair[59] which requires MutL and UvrD. It has also been reported that MutL and MutS[60], as well as UvrD[46] are required for transcription coupled repair (TCR) of DNA and it has been suggested that UvrD facilitates RNA polymerase backtracking during TCR[46]. Hence, it is possible that MutL activation of UvrD helicase activity might also play a role in these processes.
Methods
Buffers and reagents
Buffers were prepared with reagent grade chemicals using distilled, deionized (Milli-Q system; Millipore corp., Bedford, MA) water. Spectrophotometric grade glycerol was from Alfa Aesar (Ward Hill, MA). Buffer T is 10 mM Tris-HCl (pH 8.3 at 25 °C), 20 mM NaCl, 20% (v/v) glycerol, 1 mM 2-mercaptoethanol. Buffer M20/20 is 40.5 mM K2HPO4, 9.5 mM KH2PO4 (pH 7.4 at 25 °C), 20 mM NaCl, 20% (v/v) glycerol, 1 mM 2- mercaptoethanol. Imaging buffer is 10 mM Tris-HCl pH 8.3 at 25 °C, 20 mM NaCl, 20% (v/v) glycerol, 2 mM MgCl2, 3 mM Trolox, 0.8% (w/v) dextrose, 20 units/ml glucose oxidase, 20 units/ml catalase. ATP concentrations were determined spectrophotometrically using an extinction coefficient of ɛ259 = 15.4×103 M −1 cm −1.
Proteins
MutL protein was expressed and purified as described[39]. Concentrations of MutL monomer wer determined spectrophotometrically in 20 mM Tris-HCl pH 7.5 at 25 °C, 6 M Gdn-HCl using extinction coefficient of ɛ280 = 5.39 104 M−1 cm-1. MutL protein was stored in buffer M (40.5 mM K2HPO4, 9.5 mM KH2PO4 (pH 7.4 at 25 °C), 50 mM KCl, 0.1 mM EDTA, 1 mM 2-mercaptoethanol) at −80 °C. wtUvrD, biotin-UvrD, UvrDΔCys(A100C), and UvrDΔ73 were expressed and purified as described previously[19, 41, 61]. The UvrD monomer concentrations were determined spectrophotometrically in 10 mM Tris pH 8.1, 200 mM NaCl, 20% (v/v) glycerol using an extinction coefficient of ɛ280 = 1.06-105 M −1 cm −1. The UvrD protein was stored in minimal storage buffer (20 mM Tris-HCl (pH 8.3 at 25 °C), 200 mM NaCl, 50% (v/v) glycerol) at −20 °C for up to six months without any loss of helicase activity. The singlecysteine variant UvrDΔCys(A100C) was labeled with Cy3 maleimide (GE Healthcare)[41]. The Cy3 labeling efficiency of UvrDΔCys(A100C) was around 70% and was determined spectrophotometrically using extinction coefficients of 150,000 M−1 cm−1 at 550 nm for Cy3 dye. Rep protein was purified as described [62] and its concentration determined spectrophotometrically using ɛ280 = 7.68 104 M −1 cm −1.
DNA
The oligodeoxynucleotides were synthesized using an ABI model 391 (Applied Biosystems, Foster City, CA), and purified as described[63]. The concentrations of DNA strands were determined by spectrophotometric analysis as described previously[64]. The oligonucleotide sequences of DNA strands used in these studies are given in Supplementary Table 2. DNA duplexes were prepared by mixing equimolar concentrations of two complementary strands in 10 mM Tris pH 8.1, 0.1 M NaCl, followed by heating to 95 °C for five minutes and slow cooling to room temperature in water bath.
Single molecule total internal reflection fluorescence microscopy experiments
Single molecule experiments were performed on an Olympus IX71 microscope (model IX2_MPITIRTL) as described[65, 66]. Experiments were performed in imaging buffer at 25 °C. The Cy3 fluorophore was excited with a 532 nm laser and the fluorescence emissions of Cy3 and Cy5 were recorded at a 32 ms resolution (30.3 ms exposure time) for 3-4 minutes on EM-CCD camera. The FRET efficiency was calculated from the ratio of corrected Cy3 and Cy5 signals as described[66, 67].
The biotin-UvrD (20 μl of 250 μM) was immobilized onto the NeutrAvidin-PEG surface for 5 minutes and then was washed to remove the excess of biotin-UvrD. Under this condition, UvrD is monomeric as confirmed from the fluorescence intensities and photobleaching steps. Unwinding reactions were initiated by flowing 1 nM DNA (DNA I), indicated concentration of ATP, 2 mM MgCl2, and if necessary 50 nM MutL dimer in imaging buffer. Dwell times of DNA bound to a UvrD monomer were analyzed using one-step dissociation model described by the probability density function of an exponential distribution, kde-kdt, where kd is the dissociation rate constant. To analyze the distribution of DNA unwinding times, t, we used the simplest model presented in Supplementary Scheme 1, where the helicase proceeds through n irreversible rate- limiting steps to produce fully unwound ssDNA. This model is described by the probability density function of a gamma distribution[35, 37] as given in Eq. 1:
| 1 |
where k is the stepping rate and n is the number of steps. Fitting parameters were estimated utilizing MLE method implemented in the Python scipy.stats module. Standard errors were estimated using a bootstrap method. The average unwinding rate is , where L is the duplex DNA length in bp and is the average unwinding duration time and the kinetic step size is m = L/n.
Stopped-flow fluorescence experiments
All fluorescence stopped-flow experiments were carried out at 25 °C using an Applied Photophysics SX.18MV stopped-flow instrument (Applied Photophysics Ltd., Leatherhead, UK). DNA was pre-incubated with UvrD in buffer T for five minutes and then mixed with MutL if necessary and loaded into one syringe of the stopped-flow instrument. The solution of 1 mM ATP, 2 mM MgCl2 and 2 μM of 10 bp DNA hairpin possessing a 3’-(dT)40 ssDNA tail (DNA X) in buffer T was loaded into the other syringe of the stopped-flow instrument and both solutions were incubated for an additional five minutes at 25 °C prior to mixing. Cy5 fluorophore was excited using 625 nm LED (Applied Photophysics Ltd., Leatherhead, UK) and its fluorescence emission was monitored at wavelengths > 665 nm using a long-pass filter (Oriel Corp., Stradford, CT). The kinetic traces shown represent an average of at least 10 individual measurements. Trap tests were carried out by including the DNA substrate with the ATP and DNA hairpin solution. When mixed with UvrD or UvrD plus MutL, no unwinding signal was observed, indicating that DNA hairpin trapped all of the free UvrD (Supplementary Fig. 2).
To quantify the fraction of DNA molecules unwound for each time point, fss(t), the fluorescence signal, S(t), was calibrated using Eq. 2:
| 2 |
where S0 is the baseline fluorescence immediately after addition of ATP and Smax is the maximum fluorescence signal determined by mocking unwinding reaction by using DNA strand with Cy5 fluorophore in the absence of BHQ2-modified complementary strand and all the other reaction components.
Single round DNA unwinding time courses were analyzed using Supplementary Scheme 2 described by Eq. 3[37]:
| 3 |
where fss(t) is the fraction of ssDNA molecules formed, Fss(s) is the Laplace transform of fss(t), ℒ−1 is the inverse Laplace transform operator, s is the Laplace variable, AT is the total DNA unwinding amplitude, kc is the rate constant for the additional step not involved in unwinding, ku is the unwinding rate constant for n repeating steps, kd is the dissociation rate constant, L is the duplex region length, m is the kinetic step size, k0bs = ku + kd and n = L/m. Global NLLS fitting of the unwinding time courses was performed using Python lmfit module. The numerical inversion routine of the Laplace transform is based on Cody-Meinardus-Varga approximations of Talbot’s method[68].
On the basis of the mechanism in Supplementary Scheme 2 the unwinding amplitude is described by Eq. 4[37]:
where the processivity of DNA unwinding, P, is the probability that the helicase will unwind the next base pair rather than dissociate from the DNA[37]. The processivity, P, is related to the average number of base pairs unwound, (Nbp), by Eq. 5[37]:
| 5 |
where the approximation holds when ku » kd.
Experiments to examine Rep monomer translocation along ssDNA were performed in a stopped-flow apparatus as described[14, 23]. Rep (50 nM) was preincubated with 5’-F-(dT)N ssDNA (100 nM) in buffer T for five minutes at 25 °C and then rapidly mixed with buffer T containing 1 mM ATP, 2 mM MgCl2, and 8 mg/ml heparin. Fluorescein fluorescence was excited at 490 nm (Applied Photophysics Ltd., Leatherhead, UK) and its fluorescence emission was monitored at wavelengths > 520 nm using a long-pass filter (Oriel Corp., Stradford, CT). ssDNA translocation time courses were analyzed as described [23].
Analytical Ultracentrifugation
Dialyzed proteins were clarified by centrifugation at 14,000 rpm for 15 min at 4 °C. Sedimentation equilibrium and velocity experiments were performed using a ProteomeLab XL-A analytical ultracentrifuge equipped with an An50Ti rotor (Beckman Coulter, Fullerton, CA) at 25 °C. Absorbance signal for Cy3 labeled UvrDACys(A100C) was collected by scanning the sample cells at 555 nm wavelength.
Samples for sedimentation equilibrium (110 μl) were loaded into the three channels of an Epon charcoal-filled six-channel centerpieces with 130 μl of buffer M20/20 in the reference chambers. Absorbance data were collected by scanning the sample cells at intervals of 0.003 cm in the step mode with 5 averages per step. Samples were sedimented to equilibrium at the indicated rotor speeds, starting with the lowest and finishing with the highest rotor speed. The baseline offset was determined by increasing the rotor speed to 42,000 rpm at the end of the run to pellet the solutes and then measuring the residual absorbance in the solution column.
The resulting absorbance profiles, Ar, were analyzed by using NLLS fitting to Eq. 6 as implemented in SEDPHAT[69],
| 6 |
where r is the distance from the center of rotation, r0 is an arbitrary reference radius, m is angular velocity, T is absolute temperature, R is the gas constant,mi is the molecular mass of component i, is partial specific volume of component i, p is buffer density, Aro,i is the absorbance of component i at the reference position, and br is a radial- dependent baseline offset. Buffer density, p, was calculated from buffer composition using SEDNTERP[70]. Using the calculated partial specific volume of component i the molecular mass of component i (Mt) can be estimated. Alternatively, a more accurate value of the partial specific volume of macromolecule i can be determined if the true molecular mass of the macromolecule is known and constrained.
Partial specific volumes for UvrD-Cy3 (= 0.7308 ml/g) and MutL (= 0.7414 ml/g) at 25 °C were calculated from the amino acid composition according to Eq. 7 using SEDNTERP[70] as well as the weight-average partial specific volumes for complexes:
| 7 |
where Mi and vt are the molecular mass and viscosity of component i.
In sedimentation velocity experiments, the sample (380 μl) and buffer (392 μl) were loaded into each sector of an Epon charcoal-filled two-sector centerpiece. Experiments were performed at 25 °C and 42,000 rpm. Absorbance data were collected by scanning the sample cells at intervals of 0.003 cm. The continuous sedimentation coefficient distribution, c(s), was calculated using SEDFIT[71]. Calculated s values were converted to s20 w according to Eq. 8:
| 8 |
where p20w and rç20w are density and viscosity of the water at 20 °C, pexp and ijexp are density and viscosity of the buffer at 25 °C, and are partial specific volumes of macromolecule at 20 °C and 25 °C. Buffer density, p, and viscosity, rç, were calculated from buffer composition using SEDNTERP[70].
The ratio, f/f0, of the frictional coefficient of the macromolecule, f, to the frictional coefficient of an unhydrated sphere of equivalent mass, f0, was calculated using Eq. 9[72]:
| 9 |
where M and are the molecular mass and partial specific volume of the macromolecule, 𝜂 and p are the viscosity and density of the buffer, s is the measured sedimentation coefficient and NA is Avogadro’s number.
Supplementary Material
Highlights.
MutL protein regulates the helicase activity of UvrD.
A single MutL dimer is able to activate the helicase activity of a UvrD monomer.
MutL is also able to stimulate the helicase activity of a UvrD dimer.
MutL functions as a processivity factor for UvrD helicase activity.
Acknowledgements
We thank Alex Kozlov and Eric Tomko for assistance, as well as all members of the Lohman lab for comments and suggestions, and Thang Ho for oligodeoxynucleotides. This work was supported by NIH (GM045948 to TML).
Abbreviations:
- ssDNA
single stranded DNA
- smTIRF
single molecule total internal reflectance fluorescence
Footnotes
Competing interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Lohman TM, Tomko EJ, Wu CG. Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat Rev Mol Cell Biol. 2008;9:391–401. [DOI] [PubMed] [Google Scholar]
- [2].Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106:302–23. [DOI] [PubMed] [Google Scholar]
- [3].Sancar A DNA Excision Repair. AnnuRevBiochem. 1996;65:43–81. [DOI] [PubMed] [Google Scholar]
- [4].Atkinson J, McGlynn P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009;37:3475–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Heller RC, Marians KJ. Non-replicative helicases at the replication fork. DNA Repair (Amst). 2007;6:945–52. [DOI] [PubMed] [Google Scholar]
- [6].Bruand C, Ehrlich SD. UvrD-dependent replication of rolling-circle plasmids in Escherichia coli. Mol Microbiol. 2000;35:204–10. [DOI] [PubMed] [Google Scholar]
- [7].Flores MJ, Sanchez N, Michel B. A fork-clearing role for UvrD. Mol Microbiol. 2005;57:1664–75. [DOI] [PubMed] [Google Scholar]
- [8].Arthur HM, Lloyd RG. Hyper-recombination in uvrD mutants of Escherichia coli K-12. Mol Gen Genet. 1980;180:185–91. [DOI] [PubMed] [Google Scholar]
- [9].Veaute X, Delmas S, Selva M, Jeusset J, Le Cam E, Matic I, et al. UvrD helicase, unlike Rep helicase, dismantles RecA nucleoprotein filaments in Escherichia coli. EMBO J. 2005;24:180–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Petrova V, Chen SH, Molzberger ET, Tomko E, Chitteni-Pattu S, Jia H, et al. Active displacement of RecA filaments by UvrD translocase activity. Nucleic Acids Res. 2015;43:4133–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Maluf NK, Lohman TM. Self-association equilibria of Escherichia coli UvrD helicase studied by analytical ultracentrifugation. J Mol Biol. 2003;325:889–912. [DOI] [PubMed] [Google Scholar]
- [12].Maluf NK, Ali JA, Lohman TM. Kinetic mechanism for formation of the active, dimeric UvrD helicase-DNA complex. J Biol Chem. 2003;278:31930–40. [DOI] [PubMed] [Google Scholar]
- [13].Maluf NK, Fischer CJ, Lohman TM. A Dimer of Escherichia coli UvrD is the active form of the helicase in vitro. J Mol Biol. 2003;325:913–35. [DOI] [PubMed] [Google Scholar]
- [14].Fischer CJ, Maluf NK, Lohman TM. Mechanism of ATP-dependent translocation of E.coli UvrD monomers along single-stranded DNA. J Mol Biol. 2004;344:1287–309. [DOI] [PubMed] [Google Scholar]
- [15].Lee KS, Balci H, Jia H, Lohman TM, Ha T. Direct imaging of single UvrD helicase dynamics on long single-stranded DNA. Nat Commun. 2013;4:1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Comstock MJ, Whitley KD, Jia H, Sokoloski J, Lohman TM, Ha T, et al. Protein structure. Direct observation of structure-function relationship in a nucleic acid-processing enzyme. Science. 2015;348:352–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ali JA, Maluf NK, Lohman TM. An oligomeric form of E. coli UvrD is required for optimal helicase activity. J Mol Biol. 1999;293:815–34. [DOI] [PubMed] [Google Scholar]
- [18].Yokota H, Chujo YA, Harada Y. Single-molecule imaging of the oligomer formation of the nonhexameric Escherichia coli UvrD helicase. Biophys J. 2013;104:924–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nguyen B, Ordabayev Y, Sokoloski JE, Weiland E, Lohman TM. Large domain movements upon UvrD dimerization and helicase activation. Proc Natl Acad Sci U S A. 2017;114:12178–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dillingham MS, Wigley DB, Webb MR. Demonstration of unidirectional single-stranded DNA translocation by PcrA helicase: measurement of step size and translocation speed. Biochemistry. 2000;39:205–12. [DOI] [PubMed] [Google Scholar]
- [21].Dillingham MS, Wigley DB, Webb MR. Direct measurement of single-stranded DNA translocation by PcrA helicase using the fluorescent base analogue 2-aminopurine. Biochemistry. 2002;41:643–51. [DOI] [PubMed] [Google Scholar]
- [22].Niedziela-Majka A, Chesnik MA, Tomko EJ, Lohman TM. Bacillus stearothermophilus PcrA Monomer Is a Single-stranded DNA Translocase but Not a Processive Helicase in Vitro. J Biol Chem. 2007;282:27076–85. [DOI] [PubMed] [Google Scholar]
- [23].Brendza KM, Cheng W, Fischer CJ, Chesnik MA, Niedziela-Majka A, Lohman TM. Autoinhibition of Escherichia coli Rep monomer helicase activity by its 2B subdomain. Proc Natl Acad Sci U S A. 2005;102:10076–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cheng W, Hsieh J, Brendza KM, Lohman TM. E. coli Rep oligomers are required to initiate DNA unwinding in vitro. J Mol Biol. 2001;310:327–50. [DOI] [PubMed] [Google Scholar]
- [25].Ha T, Rasnik I, Cheng W, Babcock HP, Gauss GH, Lohman TM, et al. Initiation and reinitiation of DNA unwinding by the Escherichia coli Rep helicase. Nature. 2002;419:638–41. [DOI] [PubMed] [Google Scholar]
- [26].Chisty LT, Toseland CP, Fili N, Mashanov GI, Dillingham MS, Molloy JE, et al. Monomeric PcrA helicase processively unwinds plasmid lengths of DNA in the presence of the initiator protein RepD. Nucleic Acids Res. 2013;41:5010–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dao V, Modrich P. Mismatch-, MutS-, MutL-, and helicase II-dependent unwinding from the single-strand break of an incised heteroduplex. J Biol Chem. 1998;273:9202–7. [DOI] [PubMed] [Google Scholar]
- [28].Spampinato C, Modrich P. The MutL ATPase is required for mismatch repair. J Biol Chem. 2000;275:9863–9. [DOI] [PubMed] [Google Scholar]
- [29].Yamaguchi M, Dao V, Modrich P. MutS and MutL activate DNA helicase II in a mismatch- dependent manner. J Biol Chem. 1998;273:9197–201. [DOI] [PubMed] [Google Scholar]
- [30].Matson SW, Robertson AB. The UvrD helicase and its modulation by the mismatch repair protein MutL. Nucl Acids Res. 2006;34:4089–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hall MC, Jordan JR, Matson SW. Evidence for a physical interaction between the Escherichia coli methyl-directed mismatch repair proteins MutL and UvrD. EMBO J. 1998;17:1535–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mechanic LE, Frankel BA, Matson SW. Escherichia coli MutL loads DNA helicase II onto DNA. J Biol Chem. 2000;275:38337–46. [DOI] [PubMed] [Google Scholar]
- [33].Ha T, Kozlov AG, Lohman TM. Single-molecule views of protein movement on singlestranded DNA. Annu Rev Biophys. 2012;41:295–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Arslan S, Khafizov R, Thomas CD, Chemla YR, Ha T. Protein structure. Engineering of a superhelicase through conformational control. Science. 2015;348:344–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Neuman KC, Saleh OA, Lionnet T, Lia G, Allemand JF, Bensimon D, et al. Statistical determination of the step size of molecular motors. J Phys Condens Matter. 2005;17:S3811–20. [DOI] [PubMed] [Google Scholar]
- [36].Ali JA, Lohman TM. Kinetic measurement of the step size of DNA unwinding by Escherichia coli UvrD helicase. Science. 1997;275:377–80. [DOI] [PubMed] [Google Scholar]
- [37].Lucius AL, Maluf NK, Fischer CJ, Lohman TM. General methods for analysis of sequential “n-step” kinetic mechanisms: application to single turnover kinetics of helicase-catalyzed DNA unwinding. Biophys J. 2003;85:2224–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Marras SA, Kramer FR, Tyagi S. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 2002;30:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Niedziela-Majka A, Maluf NK, Antony E, Lohman TM. Self-assembly of Escherichia coli MutL and its complexes with DNA. Biochemistry. 2011;50:7868–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lee JY, Yang W. UvrD Helicase Unwinds DNA One Base Pair at a Time by a Two-Part Power Stroke. Cell. 2006;127:1349–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jia H, Korolev S, Niedziela-Majka A, Maluf NK, Gauss GH, Myong S, et al. Rotations of the 2B sub-domain of E. coli UvrD helicase/translocase coupled to nucleotide and DNA binding. J Mol Biol. 2011;411:633–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cheng W, Brendza KM, Gauss GH, Korolev S, Waksman G, Lohman TM. The 2B Domain of the Escherichia coli Rep protein is not required for DNA helicase activity. Proc Natl Acad Sci, USA. 2002;99:16006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sanders K, Lin CL, Smith AJ, Cronin N, Fisher G, Eftychidis V, et al. The structure and function of an RNA polymerase interaction domain in the PcrA/UvrD helicase. Nucleic Acids Res. 2017;45:3875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Michel B, Boubakri H, Baharoglu Z, Lemasson M, Lestini R. Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst). 2007;6:967–80. [DOI] [PubMed] [Google Scholar]
- [45].Gwynn EJ, Smith AJ, Guy CP, Savery NJ, McGlynn P, Dillingham MS. The Conserved C- Terminus of the PcrA/UvrD Helicase Interacts Directly with RNA Polymerase. PLoS One. 2013;8:e78141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Epshtein V, Kamarthapu V, McGary K, Svetlov V, Ueberheide B, Proshkin S, et al. UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature. 2014;505:372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].McGlynn P, Savery NJ, Dillingham MS. The conflict between DNA replication and transcription. Mol Microbiol. 2012;85:12–20. [DOI] [PubMed] [Google Scholar]
- [48].Tomko EJ, Fischer CJ, Niedziela-Majka A, Lohman TM. A Nonuniform Stepping Mechanism for E. coli UvrD Monomer Translocation along Single-Stranded DNA. Molecular Cell. 2007;26:335–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tomko EJ, Jia H, Park J, Maluf NK, Ha T, Lohman TM. 5’-Single-stranded/duplex DNA junctions are loading sites for E. coli UvrD translocase. EMBO J. 2010;29:3826–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Soultanas P, Dillingham MS, Papadopoulos F, Phillips SE, Thomas CD, Wigley DB. Plasmid replication initiator protein RepD increases the processivity of PcrA DNA helicase. Nucleic Acids Res. 1999;27:1421–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Atkinson J, Guy CP, Cadman CJ, Moolenaar GF, Goosen N, McGlynn P. Stimulation of UvrD helicase by UvrAB. J Biol Chem. 2009;284:9612–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Manelyte L, Guy CP, Smith RM, Dillingham MS, McGlynn P, Savery NJ. The unstructured C-terminal extension of UvrD interacts with UvrB, but is dispensable for nucleotide excision repair. DNA Repair (Amst). 2009;8:1300–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ban C, Junop M, Yang W. Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell. 1999;97:85–97. [DOI] [PubMed] [Google Scholar]
- [54].Guarne A, Ramon-Maiques S, Wolff EM, Ghirlando R, Hu X, Miller JH, et al. Structure of the MutL C-terminal domain: a model of intact MutL and its roles in mismatch repair. EMBO J. 2004;23:4134–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Robertson A, Pattishall SR, Matson SW. The DNA binding activity of MutL is required for methyl-directed mismatch repair in Escherichia coli. J Biol Chem. 2006;281:8399–408. [DOI] [PubMed] [Google Scholar]
- [56].Ban C, Yang W. Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell. 1998;95:541–52. [DOI] [PubMed] [Google Scholar]
- [57].Runyon GT, Lohman TM. Kinetics of Escherichia coli helicase II-catalyzed unwinding of fully duplex and nicked circular DNA. Biochemistry. 1993;32:4128–38. [DOI] [PubMed] [Google Scholar]
- [58].Robertson AB, Pattishall SR, Gibbons EA, Matson SW. MutL-catalyzed ATP Hydrolysis Is Required at a Post-UvrD Loading Step in Methyl-directed Mismatch Repair. J Biol Chem. 2006;281:19949–59. [DOI] [PubMed] [Google Scholar]
- [59].Tham KC, Hermans N, Winterwerp HH, Cox MM, Wyman C, Kanaar R, et al. Mismatch repair inhibits homeologous recombination via coordinated directional unwinding of trapped DNA structures. Mol Cell. 2013;51:326–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mellon I, Champe GN. Products of DNA mismatch repair genes mutS and mutL are required for transcription-coupled nucleotide-excision repair of the lactose operon in Escherichia coli. Proc Natl Acad Sci U S A. 1996;93:1292–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Runyon GT, Wong I, Lohman TM. Overexpression, purification, DNA binding, and dimerization of the Escherichia coli uvrD gene product (helicase II). Biochemistry. 1993;32:602–12. [DOI] [PubMed] [Google Scholar]
- [62].Lohman TM, Chao K, Green JM, Sage S, Runyon G. Large-scale purification and characterization of the Escherichia coli rep gene product. jBiolChem. 1989;264:10139–47. [PubMed] [Google Scholar]
- [63].Wong I, Chao KL, Bujalowski W, Lohman TM. DNA-induced dimerization of the Escherichia coli rep helicase. Allosteric effects of single-stranded and duplex DNA. J Biol Chem. 1992;267:7596–610. [PubMed] [Google Scholar]
- [64].Lucius AL, Jason Wong C, Lohman TM. Fluorescence stopped-flow studies of single turnover kinetics of E.coli RecBCD helicase-catalyzed DNA unwinding. J Mol Biol. 2004;339:731–50. [DOI] [PubMed] [Google Scholar]
- [65].Antony E, Kozlov AG, Nguyen B, Lohman TM. Plasmodium falciparum SSB tetramer binds single-stranded DNA only in a fully wrapped mode. J Mol Biol. 2012;420:284–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Nguyen B, Sokoloski J, Galletto R, Elson EL, Wold MS, Lohman TM. Diffusion of human replication protein A along single-stranded DNA. J Mol Biol. 2014;426:3246–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Joo C, Ha T. Single Molecule FRET with Total Internal Reflection Microscopy In: Selvin PR, Ha T, editors. Single Molecule techniques: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, NY; 2008. p. 3–36. [Google Scholar]
- [68].Trefethen LN, Weideman JAC, Schmelzer T. Talbot quadratures and rational approximations. BIT Numerical Mathematics. 2006;46:653–70. [Google Scholar]
- [69].Vistica J, Dam J, Balbo A, Yikilmaz E, Mariuzza RA, Rouault TA, et al. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal Biochem. 2004;326:234–56. [DOI] [PubMed] [Google Scholar]
- [70].Laue TM, Shah BD, Ridgeway TM and Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. Cambridge, UK: Royal Society of Chemistry; 1992. [Google Scholar]
- [71].Dam J, Schuck P. Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles. Methods Enzymol. 2004;384:185–212. [DOI] [PubMed] [Google Scholar]
- [72].Tanford C Physical Chemistry of Macromolecules. Wiley, NY: 1961. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.