

Abstract
BACKGROUND:
The coexistence of α-synuclein and tau aggregates in several neurodegenerative disorders, including Parkinson’s disease and Alzheimer’s disease, raises the possibility that a seeding mechanism is involved in disease progression.
METHODS:
To further investigate the role of α-synuclein in the tau aggregation pathway, we performed a set of experiments using both recombinant and brain-derived tau and α-synuclein oligomers to seed monomeric tau aggregation in vitro and in vivo. Brain-derived tau oligomers were isolated from well-characterized cases of progressive supranuclear palsy (n = 4) and complexes of brain-derived α-synuclein/tau oligomers isolated from patients with Parkinson’s disease (n = 4). The isolated structures were purified and characterized by standard biochemical methods, then injected into Htau mice (n = 24) to assess their toxicity and role in tau aggregation.
RESULTS:
We found that α-synuclein induced a distinct toxic tau oligomeric strain that avoids fibril formation. In vivo, Parkinson’s disease brain-derived α-synuclein/tau oligomers administered into Htau mouse brains accelerated endogenous tau oligomer formation concurrent with increasing cell loss.
CONCLUSIONS:
Our findings provide evidence, for the first time, that α-synuclein enhances the harmful effects of tau, thus contributing to disease progression.
Keywords: Alpha-synuclein, Oligomeric complexes, Seeding, Strain, Tau oligomers, Toxicity
The misfolding of tau and α-synuclein is a key initiation event in a large group of neurodegenerative disorders. Compelling evidence suggests that misfolded proteins may seed the conformational change of other amyloidogenic proteins in a prion-like manner. The discovery of tau and α-synuclein aggregates in the same cell suggests that protein seeding may occur in vivo (1). This could explain why tau and α-synuclein aggregates coexist in pathologies such as Alzheimer’s disease (AD) (2–4), Down syndrome (5), Parkinsonism dementia complex of Guam (6), Lewy body dementia (LBD) (7–10), and Parkinson’s disease (PD) (11).
Studies from transgenic mice support the hypothesis that α-synuclein influences tau deposition. The overexpression of mutant human α-synuclein (A53T) in mice was associated with abundant tau-positive threads, grains, spheroids, and pretangles in several brain regions (12), and mice overexpressing human wild-type α-synuclein spontaneously developed tau pathology in an age-dependent manner (13). In addition, recombinant tau and α-synuclein are capable of inducing fibril-lization of each other (12), and the exposure of cells in culture to exogenous fibrils of recombinant tau and α-synuclein leads to the development of large inclusions (14,15).
Remarkably, large inclusions composed of tau and a-synu-clein do not correlate with disease progression, and this casts doubt on the toxicity of stable fibrils and highlights the oligomeric forms of both proteins as being responsible for neuronal and synaptic loss (16,17). Moreover, both tau and α-synuclein oligomers have been described in PD and LBD (11,18,19), while tau oligomers are an established component of tauopathies, including AD (20–22) and progressive supranuclear palsy (PSP) (23). However, the pathological consequences of the synergy between tau and α-synuclein oligomers have yet to be determined.
To investigate the role of α-synuclein in the tau aggregation pathway, we performed a set of experiments using recombinant tau and α-synuclein oligomers to seed tau aggregation, brain- derived tau oligomers isolated from PSP (a pure tauopathy), and brain-derived complexes of α-synuclein/tau oligomers from PD cases. Our findings revealed that seeds of α-synuclein potentiate tau oligomer toxicity in SH-SY5Y and CV-1 cells overexpressing tau while promoting spine retraction in primary neurons. Surprisingly, seeds of α-synuclein induced a distinct toxic tau oligomeric strain that avert tau fibrillization. Meanwhile, PD-derived α-synuclein/tau oligomers administered into Htau mouse brains accelerated endogenous tau oligomer formation and neuronal loss when compared with the injection of PSP-derived tau oligomers. In summary, our findings suggest that α-synuclein shifts the tau aggregation pathway while potentiating tau oligomer toxicity, thereby driving disease progression and spread of pathology.
METHODS AND MATERIALS
Aggregation Assay
Recombinant wild-type 2N4R tau (tau 441) and α-synuclein were expressed in Escherichia coli and purified as described previously (24,25). Under standard conditions, recombinant full-length tau 441 AA (4–8 mM) was incubated with seeds of preformed tau oligomers, seeds of preformed α-synuclein oligomers, or heparin in assembly buffer (10 mM HEPES, pH 7.4, 100 mM NaCl, and 5 mM dithiothreitol) at either room temperature or 37°C for 3 to 6 hours. Three independent replications were performed for each experimental setting.
Human Samples
Brain tissue from patients with PSP and PD were provided by Juan C. Troncoso (Johns Hopkins University School of Medicine, Baltimore, MD) and the Brain Resource Center at Johns Hopkins University with patient consent and handled under protocols approved by the Johns Hopkins Institutional Review Board. PD cases were obtained from the Oregon Brain Bank at Oregon Health and Science University (Portland, OR). Tissue use conformed to Oregon Health and Science University Institutional Review Board-approved protocols. Neuropathological assessment conformed to National Institute on Aging/Reagan Institute consensus criteria.
Isolation of oligomers from human samples, cell culture, immunostaining, Western blotting, animal analyses, confocal and atomic microscopic imaging, and statistical analyses are all described in the Supplement.
RESULTS
Seeds of α-synuclein Enhance Tau Oligomer Toxicity in Cells in Culture
Unlike α-synuclein, which is prone to aggregate in the absence of inducers, monomeric tau does not spontaneously misfold (26). Therefore, to investigate the role of α-synuclein in the tau aggregation pathway, we used a homogeneous preparation of recombinant α-synuclein oligomers (α-synO) and tau oligomers (tauO) as seeds to induce monomeric tau aggregation (8 μM) in 1 × phosphate-buffered saline (PBS) at a ratio of 1:140 (weight/weight) (Figure 1A, B). Atomic force microscopy images showed that seeds from both α-synO and tauO induced the conversion of monomeric tau into oligomers. Tau seeded with preformed tauO (Tau/tauO) revealed oligomers arranged in a chain, which may represent the initial steps of protofibril assembly (dotted area in Figure 1A and Supplemental Figure S1F), whereas α-synO seeds induced a homogeneous oligomeric population (Tau/α-synO) (Figure 1B and Supplemental Figure S1G). Western blot analyses with T22 (to detect tau oligomers) and Tau5 (to visualize total tau) antibodies showed an increase in high molecular weight tau aggregates above 250 kDa when using seeds of tauO but not α-synO (p < .01, n = 3; t4 = 4.7; multiple t test nonparametric) (Supplemental Figure S1A–C).
Figure 1.
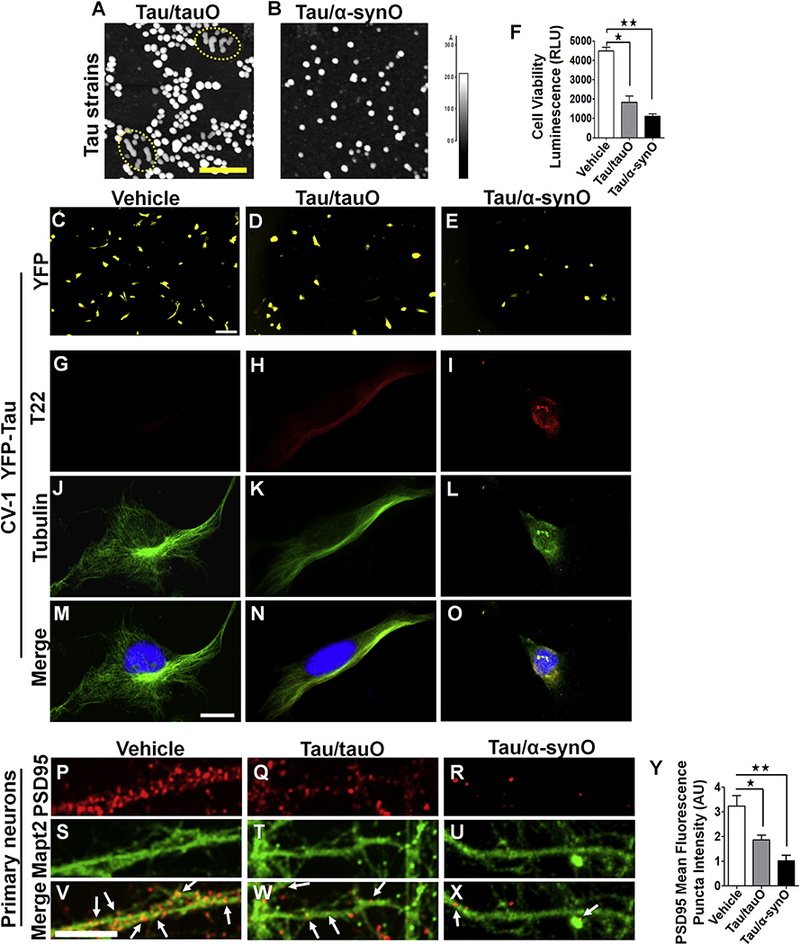
Seeds of α-synuclein enhance tau oligomer (tauO) toxicity in cells in culture. Tau strains were generated by adding seeds of preformed tauO or α-synuclein oligomers (α-synO) to 8 μM tau monomer in 1 × phosphate-buffered saline at a ratio of 1:140 (weight/weight). Atomic force microscopy images of tau seeded with (A) preformed tau oligomers (Tau/tauO) or (B) preformed α-synO (Tau/α-synO). Scale bar = 100 nm. (A) Seeded tau showed some oligomers arranged in a chain, suggesting the formation of tau protofibrils (Tau/tauO; dotted area). (C–E) Live cell imaging and (G–O) confocal images of CV-1 cells transfected with human tau linked to yellow fish (YFP-tau) plasmid treated with vehicle (phosphate-buffered saline) (C, G, J, M), or 1 μM tauO obtained by seeding (Tau/tauO) (D, H,K,N) or cross-seeding (Tau/α-synO) (E, I, L, O). Seeds of α-synuclein induced tau assembly into a distinct toxic oligomeric strain, as demonstrated by the reduced number of viable cells after treatment. The graph (F) shows the relative luminescence units (RLUs) of CellTiter Glo to cellular adenosine triphosphate. Bars represent the mean and SEM (one-way analysis of variance, Tukey multiple comparisons test; F ratio = 17.5, n = 4 independent experiments; tau/α-synO, **p < .006, F6 = 12; Tau/tauO, *p < .03, F6 = 12). (G–O) Representative confocal images showing the merge (yellow), between tubulin (green), and T22 (red) antibodies. Cross-seeded tau (Tau/α-synO) induces abnormal cell morphology. Scale bar = 12 μm. Representative images of cortical primary neurons derived from Htau mice treated with (P, S, V) vehicle, (Q, T, W) 1 μM tauO seeded with preformed tau oligomers (Tau/tauO), or (R, U, X) preformed Tau/α-synO for 6 hours. (P–X) Immunofluorescence staining using postsynaptic density protein 95 antibody to visualize synaptic spines and Mapt2 showed that cells exposed to tau cross-seeded with α-synO have a dramatic dendritic spine reduction after treatment (white arrows) compared with cells treated with (P) vehicle and (Q) seeded tau (Tau/tauO). (Y) Quantification of postsynaptic density protein 95 puncta revealed a decrease in dendrites after treatment with Tau/α-synO (**p < .001; Tau/tauO, *p < .02, F11 = 12.5; error bars, SEM, n = 5 independent experiments, one-way analysis of variance, Tukey multiple comparisons test). Scale bar = 5 μm.
To investigate tau oligomers strains’ properties, CV-1 cells that do not express endogenous tau (27) were transfected with full-length human tau linked to yellow fish. Live cell imaging of CV-1 cells revealed that α-synO seeds induced a distinct tau oligomeric strain that altered cell morphology and increased cell death compared to tau seeded with preformed tauO (Figure 1C–F). No effect was shown in cells exposed to vehicle (PBS), monomeric tau (data not shown), or α-synuclein seeds alone (Supplemental Figure S1D–E). The striking results found in CV-1 cells exposed to α-synO seeds were confirmed by measuring luminescence corresponding to adenosine triphosphate as a measure of cell toxicity (Tau/α-synO, p < .006; Tau/tauO, p < .03; error bars, SEM, n = 4; one-way analysis of variance [ANOVA], Tukey multiple comparisons test) (Figure 1F). Immunocytochemical analyses using tubulin and T22 antibodies confirmed a reduced number of cells and abnormal cellular morphology (Figure 1G–O) after treatment with tau cross-seeded with α-synO. No evidence of intracellular deposition was observed in any of the treatments.
We next investigated the toxicity of tau oligomeric strains on the human neuroblastoma cell line SH-SY5Y differentiated with retinoic acid. As expected, we found increasing cell death in cells exposed to both tau oligomer preparations (TauO/tauO, p < .0004; TauO/α-synO, p < .0001; n = 4; oneway ANOVA, Tukey multiple comparisons test) (Supplemental Figure S1I).
To further investigate the effects of tau aggregates, primary cortical neurons obtained from Htau mouse embryos were exposed to vehicle (PBS) and tau oligomeric strains (seeded with tauO or with α-synO) for 24 hours (Figure 1P–X). Live cell imaging revealed a dramatic dendritic spine retraction in cells treated with tau oligomers induced by α-synO seeds but not tauO seeds or vehicle (data not shown). Immunofluorescence staining using postsynaptic density protein 95 antibody to visualize synapses confirmed the reduction of dendritic spines (postsynaptic density protein 95 puncta; Tau/tauO, p < .02; Tau/α-synO, p < .001; n = 5; one-way ANOVA, Tukey multiple comparisons test) (Figure 1P–R, Y).
Seeds of α-synuclein Shift the Tau Aggregation Pathway, Extending the Lifespan of the Toxic Tau Conformation
To further investigate the ability of α-synuclein oligomers to induce tau fibrillization, we performed kinetic analyses using thioflavin S (Figure 2). Fluorescence intensity from thioflavin S (Figure 2A) and dot blot analyses (Figure 2B) showed that tau seeded with tauO or α-synO remained in oligomeric conformation for up to 192 hours. Surprisingly, while tau seeded with tauO continued to assemble into fibrils beyond 192 hours (Tau/tauO, p < .001; green line), tau induced with α-synO averted fibril formation (Tau/α-synO; blue line; bars represent the mean and SEM; two-way ANOVA, Bonferroni post hoc comparisons test). Dot blot analyses using T22 antibody (Figure 2B) showed a decrease in tau oligomers in Tau/tauO with time parallel to the increase in fibrils. No change was observed in tau induced with α-synO seeds. Atomic force microscopy images taken 360 hours after seeding showed that tau seeded with tauO assembled into short fibrils ranging from approximately 150 to 300 nm in length (Figure 2C, F), while tau induced with α-synO formed aggregates less than approximately 50 nm in length (Figure 2D, G). Heparin was used to induce tau fibrillization as a control (Figure 2E, H). We next compared the toxicity of tau aggregates obtained at 360 hours using CV-1 cells (human tau linked to yellow fish) (Supplemental Figure S2A–D). Surprisingly, we found that tau cross-seeded with α-synO retained its toxicity as demonstrated by the reduced number of viable CV-1 cells transfected with human tau linked to yellow fish compared with tauO or PBS (p < .006 and p < .03, respectively; one-way ANOVA, Tukey multiple comparisons test) (Supplemental Figure S2A–D). As we previously reported (28), tau fibrils had a minimum effect on cells in culture (data not shown). Furthermore, cells exposed to tau/α-synO appeared to exhibit cell membrane retraction as indicated by their compact shape, possibly preceding cell death (Supplemental Figure S2E–M). These findings suggest that seeds of oligomeric α-synuclein induce a stable distinct toxic oligomeric taustrain, thereby forming a kinetic trap and preventing tau fibril formation.
Figure 2.
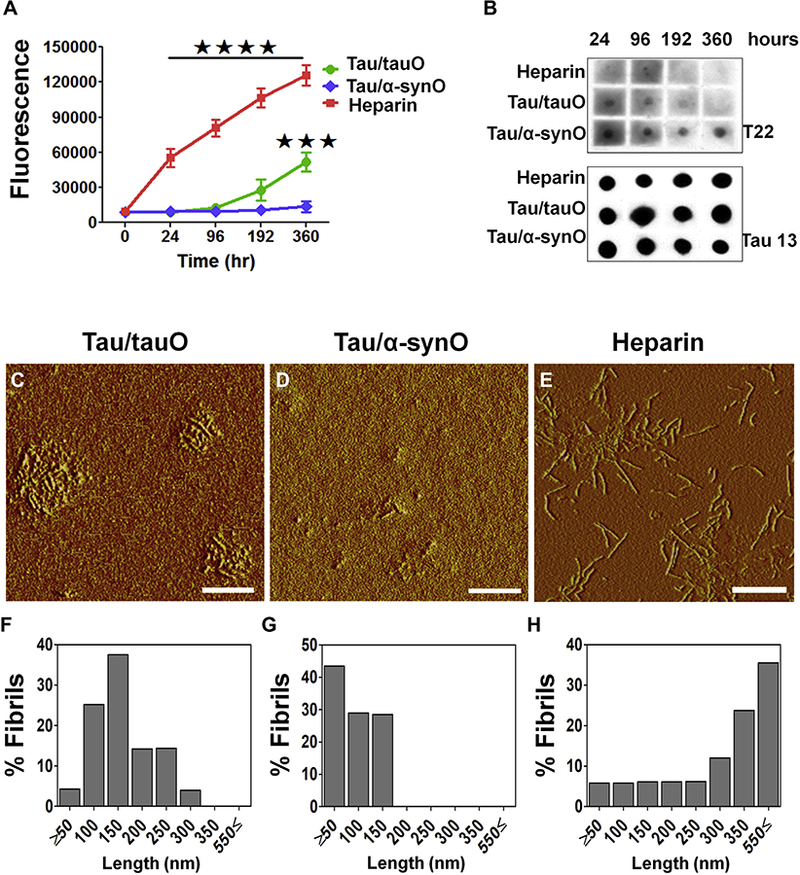
Seeds of α-synuclein shift the tau aggregation pathway, extending lifespan of tau toxic conformation. (A) Kinetic analyses of recombinant tau protein (8 μM) incubated in phosphate-buffered saline (pH 7.2) with either preformed oligomeric tau (tauO; green line) or α-synuclein (α-synO; blue line) at a ratio of 1:140 (weight/weight) or 10 μM of heparin (red) at 22°C for 360 hours. A tau strain induced by α-synO evades fibril formation (blue line), whereas tau readily fibrillizes in the presence of inducers, such as heparin (red line) or seeds of preformed tau oligomers (green line). Bars represent the mean and SEM (****p < .0001, ***p < .001; n = 3 independent experiments; two-way analysis ofvariance, Bonferroni post hoc multiple comparisons test). (B) Dot blot analyses oftauO at different time points using T22 and Tau13antibodies. Both seeds of preformed tauO (Tau/tauO) and heparin induced tau fibril formation, while cross-seeded tau (Tau/α-synO) remains oligomeric. Atomic force microscopy images of tau aggregates induced by (C) preformed tauO (Tau/tauO), (D) α-synO (Tau/α-synO), and (E) heparin. (F–H) Graphs represent the length distribution of tau aggregates. Scale bar = 400 nm.
Complexes of α-synuclein/Tau Oligomers Isolated From PD Cases Accelerate Endogenous Tau Oligomer Formation in Htau Mouse Brain
A strong argument in PD and LBD is that α-synuclein influences tau misfolding, suggesting a possible interaction between them. We have recently shown that α-synuclein and tau oligomers exist in complex in PD and LBD cases (11). However, whether these complexes can propagate tau misfolding and contribute to disease progression has not yet been investigated. To gain further insight into the synergism between tau and α-synuclein, we isolated oligomers of α-synuclein in complex with tau oligomers from PD brain cases (PD α-synO/tauO) and compared them with tau oligomers isolated from PSP, a pure tauopathy (PSP tauO) (Supplemental Table S1). Brain-derived tau oligomers were immunoprecipitated using our anti-tau oligomer antibody, T22, while PD oligomeric complexes were immunoprecipitated with the anti-α-synuclein oligomer antibody, F8H7, as well as T22, as previously described (11). One-month-old Htau mice were administered bilateral injections of 0.5 μg of PD α-synO/tauO (n = 8), PSP tauO (n = 8), or PBS (n = 8) into the hippocampus (Figure 3A). At 7 (8 months old) and 14 (15 months old) months after injection, spatial memory and strength were evaluated using Y-maze and grip strength tests. Htau mice develop cognitive deficits at around 12 months of age; however, even at 15 months of age, mice receiving PD-derived α-synO/tauO were significantly impaired compared with the PBS control group, while PSP-derived tauO did not have the same level of toxicity (spatial memory: 7 and 14 months postinjection, p < .02 and p < .01, respectively; Hind limb grip strength: 7 [p < .02] and 14 [p < .01] months postinjection for PSP tauO; p < .005 for PD α-synO/tauO; n = 8; one-way ANOVA, Bonferroni post hoc comparisons test) (Supplemental Figure 3).
Figure 3.
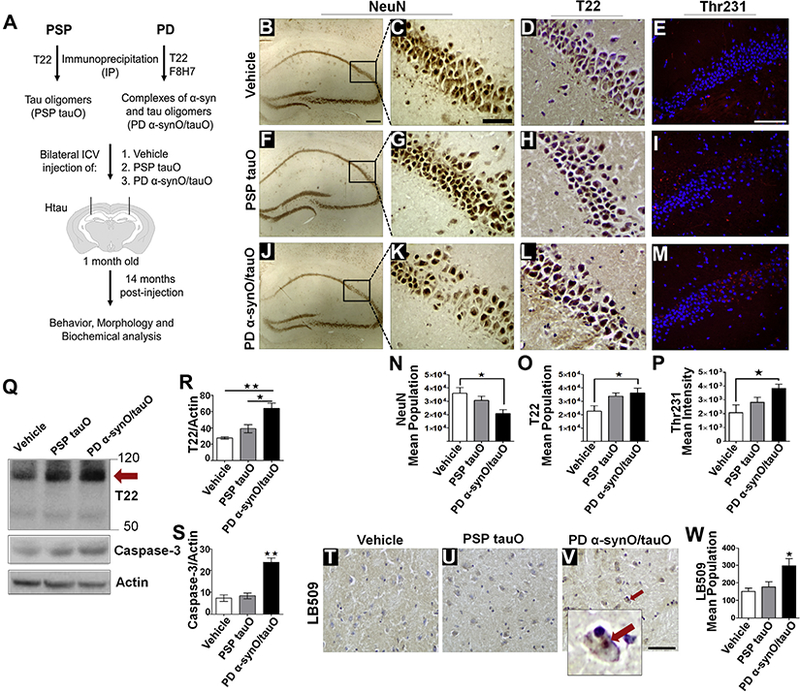
Parkinson’s disease (PD) brain-derived α-synuclein oligomers (α-synO) propagate endogenous tau misfolding and neuronal loss in Htau mice. Brain-derived tau oligomers (tauO) and oligomeric complexes of α-synO/tauO were immunoprecipitated from patients with progressive supranuclear palsy (PSP) and PD using T22 and F8H7 antibodies. One-month-old Htau mice were intracerebroventricularly injected bilaterally with (B–E) 1 × phosphate-buffered saline (PBS) (vehicle, n = 8), (F–I) 0.5 μg of PSP brain–derived tauO (n = 8), or (J–M) PD brain–derived α-synO/tauO (n = 8) into the hippocampi. Fourteen months postinjection, mouse brains were immunostained using the neuronal marker NeuN (B, C, F, G, J, K), T22 antibody (D, H, L), and Thr231(E, I, M). Images show increased neuronal loss in mice receiving PD α-synO/tauO at the injection site compared with mice injected with PSP tauO or PBS (C, G, K, N). Stereological quantification of (N) NeuN+ cells (*p < .03, F14 = 4.7, n = 6), (O) T22+ cells (*p < .03, F17 = 4.3, n = 8), and (P) Thr231+ cells (*p < .04, F9 = 3.02, n = 4). One-way analysis of variance Bonferroni post hoc comparisons test. (Q–S) Western blot analyses of brain homogenate from mice injected with PBS, PSP tauO, and PD α-synO/tauO probed with T22 and reprobed with caspase-3. (R–S) Graphs represent the protein band intensity relative to actin (T22, **p < .003, *p < .02, F6 = 15.7, n = 3 independent experiments; caspase-3, **p < .006, F6 = 14.6, n = 3 independent experiments). One-way analysis of variance Tukey multiple comparisons test. Immunohistochemistry analyses of the hypothalamic brain region from mice injected with (T) PBS (vehicle), (U) PSP tauO, and (V) PD α-synO/tauO using LB509 antibody. Mice injected with PD α-synO/tauO developed PD-like Lewy body deposits (arrow). (W) Stereological quantification of LB509+ cells (Lewy body-like deposits) (*p < .01, F12 = 5.9, n = 5; one-way analysis of variance, Tukey multiple comparisons test). Scale bar = 50 μm.
Studies from our group and others have shown that the injection of brain-derived oligomers from AD cases exacerbates tau pathology in the brain (29,30); therefore, in order to evaluate the effects of brain-derived oligomers from PSP and PD administered in the Htau mouse brain, immunohistochemical analysis was performed using the neuronal marker NeuN (Figure 3B, C, F, G, J, K) and immunofluorescence using T22 antibody (Figure 3D, H, L). Unbiased stereology analysis showed a significant neuronal loss at the hippocampus in mice receiving PD α-synO/tauO when compared with PSP tauO or PBS injection (p < .03; n = 6, one-way ANOVA, Bonferroni post hoc comparisons test) (Figure 3N). In contrast, levels of tau oligomers increased in both mice injected with PD α-synO/tauO and PSP tauO (p < .03, n = 8, one-way ANOVA, Bonferroni post hoc comparisons test) (Figure 3O). To further determine the levels of tau oligomers and total tau in mice brain, brain homogenates PBS soluble fractions were measured by enzyme-linked immunosorbent assay analysis with T22 and Tau 5 antibodies. Consistent with the stereology analysis, tau oligomers levels increased in mice receiving PD α-synO/tauO compared with vehicle (p < .03; n = 3) but not in mice receiving PSP tauO (one-way ANOVA, Tukey post hoc comparisons test) (Supplemental Figure 3). Total tau levels were similar among treatments (data not shown). Our results suggest that memory impairment and increasing cell loss depends upon PD α-synO/tauO complexes.
Tau is known to be hyperphosphorylated in Htau mice; therefore, we investigated the phosphorylation status of tau in mice injected with brain-derived oligomers or PBS. Immuno-staining analysis was performed using AT180 antibody for the detection of tau phosphorylated at the Thr231 epitope, a marker of early tau aggregation (Figure 3E, I, M). This analysis revealed increased levels of phospho-tau not only at the injection site and the hippocampus but also in the cortex (data not shown), suggesting the propagation of misfolded tau in mice injected with brain-derived oligomers compared with vehicle (p < .04; n = 4, one-way ANOVA, Bonferroni post hoc) (Figure 3P). Tau oligomers are known to be early aggregates responsible for the spread of pathology (29). To further analyze these findings, a Western blot analysis of brain homogenate using T22 antibody confirmed increased levels of tau oligomers in mice injected with PD α-synO/tauO compared with PSP tauO and control treatment (p < .003 and p < .02, respectively; one-way ANOVA, Tukey multiple comparison test) (Figure 3Q, R). We then investigated neuronal loss in mice injected with PD a-syn/tauO using Western blot by measuring active caspase-3 levels, which have been shown to be involved in the development of tau neuropathology (31). Increased caspase-3 levels were found in PD α-synO/tauO when compared with PSP tauO and PBS treatments (p < .006, n = 3; one-way ANOVA, Tukey multiple comparison test) (Figure 3Q, S). Thus, increased tau oligomers correlated with increased cell loss in PD-derived oligomer-treated mice.
PD-like Pathology in Htau Mice Injected With PD Brain-Derived α-synuclein/Tau Oligomers
Previous studies have shown that the injection of recombinant fibrils of α-synuclein induce PD-like aggregates in wild-type mice (14,32,33). These observations prompted us to investigate the formation of PD-like pathology in Htau mice injected with α-synuclein oligomers. We conducted a nonbiased stereology analysis of brain sections that were immunostained with LB509, a pan-α-synuclein antibody (Figure 3T–W). Deposits of α-synuclein were found in Htau mice receiving PD α-synO/tauO outside the injection site, the hypothalamus, resembling a Lewy body inclusion in a human diseased brain (Figure 3V). Although several sections from different brains were analyzed, few Lewy body-like deposits were found (p < .01; n = 5, one-way ANOVA, Tukey multiple comparisons test). Our findings suggest that PD brain-derived α-synO/tau oligomer complexes might initiate endogenous α-synuclein deposition into Lewy body-like inclusions. However, we cannot ignore the possibility that seeds of α-synuclein could have been dissociated from these complexes, promoting the formation of large intracellular deposits.
PD Brain-Derived α-synuclein/Tau Oligomer Complexes Delayed Neurofibrillary Tangle Formation
To evaluate fibrillar tau, we measured the neurofibrillary tangle (NFT) load in Htau mice postinjection with vehicle, PSP tauO, or PD α-synO/tauO. Immunohistochemistry and immunofluorescence analysis with AT8 antibody (Figure 4A–G) as well as Gallyas silver staining (Figure 4H–J) were performed. Mice injected with PSP tauO showed increased AT8 immunoreactivity compared with the control group. Surprisingly, mice injected with PD α-syn/tauO had fewer neurons containing NFTs in the cortex and hippocampus (p < .03; n = 5, one-way ANOVA, Bonferroni post hoc) (Figure 4D). These findings prompted us to investigate the levels of fibrillar tau in brain fractions of mice receiving vehicle, PSP tauO, or PD α-synO/tauO. PBS-insoluble material was extracted with 70% formic acid and measured using enzyme-linked immunosorbent assay analysis with PHF-13 antibody. Mice receiving PSP tauO had increased levels of fibrillar tau compared with PD α-synO/tauO (p < .04; n = 3). No difference was found between PSP tauO and vehicle (one-way ANOVA, Tukey post hoc) (Supplemental Figure 3). All of these findings suggest that while PSP tauO increased tau deposition in Htau mice brain, PD α-syn/tauO may delay or evade NFT formation in vivo.
Figure 4.
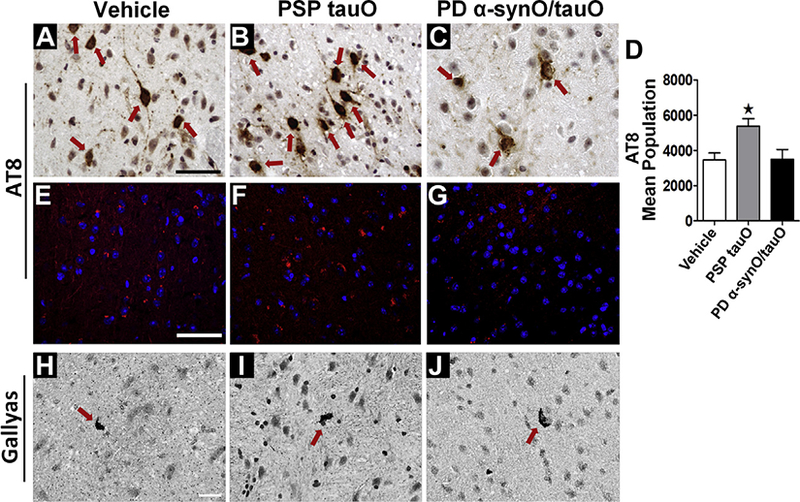
Parkinson’s disease (PD) brain-derived α-synuclein/tau oligomers (α-synO/tauO) delayed neurofibrillary tangle formation. Immunostaining of cortical brain sections from mice injected with (A) phosphate-buffered saline, (B) progressive supranuclear palsy (PSP) tauO, or (C) PD α-synO/tauO probed with AT8 antibody. (C) Mice injected with PD α-synO/tauO showed fewer neurofibrillary tangles (brown). PSP tauO showed an increased immuno-reaction to AT8 compared with mice injected with PD α-synO/tauO or phosphate-buffered saline. (D) Stereological quantification of AT8+ cells. (E–G) Immunofluorescence staining with AT8. Scale bar = 25 μm. (H–J) Gallyas silver staining for the detection of neurofibrillary tangles (red arrow) and neuropil threads (arrowhead). Scale bar = 25 mm. *p < .03, n = 5, one-way analysis of variance, Bonferroni post hoc comparisons test.
DISCUSSION
Oligomeric tau and α-synuclein appear to play an important role in the pathogenesis of a group of neurodegenerative diseases including PD and AD. The overlap between tau and a- synuclein in several brain pathologies suggests that protein seeding may be involved. While the toxicity of tau and α-synuclein is well-documented in cell culture (23,29,34) and rodents (30,35), the pathological consequences of the interaction between the two proteins remain unknown.
This study provides evidence, for the first time, that α-syn-uclein potentiates tau oligomer toxicity in vitro and in Htau mouse brain. Our findings show that oligomeric α-synuclein induces tau misfolding into a unique oligomeric toxic strain that averts tau fibril formation.
Exogenous fibrils of recombinant tau or α-synuclein can be taken up in cells and seed the fibrillization of tau and α-synuclein, respectively. Tau enhances α-synuclein toxicity in cells in culture (14,36), while having devastating effects in fruit flies (37). In addition, α-synuclein oligomers disrupt microtubule stability, affecting the transport of cargo proteins through the microtubule network (38). In our study, the deleterious effects of tau aggregates induced by α-synuclein oligomers in cultured cells do not rely on α-synuclein seeds directly, because treatment of cells with an equivalent amount of α-synuclein oligomers alone was not sufficient to alter cell morphology. A possible explanation for the differences in toxicity between both tau oligomer preparations is the fact that tau seeded with preformed tau oligomers contained some prefibrillar oligomers arranged in a chain, which is not the case in tau cross-seeded with α-synuclein oligomers, implying that self-protein interaction may facilitate fibril formation. Previous data showed that 3R-tau can seed 3R-tau but not 4R-tau fibril formation, suggesting that homogenous protein populations are prone to assemble into large aggregates, while cross-species proteins are not (39,40).
Our findings propose that a tau strain induced by α-synuclein oligomers averts fibril formation. Recently, Fang et al. (41) showed that TDP-43 oligomers can cross-seed amyloid-β mis-folding into oligomers, but not fibrils (41). Interestingly, in our study we did not observe large intracellular aggregate formation in any of the cell lines used, which may imply that fibril formation follows a different aggregation pathway or may require a longer exposure time. Remarkably, while the toxicity of fibrils is questioned, it has become more accepted that oligomers may play a role in the spread of disease pathology and neurodegeneration. A recent study using a biosensor cell line showed that tau seeding occurred before the accumulation of insoluble tau (42). Moreover, we have shown that brain-derived tau seeds can spread tau pathology throughout the brain, inducing the mis-folding of endogenous tau (29,30). These observations demonstrated that oligomeric forms of tau can propagate pathology.
In this study, we found that 14 months after intra-cerebroventricular injections of Htau mice with brain-derived PSP tau oligomers or PD oligomers containing complexes of α-synuclein and tau, the mixed oligomer populations induce neuronal loss and behavioral deficits. Our previous findings have shown that brain-derived tau oligomers induced cognitive decline and synaptic and mitochondrial dysfunction in wild-type mice while accelerating endogenous tau misfolding in Htau mice (43). Here, we found that PD brain-derived oligomers containing complexes of α-synuclein and tau induced neuronal loss and increased tau oligomer levels, coinciding with the onset of cognitive decline and motor deficits in Htau mice. Furthermore, PD brain-derived oligomeric complexes accelerate endogenous tau aggregation, implying that these brain-derived complexes are likely responsible for disease onset. While evidence has suggested that the onset of clinical symptoms in patients with AD and PSP correlate with elevated levels of tau oligomers (20,22,23), it is difficult to precisely elucidate how PD brain-derived oligomeric complexes induce tau aggregation. In support of these findings, PD patients who develop tau pathology are associated with dementia onset and poor prognosis (44), suggesting that a synergy between tau and α-synuclein may enhance clinical phenotype. In our study, α-synuclein/tau oligomer complexes seem to be more stable and pathologically relevant, given the capability to propagate endogenous tau oligomer formation and cell death in Htau mouse brain. Previous reports have found phospho-tau and α-synuclein in the same NFT, Lewy body, or neurite from patients with PD and LBD (45,46). In a recent study from our group, we demonstrated that α-synuclein and tau oligomers formed complexes in PD and LBD brains (11). In a separate study, we showed that α-synuclein interacts with oligomers of other proteins, such as amyloid-β, to form hybrid oligomers in AD brains, suggesting that protein interaction at oligomeric levels occurs broadly in the human brain (47). Our findings propose that extending the lifespan of oligomeric tau may be a key contributor to the spread of a toxic tau conformation and the misfolding of functional tau. Although oligomers have been considered intermediate to fibril formation, our data suggest that they may follow an independent pathway.
Previous studies have shown that fibrils can seed fibril formation in vitro as well as in vivo (48). Our findings demonstrated that oligomeric assemblies of α-synuclein protein templated tau oligomer formation and thus enhanced toxicity and prevented tau fibril formation in vitro. These findings are consistent with our in vivo study in which a small amount of intracerebrally infused brain-derived α-synuclein oligomers (PD α-synO/tauO) propagated tau oligomer formation while accumulating less fibrillar tau compared with Htau mice receiving PSP brain-derived oligomers. These observations suggest that PD α-synO/tauO complexes may be involved in delaying tau oligomer assembly into NFTs. The fact that PD-like α-synuclein aggregates, the so-called Lewy bodies, were found in the hypothalamus area indicated that α-synuclein/tau complexes migrated far from the injection site.
To our knowledge, this is the first demonstration that tau cross-seeded with α-synuclein oligomers induces tau aggregation and extends the toxic lifespan of tau oligomers by preventing them from undergoing transformation to a less toxic state, thus facilitating the propagation of endogenous tau protein in vivo. Overall, our findings suggest that the protein seeding mechanism is key to enhancing disease pathology; therefore, small doses of α-synuclein oligomeric seeds may have grave pathological consequences in the brain.
Supplementary Material
ACKNOWLEDGMENTS AND DISCLOSURES
This work was supported by the Michael J. Fox Foundation, the Cullen Trust, the Mitchell Center for Neurodegenerative Diseases, the Sealy Center for Vaccine Development, and National Institutes of Health Grant Nos. AG054025, RFA1AG055771, and NS094557 (to RK).
RK and DC conceived the project, designed the experiments, analyzed the results, and wrote the article. DC carried out many of the experiments and prepared the figures. MG purified the recombinant proteins and antibodies (T22 and F8H7). MG and US performed the IP and characterized the brain-derived oligomers. DC and JG performed the behavioral experiments and analysis. JG performed the stereological quantification. All authors reviewed the article.
We thank Mariana Carretero-Murillo and Salome McAllen for their excellent technical assistance and Adriana A. Paulucci-Holthauzen and Mauro Montalbano for their technical assistance with confocal microscopy. We also thank to Rabab Al Lahham, Kathleen Farmer, and Scott Shafiei for their contributions to the editing process.
Footnotes
The authors report no biomedical financial interests or potential conflicts of interest.
Supplementary material cited in this article is available online at https://doi.org/10.1016/j.biopsych.2017.12.018.
REFERENCES
- 1.Takeda A, Hashimoto M, Mallory M, Sundsumo M, Hansen L, Masliah E (2000): C-terminal alpha-synuclein immunoreactivity in structures other than Lewy bodies in neurodegenerative disorders. Acta Neuropathol 99:296–304. [DOI] [PubMed] [Google Scholar]
- 2.Hamilton RL (2000): Lewy bodies in Alzheimer’s disease: A neuro-pathological review of 145 cases using alpha-synuclein immunohis-tochemistry. Brain Pathol 10:378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lippa CF, Fujiwara H, Mann DM, Giasson B, Baba M, Schmidt ML, et al. (1998): Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol 153:1365–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raghavan R, Khin-Nu C, Brown A, Irving D, Ince PG, Day K, et al. (1993): Detection of Lewy bodies in Trisomy 21 (Down’s syndrome). Can J Neurol Sci 20:48–51. [DOI] [PubMed] [Google Scholar]
- 5.Lippa CF, Schmidt ML, Lee VM, Trojanowski JQ (1999): Antibodies to alpha-synuclein detect Lewy bodies in many Down’s syndrome brains with Alzheimer’s disease. Ann Neurol 45:353–357. [DOI] [PubMed] [Google Scholar]
- 6.Forman MS, Schmidt ML, Kasturi S, Perl DP, Lee VM, Trojanowski JQ (2002): Tau and alpha-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol 160:1725–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Obi K, Akiyama H, Kondo H, Shimomura Y, Hasegawa M, Iwatsubo T, et al. (2008): Relationship of phosphorylated alpha-synuclein and tau accumulation to Abeta deposition in the cerebral cortex of dementia with Lewy bodies. Exp Neurol 210:409–420. [DOI] [PubMed] [Google Scholar]
- 8.Popescu A, Lippa CF, Lee VM, Trojanowski JQ (2004): Lewy bodies in the amygdala: Increase of alpha-synuclein aggregates in neurodegenerative diseases with tau-based inclusions. Arch Neurol 61:1915–1919. [DOI] [PubMed] [Google Scholar]
- 9.Colom-Cadena M, Gelpi E, Charif S, Belbin O, Blesa R, Marti MJ, et al. (2013): Confluence of alpha-synuclein, tau, and beta-amyloid pathologies in dementia with Lewy bodies. J Neuropathol Exp Neurol 72:1203–1212. [DOI] [PubMed] [Google Scholar]
- 10.Colom-Cadena M, Gelpi E, Marti MJ, Charif S, Dols-Icardo O, Blesa R, et al. (2013): MAPT H1 haplotype is associated with enhanced alpha-synuclein deposition in dementia with Lewy bodies. Neurobiol Aging 34:936–942. [DOI] [PubMed] [Google Scholar]
- 11.Sengupta U, Guerrero-Muñoz MJ, Castillo-Carranza DL, LasagnaReeves CA, Gerson JE, Paulucci-Holthauzen AA, et al. (2015): Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biol Psychiatry 15:672–683. [DOI] [PubMed] [Google Scholar]
- 12.Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, et al. (2003): Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300:636–640. [DOI] [PubMed] [Google Scholar]
- 13.Haggerty T, Credle J, Rodriguez O, Wills J, Oaks AW, Masliah E, et al. (2011): Hyperphosphorylated Tau in an alpha-synuclein-overexpressing transgenic model of Parkinson’s disease. Eur J Neurosci 33:1598–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, et al. (2013): Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154:103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waxman EA, Giasson BI (2011): Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights intothe hyperphosphorylation of tau. J Neurosci 31:7604–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, et al. (2003): Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300:486–489. [DOI] [PubMed] [Google Scholar]
- 17.Bucciantini M, Calloni G, Chiti F, Formigli L, Nosi D, Dobson CM, et al. (2004): Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J Biol Chem 279:31374–31382. [DOI] [PubMed] [Google Scholar]
- 18.Sharon R, Bar-Joseph I, Frosch MP, Walsh DM, Hamilton JA, Selkoe DJ (2003): The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 37:583–595. [DOI] [PubMed] [Google Scholar]
- 19.Paleologou KE, Kragh CL, Mann DM, Salem SA, Al-Shami R, Allsop D, et al. (2009): Detection of elevated levels of soluble alpha-synuclein oligomers in post-mortem brain extracts from patients with dementia with Lewy bodies. Brain 132:1093–1101. [DOI] [PubMed] [Google Scholar]
- 20.Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A (2006): Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci Res 54:197–201. [DOI] [PubMed] [Google Scholar]
- 21.Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, et al. (2011): Characterization of prefibrillar tau oligomers in vitro and in Alzheimer disease. J Biol Chem 286:23063–23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, et al. (2012): Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J 26:1946–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerson JE, Sengupta U, Lasagna-Reeves CA, Guerrero-Munoz MJ, Troncoso J, Kayed R (2014): Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol Commun 2:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Der-Sarkissian A, Jao CC, Chen J, Langen R (2003): Structural organization ofalpha-synuclein fibrils studied by site-directed spin labeling. J Biol Chem 278:37530–37535. [DOI] [PubMed] [Google Scholar]
- 25.Margittai M, Langen R (2004): Template-assisted filament growth by parallel stacking of tau. Proc Natl Acad Sci USA 101:10278–10283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morozova OA, March ZM, Robinson AS, Colby DW (2013): Conformational features of tau fibrils from Alzheimer’s disease brain are faithfully propagated by unmodified recombinant protein. Biochemistry 52:6960–6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.King ME, Kan HM, Baas PW, Erisir A, Glabe CG, Bloom GS (2006): Tau-dependent microtubule disassembly initiated by prefibrillar beta-amyloid. J Cell Biol 175:541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R (2010): Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49:10039–10041. [DOI] [PubMed] [Google Scholar]
- 29.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Kiritoshi T, Neugebauer V, et al. (2012): Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep 2:700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R (2011): Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener 6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu J, Lauretti E, Pratico D (2017): Caspase-3-dependent cleavage of Akt modulates tau phosphorylation via GSK3beta kinase: Implications for Alzheimer’s disease. Mol Psychiatry 22:1002–1008. [DOI] [PubMed] [Google Scholar]
- 32.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, et al. (2012): Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, et al. (2013): Prion-like spreading of pathological alpha-synuclein in brain. Brain 136:1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danzer KM, Krebs SK, Wolff M, Birk G, Hengerer B (2009): Seeding induced by alpha-synuclein oligomers provides evidenceforspreading of alpha-synuclein pathology. J Neurochem 111:192–203. [DOI] [PubMed] [Google Scholar]
- 35.Rockenstein E, Nuber S, Overk CR, Ubhi K, Mante M, Patrick C, et al. (2014): Accumulation of oligomer-prone alpha-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 137:1496–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Badiola N, de Oliveira RM, Herrera F, Guardia-Laguarta C, Goncalves SA, Pera M, et al. (2011): Tau enhances alpha-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS One 6, e26609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roy B, Jackson GR (2014): Interactions between tau and alpha-synuclein augment neurotoxicity in a Drosophila model of Parkinson’s disease. Hum Mol Genet 23:3008–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prots I, Veber V, Brey S, Campioni S, Buder K, Riek R, et al. (2013): alpha-synuclein oligomers impair neuronal microtubule-kinesin interplay. J Biol Chem 288:21742–21754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M (2010): Seeded aggregation and toxicity of {alpha}-synuclein and tau: Cellular models of neurodegenerative diseases. J Biol Chem 285:34885–34898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guerrero-Munoz MJ, Castillo-Carranza DL, Krishnamurthy S, Pau-lucci-Holthauzen AA, Sengupta U, Lasagna-Reeves CA, et al. (2014): Amyloid-beta oligomers as a template for secondary amyloidosis in Alzheimer’s disease. Neurobiol Dis 71:14–23. [DOI] [PubMed] [Google Scholar]
- 41.Fang YS, Tsai KJ, Chang YJ, Kao P, Woods R, Kuo PH, et al. (2014): Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat Com-mun 5:4824. [DOI] [PubMed] [Google Scholar]
- 42.Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC, et al. (2014): Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci U S A 111:E4376–E4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shafiei SS, Guerrero-Munoz MJ, Castillo-Carranza DL (2017): Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage. Front Aging Neurosci 9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Irwin DJ, Lee VM, Trojanowski JQ (2013): Parkinson’s disease dementia: Convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci 14:626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arima K, Mizutani T, Alim MA, Tonozuka-Uehara H, Izumiyama Y, Hirai S, et al. (2000): NACP/alpha-synuclein and tau constitute two distinctive subsets of filaments in the same neuronal inclusions in brains from a family of parkinsonism and dementia with Lewy bodies: Double-immunolabeling fluorescence and electron microscopic studies. Acta Neuropathol 100:115–121. [DOI] [PubMed] [Google Scholar]
- 46.Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW (2003): Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol 62:389–397. [DOI] [PubMed] [Google Scholar]
- 47.Guerrero-Munoz MJ, Castillo-Carranza DL, Kayed R (2014): Therapeutic approaches against common structural features of toxic oligomers shared by multiple amyloidogenic proteins. Biochem Pharmacol 88:468–478. [DOI] [PubMed] [Google Scholar]
- 48.Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM (2013): Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 33:1024–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.