

Abstract
RIG-I-like receptors (RLRs) are cytoplasmic sensors of viral RNA that trigger the signaling cascade that leads to type I interferon (IFN) production. Transcriptional induction of RLRs by IFN is believed to play the role of positive feedback to further amplify viral sensing. We found that RLRs and several other IFN-stimulated genes (ISGs) are induced early in viral infection independent of IFN. Expression of these early ISGs requires IRF3/IRF7 and is highly correlated amongst them. Simultaneous detection of mRNA of IFNB1, viral replicase, and ISGs revealed distinct populations of IFNB1 expressing and non-expressing cells which are highly correlated with the levels of early ISGs but are uncorrelated with IFN-dependent ISGs and viral gene expression. Individual expression of RLRs made IFNB1 expression more robust and earlier, suggesting a causal relation between levels of RLR and induction of IFN.
Keywords: cellular heterogeneity, RIG-I, IFNB1 induction
Introduction
RIG-I-like receptors (RLRs) RIG-I, MDA5, and LGP2 constitute a family of cytoplasmic sensors of viral RNA with indispensable roles in innate immunity 1–5. Knockout mice that lack RIG-I or MDA5 are highly susceptible to infection by the respective RNA viruses that these sensors recognize 3. Knockdown of RIG-I expression in fibroblasts, epithelial cells, conventional dendritic cells and macrophages resulted in failure to activate type I IFN upon infection with various RNA viruses, highlighting the indispensable antiviral role of RLRs in a broad range of cell types 4. Type I IFN is known to increase transcription of hundreds of IFN-stimulated genes (ISGs), including RLRs, that can effectively combat the infection. A screening of more than 380 human ISGs for their ability to inhibit the replication of several clinically important human and animal viruses showed that RIG-I and MDA5 are among the top five in their antiviral activities against a broad range of viruses 6.
After viral RNA recognition, RIG-I and MDA5 interact with the mitochondrial antiviral signaling protein (MAVS), and through a series of signaling cascades, activate transcription of type I IFN which includes IFNB1 and IFNA subtypes 7–10. Type I IFN binds to the IFNα/β receptor (IFNAR) on the cell membrane, and activates transcription of ISGs through JAK-STAT pathway, resulting in an antiviral state that controls infection 11–13. Some ISGs such as Viperin, IFIT1 and OASL are not only IFN-inducible, but also induced directly by viral infection in the absence of IFN 14–16.
Induction of RLRs by IFN is believed to play the role of positive feedback to further amplify viral sensing, however, their expression kinetics during viral infection, and in particular with respect to type I IFN production, has not been systematically analyzed, especially at the early stages of infection, where highly sensitive methods are needed to capture the small changes in gene expression. Majority of the earlier works are based on bulk measurements, where results are ensemble-averaged over a large number of cells, which may hide changes in gene expression, if these changes originated from a small fraction of the cells.
In fact, it is well recognized that there is significant cell-to-cell heterogeneity in IFNB1 expression such that only a fraction of cells produces IFN-β upon viral infection17–24. Several factors have been suggested to contribute to this heterogeneity including infecting virus quasispecies, complexities of multiple transcription factor binding to the IFNB1 promoter, cellular heterogeneity in the host gene expression levels and paracrine signaling 19, 22–26. For example, components of the RIG-I signaling pathway, such as RIG-I, MDA5 and TRIM25 are expressed at higher levels in the IFN-β expressing cells 22, and the fraction of IFN-β producing cells increased when RIG-I signaling pathway components such as RIG-I, TRIM25, NF-κB, IRF3 and IRF7 were overexpressed in cells prior to viral infection 19, 22. Shalek et al have shown that cellular variation is controlled in a paracrine manner in dendritic cells26, while Rand et al and Patil et al have shown in fibroblasts and dendritic cells that heterogeneity in IFNB1 expression is predominantly cell-intrinsic at early time points post infection, which is then propagated by paracrine response23, 24.
As RLRs are major players in viral recognition and subsequent IFN induction, and have been implicated in the heterogeneous expression of IFNB1, single-cell measurements with high precision and sensitivity are needed to more accurately understand their expression kinetics and functional consequences in antiviral signaling. We studied host and viral mRNA expression in single cells using single-molecule fluorescence in situ hybridization (smFISH) 27, 28. The single cell approach with the ultimate resolution of single transcripts enabled the discovery of a virus-induced, IFN-independent up-regulation of RIG-I that is hidden in ensemble analysis. This IFN-independent mechanism operates as early as 3 hours post infection, requires the IRF3/IRF7 pathway, and induces not only RIG-I but also several other ISGs, that we will refer to as “early ISGs”, prior to IFN production.
Multi-color smFISH analysis revealed that the mRNA levels of early ISGs are highly correlated with the large cell-to-cell heterogeneity in IFNB1 expression, in contrast to the viral gene and IFN-dependent ISG levels that are uncorrelated with IFNB1 expression. Over expression of RIG-I and MDA5 made IFNB1 expression more robust and earlier, indicating that early expression of RIG-I and MDA5 in a subset of infected cells may contribute to the decision making process for turning on the paracrine IFN-dependent innate immune response.
Results
Quantification of antiviral gene expression with single-cell resolution
We performed smFISH experiments on fixed cells 27, where each mRNA is decorated with 38 to 48 different sequences of fluorescently labeled probes, each 20 bases long (Fig. 1A). The large number of fluorophores bound to a single mRNA yielded a diffraction-limited fluorescent spot clearly above the background. smFISH assay also revealed the active transcription sites in the nucleus as brighter fluorescent spots where nascent mRNA molecules accumulate. For simultaneous quantification of up to three different genes in single cells, we labeled the probes with spectrally distinguishable fluorophores (Cy3, Alexa Fluor 594 and Cy5) (Fig. 1A), and counted the fluorescent spots from the 3D images of cells 27.
Figure 1. Quantification of IFN induced gene expression in single HepG2 cells.

(A) The schematics of single-molecule fluorescence in-situ hybridization (smFISH) and simultaneous imaging of RIG-I, MXA, and MDA5 mRNA in individual HepG2 cells treated with IFN-β for 6 hours. (B) Images of RIG-I and housekeeping gene ALAS mRNA in control and IFN-β treated (6 hours) HepG2 cells. (C) Kinetics of RIG-I mRNA expression post IFN-β treatment. Transcript numbers (mean ± SD) are obtained from 10 to 20 cells at each time point.
We first examined IFN-induced RIG-I expression. We treated previously described clonal HepG2 cells 29 with IFN-β and imaged RIG-I and housekeeping gene ALAS 30 mRNA at various time points. Unstimulated cells displayed basal level of RIG-I mRNA, 27 transcripts per cell on average. IFN-β treatment changed RIG-I mRNA counts in a time-dependent manner with the average number increasing with time but the mean intensity of individual spots remaining constant (Fig. 1A, B, C, Fig. S1), supporting the detection of individual transcripts. The cytoplasmic RIG-I mRNA counts displayed a steep increase until 6 hours followed by a plateau with an average of >200 transcripts per cell (Fig. 1C). In contrast, ALAS mRNA counts stayed relatively unchanged.
RIG-I is directly induced at the early stages of viral infection, independent of IFN
To examine the virus-induced expression of RIG-I and IFNB1, we infected clonal HepG2 cells with Sendai virus (SeV), fixed the cells at various time points post infection, and imaged RIG-I and IFNB1 mRNAs. The earliest time point at which we could detect IFNB1 transcripts was 9 hours post infection (hpi) (Fig. 2A, B). Consistently, IFN-β in the culture medium was detected with ELISA from 9 hpi onwards (Fig. 2C). On the other hand, bright RIG-I transcription sites were visible in the nuclei of a small fraction of the cells as early as 3 hpi, several hours prior to IFNB1 induction (Fig. 2A, Fig. S2). These cells contained 58 cytoplasmic RIG-I mRNA on average, which is about twice as many as basal level of 27 transcripts (Fig. 2D, Table 1). Percentage of cells expressing RIG-I mRNA over basal levels reached 32% by 6 hpi with an average of 145 transcripts per cell (Fig. 2B, D), and further rose to 80% at 9 hpi. Thus, our results show that RIG-I is directly induced at the early stages of viral infection in the absence of detectable IFNB1.
Figure 2. RIG-I is induced IFN-independently at the early stages of Sendai virus infection.
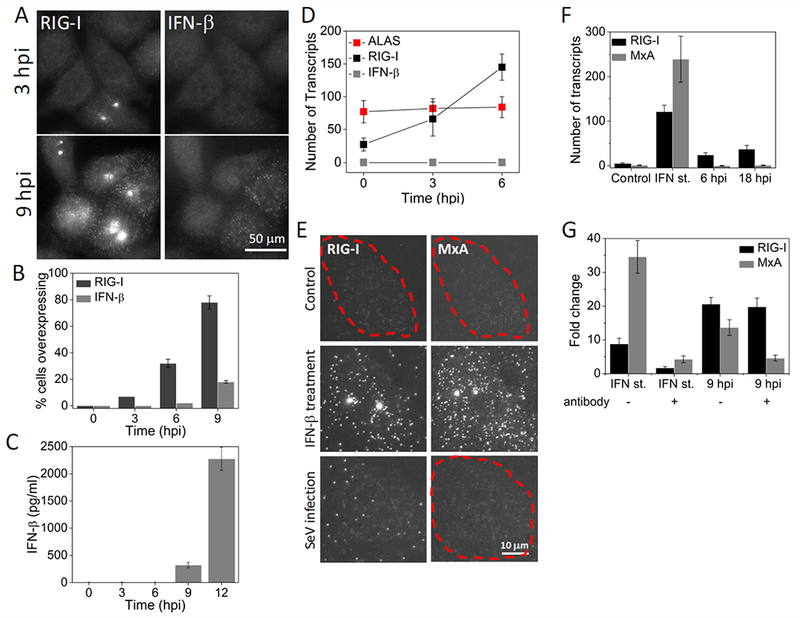
(A) Images of RIG-I and IFNB1 mRNA in HepG2 cells at 3 and 9 hours post SeV infection. (B) Percentage (mean ± SD) of HepG2 cells overexpressing RIG-I and IFNB1 mRNA as a function of time post SeV infection. 600 to 1000 cells were analyzed at each time point. (C) ELISA to detect IFN-β (mean ± SD) in the culture medium at different time points after SeV infection. Data is obtained from three independent experiments. (D) Number of RIG-I, IFNB1 and ALAS transcripts per cell (mean ± SD) in SeV infected (3 and 6 hours) HepG2 cells. Transcript numbers are obtained from 50 cells that overexpress RIG-I at each time point. (E) Images of RIG-I and MXA mRNA in control, IFN-β treated (12 hours) or SeV infected (18 hours) Vero cells. Cell borders are denoted by dashed outlines. (F) Number of RIG-I and MXA mRNA per 25 cell (mean ± SD) in control, IFN-β treated (12 hours) or SeV infected (6 and 18 hours) Vero cells. 20 cells are analyzed at each condition. (G) qRT-PCR analysis of RIG-I and MXA mRNA in IFN-β treated (2 hours) or SeV infected (9 hours) HepG2 cells in the presence or absence of neutralizing antibodies against IFN-α, IFN-β, IFN-γ, and IFNAR. Results are normalized to GAPDH, and presented as fold change relative to control cells. Error bars show standard deviation of two independent experiments.
Table 1.
Number of transcripts (mean ± SD) detected by smFISH in control, IFN-β treated (12 hours), and SeV infected (3 hours) HepG2 cells. Over-expressing cells were chosen to determine average transcript numbers at 3 hpi for genes in the dark grey rows. Cells were chosen randomly to determine the rest of the data. Data obtained from at least 20 cells for each condition and gene. Dark gray rows denote early ISGs.
| Basal | IFN-β st. | SeV (3 hpi) | |
|---|---|---|---|
| RIG-I | 27 ± 10 | 247 ± 64 | 58 ± 20 |
| MDA5 | 32 ± 12 | 231 ± 60 | 68 ± 18 |
| LGP2 | 7 ± 2 | 44 ± 12 | 31 ±7 |
| OasL | 0.4 ± 0.5 | 148 ± 44 | 255 ± 52 |
| IFIT1 | 3.5 ± 1.8 | 212 ± 68 | 303 ± 84 |
| MxA | 34 ± 7 | 273 ± 52 | 36 ± 8 |
| NLRX1 | 6 ± 5 | 68 ± 27 | 6 ± 4 |
| TRIM25 | 82 ± 52 | 334 ± 97 | 62 ± 33 |
To confirm that virus-induced early expression of RIG-I is indeed independent of IFN, first, we employed Vero cells that cannot produce any type of IFN due to genetic defects, but do have IFNAR and an intact JAK-STAT pathway, and therefore produce ISGs in response to IFN-β stimulation 31. As expected, upon IFN-β treatment, Vero cells showed a large increase in RIG-I transcription, from less than 5 to ~ 120 transcripts per cell. Upon infection with SeV, we did not detect any IFNB1 mRNA at any time point while the number of RIG-I transcripts gradually increased up to ~50 transcripts per cell, supporting our conclusion that RIG-I is induced during viral infection independent of IFN. To exclude the possibility that trace amount of IFNB1 in the virus stock may give rise to RIG-I induction, we also imaged MXA mRNA, which is strictly induced by IFN32. MXA transcript counts remained negligible during infection while externally added IFNB1 did induce MXA up to −240 transcripts per cell (Fig. 2E, F).
In another experiment, we used quantitative RT-PCR (qRT-PCR) to determine how RIG-I induction is modulated by the neutralizing antibodies against IFN-α, IFN-β, IFN-λ, and IFNAR. In the absence of neutralizing antibodies, RIG-I and MXA were induced 9- and 35-fold respectively upon treating the cells with IFN-β (Fig. 2G). The addition of the neutralizing antibodies during IFN-β treatment suppressed RIG-I and MXA induction, showing their efficacy (Fig. 2G). At 9 hours post SeV infection, RIG-I and MXA were induced 21- and 14-fold respectively in the absence of neutralizing antibodies. Addition of neutralizing antibodies during infection effectively suppressed MXA but did not affect RIG-I levels (Fig. 2G). These results further support that RIG-I is mainly regulated by an IFN-independent pathway at the early stages of viral infection, and even at 9 hpi much of RIG-I expression occurs independent of the IFN signaling pathway.
IFN-independent expression of early ISGs is strongly correlated amongst them and is dependent on IRF3/IRF7
Upon further examination using smFISH, expression kinetics of a small subset of ISGs revealed differential regulation of ISGs in response to viral infection. Antiviral genes RIG-I, MDA5, LGP2, as well as IFIT1 and OASL, already known to be induced IFN-independently, were directly induced by SeV infection as early as 3 hpi, observed as bright transcription sites in the nucleus and higher cytoplasmic mRNA copy numbers compared to control cells (Table 1). In contrast, IRF7, NLRX1, MXA, PKR, and TRIM25 did not display elevated levels of mRNA at early times post infection, even though they all are induced upon IFN-β treatment (Fig. 3A, B, Table 1) 13–15.
Figure 3. Virus-induced IFN-independent expression of early ISGs is strongly correlated and is dependent on IRF3/IRF7.

(A) Pairwise images of RIG-I and OASL, RIG-I and MDA5, LGP2 and MDA5, and RIG-I and IFIT1 transcripts in SeV infected HepG2 cells. Scale bar: 50 µm (B) Percentage of cells expressing RIG-I and/or OASL, RIG-I and/or MDA5, MDA5 and/or LGP2, and RIG-I and/or IFIT1 at indicated times post SeV infection. 200-300 cells were analyzed for each case. (C) Scatter plots of number of MDA5 vs. LGP2, and RIG-I vs. IFIT1 transcripts in single HepG2 cells at 9 hours post SeV infection. N: Number of cells analyzed, ρ: Pearson’s correlation coefficient. (D) Images of immunofluorescently labeled IRF3 at various time points post SeV infection. Scale bar: 50 µm (E) Percentage of cells (mean ± SD) displaying nuclear IRF3 as a function of time post SeV infection. 450 to 600 cells were analyzed at each time point. (F) qRT-PCR analysis of RIG-I mRNA in SeV infected (9 hours) HepG2 cells with prior siRNA knockdown (30 hours) of mock (HPRT1), IRF3, IRF7 and double knockdown of IRF3 and IRF7. Results are normalized to GAPDH, and presented as fold change relative to control cells. Error bars show standard deviation of two independent experiments.
Two-color smFISH analysis of single cells revealed that SeV-induced expressions of early ISGs are highly correlated with each other. Pairwise analyses of RIG-I vs. MDA5, MDA5 vs. LGP2, RIG-I vs. OASL and RIG-I vs. IFIT1 showed that if a cell activates transcription one of these genes, the other paired gene is also activated (Fig. 3A, B). For example, at 3 hpi, 92% of cells showed no induction of either RIG-I or OASL, 8% of cells showed both, and none showed RIG-I only or OASL only. We obtained high Pearson’s correlation coefficients for the mRNA counts for early ISGs in individual cells, for instance 0.8 between RIG-I and IFIT1, and 0.68 between LGP2 and MDA5 9 hpi (Fig. 3C).
Next, we used the Nanostring nCounter system 33 and tested a panel of 49 innate immunity genes for their early expression during infection, as well as to compare an ensemble assay to our single cell assay. nCounter data obtained from an ensemble of cells confirmed that first considerable IFNB1 expression happens at 9 hpi. (Table 2). At 6 hours post SeV infection, we identified 8 out of the 49 genes that are up-regulated (more than 2-fold increase with respect to the uninfected cells) in the absence of IFNB1: DDx60, ISG15, IFIT1, LGP2, MDA5, OASL, RIG-I, and Viperin (Table 2). Several other genes that strongly responded to IFN-β treatment, such as IFITM3, MXA, Myd88, OAS1, PKR, and TRIM25 were not induced (less than 1.5-fold increase with respect to the uninfected cells) at 6 hpi (Table 2, Table S1).
Table 2.
Number of transcripts detected by Nanostring nCounter gene expression system in control, IFN-β treated (2 hours), and SeV infected (2, 3, 6, and 9 hours) HepG2 cells. Number of transcripts is normalized to housekeeping gene GAPDH. Green rows denote viral genes, blue rows cytokines, dark gray rows early ISGs.
| Basal | IFN-β St. | 2 hpi | 3 hpi | 6 hpi | 9 hpi | |
|---|---|---|---|---|---|---|
| L gene | 4 | 15 | 240 | 466 | 1485 | 12720 |
| N gene | 1 | 13 | 1511 | 3179 | 13252 | 101475 |
| IFNA1 | 1 | 14 (14) | 1.5 (1.5) | 1 (1) | 2 (2) | 7 (7) |
| IFNA4 | 7 | 14 (2) | 6 (0.9) | 3 (0.4) | 9 (1.3) | 19 (3) |
| IFN-β | 6 | 6 (1) | 3 (0.5) | 3 (0.5) | 50 (8) | 2613 (436) |
| TNF-α | 35 | 66 (2) | 49 (1.4) | 56 (1.6) | 200 (6) | 3248 (93) |
| DDx60 | 1 | 514 (514) | 14 (14) | 17 (17) | 122 (122) | 2456 (2456) |
| IFIT1 | 62 | 17289 (278) | 107 (1.7) | 455 (7) | 3119 (50) | 41351 (667) |
| ISG15 | 1923 | 48622 (25) | 2022 (1.1) | 2862 (1.5) | 9524 (5) | 113474 (59) |
| LGP2 | 94 | 558 (6) | 107 (1.1) | 78 (0.8) | 187 (2) | 1412 (15) |
| MDA5 | 420 | 4527 (11) | 402 (1) | 509 (1.2) | 1051 (3) | 11027 (26) |
| OasL | 12 | 2157 (180) | 27 (2) | 119 (10) | 1199 (100) | 22884 (1902) |
| RIG-I | 306 | 4890 (16) | 215 (0.7) | 303 (1) | 613 (2) | 6968 (23) |
| Viperin | 1 | 3942 (3942) | 2 (1) | 6 (6) | 107 (107) | 2955 (2955) |
| IRF1 | 221 | 1994 (9) | 264 (1.2) | 238 (1.1) | 306 (1.4) | 1256 (6) |
| IRF7 | 2 | 20 (10) | 3 (1.5) | 1 (0.5) | 3 (1.5) | 9 (4.5) |
| IFITM3 | 3898 | 11812 (3) | 4092 (1) | 4595 (1.2) | 4809 (1.2) | 9809 (3) |
| MxA | 279 | 13396 (48) | 291 (1) | 318 (1.1) | 382 (1.4) | 3230 (12) |
| MyD88 | 1648 | 4590 (3) | 1744 (1.1) | 1862 (1.1) | 1711 (1) | 2381 (1.4) |
| Oas1 | 330 | 1457 (4) | 274 (0.8) | 285 (0.9) | 312 (1) | 751 (2) |
Up-regulation was also detected at 3 hpi for some of the early ISGs, such as DDX60, IFIT1, OASL and Viperin. A common property of these genes is that their basal level expression is very low, therefore when induced, they produce very high folds of increase with respect to the uninfected cells. On the other hand, induction of ISG15, LGP2, MDA5 and RIG-I was not detectable at 3 hpi, because their basal expression levels were higher, therefore, induction in a small fraction of the cells did not produce a significant fold change when ensemble averaged (Table 1 & Table 2). On the other hand, smFISH data show simultaneous induction of RIG-I, MDA5 and LGP2 with bright transcription sites and at least 2-fold increase in the number of cytoplasmic transcripts (Table 1, Fig. 3A), in the same cells that activated transcription of IFIT1 or OASL at 3 hpi, without exception. Overall, this comparison highlights the power of single cell approach with single molecule sensitivity to reveal gene expression patterns hidden in ensemble analysis.
In addition to their role in induction of IFNB1, transcription factors IRF3 and IRF7 have been shown to directly induce expression of some antiviral genes 16, 34 To test their roles in expression of the early ISGs, we immunofluorescently labeled IRF3 in SeV-infected cells, and analyzed the time-course of its nuclear translocation, which is required for its transcriptional activation role. We observed nuclear localization of IRF3 in 6 %, 25 % and 64% of the cells at 3, 6 and 9 hpi respectively, which is similar to that of RIG-I induction (Fig. 3D, E, Fig. 2B). Further, we used siRNA to knockdown IRF3 and/or IRF7. Following knockdown, we infected HepG2 cells for 9 hours with SeV, and performed qRT-PCR to measure the level of RIG-I mRNA. We found that SeV-induced expression of RIG-I is significantly reduced when both IRF3 and IRF7 are knockdown (Fig. 3F). These results together indicate that early ISGs expression is dependent on IRF3 or IRF7.
Early ISGs expression may contribute to decision making process of IFNB1 induction
smFISH analysis of IFNB1 transcripts in single cells confirmed the heterogeneous nature of IFNB1 expression among the cell population, which would otherwise be obscured in ensemble measurements. At any given time from 9 hpi onwards, we observed only a fraction of cells, typically less than 25%, expressing IFNB1 as opposed to more than 70% showing RIG-I induction (Fig. 2A, B).
Several factors may shape the decision making process of IFNB1 induction, one of which is the variation in the extent of viral replication 20, 25. We therefore performed smFISH analysis on SeV L gene mRNA which codes for the viral replicase to quantify viral replication. SeV-infected cells displayed viral mRNA while control cells did not show any signal above the background (Fig. 4A). At 9 hpi, we observed a high degree of cell-to-cell variation in L gene transcript counts, ranging from a few to several hundred regardless of whether IFNB1 is expressed or not (Fig. 4B and Fig. S3) and determined a low Pearson’s correlation coefficient of only 0.26 between L gene and IFNB1 mRNA counts (Fig. 4B), showing the amount of viral load is not a significant determinant of bimodal IFNB1 expression in single cells.
Figure 4. Early ISGs, but not viral L gene or other ISGs, are highly correlated with IFNB1 expression.

(A) Images of SeV L gene transcripts in control and SeV infected (6 hours) HepG2 cells. Cell border is denoted by dashed outline. (B) Scatter plots of number of L gene, RIG-I, IFIT1, LGP2, TRIM25, and NLRX1 against IFNB1 transcripts in single HepG2 cells at 9 hours post SeV infection. N: Number of cells analyzed, ρ: Pearson’s correlation coefficient. (C) qRT-PCR analysis of IFNB1 mRNA in SeV infected (9 hours) HepG2 cells with prior plasmid expression (12 hours) of mock, RIG-I, MDA5, LGP2, OASL, IFIT1, ISG15 and IFI6. Results are normalized to GAPDH, and presented as fold change relative to control cells. Error bars show standard deviation of two independent experiments. (D) Images of IFNB1 transcripts in HeLa cells 6 h post SeV infection with prior transfection of an empty vector, RIG-I or MDA5 (E) Percentage of HeLa cells (mean ± SD) expressing the IFNB1 gene at different time points post SeV infection, with prior transfection of an empty vector or RIG-I. (F) Percentage of HeLa cells (mean ± SD) expressing the IFNB1 gene at 6 hours post SeV infection, with prior transfection of an empty vector, RIG-I or MDA5.
In contrast to the L gene mRNA which was largely uncorrelated with IFNB1 induction, we observed that every cell that expressed IFNB1 also expressed RIG-I, as well as other early ISGs, including MDA5, LGP2, and IFIT1, over basal levels, and cells expressing higher amounts of early ISGs are more likely to be expressing IFNB1 with high Pearson’s correlation coefficients (Fig. 4B, Fig. S4). On the other hand, ISGs that require IFN activity for induction such as TRIM25 and NLRX1 did not correlate with IFNB1 induction (Fig. 4B).
The fact that RIG-I and MDA5 are major players in viral recognition and subsequent IFNB1 induction, together with our data showing early RIG-I and MDA5 expression precedes IFN, suggests that early expression of RIG-I and MDA5 may have a causal role in inducing IFNB1 in response to viral infection. In support of this hypothesis, we individually overexpressed RIG-I, MDA5, LGP2, OASL, IFIT1, IFI6 and ISG15 prior to viral infection by transfecting HepG2 cells with plasmids and monitored IFNB1 expression following SeV infection by qRT-PCR. Transfection of RIG-I or MDA5 encoding plasmids increased SeV-induced IFNB1 production compared to mock transfected cells, but not other early ISGs, consistent with previous observations (Fig. 4C)19, 22. By smFISH analysis, we also showed that the percentage of IFNB1 expressing HeLa cells significantly increased from about 20% to 60% at 6 hpi if the cells were transfected with RIG-I prior to viral infection. Additionally, we were able to detect IFNB1 in about 18% of cells at 3 hpi (Fig. 4D, E). Likewise, transfection with MDA5 increased the percentage of IFNB1 producing HeLa cells upon subsequent infection (Fig. 4D, F). Together, these experiments suggest that IFN-independent expression of RIG-I and MDA5 may have a causal role in activating type I IFN in response to viral infection.
Discussion
Although induction of ISGs through signaling triggered by type I IFN binding to the receptor is a major innate immune response, a more comprehensive picture is developing and showcases other mechanisms through which antiviral gene expression takes place 16, 34 In this study, using smFISH, we could identify a small fraction of cells showing up-regulation of RIG-I and several other genes upon viral infection, IFN-independently, whereas if we had relied on bulk measurements, such as qRT-PCR or Nanostring system, some of this information would have been lost due to the ensemble averaging over the entire population.
We exploited the multicolor nature of smFISH technology to study the well-known heterogeneity in the IFNB1 expression in single cells17–24. Our results together suggest that amplification of RIG-I and MDA5 in a fraction of cells at the early stages of infection may be a determinant of heterogeneous IFNB1 induction. Our findings are in agreement with previous reports showing that IFNB1 expressing cells have higher expression of RIG-I signaling pathway components 22 and heterogeneity in IFNB1 expression is mainly of cellular origin 23, 26. Cell-to cell variability in early ISGs’ expression may be a result of expression/phosphorylation levels of IRF3 and IRF7 transcription factors and upstream regulators, as well as differences in epigenetic changes and associated chromosomal organization.
Dendritic cells are shown to rapidly sense and respond to pathogens by expressing antiviral genes, including IFNB1, by a few early responding cells and the cellular variation is then controlled by IFN mediated paracrine signaling from these early responding cells 18, 26. Patil et al have shown that secreted factors accounted for <10% of the total variation in single cell IFNB1 responses, at the earliest time points IFNB1 was detectable (40% of cells were expressing IFNB1). The fraction of cells expressing IFNB1 then further increased over time due to paracrine signaling because treatment with drug Brefeldin A, which blocks paracrine signaling, blocked the increase in the fraction of cells expressing IFNB1 24. Because we mainly looked at the earliest time point IFNB1 was detectable, the cell-to-cell variation is likely due to cell-intrinsic factors, not paracrine signaling.
The heterogeneity in IFNB1 response has previously been shown to be relieved by increasing the concentration of various components of the pathway, such as RIG-I, TRIM25, IRF3, IRF7 and NF-κB to non-physiological levels 19, 22. In contrast, here we performed our smFISH experiments without disturbing the native system by forced expression of any of the components. We have found that RIG-I, but not TRIM25, IRF3 or IRF7, is up-regulated in the native system prior to IFNB1 induction, although over-expression of all of these components seemed to hyperactivate the IFNB1 response in previous studies.
Further studies are required to understand the mechanism with which the early expression of RIG-I and MDA5 facilitates induction of IFNB1. The obvious explanation is that the more RIG-I cells contain, the more responsive they are to viral infection. RIG-I and MDA5 have been shown to oligomerize upon viral infection, and the oligomeric forms of these proteins are highly active in inducing IFNB1 35–37. Amplification of RIG-I and MDA5 at the early stages of viral infection may promote oligomerization of these proteins, and subsequent aggregation of MAVS, and downstream signaling to IFNB1 induction. RIG-I may also be promoting IFNB1 expression by regulating NF-κB activity through binding to NF-κB 3’-UTR mRNA 38.
Finally, we still observe a fraction of IFNB1 non-expressing cells showing high expression of early ISGs. Therefore, in addition to the expression of IRF3/7 dependent RIG-I and MDA5, there must be additional steps required for IFNB1 induction, for example promoter binding and interchromosomal associations, providing further means of fine tuning the signaling process 19.
Conclusion
Using a single-cell, single molecule technique, we could study the very early stages of antiviral gene expression and demonstrated a novelty in the expression kinetics of RLRs. Our results together suggest that there may be a link between the early expression of RIG-I and MDA5 and induction of the IFNB1 gene.
Materials and Methods
Cell Culture, Virus Infection, IFN treatment, IFN neutralization and ELISA
HeLa and Vero cells were grown in DMEM supplemented with 10% FBS and 1% PENSTREP at 37°C. HepG2 cells were grown in DMEM supplemented with 10% FBS, 1% PENSTREP and lmg/ml G418 at 37°C. Sendai Virus Cantell (SeV) was grown in 10 day old embryonated chicken eggs (Charles River Laboratories). Eggs were inoculated with 100 μl of undiluted virus in order to generate Di-rich virus stocks. Cells were seeded at least 24 hours prior to infection. On the day of infection, medium is replaced by fresh medium containing SeV at MOI 5. IFN-β treatments were carried out by adding 1000 units/ml of human IFN-β (PBL InterferonSource, 11410-2). Type I IFN activity was blocked by addition of 10 μg/ml of anti-IFN-α (Abcam, ab20200), 10 μg/ml anti-IFN-β (R&D systems, AF814), 10 μg/ml anti-IFN-λ (R&D Systems, MAB15981), and 5 μg/ml anti-IFNAR chain 2 (PBL InterferonSource, 21385-1) into the culture medium during infection. Verikine human IFN beta ELISA kit (PBL InterferonSource, 41410-1B) was used to measure IFN-β from the culture medium.
Plasmids, transfections and RNAi interference
Expression plasmids for RIG-I (pEF-flagRIG-Ifull) and MDA5 (pEF-flagMDA5full) were provided by Takashi Fujita.
A custom Gateway destination vector, pT-Rex™-DEST30/FLAG was created by using QuikChange mutagenesis to create a silent mutation NotI restriction site in the attR1 site in pT-Rex™-DEST30 (Invitrogen) by introducing C825A and G826A mutations using primers manufacturer-suggested primers (Stratagene QuikChange Lightning Kit). A dsDNA insert of the sequence “gttgatgggcggccgctcgaaaacctgtattttcagggcactagtggcgacagcctgagctggctgctgagactgctgaacctgtgcacccccagccgggccgccctgctgaccggccggcctgcaggggactacaaagaccatgacggtgattataaagatcatgacatcgattacaaggatgacgatgacaagtagtaatgagggcccggagatg” was synthesized as a gBlok (IDT-DNA) and inserted via ligation into the NotI and AgeI sites of pT-Rex™-DEST30, producing a mammalian expression vector designed to append an amino acid sequence to the C-terminus of a hORF gene:
LPTFLYKVVDGRPLENLYFQGTSGDSLSWLLRLLNLCTPSRAALLTGRPAGDYKDHDGDYKDHDIDYKDDDDK-[STOP]
This sequence encodes a TEV protease recognition sequence, an SFP synthase tag, 13-aa aldehyde tag, and a 3×-FLAG-tag.
To obtain expression vectors pT-Rex™-DEST30/RIG-I/FLAG, pT-Rex™-DEST30/MDA5/FLAG, pT-Rex™-DEST30/LGP2/FLAG, pT-Rex™-DEST30/OasL/FLAG, pT-Rex™-DEST30/IFIT1/FLAG, pT-Rex™-DEST30/ISG15FLAG and pT-Rex™-DEST30/IFI6/FLAG, hORF plasmids from the CCSB Human ORFeome Collection (ThermoFisher) encoding MDA5, LGP2, OasL, IFIT1, ISG15 and IFI6 were cloned via Gateway directional LR cloning using LR Clonase II plus (Invitrogen) in 16 hours overnight reactions according to the manufacturer’s directions. LR reaction mixtures were transformed into chemically competent BL21-DH5-alpha cells and selected overnight on 100 ug/mL carbenicillin plates.
HeLa cells were transiently transfected with RIG-I (pEF-flagRIG-Ifull) and MDA5 (pEF-flagMDA5full) using JetPrime™ transfection reagent (Polyplus transfection, France).
HepG2 cell were transiently reverse transfected with pT-Rex™-DEST30/RIG-I/FLAG, pT-Rex™-DEST30/MDA5/FLAG, pT-Rex™-DEST30/LGP2/FLAG, pT-Rex™-DEST3O/OasL/FLAG, pT-Rex™-DEST30/IFITl/FLAG, pT-Rex™-DEST30/ISG15FLAG and pT-Rex™-DEST30/IFI6/FLAG using JetPrime™ transfection reagent (Polyplus transfection, France).
Plasmid transfected cells were grown for 9 to 12 hours before viral infection.
siRNAs targeting IRF3 (sc-35710) and IRF7 (sc-38011) were obtained from Santa Cruz Biotechnology. 30 pmol of each siRNA was transfected at a final concentration of ~45 nM, using Lipofectamine RNAiMAX (Invitrogen) transfection reagent. Cells transfected with siRNA were grown for 30 hours before viral infection.
RNA extractions, qRT-PCR and nCounter
Total RNA was extracted using QIAShredder and RNeasy mini kit (Qiagen) according to the manufacturer’s instructions. RNA quality was determined by Agilent 2100 bioanalyzer, and only RNA with RIN above 9 was used for reverse transcription reactions. Total RNA (900 μg) was reverse transcribed using the iScript™ cDNA Synthesis Kit (BIO-RAD). qRT-PCR analysis was carried out using SYBR-Green (Applied Biosystems).
The primer sequences used for qRT-PCR were as follows:
RIG-I forward: GGTTTAGGGAGGAAGAGGTGC, reverse: AAGTGTGGCAGCCTCCATTG;
MXA forward: ATGCTACTGTGGCCCAGAAA, reverse: GGCGCACCTTCTCCTCATA;
TNFA forward: GAGTGACAAGCCTGTAGCC, reverse: GCTGGTTATCTCTCAGCTCCA;
IFNB1 forward: GACGCCGCATTGACCATCTA, reverse: GTGACTGTACTCCTTGGCCT;
GAPDH forward: GACAGTCAGCCGCATCTTCT, reverse: AAATGAGCCCCAGCCTTCTC.
100 ng total RNA was hybridized to a custom gene expression CodeSet according to the NanoString Gene Expression Assay Manual and analyzed on an nCounter digital analyzer (NanoString Technologies). All data analysis was performed using the nSolver software analysis (freely available for download from NanoString Technologies). Briefly, counts are normalized for all target RNAs in all samples based on the positive control RNA to account for differences in hybridization efficiency and post-hybridization processing. Subsequently an mRNA content normalization is done using the housekeeping gene GAPDH.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 10 minutes and then permeabilized with 0.5% Triton-X 100 for 10 minutes. Cells were incubated with 3% BSA for 1 hour, and primary and secondary antibodies, each for 2 hours, at the recommended dilutions. Antibodies for IRF3 and NF-κB were obtained from Cell Signaling (11904P) and Abeam (ab119826) respectively. Whole procedure was performed at room temperature. Immunofluorescence images were obtained using Zeiss LSM 710 confocal scanning microscope equipped with a 40× oil-immersion objective (NA 1.3).
Single molecule FISH
DNA oligos for FISH experiments are designed using software available online (http://www.singlemoleculefish.com/). FISH probes are ordered from Biosearch Technologies.
For smFISH, cells were washed with PBS buffer, and fixed with 4% formaldehyde for 10 minutes at room temperature. After fixation, the cells were washed three times with PBS, before being permeabilized in 70% ethanol overnight. Cells were rehydrated in a solution of 10% formamide and 2× SSC for 5 minutes before hybridization. Hybridization reactions were carried out in 100 μl of hybridization buffer containing 10% dextran sulfate, 2 mM vanadyl-ribonucleoside complex, 0.02% RNAse-free BSA, 50 μg E.coli tRNA, 2× SSC, 10% formamide, and probe cocktail (5-50 ng) for overnight at 37°C. A coverslip was placed over the hybridization buffer to spread the small volume over the entire surface of the chamber, and also to prevent evaporation during the overnight incubation. After hybridization, the cells were washed twice for 30 minutes each at 37°C using a wash buffer containing 10% formamide and 2× SSC. During imaging, we used an oxygen scavenging mounting medium containing 10 mM Tris HC1 pH 8.0, 2× SSC, 1% glucose, glucose oxidase (or pyranose oxidase) and catalase.
We acquired smFISH images with a Zeiss Axiovert 200M inverted fluorescence microscope, equipped with a 100× oil-immersion objective (NA 1.45) and a Cascade 512b high sensitivity camera. Filter cubes to discriminate between Cy3, Alexa 594, and Cy5 were obtained from Semrock (SpOr-B-000-ZERO, SpRed-B-000-ZERO, and Cy5–4040C-000-ZERO respectively). 30 z-stacks were taken automatically with 0.3 microns between the z-slices, with an exposure time of 0.5 s.
Low magnification smFISH images (Supplementary Figure S2) images were obtained using Zeiss LSM 710 confocal scanning microscope equipped with a 40× oil-immersion objective (NA 1.3).
To identify and count the spots corresponding to individual mRNA molecules, we used a MATLAB program described previously 27 Briefly the program applies a Laplacian of Gaussian filter to the input TIFF images to enhance the spots. To identify spots, it applies an intensity threshold. The program counts number of spots detected for all possible threshold values in order to identify the appropriate threshold. When the number of spots identified is plotted as a function of the threshold value, a plateau region is observed, where the spot count does not vary significantly over a broad range of thresholds. The threshold is manually selected at the plateau region, based on the graph and visual feedback.
Supplementary Material
Acknowledgements:
We would like to thank Mayandi Sivaguru and Glenn Fried (microscopy), Benjamin J. Leslie (cloning) for expert help. We would like to thank Arjun Raj and Kaushik Ragunathan for helpful discussions. This research was partly funded by NIAID grants U19 AI083025 (to TH, SM and AG-S), by NIAID grant U19AI117873 (to AG-S).
References
- 1.Kato H; Sato S; Yoneyama M; Yamamoto M; Uematsu S; Matsui K; Tsujimura T; Takeda K; Fujita T; Takeuchi O; Akira S, Cell type-specific involvement of RIG-I in antiviral response. Immunity 2005, 23 (1), 19–28. [DOI] [PubMed] [Google Scholar]
- 2.Kato H; Takeuchi O; Sato S; Yoneyama M; Yamamoto M; Matsui K; Uematsu S; Jung A; Kawai T; Ishii KJ; Yamaguchi O; Otsu K; Tsujimura T; Koh CS; Reis e Sousa C; Matsuura Y; Fujita T; Akira S, Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441 (7089), 101–5. [DOI] [PubMed] [Google Scholar]
- 3.Gitlin L; Barchet W; Gilfillan S; Cella M; Beutler B; Flavell RA; Diamond MS; Colonna M, Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A 2006, 103 (22), 8459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoneyama M; Fujita T, RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev 2009, 227 (1), 54–65. [DOI] [PubMed] [Google Scholar]
- 5.Loo YM; Gale M, Immune signaling by RIG-I-like receptors. Immunity 2011, 34 (5), 680–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schoggins JW; Wilson SJ; Panis M; Murphy MY; Jones CT; Bieniasz P; Rice CM, A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472 (7344), 481–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawai T; Takahashi K; Sato S; Coban C; Kumar H; Kato H; Ishii KJ; Takeuchi O; Akira S, IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 2005, 6 (10), 981–8. [DOI] [PubMed] [Google Scholar]
- 8.Seth RB; Sun L; Ea CK; Chen ZJ, Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122 (5), 669–82. [DOI] [PubMed] [Google Scholar]
- 9.Xu LG; Wang YY; Han KJ; Li LY; Zhai Z; Shu HB, VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 2005, 19 (6), 727–40. [DOI] [PubMed] [Google Scholar]
- 10.Meylan E; Curran J; Hofmann K; Moradpour D; Binder M; Bartenschlager R; Tschopp J, Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437 (7062), 1167–72. [DOI] [PubMed] [Google Scholar]
- 11.Stetson DB; Medzhitov R, Type I interferons in host defense. Immunity 2006, 25 (3), 373–81. [DOI] [PubMed] [Google Scholar]
- 12.Taniguchi T; Takaoka A, The interferon-alpha/beta system in antiviral responses: a multimodal machinery of gene regulation by the IRF family of transcription factors. Curr Opin Immunol 2002, 14 (1), 111–6. [DOI] [PubMed] [Google Scholar]
- 13.de Veer MJ; Holko M; Frevel M; Walker E; Der S; Paranjape JM; Silverman RH; Williams BR, Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol 2001, 69 (6), 912–20. [PubMed] [Google Scholar]
- 14.Collins SE; Noyce RS; Mossman KL, Innate cellular response to virus particle entry requires IRF3 but not virus replication. J Virol 2004, 78 (4), 1706–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melchjorsen J; Kristiansen H; Christiansen R; Rintahaka J; Matikainen S; Paludan SR; Hartmann R, Differential regulation of the OASL and OAS1 genes in response to viral infections. J Interferon Cytokine Res 2009, 29 (4), 199–207. [DOI] [PubMed] [Google Scholar]
- 16.Dixit E; Boulant S; Zhang Y; Lee AS; Odendall C; Shum B; Hacohen N; Chen ZJ; Whelan SP; Fransen M; Nibert ML; Superti-Furga G; Kagan JC, Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141 (4), 668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zawatzky R; De Maeyer E; De Maeyer-Guignard J, Identification of individual interferon-producing cells by in situ hybridization. Proc Natl Acad Sci U S A 1985, 82 (4), 1136–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu J; Sealfon SC; Hayot F; Jayaprakash C; Kumar M; Pendleton AC; Ganee A; Fernandez-Sesma A; Moran TM; Wetmur JG, Chromosome-specific and noisy IFNB1 transcription in individual virus-infected human primary dendritic cells. Nucleic Acids Res 2007, 35 (15), 5232–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Apostolou E; Thanos D, Virus Infection Induces NF-kappaB-dependent interchromosomal associations mediating monoallelic IFN-beta gene expression. Cell 2008, 134 (1), 85–96. [DOI] [PubMed] [Google Scholar]
- 20.Chen S; Short JA; Young DF; Killip MJ; Schneider M; Goodbourn S; Randall RE, Heterocellular induction of interferon by negative-sense RNA viruses. Virology 2010, 407 (2), 247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu J; Nudelman G; Shimoni Y; Kumar M; Ding Y; López C; Hayot F; Wetmur JG; Sealfon SC, Role of cell-to-cell variability in activating a positive feedback antiviral response in human dendritic cells. PLoS One 2011, 6 (2), e16614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao M; Zhang J; Phatnani H; Scheu S; Maniatis T, Stochastic expression of the interferon-β gene. PLoS Biol 2012, 10 (1), e1001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rand U; Rinas M; Schwerk J; Nöhren G; Linnes M; Kröger A; Flossdorf M; Kály-Kullai K; Hauser H; Höfer T; Köster M, Multi-layered stochasticity and paracrine signal propagation shape the type-I interferon response. Mol Syst Biol 2012, 8, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patil S; Fribourg M; Ge Y; Batish M; Tyagi S; Hayot F; Sealfon SC, Single-cell analysis shows that paracrine signaling by first responder cells shapes the interferon-β response to viral infection. Sci Signal 2015, 8 (363), ral6. [DOI] [PubMed] [Google Scholar]
- 25.Killip MJ; Young DF; Ross CS; Chen S; Goodbourn S; Randall RE, Failure to activate the IFN-β promoter by a paramyxovirus lacking an interferon antagonist. Virology 2011, 415 (1), 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shalek AK; Satija R; Shuga J; Trombetta JJ; Gennert D; Lu D; Chen P; Gertner RS; Gaublomme JT; Yosef N; Schwartz S; Fowler B; Weaver S; Wang J; Wang X; Ding R; Raychowdhury R; Friedman N; Hacohen N; Park H; May AP; Regev A, Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014, 510 (7505), 363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raj A; van den Bogaard P; Rifkin SA; van Oudenaarden A; Tyagi S, Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 2008, 5 (10), 877–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Itzkovitz S; van Oudenaarden A, Validating transcripts with probes and imaging technology. Nat Methods 2011, 8 (4 Suppl), S12–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hwang SY; Hur KY; Kim JR; Cho KH; Kim SH; Yoo JY, Biphasic RLR-IFN-β response controls the balance between antiviral immunity and cell damage. J Immunol 2013, 190 (3), 1192–200. [DOI] [PubMed] [Google Scholar]
- 30.Bishop DF; Henderson AS; Astrin KH, Human delta-aminolevulinate synthase: assignment of the housekeeping gene to 3p21 and the erythroid-specific gene to the X chromosome. Genomics 1990, 7 (2), 207–14. [DOI] [PubMed] [Google Scholar]
- 31.Desmyter J; Melnick JL; Rawls WE, Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J Virol 1968, 2 (10), 955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durbin JE; Hackenmiller R; Simon MC; Levy DE, Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 1996, 84 (3), 443–50. [DOI] [PubMed] [Google Scholar]
- 33.Geiss GK; Bumgarner RE; Birditt B; Dahl T; Dowidar N; Dunaway DL; Fell HP; Ferree S; George RD; Grogan T; James JJ; Maysuria M; Mitton JD; Oliveri P; Osborn JL; Peng T; Ratcliffe AL; Webster PJ; Davidson EH; Hood L; Dimitrov K, Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 2008, 26 (3), 317–25. [DOI] [PubMed] [Google Scholar]
- 34.Noyce RS; Collins SE; Mossman KL, Identification of a novel pathway essential for the immediate-early, interferon-independent antiviral response to enveloped virions. J Virol 2006, 80 (1), 226–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang X; Kinch LN; Brautigam CA; Chen X; Du F; Grishin NV; Chen ZJ, Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36 (6), 959–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel JR; Jain A; Chou YY; Baum A; Ha T; García-Sastre A, ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type-I interferon. EMBO Rep 2013, 14 (9), 780–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hou F; Sun L; Zheng H; Skaug B; Jiang QX; Chen ZJ, MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146 (3), 448–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang HX; Liu ZX; Sun YP; Zhu J; Lu SY; Liu XS; Huang QH; Xie YY; Zhu HB; Dang SY; Chen HF; Zheng GY; Li YX; Kuang Y; Fei J; Chen SJ; Chen Z; Wang ZG, Rig-I regulates NF-κB activity through binding to Nf-Kbl 3′-UTR mRNA. Proc Natl Acad Sci U S A 2013, 110 (16), 6459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.