

Abstract
The application of frontal affinity chromatography−mass spectrometry (FAC−MS), along with molecular modeling studies, to the screening of potential drug candidates toward the recently deorphanized G-protein-coupled receptor (GPCR) GPR17 is shown. GPR17 is dually activated by uracil nucleotides and cysteinyl-leukotrienes, and is expressed in organs typically undergoing ischemic damage (i.e., brain, heart and kidney), thus representing a new pharmacological target for acute and chronic neurodegeneration. GPR17 was entrapped on an immobilized artificial membrane (IAM), and this stationary phase was used to screen a library of nucleotide derivatives by FAC−MS to select high affinity ligands. The chromatographic results have been validated with a reference functional assay ([35S]GTPγS binding assay). The receptor nucleotide-binding site was studied by setting up a column where a mutated GPR17 receptor (Arg255Ile) has been immobilized. The chromatographic behavior of the tested nucleotide derivatives together with in silico studies have been used to gain insights into the structure requirement of GPR17 ligands.
Introduction
The combination of frontal affinity chromatography (FACa) and mass spectrometry (MS) for binding studies is a rather new analytical concept that has been first described in 1998 by Schreimer and Hindsgaul.1–3 On one side, FAC allows us to study molecular interactions in a simple fashion, is amenable to automation, and provides precise and accurate binding data. On the other hand, MS allows the identification of compounds solely based on their mass-to-charge ratio (m/z) and is a label-free detection system. The principles of FAC−MS have been well described.4–8 Nonetheless, the use of FAC−MS is not yet so widespread and more work should be done to expand and validate the application of this methodology to the screening of combinatorial libraries toward important biological targets such as G-protein-coupled receptors (GPCRs).
GPCRs are important appealing pharmaceutical targets, but useful potent and selective ligands do not exist for the majority of these receptors and several of them are still “orphan”. In addition, from a drug discovery and design prospective, structural information on GPCRs can represent a fundamental tool for structural-based drug design. However, the X-ray structure has been determined for only four GPCRs (i.e., the visual receptor rhodopsin, β1 and β2 adrenoreceptors, and very recently the A2A adenosine receptor).9,10
The feasibility of the FAC approach has already been demonstrated for some GPCRs upon immobilization on artificial membranes (IAM);11,12 however, the application of these particular stationary phases to screening studies has not been yet reported. In this respect, we have recently described the development of a chromatographic stationary phase by immobilizing on IAM particles membrane fragments from a cell line expressing the recently deorphanized G-protein-coupled receptor GPR17.13
This is an interesting “dual “ receptor responding to two distinct families of inflammatory mediators, namely, uracil nucleotides and cysteinyl-leukotrienes (cysLTs), which are massively released in the brain upon different types of injury.14
We have recently reported that GPR17 is pathologically activated during acute CNS injury, thus contributing to early necrotic death inside the lesion,14 but also participates in the subsequent remodeling and repair of the lesioned area occurring in the days and weeks after injury.15 Thus, GPR17 ligands could represent a new class of neuroprotective and neuro-reparative agents in several types of human neurodegenerative diseases, such as stroke, trauma, and multiple sclerosis. Since, as mentioned above, GPR17 dually responds to two unrelated families of endogenous ligands and may represent just the first example of a series of “dual” GPRCs,16 we hypothesize that the identification of selective GPR17 ligands could lead to the development of novel dualistic pharmacological agents with previously unexplored therapeutic potential.
In this context, elucidation of the structure of GPR17 and of ligand binding mechanisms is a necessary steps to obtain selective and potent drugs for this new potential target. On this basis, a 3D molecular model of GPR17 embedded in a solvated phospholipid bilayer and refined by molecular dynamics simulations has been created.17
Molecular dynamics simulations suggest that the GPR17 nucleotide binding pocket is similar to that described for the other P2Y receptors, although only one of the three basic residues that have been typically involved in ligand recognition is conserved (Arg255).
In this work we report, for the first time, the application of FAC−MS to the screening of a small library of compounds toward GPR17 immobilized on a chromatographic support. This approach allowed the rapid and accurate ranking of the selected compounds. The chromatographic data have been validated by a reference functional assay ([35S]GTPγS binding assay). In addition, the simplicity of the method has allowed us to explore the binding mode of the nucleotide site of the receptor by extending this approach to a column where an artificial GPR17 receptor bearing a mutation in a key amino add for ligand recognition has been immobilized. Comparison of the different chromatographic behavior of the nucleotide derivatives in the library together with in silico studies has been used to gain insight into the structure requirement of GPR17 ligands. We expect that this integrated approach can contribute to GPCR-based drug discovery process.
Results and Discussion
Chemical Library Design and Synthesis.
The compound library for GPR17 was selected taking into account the chemical structures of three known receptor ligands with different potencies: the antagonists 1 (cangrelor; IC50 = 0.7 nM) and 2 (MRS 2179; IC50 = 508 nM) and the agonist 3 (UDP; EC50 = 1.14 µM) (Table l).14 Starting from the cangrelor structure and with the aim of investigating the structural requirements for ligand recognition at the nucleotide receptor active site, a series of ATP derivatives 4−7, substituted at the 2- and/or N6-position, were selected. Furthermore, in order to evaluate the importance of the 3-nitrogen purine ring and the grade of nucleotide phosphorylation, the 2,N6-disubstituted ATP 3-deaza analogue 8 and some diphosphate nucleotides 9−11 were evaluated. Furthermore, compound 12, a bisphosphate derivative analogue to MRS 2179, and the two 5-substituted UDP analogues 13 and 14 were selected. Moreover, some phosphorylated acyclonucleotides 15−21, bearing an adenine base and a four-carbon alkyl spacer in the 9-position, which can mimic the ribose moiety of nucleotide ligands, were added to the selection to obtain additional information about the structural requirements of the sugar portion that binds the receptor.27 Hence, compounds consisting of 18 members, ranging from 479 to 675 in molecular weight, have been evaluated.
The synthesis of the known tested compounds was carried out following already described procedures, which are listed in Table 1. New nucleotides 7, 8, and 14 were prepared by phosphorylation of the corresponding nucleosides 22,28 23,29 and 2630 (Scheme 1). Nucleosides were first phosphorylated to the monophosphates, and from these the corresponding di- and triphosphates were prepared.
Table 1.
Chemical Structures of the Compounds Selected for FAC—MS Studies
| compound | structure | pharmacology | compound | structure | pharmacology | |||
| Reference compound | Reference compound | |||||||
| Cangrelor(1) | 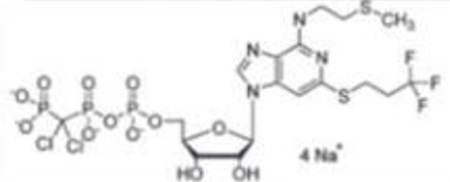 |
P2Y12antagonist IC50=0.7 nM |
UDP(3) | 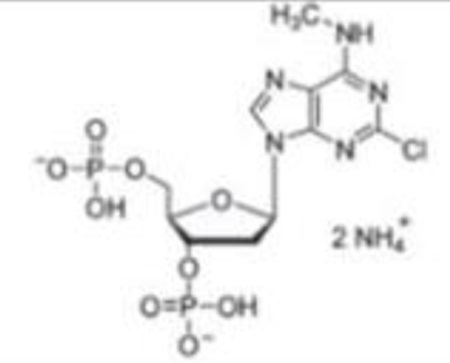 |
P2Y agonist EC50=1.14 μM |
|||
| MRS2179(2) | 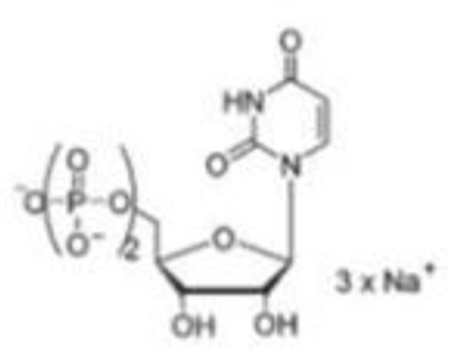 |
P2Y1 competitive Antagonist IC50=508 nM |
||||||
| Library A | ||||||||
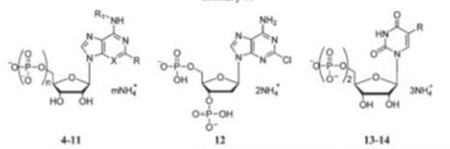 | ||||||||
| 418 | R=C=C=Ph R1=H X=N n=3, m=4 |
P2Y1,12antogonist | 8 | R=CI R1=CH X=CH n=3, m=4 |
||||
| 519,20 | R=H R1=CH3 X=N n=3, m=4 |
922 | R=CI R1=CH3 X=N n=2, m=3 |
|||||
| 621 | R=H R1=cC3H9 X=N n-3, m-4 |
1018 | R=C=C.Ph R1=H X=N n=2, m=3 |
P2Y2.12 antagonist | ||||
| 7 | R=CI R1=CH3 X=N n=3, m=4 |
1118 | R=C=C-nC4H9 R1=H X=N n=2, m=3 |
P2Y12 agonist |
||||
| 1223,24 | MRS2179 analogue | |||||||
| 1325,26 | R=1 | P2Y4agonist | ||||||
| 14 | R=C=C-nC4H9 | P2Y4agonist | ||||||
Scheme 1a:
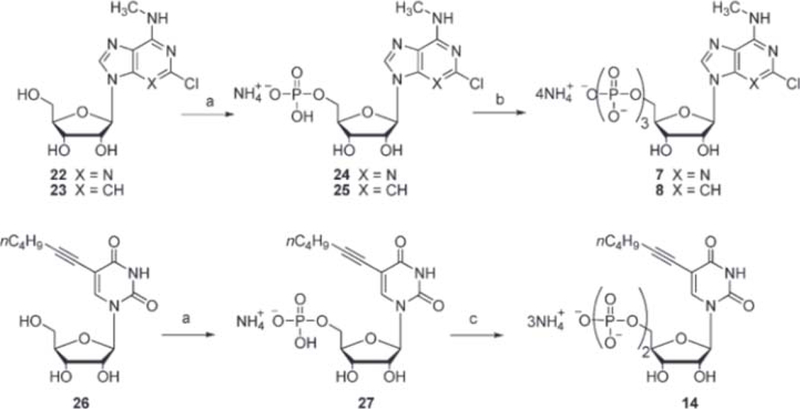
a Reaction conditions: (a) (i) POCI3, (CH3O)3 PO;(ii) NH4HCO3; b) (i) n-Bu3N, DMF; (ii) CDI, DMF;(iii) CH3OH,DMF;(iv) (n-Bu3NH)2(P2O7H2), DMF; (v) NH4HCO3; (c) (i) n-Bu3N, DMF; (ii) CDI, DMF; (iii) CH3OH, DMF; (iv) (n-Bu3NH)(HPO4), DMF; (v) NH4HCO3.
Monophosphates 24, 25, and 27 were synthesized by reacting the corresponding nucleosides 22, 23, and 26 with POCl3 in trimethylphosphate at room temperature, following the Yoshikawa method (Scheme l).31 Synthesis of triphosphates 7 and 8 and diphosphate 14 were carried out by a modification of the Hoard−Ott method (Scheme l).32 To this purpose, the tributylammonium salts of monophosphates were activated as phosphoroimidazolates by reacting with N,N’-carbonyldiimidazole (CDI). These unstable intermediates were coupled with bis(tri-n-butylammonium) pyrophosphate at room temperature to give the corresponding nucleoside triphosphates 7 and 8 (Scheme 1). The nucleoside diphosphate 14 was prepared by reacting the corresponding phosphorimidazolate with tri-n-butylammonium phosphate at room temperature (Scheme 1).
The phosphorylating reagents were freshly prepared starting from the inorganic salts, which were first converted to inorganic acid with Dowex H+ form resin and then into the tributylammonium salts by adding tri-n-butylamine. The removal of water under vacuum, followed by coevaporation with dry ethanol, furnished the dry organic salts, which were suspended in dry DMF after coevaporation of the residue ethanol with DMF.
All the new nucleotide analogues were obtained as ammonium salts after ion exchange chromatography on a Sephadex diethylaminoethyl (DEAE) column (HC03− form), eluted with a linear gradient of ammonium bicarbonate, which gives a gradual increase of ionic strength.
Synthesized nucleotides were characterized with 1H NMR and the phosphoric groups identified through 31P NMR analysis in D2O. In fact mono-, di-, and triphosphates show characteristic 31P NMR spectra. Monophosphate nucleotides show a characteristic peak at about 0 ppm; diphosphates show characteristic doublet at −9 to −10 ppm, and triphosphates show a series of peaks at −6, −11, and −22, corresponding to the γ, α, and δ phosphorus atoms, respectively. The phosphorus chemical shift is highly dependent on the pH of the solution and the counterion of the nucleotide.
FAC−MS Screening of the Chemical Libraries.
To explore the ability of FAC−MS to rank mixtures containing multiple ligands, a first column (column GPR17-IAM-I) was prepared by immobilizing crude membranes of 1321N1 cells expressing wild type GPR17, following the acquisitions from previous studies on the optimization of the GPCR stationary phase where the nonspecific interactions were minimized by analyzing the retention of the three reference compounds on a blank column only containing IAM and an additional column containing membranes from cells transfected with an empty vector.13,33 Because our previous studies demonstrated that nonspecific interactions contribute to the total retention time but do not interfere with the specific displacement study, we have reasoned that it was no longer necessary to run a blank column in parallel for every single ranking experiment. The preparation of the receptor stationary phase is described in the Experimental Section.
The selected 18 compounds were split into two series: analogues of nucleotides are included in library A, while acylic nucleotides are part of library B (see Tables 1 and 2) Solutions containing the ligands belonging to the two libraries and the three already known GPR17 ligands (UDP, MRS 2179,and cangrelor) all at 1 µM were prepared in mobile phase buffer and continuously infused through the column. The effluent was analyzed in an ESI mass spectrometer to detect each components. The mass analysis of the library was performed in negative mode to ensure efficient ionization and detection of the compounds. Detailed chromatographic experimental conditions are reported in the Experimental Section. The potential ligands were retained in the column based on their affinity, and the total run time was 55 and 20 min for libraries A and B, respectively. Because the molecular weight for each compound is known, the individual breakthrough front of each compound was easily identified as shown in Figure 1 reporting the extracted breakthrough curves for each analyte of library A and library B. The breakthrough times reported in Table 2 are the results of a single determination carried out on the freshly prepared stationary phase. To compare the chromatographic results obtained with the screening of the two libraries, the percentage of breakthrough time with respect to cangrelor was calculated for all the analytes (Table 2).
Table 2.
Frontal Affinity Chromatography—Mass Spectrometry (FAC— MS) Results for Library A and Library B Screened with Immobilized GPR17a
| Compd | tb(min) | t/tcc × 100 |
|---|---|---|
| Library A | ||
| UDP | 3.20 | 13.03 |
| 13 | 4.06 | 16.54 |
| 9 | 4.78 | 19.47 |
| 14 | 4.83 | 19.67 |
| 12 | 6.38 | 25.99 |
| MRS2179 | 7.10 | 28.92 |
| 5 | 7.53 | 3.67 |
| 11 | 8.86 | 36..9 |
| 7 | 9.22 | 37.56 |
| 8 | 11.06 | 45.05 |
| 6 | 12.25 | 49.90 |
| 10 | 16.80 | 68.43 |
| Cangrelor | 24.55 | 100.00 |
| 4 | 29.55 | 120.37 |
| Library B | ||
| UDP | 1.50 | 12.30 |
| 15 | 1.51 | 12.38 |
| 18 | 1.60 | 13.11 |
| 19 | 2.42 | 19.84 |
| 16 | 2.83 | 23.20 |
| 17 | 3.24 | 26.56 |
| MRS 2179 | 3.40 | 27.87 |
| 20 | 3.58 | 29.34 |
| 21 | 9.22 | 75.57 |
| Cangrelor | 12.20 | 100.00 |
The breakthrough times are from a single raking experiment carried out on the freshly prepared column. Column: GPR17-IAM-I (20.72 milion cells). Breakthrough times of the three reference compounds (UDP, MRS 2179, and cangrelor) are marked in bold. See text for detailed chromatographic conditions
t is the breakthrough time of the ligand with the immobilized GPR17.
tc represents the breakthrough time of the indicator cangrelor.
Figure 1.
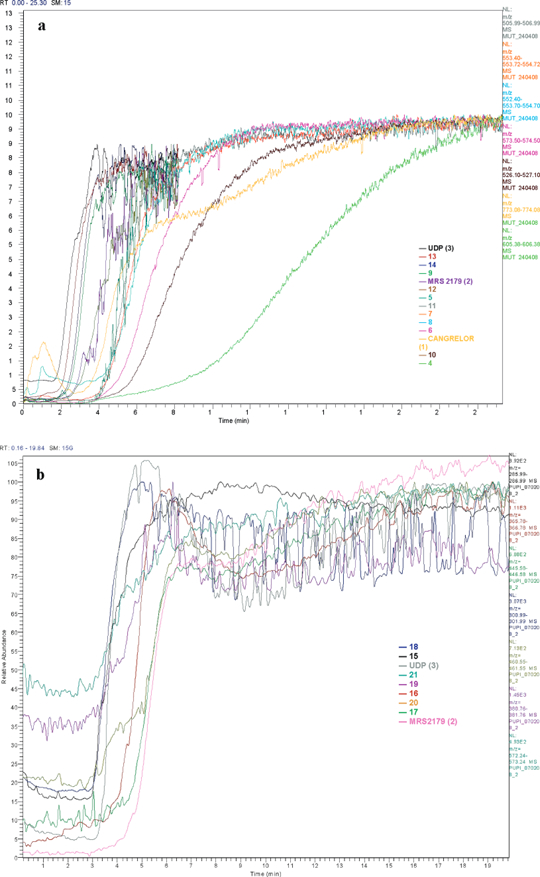
Frontal affinity chromatography—mass spectrometry of library A (a) and library B (b). Shown are extracted breakthrough curves for each analyte of library A and library B. Mixtures of ligands in the presence of the three reference compounds each at 1 fiM were infused through the GPR17-IAM-I column using the mass spectrometer in negative mode.
As expected, the elution order of the three reference compounds in the two experiments was consistent with our previous results13 and in agreement with their potency at this receptor (cangrelor > MRS 2179 > UDP). Moreover, a very good correspondence between the relative breakthrough % time of the less potent reference compounds (MRS 2179 and UDP) with respect to cangrelor was obtained in the two experiments (relative % frontal time of 29 and 13 in the first experiment and of 28 and 12 in the second experiment for MRS 2179 and UDP, respectively).
In general, the first set of compounds (library A) was significantly retained by the column, while the second set (library B) weakly interacted with GPR 17-column, with the exception of the acyclonucleotide triphosphate 21, which was eluted immediately before cangrelor with a relative % frontal time of 75, which indeed represents one of the highest values observed.
Moreover, in both series, the binding order of the phosphorylated compounds was consistent with the degree of phosphorylation; hence, in library A the binding order was 3P > 2P (compare 4 and 7 with 10 and 9, respectively) and in library B the order was 3P > 2P > IP (Tabel 2)
The large breakthrough volumes for compounds 4, 10, and 21 indicate that they can be identified as high binding candidates. In particular, for 4, which was the strongest binding ligand, a subnanomolar Kd value can be hypothesized from the chromatographic data, as it elutes after cangrelor. On the other hand, compounds 5—8 and 11 seem to be medium-high binding ligands, since they were eluted with breakthrough times between those of cangrelor and MRS 2179
Compound 12 elutes with a breakthrough time close that of MRS 2179. This finding is in agreement with the structure of the two compounds that are very similar bisphosphate analogues. The same consideration can be drawn for the 5-substituted UDP derivatives 13 and 14, which elute close to UDP. These compounds can thus be classified as weak binders toward GPR17, like UDP itself. Interestingly, cangrelor and MRS 2179 are antagonists at GPR17 while UDP acts as an agonist;14 thus, as already demonstrated for many other receptors, also for GPR17, antagonists display higher binding affinity with respect to agonists.
On the basis of these results, library A containing the most promising ligands was selected for further experiments. The reproducibility of the method was assessed by preparing and testing a second GPR17 column (GPR17-IAM-II) starting from a new batch of transfected cells. Table 3 reports the data in comparison with the data previously obtained on the GPR17-IAM-I column. An overall decrease of the breakthrough times for all the compounds analyzed with GPR17-IAM-II column was evident, but remarkably, the relative ranking of the known and potential ligands was highly consistent for both columns, with the only exception of 9 and 14, for which an apparent reversal of the breakthrough time order was observed. However, it must be emphasized that for these two analytes, the elution time difference (approximately 0.05 min) is too small to draw such a conclusion, and a more plausible explanation is that the two analytes coelute. The different run times between the two GPR17 columns can be ascribed to different numbers of the expressed receptors in the two different cell preparations, as the transfection efficiency ranged from 30% to 50% (data not shown). Globally, these results confirm that FAC—MS is a powerful tool that can be used as a qualitative and reproducible method for ligand discovery.
Table 3.
Comparison of the Breakthrough Times of Derivatives in Library A Obtained with Two Different GPR17-IAM Columnsa
| library A compd | GPR17-IAM-IIb | GPRI7-IAM-Ic | ||
|---|---|---|---|---|
| t (min) | t/tC ×100 | t (min) | t/tC × 100 | |
| UDP | 2.69 | 25.38 | 3.20 | 13.03 |
| 13 | 2.79 | 26.32 | 4.06 | 16.54 |
| 9 | 3.03 | 28.58 | 4.78 | 19.47 |
| 14 | 2.97 | 28.02 | 4.83 | 19.67 |
| 12 | 3.55 | 33.49 | 6.38 | 25.99 |
| MRS 2179 | 3.62 | 34.15 | 7.10 | 28.92 |
| 5 | 3.69 | 34.81 | 7.53 | 30.67 |
| 11 | 4.26 | 40.19 | 8.86 | 36.09 |
| 7 | 4.50 | 42.45 | 9.22 | 37.56 |
| 8 | 4.77 | 45.00 | 11.06 | 45.05 |
| 6 | 5.67 | 53.49 | 12.25 | 49.90 |
| 10 | 6.45 | 60.85 | 16.80 | 68.43 |
| cangrelor | 10.60 | 100.00 | 24.55 | 100.00 |
| 4 | 13.07 | 123.30 | 29.55 | 120.37 |
The breakthrough times are from a single ranking experiment carried out on the freshly prepared column.
Column: GPR17-IAM-II (19.5 milion cells).
Column: GPR17-IAM-I (20.72 milion cells).
Structure−Affinity Relationships (SARs).
The obtained screening results have been used to draw preliminary binding SAR speculations at the hit discovery stage that would reveal ways of designing potent and selective GPR17 ligands. The data from FAC—MS experiments suggest that lipophilic substituents on the C-2 and on the N6 amino group of purine ring can lead to a tight interaction with the hydrophobic residues in the binding pocket. In fact, in both series listed in library A and library B, compounds that strongly bind to the receptor bear lipophilic groups in the 2-position like the phenylalkynyl chain or halogens (see 4,10, and 21) and/or an alkyl/cycloalkyl substituent on the N6 amino group (see 6,7, and 8). The replacement of the phenyl group with a «-butyl moiety brings about a reduction of binding affinity to GPR17 (compare 10 and 11). On the other hand, it could be that the presence of a nitrogen in 3-position is not essential, since its isosteric replacement with a CH does not significantly change the breakthrough time (compare 7 and 8).
It is clear that in compounds with the same substituents on the purine moiety, the rank order of potency is 3P > 2P > IP (i.e., 17 > 16 > 15 and 20 > 19 > 18). On the other hand, the presence of an intact ribose seems to be crucial; in fact, compounds of library B show, in general, lower retention time compared with nucleotides analogues. It is worthwhile to note that acyclonucleotide triphosphate 21, the only compound of library B binding quite strongly to the column, bears in the 2-position an iodine atom with high lipophilic feature. Accordingly, it is clear that the nature and the position of substituents on the purine ring modulate the receptor binding affinity, the presence of rigid lipophilic groups in 2-position being the most favorable for the interaction of these compounds with the receptor
Functional Assay.
The strength of a FAC−MS method is the ability to rank multiple ligands simultaneously, offering the possibility to discriminate between low, medium, and high affinity candidates; however, it identifies compounds simply on the basis of their binding affinities, irrespective of their ability to activate the target.
In order to verify the reliability of the obtained results and to evaluate the functional activity of the studied compounds on GPR17, some derivatives were tested in a well established GPCR assay, the [35S]GTPyS binding, based on the ability of agonists to increase the binding of radioactive GTP to the activated GPCR. Antagonist activity has been assessed by the ability to counteract the increase of [35S]GTPyS binding induced by the agonist UDP-glucose.14,15
To validate the FAC—MS data, the higher binding ligand 4, some of the presumed medium-high binding ligands 6−8, the bisphosphate 12, and the UDP derivative 13 were selected for the functional study. The GPR17 antagonists cangrelor, MRS 2179, and the agonist UDP were also assayed in parallel as reference compounds.
The [35S]GTPyS binding results are reported in Table 4. The obtained potency values of the tested ligands versus wild-type GPR17 closely reflect the elution order of the analytes in the GPR17 wild-type column. A strong correlation was found by comparing these results, expressed as halfmaximal response concentrations (EC50) or half-maximal inhibition (IC5o) values for agonists and antagonists, respectively, with the data obtained with the GPR17 column by FAC−MS, thus confirming the validity of our approach in the ranking of new potential ligands for GPR17.
Table 4.
FAC−MS Data in Comparison with [35S]GTPyS Binding Results Obtained on GPR17-IAM-II and Mutated GPR17-IAMa
| GPR17-IAM-II |
mutated-GPR17-IAM |
|||||
|---|---|---|---|---|---|---|
| compd | tb (min) | EC 50 | IC50 c(nM) | tb (min) | EC50 | IC50c (nM) |
| UDP | 2.69 | 1.14 ± 0.2/µM | 2.15 | 3.0 ± 0.3 µM | ||
| 13 | 2.79 | 945 ± 48 nM | 2.51 | 1.87 ± 0.20 µM | ||
| 12 | 3.55 | 582 ± 57 | ||||
| MRS2179 | 3.62 | 508 ± 29 | 3.47 | 227 ± 22 | ||
| 5 | 3.69 | 112 ± 7 | ||||
| 7 | 4.5 | 11 ± 1 nM | ||||
| 8 | 4.77 | 1.7 ± 0.1 nM | ||||
| 6 | 5.67 | 1.4 ± 0.1 nM | ||||
| cangrelor | 10.6 | 0.7 ± 0.02 | 13.16 | 0.15 ±0.01 | ||
| 4 | 13.07 | 36 ± 3 pM | 16.07 | 25.4 ± 1.0 pM | ||
Columns: GPR17-IAM-II (19.5 million cells), mutated GPR17-IAM (19.5 million cells)
t is the breakthrough time of the ligand with the immobilized GPR17.
Versus UDP-glucose 10 µM.
The ligands 13, 6, 8, 7, and 4 were able to increase [35S]GTPyS binding, with potency values in the micromolar and subnanomolar range, showing an agonist profile (Figure 2a). In particular, the ATP analogue 4, bearing a lipophilic steric hindered substituent in the 2-position, was a very potent agonist of GPR17 with an EC50 of 36 pM, in agreement with the fact that this is the compound with the longer breakthrough time from the GPR17-column. Also, a lipophilic substituent in the Apposition of ATP favored, although to a less extent, the interaction with the GPR17 receptor. In fact, comparison of potency of 6 (EC50 =1.4 nM), bearing a N6-cyclopentyl group, and 7 (EC50 =11 nM), bearing a smaller methyl group, demonstrated clearly the contribution of the more lipophilic substituent. With regard to the purine ring, we can confirm that the presence of a nitrogen in the 3-position is not essential (see above), since its isosteric replacement by a CH induced even a 7-fold increase of activity as shown by comparing 7 (EC50 =11 nM) and 8 (EC50 = 1.7 nM).
Figure 2.

[35S]GTPyS experiments: (a) dose—response curves of some compounds of library A in 1321N1 cells expressing wild-type GPR17; (b) antagonistic effect of 12 on UDP-glucose stimulation of wild-type GPR17 in the [35S]GTPyS binding, where each point is the mean ± SEM of three independent experiments run in triplicate; (c) dose—response curves of UDP (3), 4, and 13 in 1321N1 cells expressing mutated GPR17; (d) antagonistic effect of cangrelor (1) and MRS 2179 (2) on UDP-glucose stimulation of mutated GPR17 in the [35S]GTPyS binding, where each point is the mean ± SEM of three independent experiments run in triplicate.
Compound 12 did not induce any significant increase in [35S]GTPyS binding; conversely, it was able to counteract [35S]GTPyS binding stimulation induced by the purinergic agonist UDP-glucose (used at 10 µM), with an antagonist profile (Figure 2b) and an affinity constant (IC50) in the nanomolar range, comparable to that reported for its analogue derivative MRS 2179.14
As expected, the UDP derivative 13 showed an activity comparable to that of UDP itself (EC50 = 0.945 versus 1.140 /µM, respectively).
Comparison between Wild Type and Mutated GPR17.
Binding of extracellular nucleotides to P2Y receptors is critically dependent on the basic arginine residue belonging to the conserved motif H-X-X-R/K in transmembrane domain (TM) 6 of P2Y and CysLTl and CysLT2 receptors. 34 Our previous computational studies suggest that this also holds true for Arg255 of GPR17.17 Here, to assess the actual role of this residue in receptor binding, starting from the previously published model of GPR17, we mutated this basic amino acid to isoleucine and generated an in silico mutant GPR17 receptor (R255I). Using steered molecular dynamics simulations (SMD), we then simulated a forced unbinding of the endogenous ligand UDP from both the wild type (WT) and the R255I receptor models of GPR17.
Figure 3 shows a global view of the SMD trajectories where the “extraction” of UDP from the WT and R255I receptor models is compared. We found that the energy required to unbind UDP from its pocket was higher for the WT than for the mutated R255I receptor and the exit of the ligand from its intracellular cavities occurred earlier in the R255I model with respect to the WT receptor. The different kinetics of UDP unbinding from the WT and the R255I mutated receptors are evident in our MPEG video where visual representations of the two different SMD trajectories for the WT and the mutated receptors (in purple and red, respectively) are reported as a function of time. The video is available in Supporting Information.
Figure 3.

Forced unbinding profile of UDP. To elucidate the effect of a single point mutation of the arginine residue (Arg255) of GPR17, potentially involved in nucleotide binding, simulations of the forced unbinding of UDP from the wild type (WT) and the mutant (R255I) receptor models are compared. Panel a shows the pulling energy developed to unbind UDP from WT (in magenta) and from R255I (in black). The detected significant difference in energy peak intensities suggests that the mutation actually affects the binding of UDP. The distances between the couples of atoms involved in the formation of hydrogen/electrostatic bonds occurring between the ligand and either WT or R255I receptors are reported in panels b and c. In the case of WT, for almost the whole duration of the simulation, residue Arg255 holds the ligand near the pocket, thus allowing UDP to maintain interactions with other residues, among which are Tyrl85, Tyr251, and Tyr262 (b). The same interactions are loosen up at the early stages of simulation in R255I receptor (c). In panel d, some representative frames of the unbinding pathway of UDP (red sticks) from GPR17 (blue cartoon) are reported.
To experimentally validate the above in silico simulations and to challenge the reliability of the FAC-MS analysis, we generated and expressed the mutated receptor in 132INI cells and prepared a column from these cells (mutated GPR17-IAM), in parallel with a column loaded with membranes from cells expressing the WT receptor (GPR17- IAM), containing the same number of cells (19.5 millions)/ column. An equimolar solution containing all the analytes of library A (1 µM each) and the three reference compounds (1 µM each) was then profiled by FAC-MS on both columns. As far as the comparison of the elution order on the two columns is concerned (Table 5), the same trend was observed with the exception of the structurally similar compounds 12 and MRS 2179, whose breakthrough times were reversed in mutated GPR17 column. Again, considering that the breakthrough curves of the two analytes are very close, this apparent change of elution order needs confirmation by using a column with a higher binding site density. Table 5 also reports the % variation of the breakthrough time determined on mutated GPR17-IAM with respect to the GPR17-IAM-II column. From these data, it is clear that the mutation is not silent. The ligands can be divided into two groups depending on the sign of the breakthrough time variation. It is speculated that analytes with a positive variation most likely interact with the receptor binding pocket through hydrophobic interactions, while for compounds with a negative variation electrostatic interactions are predominant. A variation % of less than ±10% has been considered as nonsignificant, since this change is linked to the experimental variability.
Table 5.
FAC-MS Results for Library a Screened on GPR17-IAM and Mutated GPR17-IAM Columnsa
| compd | GPR17-IAM-II | Mutated GPR17-IAM |
(tm/tw×100)-100b(%) |
|---|---|---|---|
| UDP | 2.69 | 2.15 | −20.1 |
| 13 | 2.79 | 2.51 | −10.0 |
| 14 | 2.97 | 2.90 | −2.4 |
| 9 | 3.03 | 2.96 | −2.3 |
| 12 | 3.55 | 3.54 | −0.3 |
| MRS 2179 | 3.62 | 3.47 | −4.1 |
| 5 | 3.69 | 4.08 | +10.6 |
| 11 | 4.26 | 4.68 | +10.1 |
| 7 | 4.50 | 5.27 | +17.1 |
| 8 | 4.77 | 5.79 | +21.4 |
| 6 | 5.67 | 7.08 | +24.9 |
| 10 | 6.45 | 8.42 | +30.5 |
| cangrelor | 10.60 | 13.16 | +24.1 |
| 4 | 13.07 | 16.07 | +23.0 |
The breakthrough times are the result of a single ranking experiment carried out on the freshly prepared column. Columns: GPR17-IAM-II and mutated GPR17-IAM (19.5 million cells).
The breakthrough time variation % was calculated on mutated GPR17-IAM (tw) with respect to GPR17-IAM-II column (tw).
Di- and triphosphate derivatives show an opposite behavior. The diphosphate compounds UDP and 13 show a lower affinity (negative variation %) on the mutated GPR 17- column: for UDP this is in line with the predicted “in silico” modeling results (Supporting Information video). Conversely, triphosphate derivatives (5–8) show an increased affinity (positive variation %). The exception is given by 10 and 11, diphosphate derivatives structurally characterized by the presence of a lipophilic chain on the adenine moiety.
We also performed in silico docking studies using a virtual library of the synthetic nucleotide derivative ligands tested in vitro to allow a theoretical interpretation of results and to get more information regarding the molecular interactions of the most interesting ligands with GPR17. Two of the most representative ligand candidates (MRS 2179 and 4, a bisphosphate and a triphosphate compound, respectively) were chosen, and their docking poses are shown in Figure 4. As evident from these docking results, while Arg255 is involved in GPR17 binding to MRS 2179, another residue (Arg87) seems to be primarily involved in binding of the receptor to the phosphate chain of 4 (see arrows in Figure 4). These different docking poses could account for the different effect of the Arg255 mutation on the behavior of bisphosphate and triphosphate ligands on the GPR17 column. Specifically, it can be hypothesized that, different from bisphosphate derivatives like MRS 2179, for triphosphate derivatives such as 4, mutation of Arg255 is not enough to reduce the compound affinity for the column, as the longer phosphate chain can interact with other arginine residues (e.g., Arg87) through electrostatic interactions.
Figure 4.
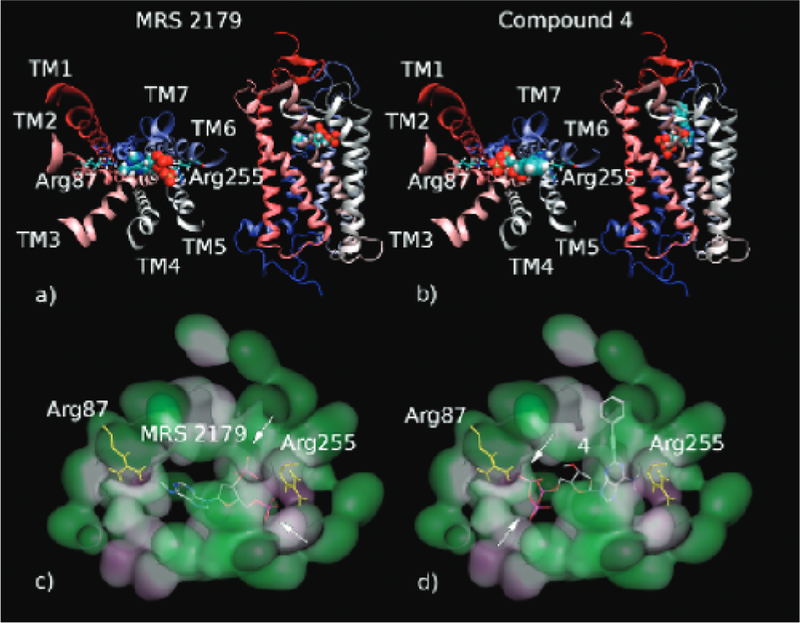
Binding pose hypothesis of nucleotide derivatives on GPR17. The picture shows the two different possible binding modalities hypothesized for bisphosphate and triphosphate compounds on GPR17. Panels a and b show a macroscopic view of a typical diphosphate compound (MRS 2179, panel a, spheres representation) and of a triphosphate one (4, panel b, spheres representation) docked to the receptor model (cartoon representation). Panels c and d show the molecular surface of the pocket identified for MRS 2179 and 4, respectively. Two possible target arginine residues of ligand phosphate moieties are highlighted (yellow, stick representation): Arg255 for MRS 2179 phosphates and Arg87 for 4 phosphates. Regions with hydrophobic, hydrophilic, and mild polar features are reported in green, purple, and gray, respectively. Phosphates of MRS 2179 and 4 involved in arginine binding are reported in magenta and highlighted with arrows. It is evident that different arginine residues (Arg255 and Arg87) participate in ligand binding in the cases of MRS 2179 and 4, respectively (see text for more detail).
Moreover, it could be hypothesized that triphosphate 4, bearing a lipophilic phenylethynyl chain, can be better accommodated into the more lipophilic pocket of mutated GPR17, thus leading to an increased breakthrough time. This effect is less evident for diphosphate derivatives where the loss of electrostatic interaction, due to the mutation, is preponderant with respect to the increased lipophilicity.
Functional Assay on Mutated Receptor.
To further confirm the data reported above, some analytes from library A (i.e., 4 and 13, demonstrating the highest and the lowest affinity toward wild-type GPR17, respectively) together with the three standard compounds (UDP, MRS 2179, and cangrelor) were also tested in parallel on wild-type and mutated GPR 17 in the [35S]GTPγS functional binding assay. All the tested compounds maintained the same pharmacological profile (agonist/antagonist) at both the wild-type (Figure 2a and Figure 2b) and the mutated GPR17 receptor (Figure 2c and Figure 2d).
No significant changes in receptor affinity (i.e., EC50) were demonstrated for all tested agonists, but a trend toward a reduced affinity was observed for UDP and compound 13, whereas a trend toward an increased affinity was evident for compound 4 (Table 5). In contrast, the antagonists cangrelor and MRS 2179 showed a significant increase in affinity toward the mutated vs the wild type receptor (P < 0.05; see Table 4).
Data from [35S]GTPγS functional experiments paralleled those obtained in affinity chromatography studies. In fact, a decrease in the breakthrough time was observed for those compounds showing a trend toward a reduced affinity for the mutated receptor (e.g., UDP and 13; Table 5), whereas a higher breakthrough time was observed for cangrelor and compound 4, showing an increased affinity toward the mutated receptor (Table 5). Among all the tested compounds, despite a significant decrease in the IC50 value toward the mutated receptor, no significant shift in the retention time was observed for MRS 2179 (Table 5).
Conclusions
In this work, we have developed a new strategy based on the FAC-MS method coupled to in silico docking studies for deeper structural information on the GPR17 receptor and for the screening of new and potent agonist and antagonist ligands acting on this recently deorphanized GPCR that plays a crucial role in the development of ischemic brain damage and in the later remodeling/repair processes.
The use of FAC-MS as a reliable method for screening compound libraries toward important pharmaceutical targets such as GPCRs is still a challenge; however, in this study, we have demonstrated the applicability of this technique to the screening of drug candidates toward GPR 17.
Unlike other affinity-based screening strategies, which are not specific enough for chemical identification and/or suffer from low throughput, FAC-MS methodology is highly specific and can be used for the discovery of classes of small molecule ligands (agonists and antagonists) in a single fast screening campaign. The strength of the FAC-MS approach has been validated using a reference functional assay ([35S]GTPγS assay), and the chromatographic data allowed the drawing of preliminary structure-activity relationships.
However, the design and synthesis of selective GPR17 agonist/ antagonist ligands would greatly benefit from the knowledge of receptor 3D structure and from the definition of its ligand binding mode. In the case of GPCRs, modeling crucially depends on biological validation that is, in turn, typically supported by site-directed mutagenesis of the binding site. Moreover, a further refinement of the 3D model also came from in silico mutagenesis of the predicted key amino add residues in or near the target binding site.
To the best of our knowledge, this is the first time that FAC—MS in combination with molecular modeling simulations is applied to the study of membrane proteins. The integration between analytical, biochemical/pharmacological, and computational assays will drive a progressive reduction of the number of potential candidate ligands by selecting only the most promising lead compounds. Moreover, the data obtained will address the design and the synthesis of new refined compounds acting as new potent and selective ligands
Experimental Section
Synthesis.
1H NMR spectra were obtained with Varian Mercury 400 MHz spectrometer; <5 is in ppm, J in Hz. All exchangeable protons were confirmed by addition of D20. 31P NMR spectra were recorded at room temperature using a Varian Mercury 400 MHz spectrometer. Elemental analyses were determined on a Fisons model EA 1108 analyzer and are within ±0.4% of theoretical values. Purity of the compounds was used g 95 % according to elemental analysis data TLCs were carried out on precoated TLC plates with silica gel 60 F-254 (Merck). For ion exchange chromatography, Sephadex DEAE A-25 (Fluka) was used. Mass analyses were performed on a quadrupole ESI mass apparatus (HP 1100 MSD).
General Procedure for the Synthesis of the Nucleoside 5’-Mono-phosphates 24,25, and 27.
To 0.56 mmol of the nucleosides 22,28 23 29, and 26 30, in turn dissolved in 3.0 mL of trimethylphosphate, an amount of 4 equiv of POCl3 (209 µL, 2.24 mmol) was added. The solution was stirred at room temperature for 3 h. H20 (3 mL) was added, and the solution was neutralized by adding triethylamine dropwise. The mixture was purified by ion exchange chromatography on Sephadex DEAE A-25 (HC03_ form) equilibrated with H20 and eluted with a linear gradient of H20/NH4HC03 0.5 M, after washing the resin with H20.
2-Choro-N6 methyladenosine monophosphate (24).
Reaction of 22 furnished 24 as white solid; yield 68%. 1H NMR (D20) δ 2.77 (s, 3H, CH3), 3.89 (m, 2H, CH −5’), 4.16 (m, 1H, H-4’), 4.28 (d, 1H, /= 4.4 Hz, H-3’), 4.50 (d, 1H, /= 5.2 Hz, H-2’), 5.76 (d, 1H, / = 5.6 Hz, H-l’), 8.16 (s, 1H, H-8). 31P NMR (D20) δ2.59 (s). Anal. Calcd for C11H18C1N607P: C, 32.01; H, 4.40; N, 20.36. Found: C, 31.79; H, 4.37; N, 20.65. ESI-MS m/z: 394.0 [M - H]”, 789.0 [2M-H] −.
2.choro-3-deaza-N6 –methyladenosine monophosphate (25).
Reaction of 23 furnished 25 as white solid; yield 88%. *H NMR (D20) 6 2.85 (s, 3H, CH3), 3.84 (m, 2H, CH2-5’), 4.19 (m, 1H, H-4’), 4.25 (m, 1H, H-3’), 4.53 (m, 1H, H-2’), 5.76 (d, 1H, J = 6.4 Hz, H-l’), 6.82 (s, 1H, H-3), 8.29 (s, 1H, H-8).31 p NMR (D20) <5 0.87 (s). Anal. Calcd for C12H19C1N507P: C, 35.01; H, 4.65; N, 17.01. Found: C, 34.86; H, 4.43; N, 17.39. ESI- MS m/z: 393.0 [M - H]”, 786.9 [2M - H]-.
5-Hexynyluridine Monophosphate (27).
Reaction of 26 furnished 27 as white solid; 45% yield. 1H NMR (D20) <3 0.75 (t, 3H, J = 7.2 Hz, CH3), 1.27 (m, 2H, CH2CH3), 1.43 (m, 2H, CH2CH2CH2), 2.27 (t, 2H, J = 6.9 Hz, CH2C=C), 3.94 (m, 2H, CH2-5’), 4.15 (m, 1H, H-4’), 4.20 (m, 2H H-2’ e H-3’), 5.70 (d, 1H, J = 5.1 Hz, H-l’), 7.86 (s, 1H, H-6). 31P NMR (D2O) δ 1.75 (s). Anal. Calcd for C15H24N309P: C, 42.76; H, 5.74; N, 9.97. Found: C, 42.62; H, 5.65; N, 10.21. ESI-MS m/z 201.1 [M- 2H]2-, 403.0 [M - H]-, 807.0 [2M - H]-
General Procedure for the Synthesis of the Nucleoside 5’- Triphosphates (7 and 8) and Nucleoside 5’-Diphosphates (14).
To 0.15 mmol of the nucleoside 5’-monophosphates 24,25, and. 27, in turn dissolved in 1 mL of dry DMF, an amount of 36 µL of tri-n-butylamine (28 mg, 0.15 mmol) was added. The solution was stirred for 20 min at room temperature and then evaporated to dryness under anhydrous conditions. After resuspension in 1.4 mL of dry DMF, N,N’-carbonyldiimidazole (122 mg, 0.75 mmol) was added and the mixture was stirred for 3 h at room temperature. Methanol (49 µL, 1.2 mmol) was added and the mixture stirred for 30 min at room temperature. Then a total of 6 mL (3 mmol) of a 0.5 M solution of bis(tri-n-butylammonium) pyrophosphate or tri-n-butylammonium phosphate in DMF was added (for the synthesis of the triphosphate or the diphosphate derivatives, respectively). The mixture was stirred for 14 h at room temperature. The solvent was removed in vacuo. The mixture, dissolved in H20, was purified by means of ion ionexchange chromatography
2.-choro-N6 - methyladenosine Triphospate (7).
Reaction of 24 furnished 7 as white solid; yield 37%. H NMR (D20) δ 2.88 (s, 3H, CH3), 4.08 (m, 2H, CH2-5’), 4.21 (m, 1H, H-4’), 4.40 (m, 1H, H-3’), 4.58 (m, 1H, H-2’), 5.85 (d, 1H,J= 6.6 Hz, H-l’), 8.25 (s, 1H, H-8). 31P NMR (D20) δ −21.50 (m), −9.23 (m). Anal. Calcd for C11H29C1N9013P3: C, 21.18; H, 4.69; N, 20.21. Found: C, 21.03; H, 4.46; N, 20.46. ESI-MS m/z: 276.5 [M - 2H]2”,554.0 [M-H]-.
2.-choro-3-deaza-N6-- methyladenosine Triphospate (8).
Reaction of 25 furnished 8 as white solid; yield 69%. 1H NMR (D20) δ 2.86 (s, 3H, CH3), 4.09 (m, 2H, CH2–5’), 4.22 (m, 1H, H- 4’), 4.41 (m, 1H, H-3’), 4.51 (m, 1H, H-2’), 5.77 (d, 1H, J = 6.6 Hz, H-l’), 6.87 (s, 1H, H-3), 8.21 (s, 1H, H-8). 31PNMR (D20) δ−21.58 (t), −10.34 (d), −7.47 (d). Anal. Calcd for C12H30- C1N8013P3: C, 23.14; H, 4.86; N, 17.99. Found: C, 22.82; H, 4.51; N, 18.36. ESI-MS m/z: 276.0 [M −2H]−2, 552.8 [M - H]-.
5-Hexynyluridine Diphosphate (14).
Reaction of 27 furnished 14 as white solid; 60% yield. 1H NMR (D20) <5 0.74 (t, 3H, J = 7.3 Hz, CH3), 1.26 (m, 2H, CH2CH3), 2.25 (t,2H,J =6.9 Hz,CH2Ct C),4.08 (m 3H, H-4’ and CH2-5’), 4.22 (m, 2H, H-2’ and H-3’), 5.77 (d, 1H, J = 4.5 Hz, H-l’), 7.85 (s, 1H, H-6). 31P NMR (D20) δ −10.49 (d), −8.21 (d). Anal. Calcd for Cl5H3lN5012P2: C, 33.65; H, 5.84; N, 13.08. Found: C, 33.37; H, 5.65; N, 13.43. ESI-MS m/z 483.0 [M-H]-
preparation of Bis (tri-n-buthylammonium ) pyrophosphate.
Sodium pyrophosphate decahydrate (3.34 g, 7.5 mmol) was dissolved in 75 mL of water and put under stirring. Dowex 50×8, 20—50 mesh, H+ form (21 g), was added, and the suspension was stirred 20 min. A solution of ethanol (30 mL) and tri-n- buthylamine (3.57 mL, 15 mmol) was putin an ice bath, and the pyrophosphate solution was filtered, leaving the filtrate to go directly into the solution. The resin on the filter was washed with water until the filtrate showed a pH of 7. The solvent was removed under vacuum (temperature below 35 °C) and co-evaporated three times with ethanol and three times with dry DMF. The residue was put in 15 mL of DMF in order to obtain a bis(tri-n-buthylammonium) pyrophosphate solution, 0.5 M in DMF. The solution was stored over molecular sieves at 4 °C.
preparation of Tri-n-buthylammonium monophosphate
Sodium phosphate monobasic monohydrate (1.38 g, lOmmol) was dissolved in 70 mL of water and put under stirring. Dowex 50×8, 20—50 mesh, H+ form (21 g), was added, and the suspension was stirred 20 min. A solution of ethanol (50 mL) and tri-w-buthyl-amine (2.40 mL, 10 mmol) was put in an ice bath and the orthophosphate solution was filtered, leaving the filtrate to go directly into the solution. The resin on the filter was washed with water until the filtrate showed a pH of 7. The solution was stirred for 30 min in an ice bath. The solvent was removed under vacuum (temperature below 35 °C) and co-evaporated three times with ethanol and three times with dry DMF. The residue was put in 20 mL of dry DMF in order to obtain a tri-n- buthylammonium monophosphate solution, 0.5 M in DMF. The solution was stored over molecular sieves at 4 °C.
Chromatographic Procedures for the Monitoring and the Purification of the Phosphorylation Reactions.
The reactions were monitored by TLC, using precoated TLC plates with silica gel 60 F-254 (Merck) and isopropanol—H20—NH40H (30%) (5.5:1:3.5) as mobile phase. The nucleotides were purified by means of ion-exchange chromatography on a Sephadex DEAE A-25 (Fluka) column (HC03- form) equilibrated with H20 and eluted with a linear gradient of H2O/0.5 M NH4HCO3.
Preparation of the DEAE-Sephadex Resin. Swelling of the Resin.
An amount of 10 g of the resin was put into a beaker with 400—500 ml of deionized water (R > 10 M Ώ ) for 24 h at 4 °C After that the resin was swelled with 500 mL of NaHC03,1 M. The resin was kept for 3 days at 4 °C, changing the supernatant buffer solution every day
Preparation of Chromatographic Columns. Cell Culture and Transfection.
Human astrocytoma cells (1321N1) were cultured as described previously.13 Cells were seeded on 100 mm Petri dishes (106 cells/dish) and transfected with pcDNA3.1 vector containing the construct encoding for either the human wild type or the R255I mutated GPR17 receptor (pcDNA3.1- wtGPR17h or pcDNA3.1-muGPR17h) with FuGENE 6 (Roche Diagnostics, Monza, Italy), according to the manufacturer’s instructions. Control cultures received in parallel the corresponding pcDNA3.1 empty vector. In selected experiments, transfection efficiency was checked under a fluorescence microscope equipped with a fluorescein filter after cotransfection of cultures with pcDNA3.1-GPR17h and a plasmid encoding for the enhanced green fluorescent protein (eGFP).
Site Directed Mutagenesis of hGPR17.
The previously constructed pcDNA3.1-hGPR17 plasmid14 was mutated using the QuikChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA). The arginine at position 255, in the DNA binding domain of GPR17, was mutated to a isoleucine, by amplification of a fragment from cloned pcDNA3.1-hGPR17 plasmid using polymerase chain reactions (PCR) primers (5’-gccctac- cacgtcaacatctccgtctacgtgctg-3’ and 5’-cagcacgtagacggagatgtt- gacgtggtagggc-3’) according to the kit manufacture’s directions. The mutated plasmids were then chemically transformed into the E. coli a-DH5 strain. The cells were grown overnight in 5 mL of Luria—Bertani (LB) medium containing ampicillin, 50 µg/mL. The overnight culture was used to prepare a miniprep by the method described in the Qiagen manual. The presence of the correct mutation and the absence of PCR-derived alterations to the coding sequence were confirmed by complete sequencing of the receptor construct.
Membrane Preparation.
For membrane preparation, the method developed for P2Y1 receptors was followed.33 Two full days (48 h) after transfection, 2 × 107 132INI cells transiently transfected with either pcDNA3.1-wtGPR17h or pcDNA3.1- muGPR17h were homogenized for 3 × 30 s at a setting of 11 on a model PT-2100 homogenizer (Kinematica, Luzern, Switzerland) in 10 mL of Hepes buffer (20 mM, pH 8.0) containing 500 mM NaCl, 5 mM 2-mercaptoethanol, 100 µ M benzamidine, 10 µ/ mL aprotinin, 10 /ig/mL leupeptin, 50 µ/mL tosylamido- 2-phenylethyl chloromethyl ketone (TPCK), 100 /µg/mL phenyl- methylsulfonyl fluoride (PMSF), and 100 µM adenosine triphos phate (ATP). The homogenate was centrifuged at 700g for 5 min, the pellet was discarded, and the supernatant was centrifuged at l00000g for 30 min at 4 °C. The resulting pellet was suspended in 10 mL of Hepes buffer (20 mM, pH 8.0) containing 2% (w/v) n-octyl-β-D-glucopyranoside, 500 mM NaCl, 5 mM 2-mercaptoethanol, 100 µM benzamidine, 10 µg/mL aprotinin, 10µg/mL leupeptin, 50 µg/mL TPCK, 100 µg/mL PMSF, 100 µM ATP, and 10% glycerol. The resulting mixture was rotated at 150 rpm using an Orbit shaker for 18 h at 4 °C and was centrifuged at l00000g for 25 min. The supernatant was collected and mixed with 70 mg of IAM particles and gently rotated at room temperature for 1 h at 150 rpm. The suspended particles were then dialyzed for 2 days against Hepes buffer (20 mM, pH 8.0) containing 500 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), and 0.01% sodium azide, changing the buffer every 24 h. The suspension was then centrifuged at 700 rpm for 3 min (4 °C), and the supernatant was discarded. The pellet was washed with 10 mL of chromatographic running buffer (5 mM ammonium acetate, pH 7.4) and the supernatant removed by centrifugation at 700 rpm for 3 min (4 °C).
The pellet was resuspended in 1 mL of chromatographic buffer and packed into an Omnifit glass column to yield a 3 mm x 6.6 mm i.d. chromatographic bed.
FAC—MS Screening.
The immobilization of wild-type and mutated GPR17 to IAM particles was carried out as previously described13,33
The frontal affinity chromatography experiments were carried out on a chromatographic system consisting of a thermos stated column oven Surveyor autosampler controlled at 20 °C (Thermo Finnigan, San Jose, CA), a quaternary gradient Surveyor MS pump (Thermo Finnigan, San Jose, CA) equipped with a diode array detector (DAD), and a LTQ linear ion trap mass spectrometer (MS) with electrospray ionization (ESI) ion source controlled by Xcalibur software 1.4 (Thermo Finnigan, San Jose, CA).
The mobile phase was composed of 90% ammonium acetate (5 mM, pH 7.4) containing 10% methanol, and the experiments were carried out at a flow rate of 500 µL/min at ambient temperature. A postcolumn mobile-phase splitting valve was also inserted so that only 200 µL/min were diverted to MS. Mass spectra were generated in negative ion mode under constant instrumental conditions: source voltage 4.0 kV, capillary voltage 46 V, sheath gas flow 38 (arbitrary units), auxiliary gas flow 8 (arbitrary units), sweep gas flow 1 (arbitrary units), capillary temperature 270 °C, tube lens voltage -85.06 V.
For the ranking experiment, MS conditions were optimized so that all the compounds were detected simultaneously as negative [M — H]- ions.
Stock solutions of each analyte were prepared in water (the concentrations were from 0.2 to 0.6 mM depending on the available amount of each compound). The stock solutions were stored at —20 °C before the preparation of the screening solution. Each screening solution was prepared to contain the library members each at 1 µM in 5 mM ammonium acetate, pH 7.4, containing 10% methanol. The mixture also contained cangrelor, MRS 2179, and UDP (1 µM) as affinity reference compounds.
Each extracted ion breakthrough curve was analyzed with a polynomial equation of degree 3 (y = ax1 + bx2 + cx + d) to fit the chromatographic data, and the inflection point, corresponding to the breakthrough time, was determined by the second derivative.
GPR17 Functional Assay.
In order to investigate the pharmacological profile and the potency of the selected compounds on purinergic GPR17 binding site, GTPyS binding assay has been performed on 1321N1 cells transiently transfected with either the wild type or the mutated human protein (see above).
The pharmacological profile of the new ligands (agonists or antagonists) toward the GPR17 receptor was evaluated by assessing the effect of different ligand concentrations to modulate GPR17 receptor-G protein coupling. In particular, the effect of the compounds alone (acting as agonist) and in the presence of the purinergic agonist UDP-glucose (behaving as an antagonist) in modulating GTPyS binding was evaluated.
In parallel, in order to investigate if compound-mediated effects were really ascribed to the interaction with GPR17 binding site, GTPyS binding was also performed in 1321N1 cells transfected with the empty vector (data not shown). Because GPCR stimulation by agonists results in increased binding of GTP to G-proteins (which can, in turn, be quantified by measuring [35S]GTPyS binding to purified membranes), 35 activation of the hGPR17 in transiently transfected 1321N1 cells was determined by testing the increase of [35S]GTPy S binding by exogenously added compounds.35–37 1321N1 cells (control and transfected cells) were homogenized in 5 mM Tris-HCl, 2 mM EDTA, pH 7.4, and centrifuged at 48000g for 15 min at 4 °C. The resulting pellets were washed in 50 mM Tris-HCl, 10 mM MgCl2, pH 7.4, and stored at 80 °C until use.
Optimal [35S]GTPγS binding conditions were determined in preliminary experiments.14 Briefly, aliquots of cell membranes (20 µg) were incubated with GDP (3 µM), [35S]GTPγS (0.15 nM, 1,250 Ci/mmol, Perkin-Elmer) and different compound concentrations (10 nM to 10 µM). After incubation at room temperature in a shaking water bath for 30 min, cells were harvested by rapid filtration and assayed for 35S radioactivity. Nonspecific binding of [35S]GTPγS was measured with 10 µM GTPγS.
Statistical Analysis.
For the analysis and graphic presentation of [35S]GTPγS binding data, a nonlinear multipurpose curve fitting computer program Graph-Pad Prism (GraphPad) was used. All data are presented as the mean ± SEM of three different experiments. Data were tested for statistical significance with the paired Student’s t test or by analysis of variance (one-way ANOVA), as appropriate. When significant differences were observed, the Newman—Keuls multiple comparison test (one-way ANOVA) was done. A value of P < 0.05 was considered significant.
Molecular Modeling. Steered Molecular Dynamics Simulation.
A previously published rhodopsin-based homology model of the human GPR17 receptor bound to its endogenous ligand UDP and refined by means of molecular dynamics (MD) simulations17 was used as a starting point for docking and SMD studies (see also the Computational Details section). In parallel, the Arg255 residue of the same receptor model was mutated to lie in order to investigate its role in ligand recognition, according to in vitro studies. The effect of this mutation on ligand binding was then studied by simulating a forced unbinding of the docked UDP from both the WT and the R255I receptor models using SMD simulations. The ligand unbinding was induced by applying an external force to the center of mass (COM) of the ligand that simulates a retracting cantilever directed along a selected pathway. Because of the action—reaction principle, the spring acts as a sensor of all interactions occurring between the protein and its ligand. The elastic force is proportional to the spring elongation relative to its equilibrium position, and it is given by the expression , where is the displacement of the restrained atom with respect to its original position, t is time elapsed from the beginning of the simulation; nm/ps corresponds to the velocity of the retracting cantilever, and k = 2000 kJ/(mol nm2) is its force constant. The combination for and k for our simulations was chosen among different values tested in a previous series of simulations (nm/ps; k = 2000—5000 kJ/(mol nm2)) so that the complete unbinding of the ligands occurs within 1 ns for each SMD simulation. For the interpretation of the results the registered pulling force was converted into energy values by applying the following expression: .
Docking studies of some representative ligands of the available chemical library of synthetic nucleotide derivatives were also performed on both the WT and the R255I receptor models in order to provide a theoretical interpretation of the results obtained by the in vitro screening. These docking studies were performed using the semiflexible docking module provided by DELOS package (http://www.delos-bio.it).
Computational Details.
All the simulations were run on a Linux cluster Blade with Xeon processors. All the simulations and concerning analysis were carried out using the Gromacs 3.3 package,17 using the Gromacs force field, modified by all the parameters necessary for the description of each component (protein, lipids, solvent, and ligands) and their reciprocal interactions, based on manufacturer’s instructions. Periodic boundary conditions were applied in all three x, y, and z dimensions. The isothermal isobaric NPT ensemble (constant number of particles, pressure, and temperature) was applied. Solvent (water molecules and chlorine ions) and nonsolvent (lipids, protein, and ligands) components of the system were separately coupled to a temperature bath at 310 K, with a coupling constant rt of 1 ps. The pressure coupling was set as independent in the x, y, and z directions (semiisotropic coupling), with a constant pressure of 1 bar and a coupling constant rp of 1 ps. A 2 fs time step was used for the integration of the equations of motions, and all bond distances involving hydrogen atoms were constrained using LINCS. Configurations were saved for every 1 ps for analysis.
Supplementary Material
Acknowledgment.
Cangrelor was a kind gift from The Medicine Company (Parsippany, NJ). This work was supported by a grant from the Italian Ministry for University and Research (Project FIRB RBN503YA3L).
a Abbreviations:
- 1P
monophosphate
- 2P
diphosphate
- 3P
triphosphate
- 3D
three-dimensional
- ATP
adenosine triphosphate
- CDI N
N’- carbonyldiimidazole
- COM
center of mass
- CysLT
cysteinyl-leukotriene
- DAD
diode array detector
- DEAE
diethylaminoethyl
- DMF N
N- dimethylformamide
- ESI
electrospray ionization
- FAC
frontal affinity chromatography
- GPCR
G-protein-coupled receptor
- GTPγS
guanosine 5’ - 0-[γ-thio] triphosphate)
- HEPES
4-(2-hydroxyethyl)piperazine-l-etha-nesulfonic sodium salt
- IAM
immobilization on artificial membranes
- LB
Luria−Bertani
- LTQ
linear trap quadrupole
- MS
mass spectrometry
- PCR
polymerase chain reactions
- PMSF
phenylmethylsulfonyl fluoride
- SEM
standard error of the mean
- SMD
steered molecular dynamics simulations
- TPCK
tosylamido-2-phenylethyl chloromethyl ketone
- UDP
uridine diphosphate
- WT
wild type
Footnotes
Supporting Information Available:
MPEG video showing the different kinetics of UDP unbinding from the WT and the R255I mutated receptors, where a visual representation of the two different SMD trajectories for the WT and the mutated receptors (in purple and red, respectively) are reported as a function of time. This material is available free of charge via the Internet at http://pubs.acs.org.
Reference
- (1).Schriemer DC; Bundle DR; Li L; Hindsgaul O Micro-scale frontal affinity chromatography with mass spectrometric detection: a new method for the screening of compound libraries. Angew. Chem 1998,110,3625.-; Angew Chem.,Int.Ed.1998,37,3383–3387. [DOI] [PubMed] [Google Scholar]
- (2).(Ng E; Schriemer D Emerging challenges in ligand discovery: new opportunities for chromatographic assay. Expert Rev. Proteomics 2005,2,891.-. [DOI] [PubMed] [Google Scholar]
- (3).Called E; Temporini C; Caccialanza G; Massolini G Target- based drug discovery: the emerging success of frontal affinity chromatography coupled to mass spectrometry. ChemMedChem 2009,4,905.-. [DOI] [PubMed] [Google Scholar]
- (4).Slon-Usakiewicz JJ; Dai JR; Ng W; Foster JE; Deretey E; Toledo-Sherman L; Redden PR; Pasternak A; Reid N Kinase screening. Applications of frontal affinity chromatography coupled to mass spectrometry in drug discovery. Anal.Chem. 2005,77,1268–1274. [DOI] [PubMed] [Google Scholar]
- (5).Ng ES; Chan NW; Lewis DF; Hindsgaul O; Schriemer DC Frontal affinity chromatography−mass spectrometry. Nat.Protoc 2007,2,1907.-. [DOI] [PubMed] [Google Scholar]
- (6).Slon-Usakiewicz JJ; Ng W; Dai J-R; Pasternak A; Redden PR Frontal affinity chromatography with MS detection(FAC−MS)in drug discovery. Drug Discovery Today 2005,10,409–416. [DOI] [PubMed] [Google Scholar]
- (7).Toledo-Sherman L; Deretey E; Slon-Usakiewicz JJ; Ng W; Dai JR; Foster JE; Redden PR; Uger MD; Liao LC; Pasternak A; Reid NL Frontal affinity chromatography with MS detection of EphB2 tyrosine kinase receptor. 2. Identification of small-molecule inhibitors via coupling with virtual screening. J Med. Chem. 2005,48,3221–3230. [DOI] [PubMed] [Google Scholar]
- (8).Ng W; Dai JR; Slon-Usakiewicz JJ; Redden PR; Pasternak A; Reid N Automated multiple ligand screening by frontal affinity chromatography−mass spectromatry (FAC−MS). J.Biomol. Screening 2007,12,167–174. [DOI] [PubMed] [Google Scholar]
- (9).Jaakola VP; Griffith MT; Hanson MA; Cherezov V; Chien EY; Lane JR; IJzerman AP; Stevens RC The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008,322,1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wame T; Serrano-Vega MJ; Baker JG; Moukhametzianov R; Edwards PC; Henderson R; Leslie AG; Tate CG; Schertler GF Structure of a betai-adrenergic G-protein-coupled receptor. Nature 2008,454,486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Pidgeon C; Marcus C; Alvarez F In Applications of Enzymes Biotechnology ; Baldwin TO,Kelly JW,Eds.;Plenum Press: New York,1992;pp 201–237. [Google Scholar]
- (12).Moaddel R; Wainer IW Development of immobilized membrane-based affinity columns for use in the online characterization of membrane bound proteins and for targeted affinity isolations. Anal. Chim. Acta 2006,564,97–105. [DOI] [PubMed] [Google Scholar]
- (13).Temporini C; Ceruti S; Calleri E; Ferrario S; Moaddel R; Abbracchio MP; Massolini G Development of an immobilized GPR17 receptor stationary phase for binding determination using frontal affinity chromatography coupled to mass spectrometry. Anal. Biochem. 2009,384,123–129. [DOI] [PubMed] [Google Scholar]
- (14).Ciana P; Fumagalli M; Trincavelli ML; Verderio C; Rosa P; Lecca D; Ferrario S; Parravicini C; Capra V; Gelosa P; Guerrini U; Belcredito S; Cimino M; Sironi L; Tremoh E; Rovati GE; Martini C; Abbracchio MP The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leuko- trienes receptor. EMBO J. 2006,25,4615–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lecca D; Trincavelli ML; Gelosa P; Sironi L; Ciana P; Fumagalli M; Villa G; Verderio C; Grumelli C; Guerrini U; Tremoli E; Rosa P; Cuboni S; Martini C; Buffo A; Cimino M; Abbracchio MP The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair PLoS One 2008,3,e3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mellor EA; Maekawa A; Austen KF; Boyce JA Cysteinyl leukotriene receptor 1 is also a pyrimidinergic receptor and is expressed by human mastcells. Proc. Natl. Acad. Sci U.S.A. 2001,98,7964–7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Parravicini C; Ranghino G; Abbracchio MP; Fantucd P GPR17: molecular modeling and dynamics studies of the 3-D structure and purinergic ligand binding features in comparison with P2Y receptors. BMC Bioinf 2008,9,263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cristalli G; Podda GM; Costanzi S; Lambertucci C; Lecchi A; Vittori S; Volpini R; Zighetti ML; Cattaneo M Effects of 5’-phosphate derivatives of 2-hexynyl adenosine and 2-phenyl- ethynyl adenosine on responses of human platelets mediated by P2Y receptors. J. Med. Chem. 2005,48,2763–2766. [DOI] [PubMed] [Google Scholar]
- (19).Fischer B; Boyer JL; Hoyle CH; Ziganshin AU; Brizzolara AL; Knight GE; Zimmet J; Burnstock G; Harden TK; Jacobson KA Identification of potent, selective P2Y-purinocep- tor agonists: structure−activity relationships for 2-thioether derivatives of adenosine 5 triphosphate. J Med. Chem 1993,36,3937–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Burnstock G; Fischer B; Hoyle CHV; Maillard M; Ziganshin AU; Brizzolara AL; von Isakovics A; Boyer JL; Harden K; Jacobson KA Structure activity relationships for derivatives of adenosine-5’-triphosphate as agonists at P2 purinoceptors: heterogeneity within P2X and P2Y subtypes. Drug Dev Res 1994,31,206–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tarun MK; Timothy JM Allele-specific activators and inhibitors for kinesin. Proc. Natl. Acad. Sci. U.S.A. 1999,96,9106–9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gough G; Maguire MH; Penglis F Analogues of adenosine 5’-diphosphate−new platelet aggregators. Influence of purine ring and phosphate chain substitutions on the platelet-aggregating potency of adenosine 5’-diphosphate. Mol. Pharmacol. 1972, 8, 170–177. [PubMed] [Google Scholar]
- (23).Camaioni E; Boyer JL; Mohanram A; Harden TK; Jacobson KA Deoxyadenosine bisphosphate derivatives as potent antagonists at P2Y1 receptors. J. Med. Chem. 1998,41,183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nandanan E; Camaioni E; Jang SY; Kim YC; Cristalli G; Herdewijn P; Secrist JA 3rd; Tiwari KN; Mohanram A; Harden TK; Boyer JL; Jacobson KA Structure−activity relationships of bisphosphate nucleotide derivatives as P2Y i receptor antagonists and partial agonists. J Med. Chem. 1999,42,1625–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Michelson AM; Dondon J; Grunberg-Manago M Action of polynucleotide phosphorylase on 5-halogenouridine 5’-pyrophosphates. Biochim.Biophys. Acta 1962,55,529–540. [DOI] [PubMed] [Google Scholar]
- (26).Besada P; Shin DH; Costanzi S; Ko H; Mathe C; Gagner- on J; Gosselin G; Maddileti S; Harden TK; Jacobson KA Structure−activity relationships of uridine 5’-diphosphate analogues at the human P2Y6 receptor. J.Med.Chem. 2006,49,5532–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Volpini R; Mishra RC; Kachare DD; Dal Ben D; Lambertucci C; Antonini I; Vittori S; Marucci G; Sokolova E; Nistri A; Cristalli G Adenine based acyclic-nucleotides as novel P2X3 receptor ligands. J.Med.Chem 2009,52,4596–4603. [DOI] [PubMed] [Google Scholar]
- (28).Venkata Palle EE; Wu Y; Dewan Zeng TM; Zablocki J 2-Pyrazolyl-JV”-Substituted adenosine derivatives as high affinity and selective adenosine A3 receptor agonists. J.Med.Chem. 2004,47,4766–4773. [DOI] [PubMed] [Google Scholar]
- (29).May JA Jr.; Townsend LB General synthesis of 4-substituted l-(yS-D-ribofuranosyl)imidazo[4,5-c]pyridines. J.Chem.Soc ,Per kin Trans. 1 1975,2 125–129. [PubMed] [Google Scholar]
- (30).Ghilagaber S; Hunter WN; Marquez R Efficient coupling of low boiling point alkynes and 5-iodo-nucleosides. Tetrahedron Lett 2006,48,483–486. [Google Scholar]
- (31).Yoshikawa M; Kato T; Takenishi T A novel method for phosphorylation of nucleosides to 5’-nucleotidase. Tetrahedraon Lett. 1967,50,5065.-. [DOI] [PubMed] [Google Scholar]
- (32).Hoard DE; Ott DGJ Conversion of mono- and oligodeoxy-ribonucleotides to 5’-triphosphates.. J Am.Chem.Soc. 1965,87,1785–1788. [DOI] [PubMed] [Google Scholar]
- (33).Moaddel R; Calleri E; Massolini G; Frazier CR; Wainer IW The synthesis and initial characterization of an immobilized purinergic receptor (P2Y1) liquid chromatography stationary phase for online screening. Anal.Biochem. 2007,364,216–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Abbracchio MP; Burnstock G; Boeynaems JM; Barnard EA; Boyer JL; Kennedy C; Knight GE; Fumagalli M; Gachet C; Jacobson KA; Weisman GA International Union of Pharmacology LVIII: Update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy Pharamacol Rev. 2006,58,281–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Marteau F; Le Poul E; Communi D; Labouret C; Savi P; Boeynaems JM; Gonzalez NS Pharmacological characterization of the human P2Y13 receptor. Mol.Pharmacol. 2003,64(1),104–112. [DOI] [PubMed] [Google Scholar]
- (36).Kotani M; Mollereau C; Detheux M; Le Poul E; Brezillon S; Vakili J; Mazarguil H; Vassart G; Zajac JM; Parmentier M Functional characterization of a human receptor for neuropeptide FF and related peptides. Br.J.Pharmacol. 2001,133,138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Fumagalli M; Trincavelli L; Lecca D; Martini C; Ciana P; Abbracchio MP Cloning, pharmacological characterisation and distribution of the rat G-protein-coupled P2Y(13) receptor. Biochem.Pharmacol. 2004,68(1),113–124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.