

Abstract
Sleep disturbances and hyperactivity are prevalent in several neurodevelopmental disorders, including autism spectrum disorders (ASDs) and attention deficit-hyperactivity disorder (ADHD). Evidence from genome-wide association studies indicates that chromosomal copy number variations (CNVs) are associated with increased prevalence of these neurodevelopmental disorders. In particular, CNVs in chromosomal region 16p11.2 profoundly increase the risk for ASD and ADHD, disorders that are more common in males than females. We hypothesized that mice hemizygous for the 16p11.2 deletion (16p11.2 del/+) would exhibit sex-specific sleep and activity alterations. To test this hypothesis, we recorded activity patterns using infrared beam breaks in the home-cage of adult male and female 16p11.2 del/+ and wildtype (WT) littermates. In comparison to controls, we found that both male and female 16p11.2 del/+ mice exhibited robust home-cage hyperactivity. In additional experiments, sleep was assessed by polysomnography over a 24-hr period. 16p11.2 del/+ male, but not female mice, exhibited significantly more time awake and significantly less time in non-rapid-eye-movement (NREM) sleep during the 24-hr period than wildtype littermates. Analysis of bouts of sleep and wakefulness revealed that 16p11.2 del/+ males, but not females, spent a significantly greater proportion of wake time in long bouts of consolidated wakefulness (greater than 42 min in duration) compared to controls. These changes in hyperactivity, wake time, and wake time distribution in the males resemble sleep disturbances observed in human ASD and ADHD patients, suggesting that the 16p11.2 del/+ mouse model may be a useful genetic model for studying sleep and activity problems in human neurodevelopmental disorders.
Keywords: autism, sleep, sex differences, ADHD, 16p11.2 deletion, copy number variation
Introduction
Sleep and activity problems are extremely prevalent in autism spectrum disorders (ASDs) and attention-deficit/ hyperactivity disorder (ADHD), yet they remain poorly understood. Disrupted sleep affects up to 80% of ASD patients and 55% of children with ADHD, compared to only 7–30% in the control population [Goldman et al., 2011; Cohen, Conduit, Lockley, Rajaratnam, & Cornish, 2014; Ivanenko & Johnson, 2008; Kirov & Brand, 2014]. The most commonly reported sleep issues in these neurodevelopmental disorders include insomnia, delayed sleep onset, and increased night awakening [Ivanenko & Johnson, 2008; Ming & Walters, 2009; Reynolds & Malow, 2011]. These subjective observations have been supported by objective polysomnography and actigraphy sleep recordings [Cortese, Faraone, Konofal, & Lecendreux, 2009; Goldman et al., 2009; Miano et al., 2007; Souders et al., 2009]. In addition, sleep problems are positively correlated with the severity of core ASD symptomology including communication deficits, withdrawal, and repetitive and stereotyped behavior [Cortesi, Giannotti, Ivanenko, & Johnson, 2010; Park et al., 2012]. Despite this, few well-controlled studies have addressed the neurobiological underpinnings of sleep problems in these disorders. The high prevalence of sleep problems in ASDs and ADHD and the impact of disrupted sleep on symptomology highlight the utility of identifying an appropriate genetic model with which to test potential treatments and elucidate mechanisms underlying these disorders.
Increasing evidence from genome-wide association studies suggests that chromosomal copy number variations (CNVs) are significantly enhanced in many neurodevelopmental disorders [Sebat et al., 2007; Grayton, Fernandes, Rujescu, & Collier, 2012]. In particular, hemideletion in chromosomal region 16p11.2 profoundly increases the risk for several neurodevelopmental disorders, including ASD and ADHD, even when controlling for the high comorbidity between these disorders [Hanson et al., 2014]. 16p11.2 hemideletion is associated with an estimated 0.6% of all ASD diagnoses [Weiss et al., 2008], and 16p11.2 hemideletion patients display cognitive deficits and other ASD symptomology even if they do not meet the criteria for ASD diagnosis [Stefansson et al., 2014]. Unlike many factors believed to contribute to neurodevelopmental disorders, the 16p11.2 chromosomal region is highly conserved in the syntenic 7qF3 region in the mouse, and thus copy number variation in this region can be accurately modeled. To date, three different mouse lines of 16p11.2 hemideletion have been created, varying in the size of the deletion as well as genetic background of the mice [Arbogast et al., 2016; Horev et al., 2011; Portmann et al., 2014]. Indeed, 16p11.2 hemideletion (16p11.2 del/+) mice have deficits in brain structure, cognition, and communication [Arbogast et al., 2016; Brunner et al., 2015; Horev et al., 2011; Portmann et al., 2014; Pucilowska et al., 2015; Yang, Lewis, Sarvi, Foley, & Crawley, 2015a; Yang et al., 2015b].
ASDs and ADHD also show significant sex bias risk, but the mechanisms contributing to this remain unknown. Males are 4 times more likely than females to be diagnosed with ASD [Werling & Geschwind, 2013] and 3 times more likely to be diagnosed with ADHD [Schneider & Eisenberg, 2006]. For this reason, developing genetic models that also demonstrate this risk bias is important for construct validity. Previous studies utilizing the 16p11.2 del/+ mouse model have not directly compared males and females [Arbogast et al., 2016; Brunner et al., 2015; Pucilowska et al., 2015; Yang et al., 2015a,b]. In this study, we assessed home-cage activity using infrared beam breaks and sleep/wake behavior using polysomnography in both male and female adult 16p11.2 del/+ mice. We found robust home-cage hyperactivity across the diurnal cycle in both male and female 16p11.2 del/+ mice compared to sex-matched wildtype littermates. In addition, we found male-specific sleep/wake decrements in total sleep time and distribution of wakefulness. In 16p11.2 del/+ males, but not females, the proportion of wake time distributed in long bouts of continuous wakefulness was significantly greater than in sex-matched WT littermates. When compared to sex-matched controls, these male-specific sleep and activity alterations parallel deficits seen in human ASD and ADHD patients. Additionally, while no systematic analysis of sleep has been performed in a population of human 16p11.2 hemideletion patients, sleep disturbances have been reported in two 16p11.2 hemideletion patients and two 16p11.2 duplication patients [Fernandez et al., 2009; Tabet et al., 2012]. To our knowledge, the present study is the first study showing male-specific sleep deficits in a rodent genetic model of neurodevelopmental disorders. These findings suggest that 16p11.2 del/+ mice are an appropriate genetic model for investigating treatment strategies and potential mechanisms underlying sleep problems, hyperactivity, and sex differences found commonly in human ASD and ADHD patients.
Methods
Animals
16p11.2 del/+ male mice on a mixed C57BL/6J and 129S1/SvImJ background purchased from The Jackson Laboratory (Stock #013128) were bred with females on a mixed C57BL/6J and 129S1/SvImJ background (Stock #101043). All available pups were used for experiments, no litters were culled, and all cages were fitted with Nestlets (Ancare, Bellmore, NY) for enrichment. Mice were weaned at 3 weeks of age and remained group housed with sex-matched littermates (4–5 mice/cage) until experimentation. Adult (2.5–4.5 month old) male and female 16p11.2 del/+ and WT littermate offspring were used for all experiments in accordance with age ranges utilized in previous mouse sleep experiments [Mang et al., 2016; Vassalli et al., 2013; Wimmer et al., 2013] and guidelines for assessment of behavior in adult mice [Crawley, 2007]. The age distribution was consistent between the 4 experimental groups (mean ± SEM; Male WT: 117.3 ± 3.9 days, Male 16p11.2 del/+: 115.7 ± 3.9 days, Female WT: 118.2 ± 4.6 days, Female 16p11.2 del/+: 117.5 ± 4.1 days). Separate cohorts of mice were used for all behavioral experiments. Animals were provided food and water ad libitum and maintained on a 12 hour light/12 hour dark cycle with light onset at 7:00 am. All animal care and experiments were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania and conducted in accordance with the National Institutes of Health guidelines.
Activity Monitoring
Activity was monitored using an infrared beam-break system (Opto M3, Columbus Instruments, Columbus, OH), which provided a scaffold of high-resolution infrared lights and detectors. The beams were spaced 0.5 inches apart and beam breaks were sampled every 10 s. Two infrared grids at 0.75 inches and 2.75 inches from the cage floor measured horizontal and vertical (rearing) activity, respectively. Mice were housed individually in noise-attenuating chambers (22″ × 16″ × 19″, Med Associates, St. Albans, VT) equipped with individual lights (250 lux) and fans. Cages were placed within the beam break system and covered with a lid to contain the mouse and to reduce brightness (80 lux at cage floor level). To mitigate the potential impact of anxiety, mice were allowed to acclimate to the lighting and social isolation of the activity chambers for 1 week before experimentation. Following 1 week of acclimation, activity data was collected for 1 week under 12-hour light/12-hour dark conditions. Beam break counts were binned into 1 hr bins and averaged over the 1 week of data collection. Lighting conditions were then switched to 24-hr constant darkness for 2 weeks and counts of beam breaks were compiled every 1 minute. Circadian period (Tau) was calculated from Day 2 to Day 14 of constant darkness using ClockLab software (Actimetrics). Because of the circadian manipulations of activity experiments, and to ensure consistent age ranges between sleep and activity experiments, separate cohorts of mice were used for activity monitoring and polysomnography sleep experiments.
Polysomnography
Animals were surgically implanted with electroencephalography (EEG) and electromyography (EMG) electrodes under isoflurane anesthesia as described previously [Wimmer et al., 2013]. Briefly, electrodes consisted of Teflon-coated wires (Cooner wires, Chatsworth, CA) soldered to gold socket contacts (Plastics One, Roanoke, VA) and pushed into a six-pin plastic plug (MS363 plug, Plastics One). Electrodes were held in place with miniature screws (J.I. Morris Co, Southbridge, MA) and dental cement (Ketac, 3M, St Paul, MN). Mice were allowed to recover from surgery for a minimum of 2 weeks. During the second week of recovery, animals were connected to amplifiers using lightweight cables (363, Plastics One) attached to a rotating commutator (SLC6, Plastics One). Mice were allowed to acclimate to the cables and to the noise-attenuating faraday recording chambers (38″ × 39″ × 33″, Med Associates, Georgia, VT) for one week before analysis of sleep/wake. All recordings were obtained using parietal (ML ± 1.5 mm, AP −2 mm from bregma) electrodes referenced to an electrode over the cerebellum (−1.5 mm from lambda). Cerebellar reference was chosen based upon a lack of signal disruption from either neck muscles or other brain areas, and has been previously validated by our lab [Wimmer et al., 2013] and others [McShane et al., 2010; Vassalli et al., 2013].
EEG/EMG signals were sampled at 256 Hertz (Hz) and filtered at 0.3–30 Hz and 1–100 Hz, respectively, with 12A5 amplifiers (Astro-Med, West Warwick, RI). Data acquisition and manual visual scoring was performed using SleepSign software (Kissei Comtec Inc, Japan). EEG/EMG data was collected for a total of 24 consecutive undisturbed hours beginning at 7:00 am (onset of the light phase). EEG/EMG data was analyzed in 4 s epochs as wake, non-rapid eye movement (NREM) sleep, or rapid eye movement (REM) sleep by a trained experimenter blind to experimental conditions. Wake was classified by increases in higher frequency waves (>10 Hz), decreases in amplitude of the EEG, and high activity in the EMG. NREM was classified by increases in delta power (0.5–4 Hz) and amplitude of the EEG, along with low amplitude activity in the EMG. REM was determined by high EEG activity in the theta range (4–8 Hz) and very low activity in the EMG. One 16p11.2 del/+ male was excluded from all analyses because its total wake time over the 24-hr period was 236 min less than the group average, >2.5 standard deviations below the group mean. For EEG spectral analysis, fast Fourier transform (FFT; Hanning window; 0.5–20 Hz, 0.25 Hz resolution) was performed across the 24-hr period on artifact-free epochs, and wake, NREM, and REM were normalized and expressed as a percentage of the total spectral power. For a subset of the animals (15 males and 5 females), spectral analysis was not appropriate due to technical error with amplifier settings in initial cohorts and/or the quality of the EEG traces.
Bout Distributions
Bouts of wake, NREM, and REM were classified as reported previously [Watson, Henson, Dorsey, & Frank, 2015]. Briefly, wake and NREM bouts were identified as 32 s (8 epochs) or greater of consecutive wake or NREM, respectively, while bouts of REM were identified as 20 s (5 epochs) or greater of consecutive REM. Bouts of NREM and REM were considered broken once interrupted by 8 epochs of any other state, while bouts of wake were terminated by a single epoch of any other state. Bouts of wake, NREM, and REM were then sorted into duration bins following a log2 pattern (30–80 s, 84–160 s, 164–320 s, 324–640 s. 644–1280 s, 1284– 2560 s, and >2560 s) in accordance with previously published analysis [Kantor, Szabo, Varga, Cuesta, & Morton, 2013; Mochizuki et al., 2004]. One WT male was excluded from all analyses due to a bout of >7 hours of consecutive wakefulness, >2.5 standard deviations above the group average.
Elevated Zero-Maze
Mice were individually placed on a 5.5 cm wide circular track with an external diameter measuring 45 cm, raised 40 cm above the floor (San Diego Instruments, San Diego, CA). The track had two open and two enclosed segments of equal dimensions. Mice were gently placed in the center of a closed segment to begin a 5-min trial. Time spent in open arms, risk assessment [Karlsson, Holmes, Heilig, & Crawley, 2005], and total distance traveled were assessed using an automated MATLAB (Mathworks) based image analysis software as described previously [Patel et al., 2014].
Open Field
Spontaneous activity was assessed in an open field arena (14″ × 14″, San Diego Instruments, San Diego, CA) fitted with photocells to detect motion. Mice were gently placed individually into the center of the open field for a 10-min trial. Total ambulation, center activity, periphery activity, and rearing behavior were recorded. Each trial was digitally recorded and analyzed with image analysis software (Mathworks). Separate cohorts of mice were used for elevated zero-maze and open field tasks.
Statistics
All statistical analyses were performed using SPSS for Windows (V. 22.0). For beam break activity, Mixed Design ANOVAs were used with genotype (WT or 16p11.2 del/+) and sex (male or female) as the between-subjects factors and time as the within-subjects factor. Two-way ANOVAs were used to compare wake, NREM and REM sleep times averaged over 24 hours, and to analyze elevated zero-maze and open field data. For 4×6-hour binned sleep state analysis, sleep state bout distribution experiments, and FFT spectral analysis, Mixed Design ANOVAs were used with genotype (WT or 16p11.2 del/+) as the between-subjects factor and time (or in the case of FFT analysis, frequency bin) as the within-subjects factor. Post hoc multiple comparisons were performed using Bonferroni’s adjustment for multiple comparisons. In instances where the assumption of sphericity was violated, Greenhouse-Geisser corrected F values are given. Mann-Whitney U-tests were performed on normalized FFT data binned into low delta, delta, theta, alpha, and beta frequency bins. Multivariate analysis of variance (MANOVA) was used on the proportion of time spent in each sleep state during the light phase and the dark phase, with alpha corrected for multiple ANOVAs and set at α = 0.05/2, followed by post hoc Bonferroni’s adjustment for multiple comparisons.
Results
Home-Cage Hyperactivity in 16p11.2 Del/+ Mice
Male and female 16p11.2 del/+ mice and WT littermates were acclimated and assessed for home-cage activity across the diurnal cycle by breaks of infrared beams in the horizontal axis (Fig. 1a) as well as the vertical axis (Fig. 1b) to measure rearing behavior. Mixed Design ANOVAs revealed that 16p11.2 del/+ mice had significantly more activity than WT mice in both the horizontal (main effect of genotype; F(1,48) =27.808, P < 0.001, Fig. 1c) and vertical axis (main effect of geno-type; F(1,48) = 34.714, P < 0.001 Fig. 1d). There was no effect of sex (F(1,48) = 0.814, P =0.371) nor a sex*genotype interaction (F(1,48) = 0.083, P = 0.774). To test whether the hyperactive behavior observed in 16p11.2 del/+ mice may be related to stress/anxiety, we performed elevated zero-maze on a separate cohort of 16p11.2 del/+ and WT mice. There were no significant differences in time spent in the open arm (Two-way ANOVA, P = 0.279, Fig. 1e) or risk assessment behavior [Karlsson et al., 2005] (P = 0.54, Fig. 1f), demonstrating that 16p11.2 del/+ mice display normal anxiety-like behavior. There were also no differences between 16p11.2 del/+ mice and WT mice in distance traveled (Two-way ANOVA, P = 0.404, Fig. 1g) or time spent in the center (P = 0.501, Fig 1h) in a 10-min novel open field task.
Figure 1.
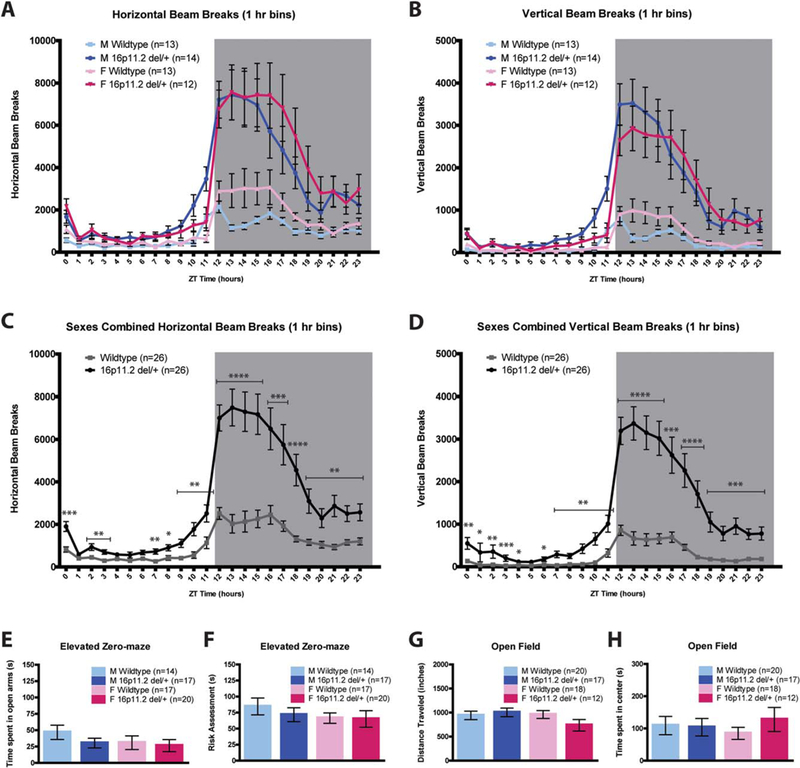
16p11.2 del/+ mice are hyperactive throughout the diurnal cycle. (A and B) Infrared beam breaks in the XY (horizontal) and Z (vertical) (B) axis plotted for all four groups, in 1 hour bins, across the 24-hr day. Gray box indicates the dark (active) period. (C and D) 16p11.2 del/+ mice have significantly greater activity relative to wildtype littermates, irrespective of sex, in both the horizontal (C) and vertical (D) axes. (E and F) There are no significant differences between 16p11.2 del/+ and wildtype mice in elevated zero-maze time spent in open arms (E) or risk assessment (F). (G and H) There are no differences in open field total ambulation (G) or time spent in center (H) between 16p11.2 del/+ mice and wildtype controls. Mean 6 standard error of the mean (s.e.m.) * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Because the hyperactivity observed in 16p11.2 del/+ males and females was most pronounced during the dark (active) phase, and circadian problems such as increased latency to sleep and early night awakenings are commonly reported in ASD patients [Glickman, 2010], we tested whether 16p11.2 del/+ mice have altered free-running circadian rhythms. Male and female 16p11.2 del/+ mice were entrained to constant darkness for 2 weeks and activity was quantified via infrared beam breaks. There were no effects of genotype on circadian period (tau) in male (WT: 23.70 ± 0.04 hrs, 16p11.2 del/+: 23.76 ± 0.05 hr; P = 0.36) or female (WT: 23.57 ± 0.05 hr, 16p11.2 del/+: 23.64 ± 0.02 hr; P 50.22) mice. This indicates that 16p11.2 del/+ mice have normal circadian rhythms and also that the hyperactivity observed in these mice is intrinsic and not a product of environmental light cues.
Male-Specific Sleep Decrements in 16p11.2 Del/+ Mice
Because 16p11.2 del/+ male and female mice exhibited robust hyperactivity, we assessed sleep and wake in 16p11.2 del/+ male and female mice using polysom-nography recordings to distinguish between activity and sleep. EEG/EMG was recorded and assessed across a 24-hr period. Female mice were awake significantly more than male mice across the 24-hr day (main effect of sex; F(1,51) = 6.318, P = 0.015) consistent with previously published results showing sex differences in sleep time and architecture between male and female mice [Koehl, Battle, & Meerlo, 2006; Paul, Dugovic, Turek, & Laposky, 2006]. There was no main effect of genotype (F(1,51) = 2.628, P =0.111) nor sex*genotype interaction (F(1,51) = 2.110, P = 0.152). Because of the inherent sleep differences between male and female mice and the main effect of sex within our dataset, male and female 16p11.2 del/+ mice were analyzed separately and compared against sex-matched WT littermates in order to focus on the biologically relevant comparisons. Male 16p11.2 del/+ mice exhibited more wake across the 24-hr day compared to sex-matched WT mice (Student’s t-test, P = 0.008; Fig. 2a). There was a significant difference in 16p11.2 del/+ male wake time compared to WTs across the light/dark cycle (MANOVA, F(2,27) = 4.628, P = 0.019; Fig. 2a). Post hoc analysis revealed that 16p11.2 del/+ males have significantly more wake time during the light phase (F(1,28) = 5.322, P =0.029), but not during the dark (active) phase (F(1,28) = 2.780, P = 0.107) than WT males. Concordant with increased total wake time, 16p11.2 del/+ male mice have less NREM sleep time than WT mice across the 24-hr period (Student’s t-test, P = 0.013; Fig. 2b). In contrast to differences in total wake and NREM time, there was no difference in REM time in 16p11.2 del/+ males relative to WT mice (Student’s t-test, P = 0.94; Fig. 2c). Sleep data was next analyzed in 6 hour time bins as previously published [Franken, Malafosse, & Tafti, 1999]. Mixed Design ANOVA revealed that the decreased NREM time observed in 16p11.2 del/+ males (main effect of genotype; F(1,28) = 7.019, P =0.013; Fig. 2d) was due primarily to the final 6 hours of the light phase (Student’s t-test, t(28) = 9.055, P = 0.005; Fig. 2d). In contrast to the males, 16p11.2 del/+ females exhibited no differences in wake (Student’s t-test, P = 0.92; Fig. 2e), NREM (Student’s t-test, P = 0.53; Fig. 2f), or REM (Student’s t-test, P =0.067; Fig. 2g) time compared to WT females across the 24-hr period. Female 16p11.2 del/+ mice also exhibited no differences in wake, NREM, or REM in the light or dark phases (wake: F(2,22) = 0.414, P = 0.67, Fig. 2e; NREM: F(2,22) = 1.633, P = 0.218, Fig. 2f; REM: F(2,22) = 3.121, P = 0.064, Fig. 2g). Analyzing female 16p11.2 del/+ NREM sleep in 6 hr time bins revealed no significant main effect (F(3,69) = 0.397, P = 0.535; Fig. 2h) nor a significant genotype*time interaction (F(3,69) = 1.272, P = 0.29; Fig. 2h). This data suggests that 16p11.2 del/+ males, but not females, have deficits in either sleep initiation or sleep maintenance.
Figure 2.
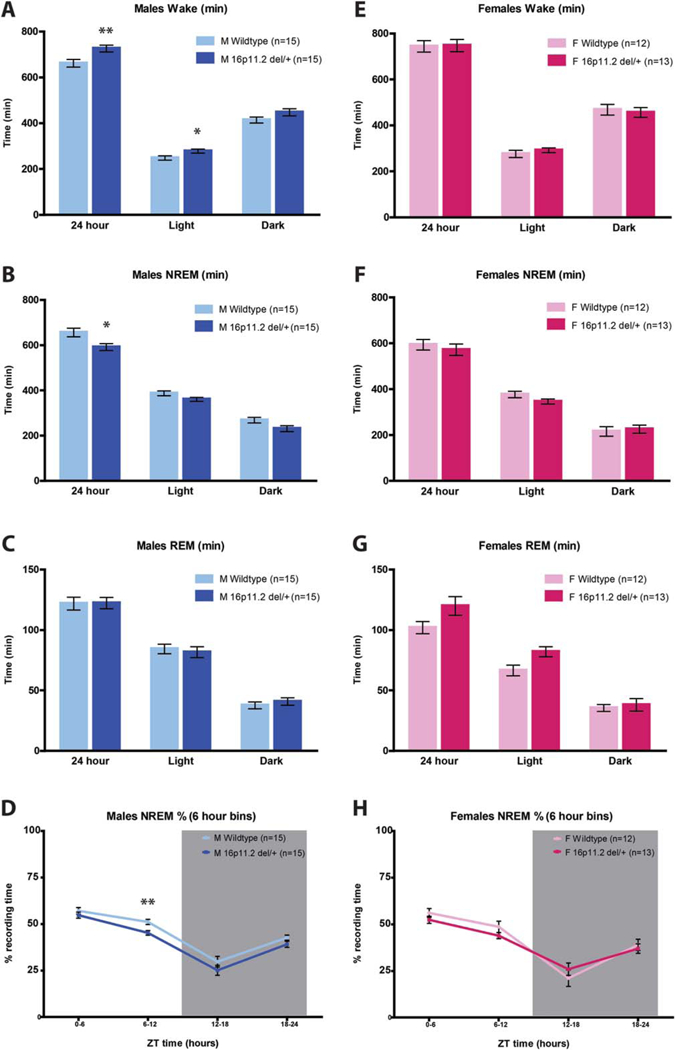
Male 16p11.2 del/+ mice sleep less than wildtype littermates. (A–C) Male 16p11.2 del/+ mice are awake significantly more (A) and spend significantly less time in NREM sleep (B) than wildtype mice over the 24-hr period. The amount of REM sleep in male 16p11.2 del/+ mice was not significantly different from wildtype (C). (D) NREM sleep time expressed in 6 hr bins reveals that males have significantly less sleep during the last 6 hours of the light in comparison to wildtype littermates. Gray box indicates the dark period. (E–F) There are no significant differences in wake (E), NREM sleep (F and H), or REM sleep (G) time between 16p11.2 del/+ females and controls. Mean ± s.e.m. *P < 0.05, **P < 0.01.
16p11.2 Del/+ Males Have Elongated Bouts of Wakefulness
Because 16p11.2 del/+ male mice have decreased sleep and increased wakefulness, we binned and analyzed the distribution of wake, NREM, and REM bout durations as described previously [Mochizuki et al., 2004; Kantor et al., 2013] to elucidate the factor(s) contributing most strongly to this phenotype. We found that 16p11.2 del/+ male mice have an altered distribution of wake bout length duration (genotype*bout time interaction; F(6,168) = 6.599, P = 0.001; Fig. 3a). Post hoc comparisons indicated that 16p11.2 del/+ males spent a higher proportion of their wake time in prolonged bouts of continuous wakefulness (>42 consecutive minutes: t(28) 511.340, P = 0.002) and less time in bouts of shorter duration (160–320 s: t(28) = 5.356, P = 0.028; 320–640 s: t(28) = 10.248, P = 0.003; 640–1280 s: t(28) = 6.480, P = 0.017). By contrast, females exhibited no differences in wake bout length distribution (F(6,138) = 1.576, P = 0.22; Fig. 3d). There were no differences in NREM bout time distribution in 16p11.2 del/+ males (F(6,168) = 0.500, P = 0.59; Fig. 3b) or females (F(6,138) = 1.924, P = 0.16; Fig. 3e). Likewise, there were no differences in REM bout length distribution in either males (F(6,168) = 0.785, P = 0.48; Fig. 3c) or females (F(6,138) = 1.816, P = 0.16; Fig. 3f) Together, this data indicates that 16p11.2 del/+ males, but not females, sleep less than WT mice due to deficits in sleep initiation, rather than sleep maintenance.
Figure 3.
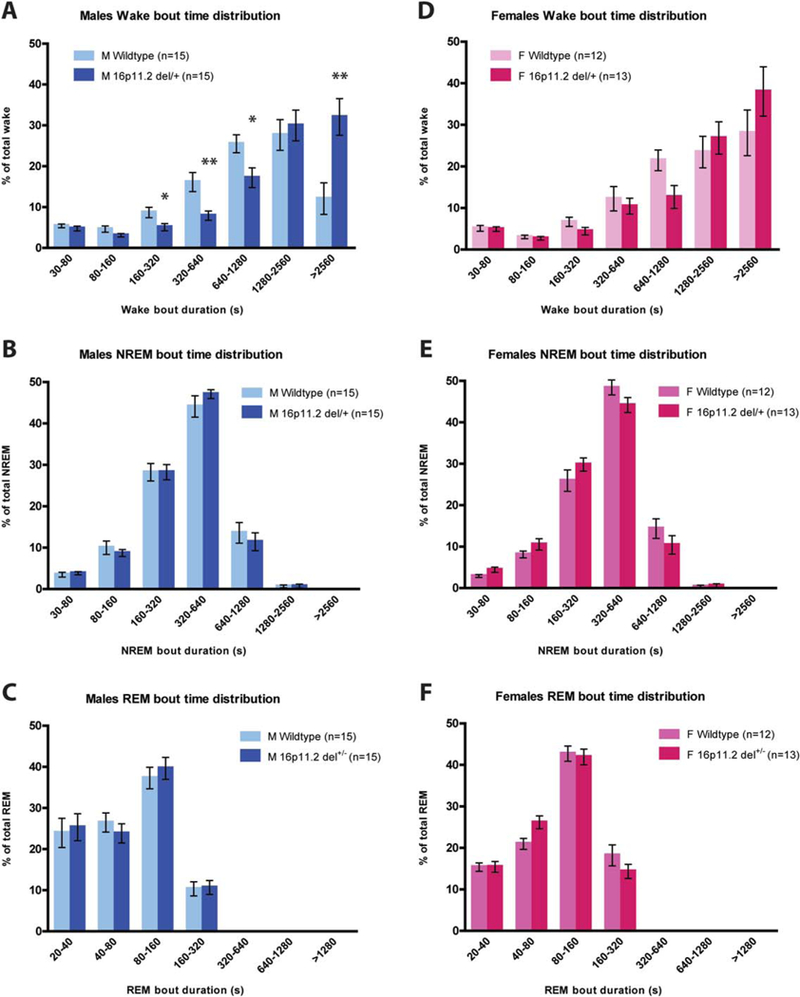
Male 16p11.2 del/+ mice spend a significantly higher proportion of wake time in prolonged bouts of wakefulness. Proportion of total wake time across the 24-hr period divided into bout lengths of 30s–80s, 80s–160s, 160s–320s, 320s–640s, 640s–1280s, 1280s– 2560s, and >2560s. (A) 16p11.2 del/+ male mice spend a significantly greater proportion of their wake time in long bouts of wakefulness (>2560s) and a significantly lower proportion of time in intermediate bouts of wakefulness (160s–1280s). (B and C) 16p11.2 del/+ males exhibit no differences in NREM (B) or REM (C) bout length distribution. (D–F) 16p11.2 del/+ female mice do not differ from wildtype littermates in distribution of wake (D), NREM (E), or REM (F) bout length. Mean ± s.e.m. *P < 0.05, **P < 0.01.
16p11.2 Del/+ Males Have Reduced Alpha Power during Wake
Next, we performed fast Fourier transform (FFT) to analyze the EEG power spectra of 16p11.2 del/+ males and females during wake, NREM, and REM. There was no main effect of genotype for any sleep state for either sex (Males wake: F(1,13) = 2.649, P = 0.13, Fig. 4a; F(1,18) = 1.680, P = 0.21, Fig. 4e; REM: F(1,18) = 0.744, P = 0.40, Fig. 4f). However, binning the data into low delta (0.5–1.5 Hz), delta (0.5–4.0 Hz), theta (4.0–8.0 Hz), alpha (8.0–12.0 Hz), and beta (12.0–20.0 Hz) frequency bands revealed that 16p11.2 del/+ males have significantly increased alpha power during wake relative to WT littermates (Mann-Whitney U = 8, P = 0.021; Fig. 4a, inset), suggesting increased arousal and vigilance during quiet wake in 16p11.2 del/+ males [Cantero, Atienza, & Salas, 2002]. Increased arousal during quiet wake, in concert with increases in total wake time and prolonged bouts of continuous wakefulness, suggests that 16p11.2 del+/males may possibly have deficits initiating wake-to-sleep transitions. In contrast, there were no differences in alpha power during wake between 16p11.2 del/+ females and WT littermates (Mann-Whitney U = 31, P = 0.18, Fig. 4d, inset). 16p11.2 del/+ females, however, had significantly increased beta power during wake (Mann-Whitney U = 22, P = 0.038, Fig. 4d, inset). There were no differences in any other frequency bands apart from a decrease in low delta power during NREM in 16p11.2 del/+ females in comparison to WT littermates (Mann-Whitney U = 16, P = 0.010, Fig. 4e, inset), which may indicate differences in sleep homeostasis.
Figure 4.
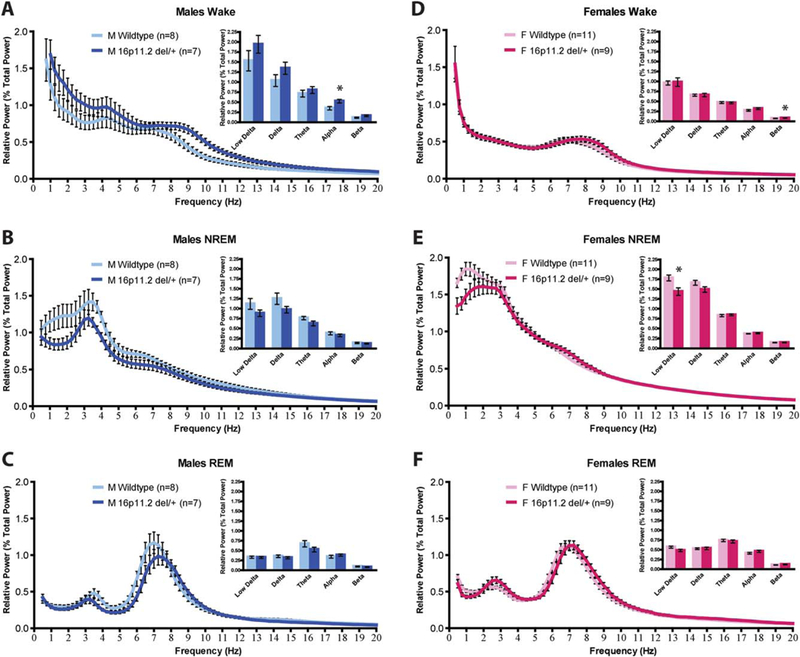
Male 16p11.2 del/+ mice have increased alpha power during wake. (A) Male 16p11.2 del/+ mice have increased alpha power during wake (A, inset). (B–C) Male 16p11.2 del/+ mice have no differences in EEG power spectra during NREM sleep (B) or REM sleep (C). (D) Female 16p11.2 del/+ mice have increased beta power during wake (D, inset). (E) Female 16p11.2 del/+ mice have decreased low delta during NREM sleep (E, inset). (F) Female 16p11.2 del/+ mice have no differences in EEG power spectra during REM sleep (F). Mean ± s.e.m. *P < 0.05.
Discussion
We investigated home-cage activity and sleep patterns in one mouse model of human 16p11.2 chromosomal hemideletion. We report robust and reliable home-cage hyperactivity across the light-dark cycle that is present in both males and females. These findings expand upon previous reports of hyperactivity in 16p11.2 del/+ mice [Arbogast et al., 2016; Brunner et al., 2015; Horev et al., 2011] by comparing males and females, assessing activity in the home-cage, and quantifying activity over a week of consecutive diurnal cycles. Importantly, there are presently three distinct mouse models of 16p11.2 hemideletion differing in deletion size and genetic background, which should be considered when comparing findings between studies. In addition, we report the first male-specific sleep decrements in a rodent model of ASD. Decreased total sleep time and prolonged bouts of wakefulness, suggesting difficulties in initiating wake-to-sleep transition, recapitulate common sleep and activity problems reported in human ASD patients [Baker & Richdale, 2015; Krakowiak, Goodlin-Jones, Hertz-Picciotto, Croen, & Hansen, 2008; Miano et al., 2007; Reynolds & Malow, 2011]. The sleep/wake deficits in 16p11.2 del/+ males, but not females relative to sex-matched controls support theories of female protectiveness from ASD given the same genetic insult Angelakos et al./Sleep and activity in an ASD model [Jacquemont et al., 2014; Robinson, Lichtenstein, Anckarsäter, Happé, & Ronald, 2013; Werling & Gesch-wind, 2013].
Additionally, compared to sex-matched controls, the sleep/wake differences observed only in male 16p11.2 del/+ mice are contrasted by activity differences in both 16p11.2 del/+ males and females. This distinction suggests disparate neurobiological mechanisms underlying the two alterations. The etiology of sleep and activity deficits in ASDs remains unknown, but numerous lines of evidence strongly support a relationship between imbalanced excitatory/inhibitory signaling, sleep, and hyperactivity. Although both ADHD and ASD are associated with imbalanced excitatory/inhibitory signaling, the neurochemical and neuroanatomical mechanisms mediating these disorders are likely distinct. The sex differences in sleep/wake, but not activity in 16p11.2 del/+ mice therefore suggest that the 16p11.2 del/+ mouse model may be a useful rodent genetic model for investigating neurobiological mechanisms mediating sex differences in neurodevelopmental disorders.
Neurochemical and Neuroanatomical Correlates in 16p11.2 Hemideletion
The mechanisms underlying altered sleep and activity in our 16p11.2 del/1 mouse model may be considered in light of recent neurochemical and neuroanatomical findings in related models. Another mouse model of 16p11.2 hemideletion was recently shown to haveincreased GABAergic medium spiny neurons (MSNs) expressing the dopamine D2 receptor in the striatum, and decreased dopamine D1 receptor neurons in the cortex [Portmann et al., 2014]. During development, D1 activation increases and D2 activation decreases GABA neuron migration from the basal forebrain to the cortex [Crandall et al., 2007]. Further, estrogen in females has been found to downregulate D2 receptor function [Bazzett & Becker, 1994]. In Drosophila, knock down of the E3 ubiquitin ligase Cul3, which interacts with the 16p11.2 region gene KCTD13 through the KCTD13-Cul3-RhoA pathway, results in increased dopa-mine signaling, reduced sleep duration, and hyperarousal [Pfeiffenberger & Allada, 2012; Stavropoulos & Young, 2011], similar to our findings in 16p11.2 del/+ mice. Hyperactivity is often thought of as a disorder of catecholamine dysfunction [Sharma & Couture, 2014], and the striatal dopaminergic system has a strong wake-promoting role [Saper, Fuller, Pedersen, Lu, & Scam-mell, 2010]. Together, these findings suggest that dysregulated dopaminergic signaling may underlie some of the sleep and activity alterations observed in 16p11.2 del/+ mice.
Imaging studies of humans and mice with 16p11.2 hemideletion reveal alterations in structure and connectivity of the striatum and frontal cortex. Magnetic resonance imaging (MRI) studies show increased striatal volume and cortical surface area in 16p11.2 hemideletion humans [Qureshi et al., 2014] and mice [Portmann et al., 2014], agreeing with structural changes reported more broadly in a meta-analysis of ASD patients [Nickl-Jockschat et al., 2012]. 16p11.2 hemideletion children show widespread white matter abnormalities, including increased axial diffusivity of the corpus callosum, external capsule, and internal capsule—the last of which is comprised largely of the corticospinal tract, carrying projections from the primary motor cortex through the striatum to the spinal cord [Owen et al., 2014]. Using diffusion tensor imaging (DTI), we have shown male-specific white matter alterations in the same fiber bundles proximal to the striatum in 16p11.2 del/+ mice [Grissom et al., 2014, Nickl-Jockschat et al., 2015]. The striatum and frontal cortex are important brain regions mediating motor control and sleep/wake. Hyperactive behavior is generally believed to relate to abnormalities in corticostriatal connectivity [Bush, 2010], and children with ASD show reduced striatal activation in response to rewards [Kohls et al., 2013; Scott-Van Zee-land, Dapretto, Ghahremani, Poldrack, & Bookheimer, 2010]. Striatal growth rate is also correlated with ASD diagnosis [Langen et al., 2014]. Together, these results may implicate corticostriatal deficits in the hyperactive behavior and increased wakefulness observed in 16p11.2 hemideletion mice.
Conclusions and Future Directions
Sleep and activity problems are among the most common complaints reported in neurodevelopmental disorders. Psychomotor stimulants are often the first-choice option for treating hyperactivity, but typically exacerbate sleep problems in ADHD patients [Lee et al., 2012; Santisteban, Stein, Bergmame, & Gruber, 2014]. Nocturnal melatonin, which is decreased in insomniacs [Rodenbeck, Huether, Ruther,€ & Hajak, 1999] and children diagnosed with ASD [Tordjman, Anderson, Pichard, Charbuy, & Touitou, 2005], has been effective for decreasing sleep latency and increasing total sleep time in some studies of ASD patients [Wright et al., 2011], but has little effect on mitigating other sleep disturbances [Cortesi, Giannotti, Sebastiani, Panunzi, & Valente, 2012; Malow et al., 2012] and may even increase night awakenings [Rossignol & Frye, 2011; Gringras et al., 2012]. Unraveling the distinct neuro-chemical mechanisms mediating the sleep and hyperactivity phenotypes observed in 16p11.2 hemideletion mice is an interesting avenue of investigation that may lead to a greater understanding of the behavioral and cognitive deficits observed in 16p11.2 hemideletion patients, as well as signaling mechanisms contributing to sex-biased ASD symptomology. Male-specific neuro-anatomical and neurochemical differences in 16p11.2 del/+ mice, such as increased white matter proximal to the striatum [Grissom et al., 2014], striatal hypertrophy [Portmann et al., 2014], and possibly increased D2 receptor expression and activity of striatal MSNs may contribute to the sex-specific differences in sleep. Conversely, neurochemical and neuroanatomical deficits shared between 16p11.2 del/+ males and females, such as decreased ERK1 protein in the striatum, or decreased white matter integrity of the corpus callosum [Grissom et al., 2014], may underlie the hyperactivity observed in both 16p11.2 del/+ males and females. Future dose-response studies of changes in sleep and activity in 16p11.2 del/+ mice in response to dopamine receptor subtype-specific drugs may reveal the contributions of these receptors and signaling pathways to the insomnia and hyperactivity observed in 16p11.2 del/+ male mice. These experiments are the first steps in elucidat-ing the signaling mechanisms involved in 16p11.2 hemideletion hyperactivity and insomnia, and may lead to more targeted brain-region or cell-type specific experiments, as well as a more complete understanding of the neurochemical mechanisms mediating behavior in a common CNV found in ASDs. Together, these findings demonstrate that the 16p11.2 del/+ mouse model will be useful for investigating the molecular basis of sex bias in ASDs, as well as distinct neural mechanisms underlying common sleep and activity problems in ASDs and related neurodevelopmental disorders.
Acknowledgments
This work was funded by a grant from the Simons Foundation Autism Research Initiative, 248429, as well as government support under and awarded by DoD, Air Force Office of Scientific Research, National Defense Science and Engineering Graduate (NDSEG) Fellowship, 32 CFR 168a. Support was also received from the IRTG 2150–The Neuroscience of Modulating Aggression and Impulsivity in Psychopathology–funded by the Deutsche Forschungsgemeinschaft (DFG). T.A. is the Brush Family Professor of Biology at the University of Penn-sylvania. Behavior procedures were performed with the assistance of the Neurobehavior Testing Core and supported by the IDDRC at CHOP/UPenn. We would like to acknowledge the helpful assistance of Dr. Sarah Ferri.
Footnotes
The authors declare no competing financial interests.
Contributor Information
Christopher C. Angelakos, Department of Neuroscience, Neuroscience Graduate Group, University of Pennsylvania, Philadelphia, PA, 19104
Adam J. Watson, Department of Biology, University of Pennsylvania, Philadelphia, PA, 19104
W. Timothy O’Brien, Department of Neuroscience, University of Pennsylvania, Philadelphia, PA, 19104.
Kyle S. Krainock, Department of Biology, University of Pennsylvania, Philadelphia, PA, 19104
Thomas Nickl-Jockschat, Department of Psychiatry Psychotherapy and Psychosomatics, RWTH Aachen University, Aachen, Germany, Jülich Aachen Research Alliance – Translational Brain Medicine, Jülich, Germany Germany and Aachen.
Ted Abel, Department of Biology, University of Pennsylvania, Philadelphia, PA, 19104.
References
- Arbogast T, Ouagazzal AM, Chevalier C, Kopanitsa M, Afinowi N, Migliavacca E, et al. (2016). Reciprocal effects on neurocognitive and metabolic phenotypes in mouse models of 16p11.2 deletion and duplication syndromes. PLoS Genetics, 12, e1005709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker EK, Richdale AL (2015). Sleep patterns in adults with a diagnosis of high-functioning autism spectrum disorder. Sleep, 38, 1765–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzett TJ, Becker JB (1994). Sex differences in the rapid and acute effects of estrogen on striatal D2 dopamine receptor binding. Brain Research, 637, 163–172. [DOI] [PubMed] [Google Scholar]
- Brunner D, Kabitzke P, He D, Cox K, Thiede L, Hanania T, et al. (2015). Comprehensive analysis of the 16p11.2 deletion and null Cntnap2 mouse models of autism spectrum disorder. PloS One, 10, e0134572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush G (2010). Attention-deficit/hyperactivity disorder and attention networks. Neuropsychopharmacology, 35, 278– 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantero JL, Atienza M, Salas RM (2002). Human alpha oscillations in wakefulness, drowsiness period, and REM sleep: Different electroencephalographic phenomena within the alpha band. Clinical Neurophysiology, 32, 54–71. [DOI] [PubMed] [Google Scholar]
- Cohen S, Conduit R, Lockley SW, Rajaratnam SM, Cornish KM (2014). The relationship between sleep and behavior in autism spectrum disorder (ASD): A review. Journal of Neurodevelopmental Disorders, 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese S, Faraone SV, Konofal E, Lecendreux M (2009). Sleep in children with attention-deficit/hyperactivity disorder: Meta-analysis of subjective and objective studies. Journal of the American Academy of Child and Adolescent Psychiatry, 48, 894–908. [DOI] [PubMed] [Google Scholar]
- Cortesi F, Giannotti F, Ivanenko A, Johnson K (2010). Sleep in children with autistic spectrum disorder. Sleep Medicine, 11, 659–664. [DOI] [PubMed] [Google Scholar]
- Cortesi F, Giannotti F, Sebastiani T, Panunzi S, Valente D (2012). Controlled-release melatonin, singly and combined with cognitive behavioural therapy, for persistent insomnia in children with autism spectrum disorders: A randomized placebo-controlled trial. Journal of Sleep Research, 21, 700–709. [DOI] [PubMed] [Google Scholar]
- Crandall JE, McCarthy DM, Araki KY, Sims JR, Ren JQ, Bhide PG (2007). Dopamine receptor activation modulates GABA neuron migration from the basal forebrain to the cerebral cortex. The Journal of Neuroscience, 27, 3813–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN (2007). What’s wrong with my mouse: behavioral phenotyping of transgenic and knockout mice 2nd ed. Hoboken, NJ: Wiley, p 32. [Google Scholar]
- Fernandez BA, Roberts W, Chung B, Weksberg R, Meyn S, Szatmari P, et al. (2009). Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11. 2 in individuals ascertained for diagnosis of autism spectrum disorder. Journal of Medical Genetics, 47, 195–203. [DOI] [PubMed] [Google Scholar]
- Franken P, Malafosse A, Tafti M (1999). Genetic determinants of sleep regulation in inbred mice. Sleep, 22, 155– 169. [PubMed] [Google Scholar]
- Glickman G (2010). Circadian rhythms and sleep in children with autism. Neuroscience & Biobehavioral Reviews, 34, 755–768. [DOI] [PubMed] [Google Scholar]
- Goldman SE, McGrew S, Johnson KP, Richdale AL, Clemons T, Malow BA (2011). Sleep is associated with problem behaviors in children and adolescents with Autism Spectrum Disorders. Research in Autism Spectrum Disorders, 5, 1223–1229. [Google Scholar]
- Goldman SE, Surdyka K, Cuevas R, Adkins K, Wang L, Malow BA (2009). Defining the sleep phenotype in children with autism. Developmental Neuropsychology, 34, 560–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayton HM, Fernandes C, Rujescu D, Collier DA (2012). Copy number variations in neurodevelopmental disorders. Progress in Neurobiology, 99, 81–91. [DOI] [PubMed] [Google Scholar]
- Gringras P, Gamble C, Jones AP, Wiggs L, Williamson PR, Sutcliffe A, et al. (2012). Melatonin for sleep problems in children with neurodevelopmental disorders: Randomised double masked placebo controlled trial. BMJ, 345, e6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissom N, McKee S, Lidsky-Everson J, Schoch H, Havekes R, Kumar M, et al. (2014). Male-specific deficits in reinforcement learning, motivation, striatal volume, and white-matter integrity in the 16p11.2del/1 mouse model of autism [abstract]. Society for Neuroscience, Washington, DC: SfN; 2014. Abstract nr 603.10/H1. [Google Scholar]
- Hanson E, Bernier R, Porche K, Jackson FI, Goin-Kochel RP, Snyder LG, et al. (2014). The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biological Psychiatry, 77, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horev G, Ellegood J, Lerch JP, Son YEE, Muthuswamy L, Vogel H, et al. (2011). Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proceedings of the National Academy of Sciences of the United States of America, 108, 17076–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanenko A, Johnson K (2008). Sleep disturbances in children with psychiatric disorders. Seminars in Pediatric Neurology, 15, 70–78. [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Coe BP, Hersch M, Duyzend MH, Krumm N, Bergmann S, et al. (2014). A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. The American Journal of Human Genetics, 94, 415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor S, Szabo L, Varga J, Cuesta M, Morton AJ (2013). Progressive sleep and electroencephalogram changes in mice carrying the Huntington’s disease mutation. Brain, 136, 2147–2158. [DOI] [PubMed] [Google Scholar]
- Karlsson RM, Holmes A, Heilig M, Crawley JN (2005). Anxiolytic-like actions of centrally-administered neuropeptide Y, but not galanin, in C57BL/6J mice. Pharmacology Biochemistry and Behavior, 80, 427–436. [DOI] [PubMed] [Google Scholar]
- Kirov R, Brand S (2014). Sleep problems and their effect in ADHD. Expert Review of Neurotherapeutics, 14, 287–299. [DOI] [PubMed] [Google Scholar]
- Koehl M, Battle S, Meerlo P (2006). Sex differences in sleep: The response to sleep deprivation and restraint stress in mice. Sleep, 29, 1224–1231. [DOI] [PubMed] [Google Scholar]
- Kohls G, Perino MT, Taylor JM, Madva EN, Cayless SJ, Troiani V, et al. (2013). The nucleus accumbens is involved in both the pursuit of social reward and the avoidance of social punishment. Neuropsychologia, 51, 2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakowiak P, Goodlin-Jones B, Hertz-Picciotto I, Croen LA, Hansen RL (2008). Sleep problems in children with autism spectrum disorders, developmental delays, and typical development: a population-based study. Journal of Sleep Research, 17, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langen M, Bos D, Noordermeer SDS, Nederveen H, van Engeland H, Durston S (2014). Changes in the development of striatum are involved in repetitive behavior in autism. Biological Psychiatry, 76, 405–411. [DOI] [PubMed] [Google Scholar]
- Lee SH, Seo WS, Sung HM, Choi TY, Kim SY, Choi SJ, et al. (2012). Effect of methylphenidate on sleep parameters in children with ADHD. Psychiatry Investigation, 9, 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malow B, Adkins KW, McGrew SG, Wang L, Goldman SE, Fawkes D, et al. (2012). Melatonin for sleep in children with autism: a controlled trial examining dose, tolerability, and outcomes. Journal of Autism and Developmental Disorders, 42, 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mang GM, La Spada F, Emmenegger Y, Chappuis S, Ripperger JA, Albrecht U, et al. (2016). Altered sleep homeostasis in reverba knockout mice. Sleep, 39, 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane BB, Galante RJ, Jensen ST, Naidoo N, Pack AI, Wyner A (2010). Characterization of the bout durations of sleep and wakefulness. Journal of Neuroscience Methods, 193, 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano S, Bruni O, Elia M, Trovato A, Smerieri A, Verrillo E, et al. (2007). Sleep in children with autistic spectrum disorder: a questionnaire and polysomnographic study. Sleep Medicine, 9, 64–70. [DOI] [PubMed] [Google Scholar]
- Ming X, Walters AS (2009). Autism spectrum disorders, attention deficit/hyperactivity disorder, and sleep disorders. Current Opinion in Pulmonary Medicine, 15, 578–584. [DOI] [PubMed] [Google Scholar]
- Mochizuki T, Crocker A, McCormack S, Yanagisawa M, Sakurai T, Scammell TE (2004). Behavioral state instability in orexin knock-out mice. The Journal of Neuroscience, 24, 6291–6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickl-Jockschat T, Habel U, Michel TM, Manning J, Laird AR, Fox PT, et al. (2012). Brain structure anomalies in autism spectrum disorder–a meta-analysis of VBM studies using anatomic likelihood estimation. Human Brain Mapping, 33, 1470–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick-Jockschat T, Jangir VK, Grissom N, McKee SE, Schoch H, Bowman N, et al. (2015). Brain structure changes in 16p11.2 deletion mouse model [abstract]. American College of Neuropsychopharmacology, Hollywood, FL: ACNP; 2015. Abstract nr T25. [Google Scholar]
- Owen JP, Chang YS, Pojman NJ, Bukshpun P, Wakahiro MLJ, Marco EJ, et al. (2014). Aberrant white matter microstructure in children with 16p11.2 deletions. Journal of Neuroscience, 34, 6214–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Cho SC, Cho IH, Kim BN, Kim JW, Shin MS, et al. (2012). Sleep problems and their correlates and comorbid psychopathology of children with autism spectrum disorders. Research in Autism Spectrum Disorders, 6, 1068–1072. [Google Scholar]
- Patel TP, Gullotti DM, Hernandez P, O’brien WT, Capehart BP, Morrison B III., et al. (2014). An open-source toolbox for automated phenotyping of mice in behavioral tasks. Frontiers in Behavioral Neuroscience, 8, 1– 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul KN, Dugovic C, Turek FW, Laposky AD (2006). Diurnal sex differences in the sleep-wake cycle of mice are dependent on gonadal function. Sleep, 29, 1211–1223. [DOI] [PubMed] [Google Scholar]
- Pfeiffenberger C, Allada R (2012). Cul3 and the BTB adaptor insomniac are key regulators of sleep homeostasis and a dopamine arousal pathway in Drosophila. PLoS Genetics, 8, e1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portmann T, Yang M, Mao R, Panagiotakos G, Ellegood J, Dolen G, et al. (2014). Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Reports, 7, 1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucilowska J, Vithayathil J, Tavares EJ, Kelly C, Karlo JC, Landreth GE (2015). The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. The Journal of Neuroscience, 35, 3190–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi AY, Mueller S, Snyder AZ, Mukherjee P, Berman JI, Roberts TPL, et al. (2014). Opposing brain differences in 16p11.2 deletion and duplication carriers. The Journal of Neuroscience, 34, 11199–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AM, Malow BA (2011). Sleep and autism spectrum disorders. Pediatric Clinics of North America, 58, 685–698. [DOI] [PubMed] [Google Scholar]
- Robinson EB, Lichtenstein P, Anckars€ater H, Happe F, Ronald A (2013). Examining and interpreting the female protective effect against autistic behavior. Proceedings of the National Academy of Sciences of the United States of America, 110, 5258–5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenbeck A, Huether G, Ruther €E, Hajak G (1999). Nocturnal melatonin secretion and its modification by treatment in patients with sleep disorders. Advances in Experimental Medicine and Biology, 467, 89–93. [DOI] [PubMed] [Google Scholar]
- Rossignol DA, Frye RE (2011). Melatonin in autism spectrum disorders: A systematic review and meta-analysis. Developmental Medicine and Child Neurology, 53, 783–792. [DOI] [PubMed] [Google Scholar]
- Santisteban JA, Stein MA, Bergmame L, Gruber R (2014). Effect of extended-release dexmethylphenidate and mixed amphetamine salts on sleep: A double-blind, randomized, crossover study in youth with attention-deficit hyperactivity disorder. CNS Drugs, 28, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE (2010). Sleep state switching. Neuron, 68, 1023–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider H, Eisenberg D (2006). Who receives a diagnosis of attention-deficit/hyperactivity disorder in the United States elementary school population? Pediatrics, 117, e601–e609. [DOI] [PubMed] [Google Scholar]
- Scott-Van Zeeland AA, Dapretto M, Ghahremani DG, Poldrack RA, Bookheimer SY (2010). Reward processing in autism. Autism Research, 3, 53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. (2007). Strong association of de novo copy number mutations with autism. Science, 316, 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Couture J (2014). A review of the pathophysiology, etiology, and treatment of attention-deficit hyperactivity disorder (ADHD). The Annals of Pharmacotherapy, 48, 209–225. [DOI] [PubMed] [Google Scholar]
- Souders MC, Mason TBA, Valladares O, Bucan M, Levy SE, Mandell DS, et al. (2009). Sleep behaviors and sleep quality in children with autism spectrum disorders. Sleep, 32, 1566–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavropoulos N, Young MW (2011). Insomniac and Cullin-3 regulate sleep and wakefulness in Drosophila. Neuron, 72, 964–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S, et al. (2014). CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature, 505, 361–366. [DOI] [PubMed] [Google Scholar]
- Tabet AC, Pilorge M, Delorme R, Amsellem F, Pinard JM, Leboyer M, et al. (2012). Autism multiplex family with 16p11. 2p12. 2 microduplication syndrome in monozygotic twins and distal 16p11. 2 deletion in their brother. European Journal of Human Genetics, 20, 540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tordjman S, Anderson GM, Pichard N, Charbuy H, Touitou Y (2005). Nocturnal excretion of 6-sulphatoxymelatonin in children and adolescents with autistic disorder. Biological Psychiatry, 57, 134–138. [DOI] [PubMed] [Google Scholar]
- Vassalli A, Dellepiane JM, Emmenegger Y, Jimenez S, Vandi S, Plazzi G, et al. (2013). Electroencephalogram paroxysmal theta characterizes cataplexy in mice and children. Brain, 136, 1592–1608. [DOI] [PubMed] [Google Scholar]
- Watson AJ, Henson K, Dorsey SG, Frank MG (2015). The truncated TrkB receptor influences mammalian sleep. American Journal of Physiology— Regulatory, Integrative and Comparative Physiology, 308, R199–R207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. (2008). Association between microdeletion and microduplication at 16p11.2 and autism. The New England Journal of Medicine, 358, 667–675. [DOI] [PubMed] [Google Scholar]
- Werling DM, Geschwind DH (2013). Sex differences in autism spectrum disorders. Current Opinion in Neurology, 26, 146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer ME, Rising J, Galante RJ, Wyner A, Pack AI, Abel T (2013). Aging in mice reduces the ability to sustain sleep/wake states. PloS One, 8, e81880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright B, Sims D, Smart S, Alwazeer A, Alderson-Day B, Allgar V, et al. (2011). Melatonin versus placebo in children with autism spectrum conditions and severe sleep problems not amenable to behaviour management strategies: a randomised controlled crossover trial. Journal of Autism and Developmental Disorders, 41, 175–184. [DOI] [PubMed] [Google Scholar]
- Yang M, Lewis FC, Sarvi MS, Foley GM, Crawley JN (2015a). 16p11.2 deletion mice display cognitive deficits in touchscreen learning and novelty recognition tasks. Learning & Memory, 22, 622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Mahrt EJ, Lewis F, Foley G, Portmann T, Dolmetsch RE, et al. (2015b). 16p11.2 deletion syndrome mice display sensory and ultrasonic vocalization deficits during social interactions. Autism Research, 8, 507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]