

Abstract
Background
Abnormal cardiac ion channels current, including transient outward potassium current (Ito), is associated with early repolarization syndrome (ERS). Previous studies showed that mutations in SCN1Bβ both to increase the Ito current and to decrease the sodium current. Yet its role in ERS remains unknown.
Objective
To determine the role of mutations in the SCN1Bβ subunits in ERS.
Methods
We screened for mutations in the SCN1B genes from four families with ERS. Wild‐type and mutant SCN1Bβ genes were co‐expressed with wild‐type KCND3 in human embryonic kidney cells (HEK293). Whole‐cell patch‐clamp technique and co‐immunoprecipitation were used to study the electrophysiological properties and explore the underlying mechanisms.
Results
S248R and R250T mutations in SCN1Bβ were detected in 4 families’ probands. Neither S248R nor R250T mutation had significant influence on the sodium channel current density (IN a) when co‐expressed with SCN5A/WT. Co‐expression of KCND3/WT and SCN1Bβ/S248R or SCN1Bβ/R250T increased the transient outward potassium current Ito by 27.44% and 199.89%, respectively (P < 0.05 and P < 0.01, respectively) when compared with SCN1Bβ/WT. Electrophysiological properties showed that S248R and R250T mutations decreased the steady‐state inactivation and recovery from inactivation of Ito channel. Co‐immunoprecipitation study demonstrated an increased association between SCN1Bβ mutations and Kv4.3 compared with SCN1Bβ/WT (P < 0.05 and P < 0.01, respectively).
Conclusion
The S248R and R250T mutations of SCN1Bβ gene caused gain‐of‐function of Ito by associated with Kv4.3, which maybe underlie the ERS phenotype of the probands.
Keywords: early repolarization syndrome, SCN1Bβ, transient outward potassium current
1. INTRODUCTION
The early repolarization pattern (ERP) is characterized by an elevation of J‐wave >0.1 mV and sometimes involving an ST‐segment elevation in at least two contiguous leads. Recent studies have revealed that ERP is associated with a higher risk of malignant ventricular arrhythmia and sudden cardiac death (SCD).1, 2, 3, 4, 5 When a subject with ERP and malignant ventricular arrhythmias, that often known as early repolarization syndrome (ERS). Since the high prevalence of ERP in the general population (1.3%‐9.2%),6, 7, 8, 9 especially in younger physically active individuals10, 11 and male sex,12 it is significant to determine whose individuals with such common electrophysiological pattern are at risk of sudden cardiac death.
Some studies revealed that cardiac ion channel mutations play a major role in the pathogenesis of malignant ERPs. Mutations in the inward‐rectifier ATP‐dependent K+ channel current (IKATP),13, 14, 15 L‐type calcium current (ICaL)16, 17 and transient outward potassium current (Ito)18, 19 can lead to ERPs. In inherited families, mutations in seven different genes (KCNJ8, CACNA1C, KCND3, KChip2, SCN5A, ABCC9, Ankyrin‐2) have been associated with ERPs.13, 14, 15, 16, 17, 20, 21, 22, 23
The transient outward potassium channel (Ito) is a multi‐subunit protein complex comprised of pore‐forming α‐subunits and auxiliary β‐subunits.24 In humans, Kv4.3 encodes the α‐subunits of Ito. 25 In human ventricular cells, Ito currents mediate the early phase of action potential repolarization,18 which can reduce the action potential duration by accelerating the early repolarization velocity and progressively suppressing the voltage of plateau phase.19 Increasing of Ito currents in region during initial ventricular repolarization can result in a J‐wave on the ECG.26 Therefore, disorders in Ito currents might be underlying mechanism of ERS.
SCN1B encodes the cardiac sodium channel β‐subunit.27 It is comprised of large extracellular immunoglobulin‐like domains, a single transmembrane‐spanning segment, and intracellular C‐terminal domains. SCN1B has two kinds of transcripts, SCN1B and SCN1Bβ, which encodes Navβ1 and Navβ1b, respectively.27 Functional analysis indicated that β‐subunit encodes by SCN1B involved in modulation of sodium channel gating and voltage dependence,28, 29 expression of sodium channel at the cell surface,30 and cell adhesion.31
Here, we reported a mutation in the SCN1Bβ gene, which encodes the regulatory β‐subunits of the transient outward potassium current (Ito), identified in four families with ERS. We studied whether mutant in the SCN1Bβ was associated with ERS and explore the possible mechanisms. Electrophysiology modification by SCN1Bβ mutant was evaluated using patch‐clamp technology.
2. METHODS
2.1. Genetic analysis
Genomic DNA used for genetic analysis was isolated from peripheral blood samples. The protein coding sequences of the SCN1B genes were amplified by polymerase chain reaction (PCR) and directed sequenced. The DNA sequence was then compared with the reference sequence of NM_001037 (SCN1B isoform a), NM_199037 (SCN1B isoform b) and NM_001321605 (SCN1B isoform c).
2.2. Plasmid constructions and cell transfection
Human embryonic kidney cells (HEK293) were obtained from Type Culture Collection of Chinese Academic of Sciences. HEK293 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS). cDNA encoding Nav1.5 (SCN5A, NM_198056) was amplified by PCR and cloned into mammalian expression vector pENTER. SCN1bβ (NM_199037) cDNA was subcloned into pIRES2‐EGFP. The p.S248R and p.R250T mutations were introduced by site‐directed mutagenesis using the QuikChange II kit (Stratagene, CA, USA). Kv4.3 (KCND3, NM_004980) cDNA was subcloned into pcDNA3.1. Nav1.5 and SCN1Bβ or Kv4.3 and SCN1Bβ plasmids were transiently co‐transfected (1:1 molar ratio) into HEK293 cells using Lipofectamine 2000 (Invitrogen Life Technologies Inc., CA, USA) according to the manufacturer's instructions.
2.3. Electrophysiology
Electrophysiological studies were performed 48 hours after transfection. Membrane currents were recorded by whole‐cell patch‐clamp technologies using a EPC‐10 amplifier (HEKA Instruments, Lambrecht, Germany) at room temperature (20‐25°C). The extracellular solution contained (in mmol/L): 140 NaCl, 5 KCl, 1 MgCl2, 1.8 CaCl2, 10 HEPES, 10 Glucose (PH 7.40 with NaOH). Pipette solution for INa contained (in mmol/L): 120 CsF, 20 CsCl, 2 EGTA, 5 HEPES, 5 Na2‐ATP (PH 7.20 with CsOH). For Ito currents, pipette solution contained (in mmol/L): 110 K‐aspartate, 20 KCl, 2 MgCl2, 10 HEPES, 5 EGTA, 5 Na2‐ATP (PH 7.20 with KOH). Pipettes were pulled from borosilicate glass capillaries using a micropipette puller (P‐97, Sutter Instruments, CA, USA). The tip resistances of patch pipettes ranged from 3‐5MΩ when filled with the pipette solution. Currents were filtered with a four pole Bessel filter at 5 kHz and digitized at 50 kHz. Series resistance was electronically compensated at around 80%.
INa currents were elicited by depolarizing pulses ranging from −90 mV to +40 mV in 10 mV increments with a holding potential (HP) at −120 mV. Peak currents were measured and INa densities (pA/pF) were obtained by dividing the peak INa by the cell capacitance obtained. The plots of voltage dependent steady state activation and inactivation were fitted by Boltzmann equation: y = 1/[1 + exp(V−V 1/2)/k], where V 1/2 is the voltage at which sodium current is half‐maximally activated, and k was the slope factor. To assess the time course of recovery from inactivation, a prepulse to 0 mV for 20 ms was followed by a recovery interpulse of variable duration (from 0.25 to 750 ms) to −120 mV and then a 25 ms test pulse to 0 mV to determine the fraction of recovered channels. To analyse the kinetics of recovery from inactivation, the time constants were obtained by fitting to a double‐exponential equation: y = A*[1‐exp(−t/τf)]+(1‐A)*[1‐exp(−t/τs)].
Ito currents were elicited from a HP of −80 mV with depolarizing voltage pulses from −80 mV to +80 mV for 400 ms. Current density (pA/pF) was calculated from the ratio of current amplitude to cell capacitance. Peak currents were normalized to the maximum peak Ito amplitude. Normalized activation and inactivation curves were fit with a Boltzmann equation: y = 1/[1 + exp(V−V 1/2)/k], where V 1/2 is the voltage at which sodium current is half‐maximally activated, and k was the slope factor. Recovery from inactivation was assessed by a standard paired pulse protocol: a 500 ms test pulse to +50 mV (P1) was followed by a variable recovery interval at 380 mV, then by a second test pulse to +50 mV (P2). The plot of P2/P1 was then fit with two exponential to determine the time constants for recovery, using the equation: y = A*[1‐exp(−t/τf)]+(1‐A)*[1‐exp(−t/τs)].
2.4. Co‐immunoprecipitation and western blot
48 hours after transfection, HEK293 cells were scraped off from 100 mm dishes. The membrane proteins were extracted using a Membrane Protein Extraction Kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. Cell membrane lysates were then incubated with anti‐Kv4.3 antibodies (Sigma, USA) overnight at 4°C with rotation. Immune complexes were incubated with Protein G Dynabeads (Life technologies, USA) for 1 hour at room temperature with rotation and washed three time with PBS including 0.02% Tween‐20 to removed unbound material. The final pellet was boiled in 1 × SDS buffer and separated by 10% SDS‐PAGE. Western blot analysis was carried out using anti‐Kv4.3 antibodies (1:1000, Sigma, USA) and anti‐SCN1Bβ antibodies (1:500, Abcam, USA). The density of each band was quantified using Image J software.
2.5. Statistical analysis
Data were presented as Mean ± SEM. Statistical comparisons were analysed using two‐tailed Student's t‐test and ANOVA with Student‐Newman‐Keuls test. A P value less than 0.05 was considered statistically significant.
3. RESULTS
3.1. Clinical data and genetic analysis
Clinical data of the four families was showed in Table 1. Four family pedigrees with ERS were showed in Figure 1A. Figure 1B showed a 12‐lead ECG of a 14‐year‐old boy from Family 1 (arrow in Figure 1A). The ECG showed J‐point elevation in leads II, III and aVF. His father experienced sudden cardiac arrest at the age of 37 while chatting with others at afternoon. The emergency team recorded a ventricular fibrillation ECG from him and he was defibrillated immediately. There was no family history of SCD or syncope. The patient denied coronary artery disease, hypertension and diabetes mellitus. The echocardiogram was normal with no structural cardiac disease. No subsequent cardiac events have occurred over the next 8 years.
Table 1.
Clinical characteristics of probands
| ID | Sex | Age (y) | HR (bpm) | J‐wave location | J‐wave amplitude (mV) | ST elevation (mV) | R (mV) | QRS (ms) | QTc (ms) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 25 | 66 | V 1‐V 4 | 0.35 | 0.25 | 0.85 | 100 | 432 |
| 2 | M | 37 | 76 | II, III, AVF, V 1‐V 3 | 0.1 | 0 | 1.5 | 80 | 360 |
| 3 | M | 32 | 72 | I, aVL | 0.1 | 0.1 | 0.9 | 60 | 394 |
| 4 | M | 71 | 75 | II, III, AVF | 0.1 | 0 | 0.8 | 70 | 391 |
Figure 1.
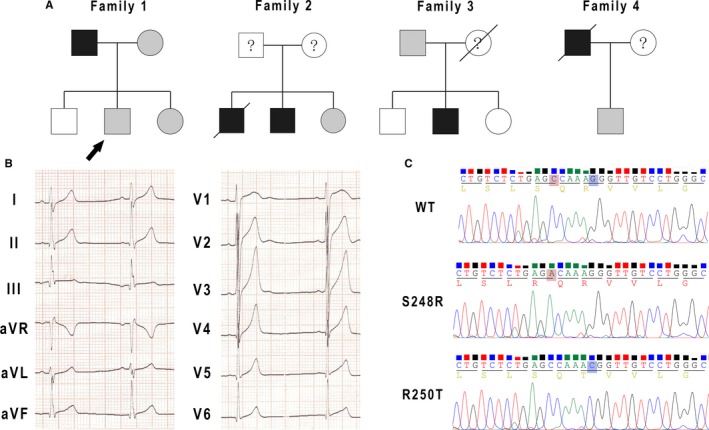
Genetic analyses and ECG in four families with early repolarization syndrome. A, Four family pedigrees with early repolarization syndrome (ERS). Black symbol: early repolarization pattern with ventricular fibrillation events. Grey symbol: early repolarization pattern without ventricular fibrillation events. B, Twelve‐lead ECG in a 14‐year‐old boy in Family 1 (arrow). C, DNA sequencing traces (chromatograms) for S248R and R250T variants identified in SCN1Bβ
Screening of SCN1Bβ in the four families revealed four mutations. Two were in UTR three of SCN1Bβ and two (c.C744A and c.G749C) were non‐synonymous mutation in exon three. Polymerase chain reaction (PCR) based sequencing analysis revealed a C‐to‐A replacement at nucleotide 744 and a G‐to‐C replacement at nucleotide 749 (Figure 1C), which result in a serine (S) to arginine (R) at residue 248 (S248R) and an arginine (R) to threonine (T) at residue 250 (R250T).
3.2. Electrophysiological characterization of SCN5A co‐expressed with SCN1Bβ/WT, SCN1Bβ/S248R and SCN1Bβ/R250T
In order to assess the effects of S248R and R250T mutation on Nav1.5 channel function, SCN5A/WT + SCN1Bβ/WT, SCN5A/WT + SCN1Bβ/S248R and SCN5A/WT + SCN1Bβ/R250T were expressed in HEK293 cells. Electrophysiological parameters of different channels were addressed by whole cell patch‐clamp technique. Figure 2A‐C showed macroscopic currents recorded at voltage in the range −120 to +80 mV from a holding potential of −120 mV. Figure 2D showed the voltage dependence of the averaged current density. S248R showed a decrease in the peak current density compared with WT (P < 0.05). No significant difference in peak current density was observed in R250T compared with WT. Some minor differences were observed in steady‐state activation, inactivation and recovery from inactivation (Table 2). Both S248R and R250T showed a negative voltage shift of steady‐state inactivation compared with WT (Figure 2F. V 1/2 = −72.62 ± 0.64 mV, n = 9 for WT; −75.94 ± 0.62 mV, n = 13 for S248R; −75.04 ± 0.37 mV, n = 12 for R250T; P < 0.01 respectively.). S248R gave a minor positive voltage shift of steady‐state activation (V 1/2 = −54.10 ± 1.06 mV, n = 7 for WT; −51.81 ± 0.99 mV, n = 9 for S248R; P < 0.01) (Figure 2E). Recovery from inactivation was obtained using double pulse protocol and fitted with two‐exponential equation. τf was similar among WT and mutants, whereas S248R had a decreased in τs (18.02 ± 8.17, n = 13 for WT; 6.23 ± 0.86, n = 14 for S248R; P < 0.01) (Figure 2G).
Figure 2.
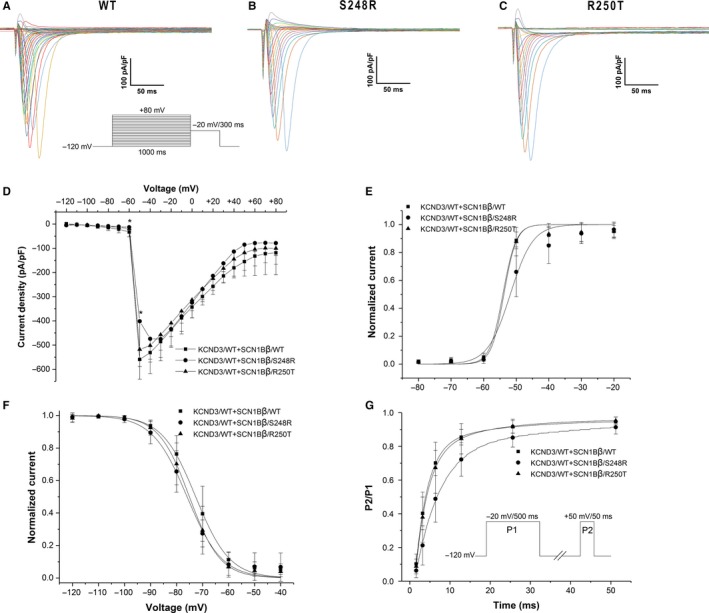
Sodium Currents recorded on HEK293 cells co‐expressed of SCN5A/WT and SCN1Bβ. A‐C, macroscopic currents recorded at Vm in the range −120 to +80 mV from a holding potential of −120 mV. Protocol used was shown in inset. D, Current‐voltage (I‐V) relationship of WT, S248R and R250T. (n = 9, 13 and 14, respectively, *P < 0.05 vs WT). E, Voltage dependent steady‐state activation (V 1/2 = −54.10 ± 1.06 mV, n = 7 for WT; −51.81 ± 0.99 mV, n = 9 for S248R; −53.79 ± 1.02 mV, n = 6 for R250T). F: Voltage dependent steady‐state inactivation (V 1/2 = −72.62 ± 0.64 mV, n = 9 for WT; −75.94 ± 0.62 mV, n = 13 for S248R; −75.04 ± 0.37 mV, n = 12 for R250T). E and F plots were fitted by Boltzmann equation: y = 1/[1 + exp(V−V 1/2)/k], V 1/2 = voltage at which sodium current is half‐maximally activated, k = slope factor. G, Recovery from inactivation (n = 13, 14 and 14 for WT, S248R and R250T, respectively). Protocol used was shown in inset. The currents recorded at P2 were normalized to that at P1. Two‐exponential equation was used to fit the plot
Table 2.
Gating kinetics parameters of INa in HEK293 cells co‐expressed of SCN5A/WT and SCN1Bβ
| Groups | Activation | Inactivation | Recovery | ||||||
|---|---|---|---|---|---|---|---|---|---|
| V 1/2 (mV) | k | n | V 1/2 (mV) | k | n | τf (ms) | τs (ms) | n | |
| SCN5A/WT + SCN1Bβ/WT | −54.10 ± 1.06 | 2.01 ± 0.48 | 7 | −72.62 ± 0.45 | 6.50 ± 0.40 | 9 | 2.98 ± 0.19 | 18.02 ± 8.17 | 13 |
| SCN5A/WT + SCN1Bβ/S248R | −51.81 ± 0.99a | 3.47 ± 1.00a | 9 | −75.94 ± 0.62a | 6.67 ± 0.55 | 13 | 2.31 ± 1.99 | 6.23 ± 0.86a | 14 |
| SCN5A/WT + SCN1Bβ/R250T | −53.79 ± 1.02 | 1.93 ± 0.48 | 6 | −75.04 ± 0.37a | 5.91 ± 0.32a | 12 | 3.04 ± 0.07 | 14.56 ± 1.45 | 14 |
Values are Mean ± SEM, V 1/2: voltage of half‐maximally activated or inactivation, k: slope factor.
P < 0.01 vs WT.
3.3. Electrophysiological characterization of KCND3/WT co‐expressed with SCN1Bβ/WT, SCN1Bβ/S248R and SCN1Bβ/R250T
To evaluate whether SCN1Bβ has an effect in modulating Ito, we compared the electrophysiological properties of KCND3/WT co‐expressed with SCN1Bβ/WT, SCN1Bβ/S248R and SCN1Bβ/R250T in HEK293 cells. Figure 3A‐C displayed the representative current traces of WT and mutants. Compared with KCND3/WT + SCN1Bβ/WT, co‐expressed with mutants of SCN1Bβ showed an increase in peak current density (238.28 ± 39.12 pA/pF, n = 9 for WT; 303.67 ± 76.30 pA/pF, n = 8 for S248R; 714.57 ± 90.43 pA/pF, n = 13 for R250T. P < 0.05 between WT and S248R. P < 0.01 between WT and R250T). At ‐80 mV, co‐expressed of KCND3/WT with SCN1Bβ/S248R and SCN1Bβ/R250T result in an increase in Ito density by 27.44% and 199.89%, respectively (P < 0.05 and P < 0.01, respectively.) (Figure 3E). Steady‐state activation and inactivation were fitting with Boltzman equation. The V 1/2 and slope factors were not statistically significant among WT and mutants (Table 3). Both S248R and R250T gave a positive shift in the steady‐state inactivation curve when compared to WT. (−49.52 ± 0.41 mV, n = 9 for WT; −39.25 ± 0.61 mV, n = 8 for S248R; −40.39 ± 0.59 mV, n = 12 for R250T; P < 0.01 respectively). (Figure 3G). Figure 3H showed recovery curves of WT and mutants. Co‐expressed with SCN1Bβ/S248R gave slightly decrease in time constants when compared to SCN1Bβ/WT (295.11 ± 3.44, n = 8 for WT; 223.75 ± 2.04, n = 6 for S248R; P < 0.01) (Table 3), whereas co‐expressed with SCN1Bβ/R250T result in a markedly reduce in time constants as compared with WT group (295.11 ± 3.44, n = 8 for WT; 150.18 ± 2.91, n = 10 for R250T, P < 0.01).
Figure 3.
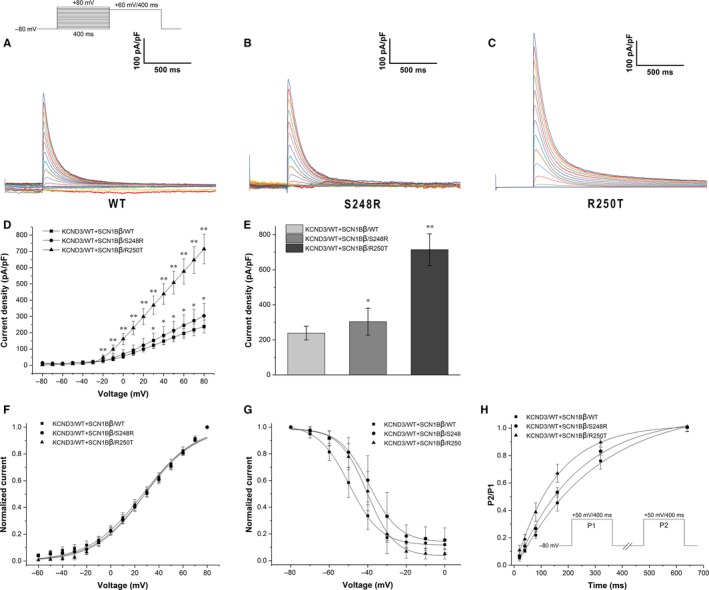
Transient outward potassium current recorded on HEK293 cells co‐expressed of KCND3/WT and SCN1Bβ. A‐C, macroscopic currents recorded at Vm in the range −80 to +80 mV from a holding potential of −80 mV. Protocol used was shown upside. D, Current‐voltage (I‐V) relationship of WT, S248R and R250T. (n = 9, 8 and 12, respectively. *P < 0.05 vs WT. **P < 0.01 vs WT). E, the mean peak Ito current densities recorded on repolarization to −80 mV from each group. *P < 0.05 vs WT; **P < 0.01 vs WT. F, Voltage dependent steady‐state activation (V 1/2 = 26.58 ± 1.15 mV, n = 9 for WT; 28.29 ± 1.05 mV, n = 7 for S248R; 28.24 ± 1.04 mV, n = 12 for R250T). G, Voltage dependent steady‐state inactivation (V 1/2 = −49.52 ± 0.41 mV, n = 9 for WT; −39.25 ± 0.61 mV, n = 8 for S248R; −40.39 ± 0.59 mV, n = 12 for R250T). F and G plots were fitted by Boltzmann equation: y = 1/[1 + exp(V−V 1/2)/k], V 1/2: voltage at which Ito is half‐maximally activated, k = slope factor. H, Recovery from inactivation (n = 8, 6 and 10 for WT, S248R and R250T respectively). Protocol used was shown in inset. The currents recorded at P2 were normalized to that at P1. Two‐exponential equation was used to fit the plot
Table 3.
Gating kinetics parameters of Ito in HEK293 cells co‐expressed of KCND3/WT and SCN1Bβ
| Groups | Activation | Inactivation | Recovery | ||||||
|---|---|---|---|---|---|---|---|---|---|
| V 1/2 (mV) | k | n | V 1/2 (mV) | k | n | τf (ms) | τs (ms) | n | |
| KCND3/WT + SCN1Bβ/WT | 26.58 ± 1.15 | 21.98 ± 1.07 | 9 | −49.52 ± 0.41 | 8.12 ± 0.38 | 9 | 295.11 ± 3.44 | 295.11 ± 3.44 | 8 |
| KCND3/WT + SCN1Bβ/S248R | 28.29 ± 1.05 | 21.39 ± 0.98 | 7 | −39.25 ± 0.61a | 7.76 ± 0.57 | 8 | 223.75 ± 2.04a | 223.75 ± 2.04a | 6 |
| KCND3/WT + SCN1Bβ/R250T | 28.24 ± 1.04 | 20.41 ± 0.95 | 12 | −40.39 ± 0.59a | 7.10 ± 0.54a | 12 | 150.18 ± 2.91a | 150.18 ± 2.91a | 10 |
Values are Mean ± SEM. V 1/2: voltage of half‐maximally activated or inactivation, k: slope factor.
P < 0.01 vs WT.
3.4. Co‐IP study
To test whether SCN1Bβ has some direct effects on Kv4.3, we next used Co‐IP to assess the relationship. KCND3/WT was co‐expressed with SCN1Bβ/WT, SCN1Bβ/S248R or SCN1Bβ/R250T in HEK293 cells and isolated by pull‐down using an antibody to Kv4.3. Figure 4A showed the association between Kv4.3 (~75 kD, top) and SCN1Bβ (~30 kD, bottom) when co‐expressed. Compared with KCND3/WT + SCN1Bβ/WT, co‐expressed with SCN1Bβ/R250T resulted in a significant increase of SCN1Bβ to Kv4.3. However, the amount of SCN1Bβ interact with Kv4.3 was not significant different between WT and S248R (Figure 4B).
Figure 4.
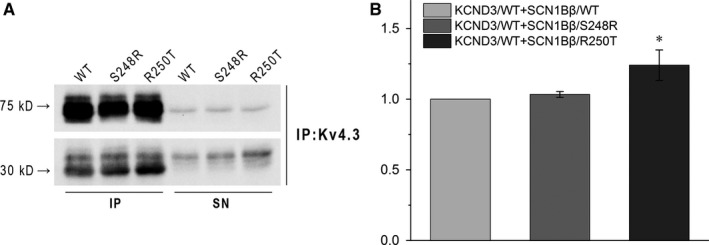
Co‐immunoprecipitation study indicated direct interaction of KCND3 and SCN1Bβ subunits. HEK293 cells were co‐transfected with KCND3/WT and SCN1Bβ (WT, S248R, R250T). Cells were lysed and total protein extracts were immunoprecipitated using anti‐KCND3 and then immunoblot with anti‐KCND3 and anti‐SCN1Bβ. A: Representative western blots of KCND3 (75 kD arrow) and SCN1Bβ (30 kD arrow). IP: immunoprecipitated pellet; SN: supernatant. B: Percentage of SCN1Bβ (WT, S248R, R250T) co‐immunoprecipitation related to the total amount of KCND3/WT immunoprecipitated. *P < 0.05 vsWT
4. DISCUSSION
In the present study, we characterized two mutations in SCN1Bβ among four families with ERS using patch‐clamp technique and Co‐IP. Electrophysiological study showed that except some minor changes in sodium current, SCN1Bβ/R250T co‐expressed with KCND3 resulted in markedly greater Ito current density when compared with SCN1Bβ/WT + KCND3. Electrophysiological properties showed that co‐expressed with SCN1Bβ/S248R or SCN1Bβ/R250T produced Ito current with altered kinetics of steady‐state inactivation and recovery from inactivation.
Ito plays an important role in the early repolarization phase and abbreviates action potential duration.19 Inhibition of Ito exerts an ameliorative effect in the setting of ERS by producing an inward shift in the balance of current during the early phases of the epicardial action potential.32 Our results showed that S248R and R250T mutations in SCN1Bβ could increase Ito channel activities when co‐expressed with KCND3. As both of the mutations of SCN1Bβ we described resulted in an increase of Ito channel current, we recognized that these mutations might contribute to ERS. However, whether the degree of augment in Ito current is correlate with the clinical presentation and prognosis of ERS is still unknown.
SCN1B gene encodes sodium channel beta1 subunit (Navβ1) and sodium channel beta1b subunit (Navβ1b), which serves as auxiliary subunits of Nav1.5. In 2002, Deschenes et al33 found that Navβ1 increased the current density of Ito by modulating the gating kinetics of Kv4.3. Silencing of Navβ1 produced a reduction in Kv4.2, Kv4.3 and KChIP2 mRNA and protein.34 These findings suggested a structural and functional association between Nav1.5 and Kv4.3 via Navβ1. Recently a study reported that R214Q mutation in SCN1Bβ linked to Brugada syndrome and sudden infant death syndrome via a combined loss‐of‐function of Nav1.5 current and gain‐of‐function of Ito current,35 which indicated a similar modulation effect of Navβ1b on Kv4.3 as Navβ1 does.
In the present study, our results showed that co‐expressed with SCN1Bβ/S248R or SCN1Bβ/R250T increased Ito current density by slowing down steady‐state inactivation and accelerating recovery from inactivation compared with co‐expressed with SCN1Bβ/WT as previous study. However, no significant sodium current density was observed in this study thought some minor changes in channel gating kinetics. The probands included in our study showed slurred or notched J‐wave on surfaced electrocardiogram. One of underlying mechanism of J‐wave is prominent voltage gradients in early repolarization between endocardium and epicardium,36 which partly due to significant downregulation of Ito in endocardium in comparison with that of midmyocardium and epicardium.37 Thus, gain‐of‐function mutation of Ito channel current in epicardium was expected to produce similar voltage gradients and J‐wave.
The effects of SCN1Bβ variants on Nav1.5 currents had been described earlier. Some studied reported that SCN1Bβ variants reduced sodium currents by accelerating recovery from inactivation and decreasing the slow inactivation rate.29, 38 In contrast, the mutations we reported in the present study could not alter Nav1.5 channel currents significantly. We have noted a previously study identified the same mutations of SCN1Bβ in three asymptomatic members from a family with Brugada syndrome and sick sinus syndrome.39 Together, it suggested that the S248R and R250T mutations of SCN1Bβ maybe not pathogenic to Nav1.5 function.
Both of the mutations, S248R and R250T, located at the C‐terminal of SCN1Bβ. This hydrophobic region included residues from 243 to 262, which serve as a transmembrane domain.40 To date, few mutations in this area were characterized by functional analysis. In present study, we observed an influence in steady‐state inactivation and recovery from inactivation of Ito currents. Previously study reported that co‐expressed with SCN1Bβ/WT slowed recovery of Ito from inactivation when compared with that of KCND3/WT alone.35 Besides, Co‐IP study showed an impaired interaction between KCND3 and SCN1Bβ/R250T. Therefore, we hypothesis that mutation in this area may affect association between KCND3 and SCN1Bβ, either in direct or indirect manners.
In summary, we found that the S248R and R250T mutations in SCN1Bβ gene causes gain‐of‐function of Ito by associated with Kv4.3. Our findings suggested that the mutations maybe underlie the ERS phenotype of the probands, and SCN1Bβ maybe one of the possible modulatory genes associated to ERS.
CONFLICT OF INTEREST
All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.
Yao H, Fan J, Cheng Y‐J, et al. SCN1Bβ mutations that affect their association with Kv4.3 underlie early repolarization syndrome. J Cell Mol Med. 2018;22:5639–5647. 10.1111/jcmm.13839
Funding information
This work was supported by National Natural Science Foundation of China (No. 81370285), and Guangzhou City Science and Technology Program (No. 201508020057) to Dr. Wu.
Yao and Fan contributed equally to this work.
REFERENCES
- 1. Steinfurt J, Odening KE. Early repolarization: a risk factor in Brugada syndrome. J Am Coll Cardiol. 2015;66(2):205‐206. [DOI] [PubMed] [Google Scholar]
- 2. Mahida S, Derval N, Sacher F, et al. Role of electrophysiological studies in predicting risk of ventricular arrhythmia in early repolarization syndrome. J Am Coll Cardiol. 2015;65(2):151‐159. [DOI] [PubMed] [Google Scholar]
- 3. Wu SH, Lin XX, Cheng YJ, Qiang CC, Zhang J. Early repolarization pattern and risk for arrhythmia death: a meta‐analysis. J Am Coll Cardiol. 2013;61(6):645‐650. [DOI] [PubMed] [Google Scholar]
- 4. Junttila MJ, Sager SJ, Tikkanen JT, Anttonen O, Huikuri HV, Myerburg RJ. Clinical significance of variants of J‐points and J‐waves: early repolarization patterns and risk. Eur Heart J. 2012;33(21):2639‐2643. [DOI] [PubMed] [Google Scholar]
- 5. Tikkanen JT, Junttila MJ, Anttonen O, et al. Early repolarization: electrocardiographic phenotypes associated with favorable long‐term outcome. Circulation. 2011;123(23):2666‐2673. [DOI] [PubMed] [Google Scholar]
- 6. Barakoti MP, Karki A, Chaulagain MK, Karki DB. Prevalence of early repolarization patterns in adults. Kathmandu Univ Med J (KUMJ). 2016;14(55):235‐238. [PubMed] [Google Scholar]
- 7. Rohel G, Perrier E, Delluc A, et al. Progression of early repolarization patterns at a four year follow‐up in a female flight crew population: implications for aviation medicine. Ann Noninvasive Electrocardiol. 2017;22(6):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun GZ, Ye N, Chen YT, Zhou Y, Li Z, Sun YX. Early repolarization pattern in the general population: prevalence and associated factors. Int J Cardiol. 2017;230:614‐618. [DOI] [PubMed] [Google Scholar]
- 9. O'Neal WT, Wang YG, Wu HT, et al. Electrocardiographic J wave and cardiovascular outcomes in the general population (from the Atherosclerosis Risk In Communities Study). Am J Cardiol. 2016;118(6):811‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klatsky AL, Oehm R, Cooper RA, Udaltsova N, Armstrong MA. The early repolarization normal variant electrocardiogram: correlates and consequences. Am J Med. 2003;115(3):171‐177. [DOI] [PubMed] [Google Scholar]
- 11. Walsh JR, Ilkhanoff L, Soliman EZ, et al. Natural history of the early repolarization pattern in a biracial cohort: CARDIA (Coronary Artery Risk Development in Young Adults) Study. J Am Coll Cardiol. 2013;61(8):863‐869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tikkanen JT, Anttonen O, Junttila MJ, et al. Long‐term outcome associated with early repolarization on electrocardiography. N Engl J Med. 2009;361(26):2529‐2537. [DOI] [PubMed] [Google Scholar]
- 13. Barajas‐Martinez H, Hu D, Ferrer T, et al. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm. 2012;9(4):548‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Medeiros‐Domingo A, Tan BH, Crotti L, et al. Gain‐of‐function mutation S422L in the KCNJ8‐encoded cardiac K (ATP) channel Kir6.1 as a pathogenic substrate for J‐wave syndromes. Heart Rhythm. 2010;7(10):1466‐1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Delaney JT, Muhammad R, Blair MA, et al. A KCNJ8 mutation associated with early repolarization and atrial fibrillation. Europace. 2012;14(10):1428‐1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Watanabe H, Minamino T. Role of mutations in L‐type calcium channel genes in Brugada syndrome, early repolarization syndrome, and idiopathic ventricular fibrillation associated with right bundle branch block. Circ J. 2013;77(7):1689‐1690. [DOI] [PubMed] [Google Scholar]
- 17. Liu X, Shen Y, Xie J, et al. A mutation in the CACNA1C gene leads to early repolarization syndrome with incomplete penetrance: a Chinese family study. PLoS One. 2017;12(5):e177532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Decher N, Barth AS, Gonzalez T, Steinmeyer K, Sanguinetti MC. Novel KChIP2 isoforms increase functional diversity of transient outward potassium currents. J Physiol. 2004;557(Pt 3):761‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hoppe UC, Johns DC, Marban E, O'Rourke B. Manipulation of cellular excitability by cell fusion: effects of rapid introduction of transient outward K+ current on the guinea pig action potential. Circ Res. 1999;84(8):964‐972. [DOI] [PubMed] [Google Scholar]
- 20. Krogh BA, Pedersen LN, Nielsen JC, Jensen HK. Ankyrin‐2 variants associated with idiopathic ventricular fibrillation storm in patients with intermittent early repolarization pattern. HeartRhythm Case Rep. 2015;1(5):337‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hu D, Barajas‐Martinez H, Terzic A, et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol. 2014;171(3):431‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo Q, Ren L, Chen X, et al. A novel mutation in the SCN5A gene contributes to arrhythmogenic characteristics of early repolarization syndrome. Int J Mol Med. 2016;37(3):727‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li N, Wang R, Hou C, Zhang Y, Teng S, Pu J. A heterozygous missense SCN5A mutation associated with early repolarization syndrome. Int J Mol Med. 2013;32(3):661‐667. [DOI] [PubMed] [Google Scholar]
- 24. Kuo HC, Cheng CF, Clark RB, et al. A defect in the Kv channel‐interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell. 2001;107(6):801‐813. [DOI] [PubMed] [Google Scholar]
- 25. Dixon JE, Shi W, Wang HS, et al. Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ Res. 1996;79(4):659‐668. [DOI] [PubMed] [Google Scholar]
- 26. Yan GX, Lankipalli RS, Burke JF, Musco S, Kowey PR. Ventricular repolarization components on the electrocardiogram: cellular basis and clinical significance. J Am Coll Cardiol. 2003;42(3):401‐409. [DOI] [PubMed] [Google Scholar]
- 27. Meadows LS, Isom LL. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res. 2005;67(3):448‐458. [DOI] [PubMed] [Google Scholar]
- 28. Nguyen HM, Miyazaki H, Hoshi N, et al. Modulation of voltage‐gated K+ channels by the sodium channel beta1 subunit. Proc Natl Acad Sci U S A. 2012;109(45):18577‐18582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Riuro H, Campuzano O, Arbelo E, et al. A missense mutation in the sodium channel beta1b subunit reveals SCN1B as a susceptibility gene underlying long QT syndrome. Heart Rhythm. 2014;11(7):1202‐1209. [DOI] [PubMed] [Google Scholar]
- 30. Baroni D, Picco C, Barbieri R, Moran O. Antisense‐mediated post‐transcriptional silencing of SCN1B gene modulates sodium channel functional expression. Biol Cell. 2014;106(1):13‐29. [DOI] [PubMed] [Google Scholar]
- 31. Patino GA, Brackenbury WJ, Bao Y, et al. Voltage‐gated Na+ channel beta1B: a secreted cell adhesion molecule involved in human epilepsy. J Neurosci. 2011;31(41):14577‐14591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Patocskai B, Barajas‐Martinez H, Hu D, Gurabi Z, Koncz I, Antzelevitch C. Cellular and ionic mechanisms underlying the effects of cilostazol, milrinone, and isoproterenol to suppress arrhythmogenesis in an experimental model of early repolarization syndrome. Heart Rhythm. 2016;13(6):1326‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Deschenes I, Tomaselli GF. Modulation of Kv4.3 current by accessory subunits. FEBS Lett. 2002;528(1–3):183‐188. [DOI] [PubMed] [Google Scholar]
- 34. Deschenes I, Armoundas AA, Jones SP, Tomaselli GF. Post‐transcriptional gene silencing of KChIP2 and Navbeta1 in neonatal rat cardiac myocytes reveals a functional association between Na and Ito currents. J Mol Cell Cardiol. 2008;45(3):336‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu D, Barajas‐Martinez H, Medeiros‐Domingo A, et al. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Na(v)1.5 and K(v)4.3 channel currents. Heart Rhythm. 2012;9(5):760‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93(2):372‐379. [DOI] [PubMed] [Google Scholar]
- 37. Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002;283(3):H1031‐H1041. [DOI] [PubMed] [Google Scholar]
- 38. Yuan L, Koivumaki JT, Liang B, et al. Investigations of the Navbeta1b sodium channel subunit in human ventricle; functional characterization of the H162P Brugada syndrome mutant. Am J Physiol Heart Circ Physiol. 2014;306(8):H1204‐H1212. [DOI] [PubMed] [Google Scholar]
- 39. Aoki H, Nakamura Y, Ohno S, Makiyama T, Horie M. Cardiac conduction defects and Brugada syndrome: a family with overlap syndrome carrying a nonsense SCN5A mutation. J Arrhythm. 2017;33(1):35‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qin N, D'Andrea MR, Lubin ML, Shafaee N, Codd EE, Correa AM. Molecular cloning and functional expression of the human sodium channel beta1B subunit, a novel splicing variant of the beta1 subunit. Eur J Biochem. 2003;270(23):4762‐4770. [DOI] [PubMed] [Google Scholar]