

Abstract
Background
Anticoagulation in patients with malignancy and atrial fibrillation is challenging because of enhanced risks for thrombosis and bleeding and the frequent need for invasive procedures. Data on direct oral antagonists in such patients are sparse.
Methods and Results
The ENGAGE AF‐TIMI 48 (Effective Anticoagulation With Factor Xa Next Generation in Atrial Fibrillation–Thrombolysis in Myocardial Infarction Study 48) trial randomized 21 105 patients with atrial fibrillation to edoxaban or warfarin. Patients with malignancy, defined as a postrandomization new diagnosis or recurrence of remote cancer, were followed up over a median of 2.8 years. Adjusted Cox proportional hazard models were used to evaluate the safety and efficacy of edoxaban versus warfarin. Over a median of 495 days (interquartile range, 230–771 days), 1153 patients (5.5%) were diagnosed with new or recurrent malignancy, most commonly involving the gastrointestinal tract (20.6%), prostate (13.6%), and lung (11.1%). Malignancy was associated with increased risk of death (adjusted hazard ratio [HR], 3.12; 95% confidence interval [CI], 2.78–3.50) and major bleeding (adjusted HR, 2.45; 95% CI, 2.07–2.89), but not stroke/systemic embolism (adjusted HR, 1.08; 95% CI, 0.83–1.42). Relative outcomes with higher‐dose edoxaban versus warfarin were consistent regardless of malignancy status for stroke/systemic embolism (HR, 0.60 [95% CI, 0.31–1.15] for malignancy versus HR, 0.89 [95% CI, 0.76–1.05] for no malignancy; interaction P=0.25) and major bleeding (HR, 0.98 [95% CI, 0.69–1.40] for malignancy versus HR, 0.79 [95% CI, 0.69–1.05] for no malignancy; interaction P=0.31). There was, however, a significant treatment interaction for the composite ischemic end point (ischemic stroke/systemic embolism/myocardial infarction), with greater efficacy of higher‐dose edoxaban versus warfarin in patients with malignancy (HR, 0.54; 95% CI, 0.31–0.93) compared with no malignancy (HR, 1.02; 95% CI, 0.88–1.18; interaction P=0.026).
Conclusions
In patients with atrial fibrillation who develop malignancy, the efficacy and safety profile of edoxaban relative to warfarin is preserved, and it may represent a more practical alternative.
Keywords: anticoagulants, atrial fibrillation, cancer, edoxaban, malignancy, warfarin
Subject Categories: Atrial Fibrillation, Cerebrovascular Disease/Stroke, Thrombosis, Clinical Studies
Clinical Perspective
What Is New?
Patients with malignancy and atrial fibrillation have enhanced thrombotic and bleeding risks, making anticoagulation management challenging, and data on direct oral antagonists in these patients are scarce.
We determined that in patients with atrial fibrillation who develop malignancy, edoxaban is similar to warfarin in the prevention of stroke and systemic embolic events and has a similar bleeding profile.
A benefit of edoxaban over warfarin was greater in patients with malignancy for the composite ischemic end point (ischemic stroke, systemic embolic event, or myocardial infarction) compared with those without malignancy.
What Are the Clinical Implications?
In patients with atrial fibrillation who develop malignancy, edoxaban may present as a practical alternative to warfarin in the prevention of stroke, with a preserved safety profile.
Our additional findings support studies evaluating the benefit of edoxaban over warfarin in reducing ischemic end points in patients with atrial fibrillation and active malignancy.
Introduction
Atrial fibrillation (AF) is the most common sustained arrhythmia in the general population, with a prevalence that increases with age.1 Although data on patients with AF with malignancy are limited, observational studies suggest an association between AF and cancer,2, 3, 4, 5 likely because of systemic inflammation, shared risk factors, and common disease states underlying both conditions in aging populations.4, 6 Although 20‐year incident malignancy in patients with new‐onset AF has been reportedly low at 10%,4 incident AF may occur in up to 30% of patients with certain types of malignancy (eg, thoracic).6 Cancer‐associated thrombosis has been well described,7, 8 and AF in patients with malignancy may independently double the risk of venous or arterial thromboembolism compared with either condition alone.9 Some malignancies may also inherently increase the risk for major bleeding in patients with AF (eg, hematologic cancer),6 which is potentiated further with anticoagulant therapy.10, 11
Although the direct oral anticoagulants (DOACs) are recommended as alternatives to warfarin for use in patients with AF,12, 13 data in the use of these drugs in patients with both AF and malignancy are scarce. Patients with active malignancies were mostly excluded in trials of DOACs versus warfarin in AF given concerns of increased bleeding and thrombotic risk, rapid changes in renal and hepatic function, hematologic derangements, and potential for chemotherapeutic agent interactions via CYP3A4 and P‐glycoprotein.6, 14, 15 Malignancy is not incorporated in the CHADS2 or CHA2DS2VASc thromboembolic risk prediction score to guide anticoagulant therapy,16, 17 and thus the approach to anticoagulant management in patients with AF with malignancy remains highly individualized.18 Cohort studies report vitamin K antagonists (VKAs) as the most commonly prescribed oral anticoagulant in clinical practice.19 However, because an increasing number of patients with AF taking DOACs may present with a new malignancy or a recurrence of a remote cancer, there is an increased need for outcomes data in these patients.
Edoxaban, an oral direct factor Xa inhibitor with a short half‐life (10–14 hours) and minimal interaction with the cytochrome P‐450 system,20 was evaluated in the ENGAGE AF‐TIMI 48 (Effective Anticoagulation With Factor Xa Next Generation in Atrial Fibrillation–Thrombolysis in Myocardial Infarction Study 48) trial.21 Both higher‐dose (HDER) and lower‐dose (LDER) edoxaban regimens were noninferior to well‐managed warfarin in preventing stroke or systemic embolic events (SEEs) in patients with AF, and both regimens reduced bleeding and cardiovascular death.22 To date, data are sparse in patients treated with edoxaban who have both AF and malignancy, although a recent randomized trial in 1046 patients with venous thromboembolism and malignancy showed that edoxaban was noninferior to dalteparin in the prevention of recurrent venous thromboembolism but was associated with a 2.9% absolute increase in the rate of major bleeding.23 In the present analysis, we evaluate the safety and efficacy of edoxaban compared with warfarin in patients with AF who have developed a new or recurrent malignancy, with particular focus on the HDER because this is the regimen approved for clinical use.
Methods
Study Design and Population
The authors support the spirit and intent of sharing of clinical trial data. We encourage interested parties to contact the corresponding author directly for further discussions. The ENGAGE AF‐TIMI 48 trial design and results have been previously described.21, 22 In brief, this was a randomized double‐blind trial of edoxaban versus warfarin. The trial was approved by an institutional review committee, and all participants gave informed consent. Patients with AF and a CHADS2 score of ≥2 were eligible for inclusion and randomized 1:1:1 to HDER (60 mg once daily), LDER (30 mg once daily), or warfarin (international normalized ratio, 2.0–3.0).21 Patients with creatinine clearance ≤50 mL/min, weight of ≤60 kg, or the concomitant use of P‐glycoprotein inhibitors verapamil, quinidine, or dronedarone received a 50% dose reduction of edoxaban.21, 22
Patients with active cancer (except nonmelanoma skin cancer or adequately treated noninvasive or in situ neoplasm) and patients who received anticancer therapy (drugs, radiation, or surgery) within 5 years were excluded from enrollment into the overall trial.21 Other key exclusion criteria were creatinine clearance <30 mL/min, active liver disease, high risk of bleeding, and use of potent P‐glycoprotein inhibitors (eg, ketoconazole, erythromycin, cyclosporine, or ritonavir).21
Study Definition of New or Recurrent Cancer
Investigator‐reported data on malignancy were captured in specific malignancy‐related electronic case‐report forms. After randomization, data collected on new or recurrent malignancy included type, site, date of first signs or symptoms and date of diagnosis, benign versus malignant neoplasm status, presence of metastases, and recurrent status of a prior remote malignancy. A review of all postrandomization malignancies was conducted by 2 physicians (C.L.F., R.P.G.), who were blinded to treatment assignment. The following new or recurrent malignancies were not included in this analysis: nonmelanoma localized skin cancer, benign tumors, and in situ precancerous lesions (eg, high‐grade cervical dysplasia). They were not included given their anticipated low clinical relevance during the trial horizon.
Measures of Pharmacokinetics and Pharmacodynamics
Blood samples were collected at 29 days postrandomization at trough. Edoxaban plasma concentrations were measured by Quintiles Bioanalytical and ADME Laboratories (Ithaca, NY) using turbo ion spray liquid chromatography–tandem mass spectrometry, with a lower limit of quantification of 0.764 ng/mL. Antifactor Xa activity was measured by the Rotachrome Heparin assay on the Stago STAR Evolution platform (TIMI Clinical Trials Laboratory, Boston, MA), with a lower limit of quantitation of 0.10 IU/mL.
Blood specimens to measure the international normalized ratio were obtained on days 8, 15, 29, and at least monthly thereafter, with the use of an encrypted point‐of‐care device to maintain blinding. The time in the therapeutic range in the warfarin group was calculated by means of linear interpolation.24
Study End Points
The primary efficacy end point of the ENGAGE AF‐TIMI 48 trial was the composite of stroke (ischemic or hemorrhagic) or SEE, which was assessed from randomization to the occurrence of the first event. A secondary composite end point of major adverse cardiovascular events was assessed, which included myocardial infarction, stroke, or cardiovascular death. The principal safety end point was major bleeding, defined by the International Society on Thrombosis and Haemostasis.25 Other safety end points analyzed included major plus clinically relevant nonmajor bleeding, life‐threatening or fatal bleeding, and major gastrointestinal bleeding. The above end points, in addition to malignancy‐related mortality, were adjudicated by an independent Clinical Events Committee blinded to treatment allocation. Deep venous thrombosis and pulmonary embolism were ascertained by review of the adverse event data and were considered exploratory end points. The TIMI Study Group developed and conducted this analysis independent of the sponsor, Daiichi Sankyo (Edison, NJ).
Statistical Analyses
Baseline characteristics are presented by postrandomization malignancy status, with categorical variables as frequencies and percentages and continuous variables as medians and interquartile ranges. Comparisons of baseline data for those with and without malignancy were performed using the χ2 test for categorical variables and the nonparametric Kruskal‐Wallis test for continuous variables. Frequencies of malignancy types were reported for the overall study population and by treatment arm. Annualized event rates of major end points using person‐time of follow‐up were calculated. Adjusted risk estimates were calculated using Cox proportional hazard modeling, which included age, body mass index, sex, race, region, prior VKA experience, creatinine clearance, prior stroke or transient ischemic attack, hypertension, coronary artery disease, peripheral artery disease, congestive heart failure, dyslipidemia, diabetes mellitus, smoking status, and randomized treatment arm. Assumptions of proportionality of the hazards for all Cox models were assessed using Schoenfeld residuals and were not violated.
For the evaluation of treatment effect within the malignancy subgroups, efficacy end points occurring during the study period were analyzed by the intention‐to‐treat principle using Cox proportional hazards modeling with the randomized treatment arm as a covariate. Bleeding end points were analyzed during the on‐treatment period from first dose to last dose plus 3 days inclusive. Cox models for treatment effect included the stratification factors of CHADS2 score and dose adjustment at randomization, as well as the subgroup interaction term. Kaplan‐Meier curves through 3 years were generated by treatment arm for both efficacy and safety end points, as well as for new or recurrent malignancy, and were compared with a log‐rank test. In sensitivity analyses, end points were also tested in the modified intention‐to‐treat population while receiving treatment, which included interval censoring of events that occurred during study drug interruptions of >3 days.
Primary efficacy data were analyzed with the assumption that malignancy was present at randomization, in line with the hypothesis that occult malignancy, attributable to heightened systemic inflammation, may lead to increased arterial thrombotic risk, similar to that which is seen in venous thromboembolism. The asymptomatic phase of common malignancies may last several years, and even after symptoms begin, there is a delay in final diagnosis because of scheduling of tests or procedures, referrals, and other social factors.26, 27 Despite these data, an additional sensitivity analysis was performed, which censored stroke/SEE events before the malignancy diagnosis. The time to diagnosis by treatment arm analysis was also evaluated.
Statistical significance for all analyses was assessed using a 2‐sided α level of 0.05, without adjustment for multiple comparisons, using Stata v14.2 (StataCorp, College Station, TX).
Results
Patient Population
Of 21 105 total patients randomized in the ENGAGE AF‐TIMI 48 trial, 1153 (5.5%) developed a malignancy during a median follow‐up period of 2.8 years, of which 991 (85.9%) were considered a new incident cancer and 162 (14.1%) were considered a recurrence of a remote (>5 years prior) cancer. Of these 1153 patients with malignancy, 395 (34.3%) were assigned to warfarin, 390 (33.8%) were assigned to HDER, and 368 (31.9%) were assigned to LDER (P=0.80 and P=0.27 for HDER and LDER versus warfarin, respectively). Baseline characteristics by malignancy status are presented in Table 1. In general, those with a new or recurrent malignancy were more often older, men, white, current smokers, and from North American or Western Europe; they were less frequently VKA naïve, with a slightly higher mean CHA2DS2VASc score at baseline and more prevalent coronary disease. Cancer characteristics of the malignancy cohort are summarized in Table 2. The most common malignancy types were gastrointestinal (20.6%), prostate (13.6% overall, 20.0% of cancers in men), lung (11.1%), bladder (7.7%), and breast (6.7% overall, 21.0% of cancers in women), which did not vary by treatment arm. The median time to diagnosis of new or recurrent malignancy was 495 days (interquartile range, 230–771 days), which also did not differ by treatment arm (3‐way P=0.21). The Kaplan‐Meier analysis of time to diagnosis of malignancy during follow‐up is shown in Figure 1, which demonstrated no difference by treatment arm (log‐rank P=0.27 for LDER and P=0.80 for HDER).
Table 1.
Baseline Characteristics by New or Recurrent Malignancy Status
| Characteristic | No Malignancy (N=19 952; 94.5%) | Malignancy (N=1153; 5.5%) | P Value |
|---|---|---|---|
| Age, median (IQR), y | 72 (64–77) | 75 (68–79) | <0.001 |
| Body mass index, median (IQR), kg/m2 | 28.7 (25.4–32.6) | 28.8 (25.6–32.3) | 0.84 |
| Male sex | 12 271 (61.5) | 794 (68.9) | <0.001 |
| Race | <0.001 | ||
| White | 16 075 (80.6) | 992 (86.0) | |
| Asian | 2786 (14.0) | 123 (10.7) | |
| Black | 264 (1.3) | 14 (1.2) | |
| Other | 826 (4.1) | 24 (2.1) | |
| Region | <0.001 | ||
| North America | 4336 (21.7) | 345 (29.9) | |
| Latin America | 2572 (12.9) | 89 (7.7) | |
| Western Europe | 3002 (15.0) | 234 (20.3) | |
| Eastern Europe | 6816 (34.2) | 328 (28.4) | |
| Asia or South Africa | 3226 (16.2) | 157 (13.6) | |
| History of remote malignancya | 1223 (6.1) | 162 (14.1) | <0.001 |
| Paroxysmal atrial fibrillation | 5076 (25.4) | 290 (25.2) | 0.24 |
| Valvular heart disease | 4172 (20.9) | 267 (23.2) | 0.07 |
| VKA naïve | 8276 (41.5) | 387 (33.6) | <0.001 |
| CHADS2 score, mean (SD)b | 2.8 (1.0) | 2.8 (1.0) | 0.83 |
| CHA2DS2VASc score, mean (SD)b | 4.3 (1.4) | 4.4 (1.3) | 0.011 |
| HASBLED score, mean (SD) | 2.5 (1.0) | 2.7 (0.9) | <0.001 |
| Creatinine clearance, median (IQR), mL/min | 70.5 (53.9–92.2) | 66.8 (51.7–86.1) | <0.001 |
| Prior stroke or TIA | 5690 (28.5) | 283 (24.5) | 0.004 |
| Hypertension | 18 663 (93.5) | 1091 (94.6) | 0.14 |
| Coronary artery disease | 6602 (33.1) | 421 (36.5) | 0.017 |
| Peripheral artery disease | 773 (3.9) | 68 (5.9) | <0.001 |
| Congestive heart failure | 11 530 (57.8) | 594 (51.5) | <0.001 |
| Dyslipidemia | 10 399 (52.1) | 659 (57.2) | <0.001 |
| Diabetes mellitus | 7179 (36.0) | 445 (38.6) | 0.072 |
| Current smoking status | 1449 (7.3) | 103 (8.9) | 0.034 |
| History of non‐ICH bleed | 1930 (9.7) | 151 (13.1) | <0.001 |
| Dose reduction at randomizationc | 5014 (24.8) | 311 (27.0) | 0.20 |
Continuous variables are presented as medians (IQRs) or means (SDs), and categorical variables are presented as frequencies (percentages). Baseline characteristics by randomized treatment, stratified by malignancy status, were generally well balanced; there were small differences in prior congestive heart failure (58.1%, 58.7%, and 56.6%) and valvular heart disease (21.2%, 19.8%, and 21.7%) in the cohort without cancer for the warfarin, higher‐dose edoxaban regimen, and lower‐dose edoxaban regimen treatment groups, respectively (P=0.043 and P=0.021, respectively). ICH indicates intracranial hemorrhage; IQR, interquartile range; TIA, transient ischemic attack; VKA, vitamin K antagonist.
Remote malignancy defined as a history of cancer that was not active or receiving treatment during the 5‐year period before enrollment; inclusion into active malignancy cohort was met for those with remote malignancy who either had a new type diagnosed during follow‐up period or who had recurrence of remote type.
CHADS2 score assigns 1 point for congestive heart failure, hypertension, age of at least 75 years, and diabetes mellitus, and 2 points for prior stroke or transient ischemic attack; CHA2DS2VASc score assigns 1 point for congestive heart failure, hypertension, age of 65 to 74 years, diabetes mellitus, vascular disease history, and female sex, and 2 points for age of at least 75 years and prior stroke or transient ischemic attack.
Patients with a creatinine clearance of ≤50 mL/min, those with a body weight of ≤60 kg, and those who were receiving the concomitant potent P‐glycoprotein inhibitor verapamil or quinidine at randomization received a 50% dose reduction of edoxaban.
Table 2.
Malignancy Characteristics of Patients With New or Recurrent Malignancy
| Characteristic | New or Recurrent Malignancy (N=1153) |
|---|---|
| Type of new or recurrent malignancy | |
| Solid | |
| Gastrointestinal | 236 (20.5) |
| Prostate | 158 (13.7) |
| Lung or pleura | 127 (11.0) |
| Bladder | 87 (7.5) |
| Breast | 75 (6.5) |
| Skina | 68 (5.9) |
| Pancreatic | 44 (3.8) |
| Liver, gallbladder, or bile ducts | 44 (3.8) |
| Esophageal | 29 (2.5) |
| Oropharyngeal | 30 (2.6) |
| Renal | 29 (2.5) |
| Uterine | 24 (2.1) |
| Brain | 24 (2.1) |
| Genital | 15 (1.3) |
| Thyroid | 13 (1.1) |
| Hematologic | |
| Leukemia | 32 (2.8) |
| Lymphoma | 25 (2.2) |
| Multiple sites | 30 (2.6) |
| Otherb | 46 (4.0) |
| Unspecified | 17 (1.5) |
| Time to diagnosis of malignancy, median (IQR), d | 495 (230–771) |
| Patients with remote malignancyc | 162 (14.1) |
| Type of remote malignancy | |
| Hematologic | 10 (6.2) |
| Solid tumor | 152 (93.8) |
| Extent of remote malignancy | |
| Spread to contiguous structures | 4 (2.5) |
| Local disease only without spread | 134 (82.7) |
| Metastatic | 1 (0.6) |
| Unknown | 18 (11.1) |
| Recurrence of remote malignancy | 41 (13.6) |
Data are given as number (percentage) unless otherwise specified. IQR indicates interquartile range.
Skin cancers included melanoma or other malignant skin cancers; localized basal or squamous cell carcinoma was excluded from the active malignancy cohort.
Includes rare types of solid or hematologic malignancies not already described for which incidence was <1.0%.
A total of 162 patients (14.1%) who were included in the active malignancy cohort for this analysis had a history of remote cancer, defined as history of cancer no longer active or receiving treatment during the 5‐year period before randomization; 41 patients in the overall analysis had recurrence of a remote malignancy, defined as a match in type for remote cancer and active cancer according to the electronic case record form.
Figure 1.

Kaplan‐Meier curve of time to diagnosis of malignancy by treatment arm. HDER indicates higher‐dose edoxaban regimen; LDER, lower‐dose edoxaban regimen.
Time‐in‐therapeutic range for those assigned to the warfarin arm also did not differ according to malignancy status (median, 68.2% [95% confidence interval {CI}, 55.9%–76.6%] versus 68.4% [95% CI, 56.6%–77.5%] for patients with malignancy versus no malignancy; P=0.29). However, interruption of anticoagulation >3 days occurred more commonly in patients with malignancy. Overall, 62.9% of patients with malignancy had at least 1 study drug interruption compared with 51.0% of those without malignancy (P<0.001). Clinical procedures caused these interruptions in 38.1% versus 23.2% of patients with and without malignancy, respectively (P<0.001). In patients with temporary drug interruptions during trial follow‐up, the mean number of interruptions for patients with malignancy compared with those without malignancy was 2.3±1.6 versus 1.7±1.2, respectively (P<0.001).
Laboratory Outcomes in Patients With Active Malignancy
Kidney dysfunction, defined as a decline in creatinine clearance to <30 mL/min during the trial, was more likely to occur in those with active malignancy versus those without active malignancy (8.0% versus 6.3%; P=0.02). Similarly, new‐onset anemia (hemoglobin, <8 g/dL) was more frequent in patients with cancer (2.7% versus 0.79%; P<0.001). The incidence of severe thrombocytopenia (platelet count, <50 000) was rare overall, but more common in patients with malignancy (0.53% versus 0.11%; P<0.001). Median trough concentrations of edoxaban as well as factor Xa activity, on the basis of treatment regimen and dose reduction at 29 days, are presented in Table 3. There was no statistically significant difference in drug concentration or factor Xa activity at trough in patients with malignancy compared with those without across all edoxaban treatment regimens.
Table 3.
Median Trough Edoxaban Drug Concentration and Factor Xa Activity by Treatment Regimen and Malignancy Status at Day 29
| Treatment Regimen and Dose | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HDER (60 mg) | HDER‐DR (30 mg) | LDER (30 mg) | LDER‐DR (15 mg) | ||||||||
| Cancer | No Cancer | P Value | Cancer | No Cancer | P Value | Cancer | No Cancer | P Value | Cancer | No Cancer | P Value |
| Edoxaban concentration, trough level at day 29, median (interquartile range), ng/mL | |||||||||||
| 38.4 (23.1–55.8) | 35.9 (19.1–61.9) | 0.57 | 22.9 (13.9–44.0) | 28.2 (15.1–46.6) | 0.40 | 17 (9.9–31.6) | 18.4 (10.0–32.4) | 0.44 | 11.1 (8.7–17.0) | 12.8 (7.3–21.6) | 0.95 |
| Factor Xa activity, trough level at day 29, median (interquartile range), IU/mL | |||||||||||
| 0.79 (0.49–1.16) | 0.63 (0.37–1.10) | 0.25 | 0.52 (0.29–0.94) | 0.52 (0.31–0.84) | 0.97 | 0.31 (0.21–0.48) | 0.35 (0.21–0.58) | 0.29 | 0.25 (0.19–0.52) | 0.27 (0.18–0.46) | 0.93 |
HDER indicates high‐dose edoxaban regimen (60 mg); HDER‐DR, HDER dose reduction (30 mg); LDER, low‐dose edoxaban regimen (30 mg); LDER‐DR, LDER dose reduction (15 mg).
*P<0.05 is significant for difference in median trough levels by Wilcoxon rank‐sum test.
Clinical Outcomes in Patients With Active Malignancy
Compared with patients without malignancy during the follow‐up period, those with malignancy had a statistically significant increased risk of major bleeding (7.4%/year versus 2.5%/year; adjusted hazard ratio [HR], 2.45; 95% CI, 2.07–2.89; P<0.001). Malignancy was also associated with statistically significant higher all‐cause mortality (12.0%/year versus 3.6%/year; adjusted HR, 3.12; 95% CI, 2.78–3.50; P<0.001), of which 273 deaths (75.1%) were directly related to malignancy (8.98%/year). The rates of stroke or SEE were similar between those with malignancy and those without malignancy (2.0%/years versus 1.8%/year; adjusted HR, 1.08; 95% CI, 0.83–1.42; P=0.55) (Figure 2).
Figure 2.

Adjusted (Adj.) risk of major end points in patients with vs without an active malignancy. Risk presented as Adj. hazard ratio (HR) with 95% confidence interval (CI). Malignancy status defined as those with new or recurrent advanced malignancy during a median 2.8‐year follow‐up period. MACE indicates major adverse cardiovascular event (myocardial infarction, stroke, or death attributable to cardiovascular cause or bleeding); SEE, systemic embolic event.
After subcategorizing malignancies as solid versus nonsolid (hematologic or skin), solid type tumors were associated with higher adjusted risk of stroke/SEE (2.2%/year versus 0.6%/year for solid versus nonsolid; adjusted HR, 3.92; 95% CI, 1.21–12.69; P=0.022). Although rates for major bleeding were numerically higher with solid type tumors, the adjusted risk was not statistically significant (7.9%/year versus 4.8%/year for solid versus nonsolid; adjusted HR, 1.56; 95% CI, 0.96–2.53; P=0.07).
HDER Versus Warfarin in Patients With Malignancy
The efficacy profile of HDER was not affected by the presence or absence of malignancy across major end points (Figure 3). Although the treatment interaction was not statistically significant, patients with malignancy randomized to HDER exhibited a greater numerical reduction in the primary end point of stroke or SEE compared with those without malignancy (malignancy: 1.43%/year versus 2.38%/year; HR, 0.60; 95% CI, 0.31–1.15; no malignancy: 1.58%/year versus 1.77%/year; HR, 0.89; 95% CI, 0.76–1.05, for HDER versus warfarin; interaction P=0.25).
Figure 3.
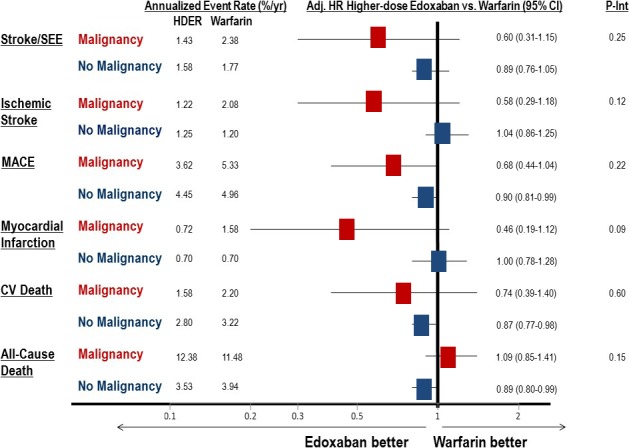
Efficacy end points by malignancy status in the higher‐dose edoxaban regimen vs warfarin groups. All efficacy end points are analyzed from the intention‐to‐treat study population. Adj. indicated adjusted; CI, confidence interval; CV, cardiovascular; HR, hazard ratio (adjusted for trial stratification factors); MACE, major adverse cardiovascular event (including myocardial infarction, stroke, or death attributable to CV cause or bleeding); P‐Int, interaction P value; SEE, systemic embolic event.
For the secondary composite end point of myocardial infarction, stroke, or cardiovascular death (major adverse cardiovascular event), no significant treatment interaction was demonstrated by malignancy status (interaction P=0.22). However, there was a large numerical reduction in myocardial infarction in patients with malignancy receiving HDER compared with warfarin (0.72%/year versus 1.58%/year; HR, 0.46; 95% CI, 0.19–1.12), as well as in cardiovascular death (1.58%/year versus 2.20%/year; HR, 0.74; 95% CI, 0.39–1.40). When limiting the analysis to ischemic events (ischemic stroke/SEE/myocardial infarction), HDER demonstrated a statistically significant 46% risk reduction compared with warfarin and an interaction by malignancy status (malignancy versus no malignancy: HR, 0.54 [95% CI, 0.31–0.93] versus 1.02 [95% CI, 0.88–1.18]; interaction P=0.026). An analysis of a broad composite of ischemic events that also included venous events (acute deep venous thrombosis and pulmonary embolism) in addition to the arterial events above also demonstrated a greater reduction with HDER compared with warfarin in patients with versus without malignancy (malignancy versus no malignancy: HR, 0.51 [95% CI, 0.30–0.86] versus 1.03 [95% CI, 0.89–1.19]; interaction P=0.011). No statistically significant difference in malignancy‐related deaths occurred between HDER and warfarin through 3 years of follow‐up (95 versus 85; log‐rank P=0.34) (Figure 4).
Figure 4.
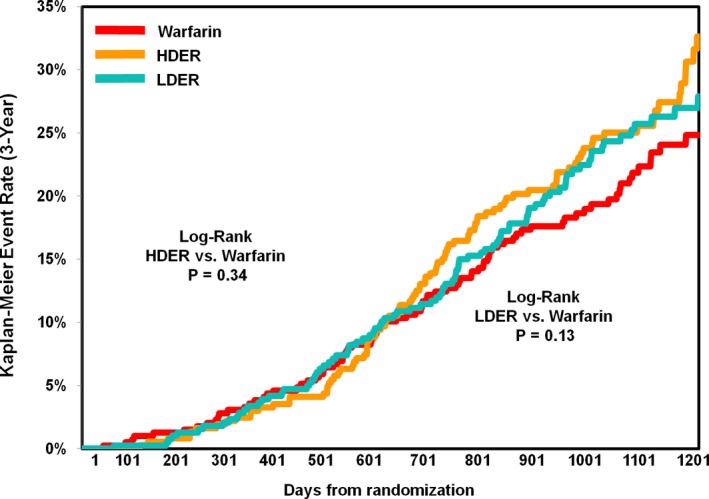
Kaplan‐Meier curve of malignancy‐related death by treatment arm in patients with malignancy. Analysis performed with the intent‐to‐treat population over entire study period. HDER indicates higher‐dose edoxaban regimen; LDER, lower‐dose edoxaban regimen.
The overall safety profile of HDER was not affected by the presence of malignancy (all interaction P values >0.05) (Figure 5). Rates of major bleeding were higher in patients with malignancy compared with no malignancy, but no statistically significant difference was seen between HDER versus warfarin (malignancy: 7.92%/year versus 8.18%/year; HR, 0.98; 95% CI, 0.69–1.40; no malignancy: 2.62%/year versus 3.34%/year; HR, 0.79; 95% CI, 0.69–1.05; interaction P=0.31). The Kaplan‐Meier curve for major bleeding on treatment over the entire study period is shown in Figure 6. The rates of fatal or life‐threatening bleeding were low, with no statistically significant difference according to malignancy group and treatment arm (malignancy: 1.06%/year versus 0.99%/year; HR, 1.06; 95% CI, 0.4–2.82; no malignancy: 0.60%/year versus 1.18%/year; HR, 0.51; 95% CI, 0.40–0.66 for HDER versus warfarin, respectively; interaction P=0.16).
Figure 5.

Safety end points by malignancy status in the higher‐dose edoxaban regimen (HDER) vs warfarin groups. All bleeding end points are analyzed in the on‐treatment study population, beginning with first dose of study treatment and ending with the last dose plus 3 days, inclusive. Major bleeding was defined by the International Society of Thrombosis and Haemostasis. Adj. indicated adjusted; CI, confidence interval; CRNM, clinically relevant nonmajor; GI, gastrointestinal; HR, hazard ratio; LT, life threatening; P‐Int, interaction P value.
Figure 6.

Kaplan‐Meier curve of major bleeding events according to treatment arm and malignancy status. Analysis performed in the on‐treatment population, defined as first day of dose administered to last dose plus 3 days. HDER indicates higher‐dose edoxaban regimen; LDER, lower‐dose edoxaban regimen.
LDER Versus Warfarin in Patients With Malignancy
Overall, the primary efficacy and safety profile of LDER was not significantly modified by the presence of malignancy (Tables 4 and 5), and all P interactions were >0.05. In patients with malignancy, the event rates of stroke or SEE in LDER and warfarin arms were 2.04%/year versus 2.38%/year (HR, 0.87; 95% CI, 0.47–1.59), respectively, compared with 2.04%/year versus 1.77%/year (HR, 1.15; 95% CI, 0.99–1.34) in patients without malignancy (interaction P=0.38). Although the interaction term was not statistically significant for major bleeding (interaction P=0.058), there was a trend toward an even greater reduction in bleeding events with LDER compared with warfarin in patients without a malignancy (malignancy: 5.95%/year versus 8.18%/year; HR, 0.73; 95% CI, 0.40–1.07; no malignancy: 1.63%/year versus 3.34%/year; HR, 0.49; 95% CI, 0.42–0.57). Similar to HDER, there was no difference in malignancy‐related death with LDER compared with warfarin (Figure 4; 96 versus 85; log‐rank P=0.13).
Table 4.
Efficacy End Points by Malignancy Status in LDER and HDER Versus Warfarin
| End Point | Malignancy | No Malignancy | Interaction P Value | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Warfarin (N=395)a | LDER (N=368)a | LDER vs Warfarin HR (95% CI) | HDER (N=390)a | HDER vs Warfarin HR (95% CI) | Warfarin (N=6641)a | LDER (N=6666)a | LDER vs Warfarin HR (95% CI) | HDER (N=6645)a | HDER vs Warfarin HR (95% CI) | LDER vs Warfarin | HDER vs Warfarin | |
| Stroke/SEE | 24 (2.38) | 19 (2.04) | 0.87 (0.47–1.59) | 14 (1.43) | 0.60 (0.31–1.15) | 313 (1.77) | 364 (2.04) | 1.15 (0.99–1.34) | 282 (1.58) | 0.89 (0.76–1.05) | 0.38 | 0.25 |
| Ischemic stroke | 21 (2.08) | 16 (1.72) | 0.84 (0.43–1.62) | 12 (1.22) | 0.58 (0.29–1.18) | 214 (1.20) | 317 (1.77) | 1.47 (1.23–1.74) | 224 (1.25) | 1.04 (0.86–1.25) | 0.10 | 0.12 |
| MACE | 53 (5.33) | 40 (4.33) | 0.82 (0.54–1.24) | 35 (3.62) | 0.68 (0.44–1.04) | 873 (4.96) | 873 (4.93) | 0.99 (0.90–1.09) | 792 (4.45) | 0.90 (0.81–0.99) | 0.39 | 0.22 |
| MI | 16 (1.58) | 12 (1.28) | 0.83 (0.39–1.75) | 7 (0.72) | 0.46 (0.19–1.12) | 125 (0.70) | 157 (0.87) | 1.24 (0.98–1.57) | 126 (0.70) | 1.00 (0.78–1.28) | 0.29 | 0.09 |
| Cardiovascular death | 23 (2.20) | 18 (1.87) | 0.88 (0.48–1.64) | 16 (1.58) | 0.74 (0.39–1.40) | 588 (3.22) | 509 (2.76) | 0.85 (0.76–0.96) | 514 (2.80) | 0.87 (0.77–0.98) | 0.93 | 0.60 |
| All‐cause death | 120 (11.5) | 116 (12.1) | 1.07 (0.83–1.38) | 125 (12.4) | 1.09 (0.85–1.41) | 719 (3.94) | 621 (3.37) | 0.85 (0.76–0.95) | 648 (3.53) | 0.89 (0.80–0.99) | 0.08 | 0.15 |
All efficacy end points are analyzed in the intention‐to‐treat study population. CI indicates confidence interval; HDER, higher‐dose edoxaban regimen; HR, hazard ratio; LDER, lower‐dose edoxaban regimen; MACE, major adverse cardiovascular event (including myocardial infarction, stroke, or death attributable to cardiovascular cause or bleeding); MI, myocardial infarction; SEE, systemic embolic event.
Data are given as number (percentage/year).
Table 5.
Major Safety End Points by Malignancy Status in LDER and HDER Versus Warfarin
| End Point | Malignancy | No Malignancy | Interaction P Value | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Warfarin (N=395)* | LDER (N=368)* | LDER vs Warfarin HR (95% CI) | HDER (N=390)* | HDER vs Warfarin HR (95% CI) | Warfarin (N=6641)* | LDER (N=6666)* | LDER vs Warfarin HR (95% CI) | HDER (N=6645)* | HDER vs Warfarin HR (95% CI) | LDER vs Warfarin | HDER vs Warfarin | |
| Major bleeding† | 63 (8.18) | 42 (5.95) | 0.73 (0.40–1.07) | 56 (7.92) | 0.98 (0.68–1.40) | 494 (3.34) | 250 (1.63) | 0.49 (0.42–0.57) | 388 (2.62) | 0.79 (0.69–0.90) | 0.058 | 0.31 |
| Major/CRNMB | 174 (27.94) | 126 (20.56) | 0.74 (0.59–0.93) | 170 (29.21) | 1.04 (0.84–1.29) | 1636 (12.49) | 1109 (7.84) | 0.63 (0.59–0.68) | 1405 (10.50) | 0.85 (0.79–0.91) | 0.14 | 0.06 |
| Fatal or life‐threatening bleeding | 8 (0.99) | 5 (0.68) | 0.69 (0.23–2.06) | 8 (1.06) | 1.06 (0.40–2.82) | 178 (1.18) | 57 (0.37) | 0.31 (0.23–0.42) | 91 (0.60) | 0.51 (0.40–0.66) | 0.17 | 0.16 |
| All bleeding | 195 (33.82) | 135 (23.01) | 0.69 (0.56–0.86) | 187 (34.01) | 1.00 (0.82–1.23) | 1969 (15.77) | 1437 (10.56) | 0.68 (0.63–0.73) | 1724 (13.46) | 0.86 (0.81–0.92) | 0.78 | 0.14 |
| Major gastrointestinal bleeding | 38 (4.82) | 27 (3.78) | 0.78 (0.47–1.27) | 34 (4.70) | 0.98 (0.61–1.55) | 160 (1.06) | 114 (0.74) | 0.70 (0.55–0.89) | 206 (1.38) | 1.30 (1.06–1.60) | 0.69 | 0.25 |
All bleeding end points are analyzed in the on‐treatment study population, which began with first dose of study treatment through last dose plus 3 days. CI indicates confidence interval; CRNMB, clinically relevant nonmajor bleeding; HDER, higher‐dose edoxaban regimen; HR, hazard ratio; LDER, lower‐dose edoxaban regimen.*Data are given as number (percentage/year).
†Major bleeding defined by International Society of Thrombosis and Haemostasis.
Sensitivity Analyses
Analyses were also performed while receiving treatment in the modified intention‐to‐treat population, which excluded events that occurred after study drug interruption of >3 days. Results were consistent with primary findings. No statistically significant treatment interaction was demonstrated by malignancy status with HDER versus warfarin for the primary efficacy end point of stroke/SEE (interaction P=0.33). This also held true for major bleeding (interaction P=0.33). Similarly, there were no statistically significant treatment interactions by malignancy status with LDER versus warfarin for the end points of stroke/SEE and major bleeding (interaction P=0.23 and P=0.12, respectively).
In an additional sensitivity analysis, we censored 19 stroke or SEE events that occurred between time of randomization and final confirmation of malignancy diagnosis. Results by treatment group were qualitatively similar, with no statistically significant effect modification of malignancy status on the treatment effect of HDER versus warfarin in the prevention of stroke or SEE (malignancy: HR, 0.54; 95% CI, 0.24–1.23; and no malignancy: HR, 0.89; 95% CI, 0.76–1.05; interaction P=0.25). Similarly, no effect modification on efficacy was demonstrated in the comparison of LDER versus warfarin (malignancy: HR, 0.86; 95% CI, 0.41–1.80; and no malignancy: HR, 1.15; 95% CI, 0.99–1.34; interaction P=0.42).
Discussion
In this analysis of 1153 patients with AF and new or recurrent malignancy after entry into the trial, we found that the most common incident tumors were gastrointestinal, prostate, and lung, with a low incidence of hematologic malignancies. After adjusting for baseline covariates, demographic factors, and randomized treatment arm, the presence of an active malignancy increased the risk for major bleeding and death, but not ischemic events. However, when malignancies were further categorized, solid‐type tumors in adjusted analyses were associated with increased risk of stroke or SEE (compared with no malignancy), whereas nonsolid malignancies were not. Overall, the efficacy and safety profiles of edoxaban compared with warfarin were not significantly modified by the presence of malignancy during the 2.8 median year trial follow‐up.
The independent contribution of malignancy to death and major bleeding in patients receiving oral anticoagulant therapy has previously been described.10, 28, 29 Our findings confirm that patients with AF and active malignancy constitute a higher‐risk population for major bleeding that warrants careful risk‐benefit assessment in stroke prevention therapies. This increased risk may be especially true for solid‐type tumors, and our analysis supports existing data that ischemic risk may vary according to cancer subtypes.30 Of 1236 patients with AF and a remote history of malignancy enrolled in the ARISTOTLE (Apixaban for Reduction in Stroke and Other Thromboembolic Events in Atrial Fibrillation) trial, the adjusted HR of myocardial infarction was 1.45 (95% CI, 0.93–2.26; P=0.1) and the adjusted HR of ischemic stroke was 1.02 (95% CI, 0.66–1.58; P=0.9).31 Only 157 (12.7%) of these patients had an active malignancy during the course of the ARISTOTLE trial.
In patients with malignancy, HDER compared with well‐managed warfarin (median time‐in‐therapeutic range, 68.2%) demonstrated similar rates of all stroke or SEE, major adverse cardiovascular event, all‐cause death, and malignancy‐related death. However, when the composite end point was restricted to ischemic events (ischemic stroke, SE, or myocardial infarction), HDER demonstrated a statistically significant reduction in events. A high proportion (66.4%) of patients with malignancy had been treated with a VKA before the trial, and we previously reported that the treatment benefit of HDER relative to warfarin is attenuated in VKA‐experienced patients.32 There was no effect modification by malignancy status on bleeding end points, including major bleeding, major or clinically relevant nonmajor bleeding, or the composite of life‐threatening or fatal bleeding, which in the overall trial population were significantly reduced with edoxaban compared with warfarin.22
This study demonstrated similar efficacy in stroke/SEE reduction with LDER compared with warfarin in patients with malignancy, with a trend toward reduced major bleeding in patients both with and without malignancy. Although the LDER is not approved for clinical use, these data support further research of less intensive anticoagulation in patients with malignancy and AF who cannot tolerate standard doses of anticoagulation (eg, because of bleeding).
To the best of our knowledge, this analysis is the first large study using prospective clinical trial data and blinded adjudicated events on the effects of a DOAC in a population of patients with AF and an active malignancy. The subgroup analysis from the ARISTOTLE trial demonstrated preserved efficacy and safety of apixaban in patients with AF and a remote history of cancer.31 However, unlike the analysis from the ARISTOTLE trial, we analyzed patients who developed active malignancy after randomization while receiving study drug. A small single‐center observational analysis of 163 patients with active malignancy and AF treated with rivaroxaban demonstrated overall low 1‐year cumulative incidence rates of ischemic stroke (1.4%) and major bleeding (1.2%) after adjustment for competing risks.33 Although the low event rates were encouraging, patients with gastrointestinal and genitourinary cancers, which are 2 subtypes with potential for increased bleeding, were excluded. In addition, patients receiving warfarin were not analyzed for comparison.33
Current AF guidelines do not provide specific recommendations for oral anticoagulation in patients with active malignancy.12, 13 Although robust randomized data in patients with malignancy and venous thromboembolism exist,34, 35 generalizability of these results to patients with AF has its limitations given different pathophysiological and risk profiles. The European Society of Cardiology recommends consideration of a DOAC as first‐line therapy in patients with AF for stroke prevention.13 Use of DOACs in patients with AF is increasing in clinical practice and the aging population.36, 37 Older patients receiving DOACs may develop a new or recurrent malignancy because of shared risk factors with aging.3, 4, 5, 6 We anticipate, therefore, an increase in use of DOACs in patients with cancer and a need for efficacy and safety data.
Because factor Xa has demonstrated procoagulant activity in cancer,38 the factor Xa inhibitors especially warrant special consideration in these patients. All DOACs may interact with certain chemotherapeutic agents through the P‐glycoprotein transporter;15 however, unlike rivaroxaban and apixaban, edoxaban has little inhibitory effect on CYP3A4.20 This may be particularly relevant in patients undergoing chemotherapy because CYP3A4 is responsible for metabolism of several anticancer therapies, including tyrosine kinase inhibitors.39 The lower potential for drug‐drug interactions combined with a fixed‐dosing regimen make edoxaban an attractive alternative to VKAs in patients with an active malignancy. Finally, the rapid onset (1–2 hours) and shorter half‐life of DOACs (6–12 hours) compared with warfarin (≈40 hours) offer additional practical advantages over VKAs in patients with active malignancy, because these patients often undergo frequent medication changes and invasive procedures.
Limitations
This is an analysis from a randomized clinical trial of a postrandomization subgroup. Power was limited to detect small differences in efficacy and safety, and we did not adjust for multiple comparisons given the exploratory nature of this analysis, thus increasing the chances for both type I and II errors, respectively. Therefore, results presented should be considered hypothesis generating. In addition, patients with malignancy from this study population may not be completely generalizable to patients with cancer in clinical practice. For example, patients with elevated liver function test results or creatinine clearance <30 mL/min were excluded from the trial,21 2 conditions that may indicate potential complications of malignancy. Last, malignancy was an investigator‐reported event, and the precise time that a malignancy becomes clinically evident was not adjudicated. We acknowledge that some patients may have had subclinical disease at the time of randomization, whereas others developed the disease at a later date. However, to address this, we performed a sensitivity analysis censoring the 19 clinical events that occurred before the date of cancer diagnosis, and results were consistent with the main analysis.
Conclusions
In a large trial population of patients with AF receiving oral anticoagulation, malignancy after randomization was not uncommon, and was independently associated with an increased risk of all‐cause death and major bleeding, but not stroke or SEE. The efficacy and safety profiles of edoxaban compared with warfarin were similar in those who developed active malignancy compared with those who did not. With an increasing number of aging patients with AF receiving DOACs with the potential to develop a malignancy, edoxaban may be a more practical alternative to a VKA, with a comparable efficacy and safety profile. Future randomized trials in patients with AF and malignancy are warranted to confirm findings.
Sources of Funding
The ENGAGE AF‐TIMI 48 (Effective Anticoagulation With Factor Xa Next Generation in Atrial Fibrillation–Thrombolysis in Myocardial Infarction Study 48) trial was funded from a research grant by Daiichi Sankyo. No support was provided for the current publication. The TIMI Study Group has received significant research grant support from Amgen, Astra‐Zeneca, Athera, Beckman Coulter, BG Medicine, Bristol‐Myers Squibb, Buhlmann Laboratories, Daiichi Sankyo, Eli Lilly and Co, Eisai, GlaxoSmithKline, Johnson & Johnson, Merck and Company, Nanosphere, Novartis Pharmaceuticals, Ortho‐Clinical Diagnostics, Pfizer, Randox, Roche Diagnostics, Sanofi‐Aventis, Siemens, and Singulex.
Disclosures
Ruff is a member of the Thrombolysis in Myocardial Infarction (TIMI) Study Group and reports grant support through his institution (Brigham and Women's Hospital) from Daiichi Sankyo and has served as a consultant and received honoraria from Daiichi Sankyo, Boehringer Ingelheim, Bayer, and Portola; and has received grant support through his institution outside the submitted work from AstraZeneca, Eisai, Intarcia, and Glaxo Smith Kline. Jin is an employee of Daiichi Sankyo. Duggal is an employee of Daiichi Sankyo. Babilonia has no relevant disclosures. Sritara has no relevant disclosures. Mercuri was an employee of Daiichi Sankyo. Kamphuisen reports research grant support from Daiichi Sankyo and Boehringer Ingelheim. Antman is a member of the TIMI Study Group and reports grant support through his institution from Daiichi Sankyo. Braunwald is a member of the TIMI Study Group and reports other support from Merck during the conduct of the study, and personal fees from Daiichi‐Sankyo, Sanofi Aventis, The Medicines Company, Menarini International, Bayer, and Medscape. Giugliano is a member of the TIMI Study Group, reports that his institution received research grant support from Daiichi Sankyo for his role as Principal Investigator of the ENGAGE AF‐TIMI 48 (Effective Anticoagulation With Factor Xa Next Generation in Atrial Fibrillation–Thrombolysis in Myocardial Infarction Study 48) trial, and that he has received honoraria for continuing medical education lectures and/or consulting from Bristol Myers Squibb, Daiichi Sankyo, Janssen, Pfizer, and Portola. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2018;7:e008987 DOI: 10.1161/JAHA.118.008987.)
References
- 1. Rahman F, Kwan GF, Benjamin EJ. Global epidemiology of atrial fibrillation. Nat Rev Cardiol. 2014;11:639–654. [DOI] [PubMed] [Google Scholar]
- 2. Erichsen R, Christiansen CF, Mehnert F, Weiss NS, Baron JA, Sørensen HT. Colorectal cancer and risk of atrial fibrillation and flutter: a population‐based case‐control study. Intern Emerg Med. 2012;7:431–438. [DOI] [PubMed] [Google Scholar]
- 3. O'Neal WT, Lakoski SG, Qureshi W, Judd SE, Howard G, Howard VJ, Cushman M, Soliman EZ. Relation between cancer and atrial fibrillation (from the REasons for Geographic And Racial Differences in Stroke Study). Am J Cardiol. 2015;115:1090–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Conen D, Wong JA, Sandhu RK, Cook NR, Lee IM, Buring JE, Albert CM. Risk of malignant cancer among women with new‐onset atrial fibrillation. JAMA Cardiol. 2016;1:389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ostenfeld EB, Erichsen R, Pedersen L, Farkas DK, Weiss NS, Sørensen HT. Atrial fibrillation as a marker of occult cancer. PLoS One. 2014;9:e102861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Farmakis D, Parissis J, Filippatos G. Insights into onco‐cardiology: atrial fibrillation in cancer. J Am Coll Cardiol. 2014;63:945–953. [DOI] [PubMed] [Google Scholar]
- 7. Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer‐associated venous thrombosis. Blood. 2013;122:1712–1723. [DOI] [PubMed] [Google Scholar]
- 8. Stein PD, Beemath A, Meyers FA, Skaf E, Sanchez J, Olson RE. Incidence of venous thromboembolism in patients hospitalized with cancer. Am J Med. 2006;119:60–68. [DOI] [PubMed] [Google Scholar]
- 9. Hu YF, Liu CJ, Chang PM, Tsao HM, Lin YJ, Chang SL, Lo LW, Tuan TC, Li CH, Chao TF, Chung FP, Liao JN, Chen TJ, Chen SA. Incident thromboembolism and heart failure associated with new‐onset atrial fibrillation in cancer patients. Int J Cardiol. 2013;165:355–357. [DOI] [PubMed] [Google Scholar]
- 10. Gage BF, Yan Y, Milligan PE, Waterman AD, Culverhouse R, Rich MW, Radford MJ. Clinical classification schemes for predicting hemorrhage: results from the National Registry of Atrial Fibrillation (NRAF). Am Heart J. 2006;151:713–719. [DOI] [PubMed] [Google Scholar]
- 11. Kamphuisen PW, Beyer‐Westendorf J. Bleeding complications during anticoagulant treatment in patients with cancer. Thromb Res. 2014;133:S49–S55. [DOI] [PubMed] [Google Scholar]
- 12. January CT, Wann LS, Alpert JS, Calkins H, Cigarroa JE, Cleveland JC Jr, Conti JB, Ellinor PT, Ezekowitz MD, Field ME, Murray KT, Sacco RL, Stevenson WG, Tchou PJ, Tracy CM, Yancy CW. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation. 2014;130:2071–2104. [DOI] [PubMed] [Google Scholar]
- 13. Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, Castella M, Diener HC, Heidbuchel H, Hendriks J, Hindricks G, Manolis AS, Oldgren J, Popescu BA, Schotten U, Van Putte B, Vardas P. 2016 ESC guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016;37:2893–2962. [DOI] [PubMed] [Google Scholar]
- 14. Suter TM, Ewer MS. Cancer drugs and the heart: importance and management. Eur Heart J. 2012;34:1102–1111. [DOI] [PubMed] [Google Scholar]
- 15. Wessler JD, Grip LT, Mendell J, Giugliano RP. The P‐glycoprotein transport system and cardiovascular drugs. J Am Coll Cardiol. 2013;61:2495–2502. [DOI] [PubMed] [Google Scholar]
- 16. Gage B, Waterman A, Shannon W, Boechler M, Rich M, Radford M. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA. 2001;285:2864–2870. [DOI] [PubMed] [Google Scholar]
- 17. Lip GYH, Nieuwlaat R, Pisters R, Lane DA, Crijns HJGM. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor‐based approach: the Euro Heart Survey on atrial fibrillation. Chest. 2010;137:263–272. [DOI] [PubMed] [Google Scholar]
- 18. Potpara TS, Lip GYH. Oral anticoagulant therapy in atrial fibrillation patients at high stroke and bleeding risk. Prog Cardiovasc Dis. 2015;58:177–194. [DOI] [PubMed] [Google Scholar]
- 19. Larsen TB, Skjøth F, Nielsen PB, Kjældgaard JN, Lip GYH. Comparative effectiveness and safety of non‐vitamin K antagonist oral anticoagulants and warfarin in patients with atrial fibrillation: propensity weighted nationwide cohort study. BMJ. 2016;353:i3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parasrampuria DA, Truitt KE. Pharmacokinetics and pharmacodynamics of edoxaban, a non‐vitamin K antagonist oral anticoagulant that inhibits clotting factor Xa. Clin Pharmacokinet. 2016;55:641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ruff CT, Giugliano RP, Antman EM, Crugnale SE, Bocanegra T, Mercuir M, Hanyok J, Patel I, Shi M, Salazar D, McCabe CH, Braunwald E. Evaluation of the novel factor Xa inhibitor edoxaban compared with warfarin in patients with atrial fibrillation: design and rationale for the Effective aNticoaGulation with factor xA next GEneration in Atrial Fibrillation‐Thrombolysis In Myocardial Infarction. Am Heart J. 2010;160:635–641. [DOI] [PubMed] [Google Scholar]
- 22. Giugliano RP, Ruff CT, Braunwald E, Murphy SA, Wiviott SD, Halperin JL, Waldo AL, Ezekowitz MD, Weitz JI, Spinar J, Ruzyllo W, Ruda M, Koretsune Y, Betcher J, Shi M, Grip LT, Patel SP, Patel I, Hanyok JJ, Mercuri M, Antman EM. Edoxaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2013;369:2093–2104. [DOI] [PubMed] [Google Scholar]
- 23. Raskob GE, van Es N, Verhamme P, Carrier M, Di Nisio M, Garcia D, Grosso MA, Kakkar AK, Kovacs MJ, Mercuri MF, Meyer G, Segers A, Shi M, Wang TF, Yeo E, Zhang G, Zwicker JI, Weitz JI, Buller HR. Edoxaban for the treatment of cancer‐associated venous thromboembolism. N Engl J Med. 2018;378:615–624. [DOI] [PubMed] [Google Scholar]
- 24. Rosendaal FR, Cannegieter SC, van der Meer FJ, Briët E. A method to determine the optimal intensity of oral anticoagulant therapy. Thromb Haemost. 1993;69:236–239. [PubMed] [Google Scholar]
- 25. Schulman S, Kearon C; and the Subcommittee on Control of Anticoagulation of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis . Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non‐surgical patients. J Thromb Haemost. 2005;3:692–694. [DOI] [PubMed] [Google Scholar]
- 26. Etzioni R, Cha R, Feuer EJ, Davidov O. Asymptomatic incidence and duration of prostate cancer. Am J Epidemiol. 1998;148:775–785. [DOI] [PubMed] [Google Scholar]
- 27. Evans WK. Prognostic implications of treatment delays in the surgical resection of lung cancer. Thorac Surg Clin. 2013;23:225–232. [DOI] [PubMed] [Google Scholar]
- 28. Palareti G, Legnani C, Lee A, Manotti C, Hirsch J, D'Angelo A, Pengo V, Moia M, Coccheri S. A comparison of the safety and efficacy of oral anticoagulation for the treatment of venous thromboembolic disease in patients with or without malignancy. Thromb Haemost. 2000;84:805–810. [PubMed] [Google Scholar]
- 29. Kuijer PM, Hutten BA, Prins MH, Büller HR. Prediction of the risk of bleeding during anticoagulant treatment for venous thromboembolism. Arch Intern Med. 1999;159:457–460. [DOI] [PubMed] [Google Scholar]
- 30. Navi BB, Singer S, Merkler AE, Cheng NT, Stone JB, Kamel H, Iadecola C, Elkind MS, DeAngelis LM. Recurrent thromboembolic events after ischemic stroke in patients with cancer. Neurology. 2014;83:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Melloni C, Dunning A, Granger CB, Thomas L, Khouri MG, Garcia DA, Hylek EM, Hanna M, Wallentin L, Gersh BJ, Douglas PS, Alexander JH, Lopes RD. Efficacy and safety of apixaban versus warfarin in patients with atrial fibrillation and a history of cancer: insights from the ARISTOTLE Trial. Am J Med. 2017;130:1440–1448.e1. [DOI] [PubMed] [Google Scholar]
- 32. O'Donoghue ML, Ruff CT, Giugliano RP, Murphy SA, Grip LT, Mercuri MF, Rutman H, Shi M, Kania G, Cermak O, Braunwald E, Antman EM. Edoxaban vs. warfarin in vitamin K antagonist experienced and naive patients with atrial fibrillation. Eur Heart J. 2015;36:1470–1477. [DOI] [PubMed] [Google Scholar]
- 33. Laube ES, Yu A, Gupta D, Miao Y, Samedy P, Wills J, Harnicar S, Soff GA, Mantha S. Rivaroxaban for stroke prevention in patients with non‐valvular atrial fibrillation and active cancer. Am J Cardiol. 2017;2:213–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Terrenato I, Sperati F, Muti P, Schünemann H. Anticoagulation for the long‐term treatment of venous thromboembolism in patients with cancer. Cochrane Database Syst Rev. 2011;6:CD006650. [DOI] [PubMed] [Google Scholar]
- 35. Lee AYY, Levine MN, Baker RI, Bowden C, Kakkar AK, Prins M, Rickles FR, Julian JA, Haley S, Kovacs MJ, Gent M. Randomized comparison of low‐molecular‐weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med. 2003;349:146–153. [DOI] [PubMed] [Google Scholar]
- 36. Olesen JB, Sorensen R, Hansen ML, Lamberts M, Weeke P, Mikkelsen AP, Køber L, Gislason GH, Torp‐Pedersen C, Fosbøl EL. Non‐vitamin K antagonist oral anticoagulation agents in anticoagulant naive atrial fibrillation patients: Danish nationwide descriptive data 2011–2013. Europace. 2015;17:187–193. [DOI] [PubMed] [Google Scholar]
- 37. Yu AYX, Malo S, Svenson LW, Wilton SB, Hill MD. Temporal trends in the use and comparative effectiveness of direct oral anticoagulant agents versus warfarin for nonvalvular atrial fibrillation: a Canadian population‐based study. J Am Heart Assoc. 2017;6:e007129 DOI: 10.1161/JAHA.117.007129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Falanga A, Panova‐Noeva M, Russo L. Procoagulant mechanisms in tumour cells. Best Pract Res Clin Haematol. 2009;22:49–60. [DOI] [PubMed] [Google Scholar]
- 39. Teo YL, Ho HK, Chan A. Metabolism‐related pharmacokinetic drug‐drug interactions with tyrosine kinase inhibitors: current understanding, challenges and recommendations. Br J Clin Pharmacol. 2015;79:241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]