

Abstract
Background
Congenital long‐QT syndrome (LQTS) is a genetic disorder characterized by prolongation of the corrected QT interval (QTc) on an ECG. The aim of the present study was to estimate the prevalence of pathogenic and likely pathogenic sequence variants in patients who had at least 1 ECG with a QTc ≥500 ms.
Methods and Results
Telemark Hospital Trust is a community hospital within the Norwegian national health system, serving ≈173 000 inhabitants. We searched the ECG database at Telemark Hospital Trust, Norway, from January 2004 to December 2014, and identified 1531 patients with at least 1 ECG with a QTc ≥500 ms. At the time of inclusion in this study (2015), 766 patients were alive. A total of 733 patients were invited to participate, and 475 accepted. The 17 genes that have been reported to cause monogenic LQTS were sequenced among the patients. Pro‐QTc score was calculated for each patient. A molecular genetic cause of LQTS was detected in 31 (6.5%) of 475 patients. These patients had a lower pro‐QTc score than those without pathogenic or likely pathogenic variants (1.7±1.0 versus 2.8±1.6; P<0.001).
Conclusions
Compared with the general population, hospitalized patients with a QTc ≥500 ms in at least 1 ECG recording had an increased likelihood for pathogenic and likely pathogenic variants in LQTS genes. We recommend increased awareness of the possibility of LQTS in patients with at least 1 ECG with a QTc ≥500 ms.
Keywords: genetic testing, inherited arrhythmia, long‐QT syndrome
Subject Categories: Arrhythmias, Sudden Cardiac Death, Genetics
Clinical Perspective
What Is New?
A molecular genetic cause of long‐QT syndrome was detected in 6.5% of patients with at least 1 ECG with a corrected QT interval ≥500 ms.
In most patients with a pathogenic or likely pathogenic variant, the prolonged corrected QT interval had not been acknowledged in the medical records.
What Are the Clinical Implications?
We recommend increased awareness of the possibility of congenital long‐QT syndrome in patients with at least 1 ECG with a corrected QT interval ≥500 ms.
Introduction
Congenital long‐QT syndrome (LQTS) is a genetic disorder characterized by prolongation of the corrected QT interval (QTc) on an ECG. It is associated with increased risk of torsade de pointes ventricular tachycardia and sudden cardiac death. The estimated prevalence of LQTS is 1:2000 live births.1
QTc ≥500 ms is considered to be highly abnormal and associated with increased risk of torsade de pointes ventricular tachycardia,2 but also carriers of LQTS mutations with a QTc ≤440 ms have an increased risk of life‐threatening cardiac events.3 Treatment with β‐blocker medication significantly reduces the risk of adverse outcomes.4
More than 1500 sequence variants in 17 genes have previously been reported to be pathogenic or likely pathogenic for LQTS, although the evidence for some of these is limited.5 Autosomal dominant is the most common inheritance pattern, and KCNQ1 (LQT1) harbors most genetic defects, followed by KCNH2 (LQT2) and SCN5A (LQT3).6, 7 Penetrance is incomplete, and expression is variable within families; these factors complicate both interpretation of pathogenicity of sequence variants in LQTS genes and genetic counseling.8
Most molecular genetic studies of LQTS have been conducted in cohorts clinically diagnosed with LQTS or ascertained through cascade screening of relatives for a specific causative variant, and 80% of families meeting clinical diagnostic criteria have detectable pathogenic or likely pathogenic sequence variants in the 17 known LQTS genes.9
Current guidelines recommend genetic testing of asymptomatic patients if repetitive ECGs show a QTc ≥480 ms (European Society of Cardiology guidelines2) or a QTc >500 ms (Heart Rhythm Society/European Heart Rhythm Association Expert Consensus Statement7) in the absence of secondary causes that may prolong the QT interval. An LQTS risk score (Schwartz) >3 is also considered diagnostic.2, 7, 10 In accordance with current clinical guidelines for LQTS, genetic analyses have often been limited to the 3 main LQTS genes. The prevalence and spectrum of pathogenic and likely pathogenic sequence variants in the remaining LQTS genes are less well documented.
The aim of the present study was to estimate the prevalence of pathogenic and likely pathogenic sequence variants in patients who had at least 1 ECG with a QTc ≥500 ms admitted to a community hospital. Targeted sequencing was used to analyze all 17 known LQTS genes. We further wanted to determine if these patients fulfilled the clinical criteria for LQTS, according to current guidelines.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Study Population and Selection of ECGs
The patients in this study have previously been described (Figure 1).11 Briefly, the ECG database at Telemark Hospital Trust contained 225 117 ECGs from 63 286 unique patients collected throughout a period of 11 years (January 2004–December 2014). The ECG database was searched with the following criteria: QTc (Bazett's formula) ≥500 ms, QRS width ≤120 ms, age ≥15 years, heart rate (beats per minute) >30 and ≤100 (because of the limitations of Bazett's formula), no acute ST‐segment–elevation infarction, and no atrial fibrillation or atrial flutter. All ECGs with a QTc ≥500 ms were manually reviewed, and a total of 1531 patients with at least 1 ECG with a QTc ≥500 ms were included in the previous study. These patients’ first ECG with a QTc ≥500 ms is further referred to as “ECG 1.”
Figure 1.
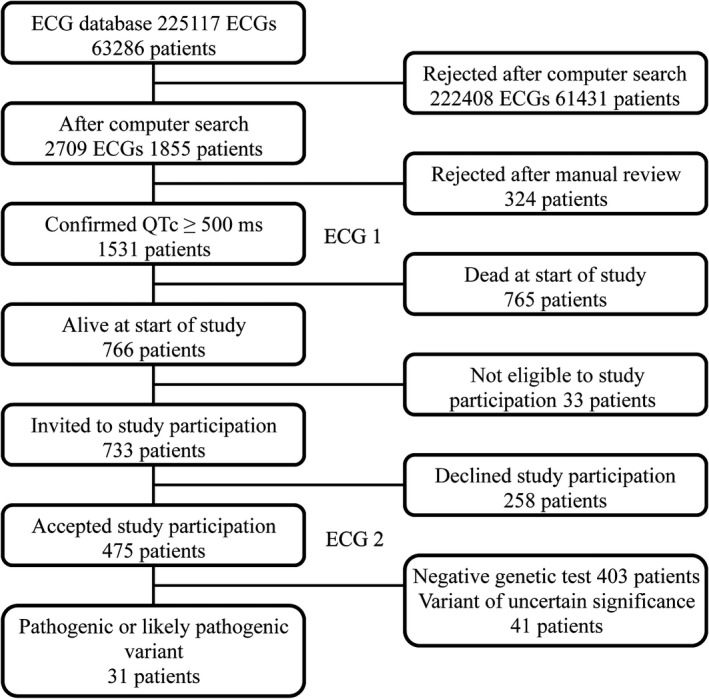
Flow chart of the inclusion process. QTc indicates corrected QT interval.
The QT interval was measured manually in the lead showing the longest QT interval as the mean of 3 consecutive beats. We determined the end of the T wave by the tangent method, and U waves were not included if distinct from the T wave.11, 12 The average heart rate over the entire recording was used if the rhythm was regular. ECGs with frequent premature ventricular beats or short runs of supraventricular tachycardia were excluded.11 The QTc was calculated according to Bazett's formula.
At the time of inclusion in the present study (2015), 766 patients were alive, but 33 of these were excluded because of severe health problems, such as advanced dementia. A total of 733 patients were invited to participate in the study, and those who consented delivered blood for genetic testing and had a new ECG. This ECG is referred to as “ECG 2.”
Clinical Data
Clinical data, including whether the QT prolongation were acknowledged by the clinician, were obtained from the medical records at time of ECG 1, and pro‐QTc risk score was calculated for each patient.12 QT‐prolonging conditions included in the pro‐QTc score are listed in Table 1. QT‐prolonging medication was defined as any medication on the Arizona CredibleMeds QTdrugs lists.13 Hypomagnesemia was defined as serum magnesium ≤0.71 mmol/L, and hypokalemia was defined as serum potassium ≤3.6 mmol/L. Hypocalcemia was defined as corrected serum calcium ≤2.17 mmol/L or ionized serum calcium ≤1.18 mmol/L.
Table 1.
Medical Conditions and Factors Known to Prolong QTc at the Time of ECG 1 and ECG 2 for 31 Patients With Pathogenic or Likely Pathogenic Variants and 41 Patients With VUS
| QT‐Prolonging Medical Conditions | Pathogenic or Likely Pathogenic Variants (n=31) | VUS (n=41) | ||
|---|---|---|---|---|
| ECG 1 | ECG 2 | ECG 1 | ECG 2 | |
| Acute coronary syndrome within 7 d | 1 (3) | 0 (0) | 9 (22) | 0 (0) |
| Anorexia or starvation | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Heart rate <45 bpm | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Diabetes mellitus 1 and 2 | 4 (13) | 4 (13) | 8 (20) | 8 (20) |
| Ejection fraction <40% | 0 (0) | 0 (0) | 7 (17) | 1 (2) |
| Female sex | 20 (65) | 20 (65) | 21 (51) | 21 (51) |
| Hypertrophic cardiomyopathy | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Hypoglycemia (in the absence of diabetes mellitus) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Intoxication with QT‐prolonging drugs | 0 (0) | 0 (0) | 2 (5) | 0 (0) |
| Known genetic LQTS | 1 (3) | 1 (3) | 0 (0) | 0 (0) |
| Pheochromocytoma | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Renal dialysis | 0 (0) | 0 (0) | 1 (2) | 1 (2) |
| Status <7 d after AF conversion | 1 (3) | 0 (0) | 1 (2) | 0 (0) |
| Status <24 h after cardiac arrest | 0 (0) | 0 (0) | 1 (2) | 0 (0) |
| Status <24 h after syncope or seizure | 1 (3) | 0 (0) | 4 (10) | 0 (0) |
| Status <7 d after stroke, subarachnoid hemorrhage, or head trauma | 1 (3) | 0 (0) | 1 (2) | 0 (0) |
| Serum electrolyte disturbances | 3 (10) | 1 (3) | 17 (41) | 0 (0) |
| Drugs with known risk of TdPa | 4 (13) | 3 (10) | 9 (22) | 9 (22) |
| Drugs with possible risk of TdPa | 2 (6) | 1 (3) | 4 (10) | 8 (20) |
| Drugs with conditional risk of TdPa | 11 (35) | 10 (32) | 18 (44) | 19 (46) |
| Drugs with special risk for patients with LQTSa | 2 (6) | 1 (3) | 6 (15) | 2 (5) |
Data are given as number (percentage) of patients. AF indicates atrial fibrillation; bpm, beats per minute; LQTS, long‐QT syndrome; QTc, corrected QT interval; TdP, torsade de pointes ventricular tachycardia; VUS, variants of uncertain significance.
The QT‐prolonging drug categories are according to AZCERT, Inc.13
QT Interval and LQTS Risk Score (Schwartz)
It was noted whether the patients with a pathogenic or likely pathogenic variant and the patients with variants of uncertain significance (VUS) met the criteria for a clinical diagnosis of LQTS on the basis of current guidelines.2, 7 The LQTS risk score (Schwartz) was based on QTc from ECG 2 because it was least affected by environmental factors.2, 10 The difference in QTc between ECG 1 and ECG 2 was also calculated.
DNA Sequencing
The 17 genes in which mutations are known to cause monogenic LQTS were sequenced: AKAP9 (NM_005751.4), ANK2 (NM_1148.4), CACNA1C (NM_000719.6), CALM1 (NM_006888.4), CALM2 (NM_001743.4), CALM3 (NM_005184.2), CAV3 (NM_033337.2), KCNE1 (NM_000219.5), KCNE2 (NM_172201.1), KCNH2 (NM_000238.3), KCNJ2 (NM_000891.2), KCNJ5 (NM_000890.3), KCNQ1 (NM_000218.2), SCN4B (NM_174934.3), SCN5A (NM_198056.2), SNTA1 (NM_003098.2), and TRDN (NM_006073.3). The patients were not referred to testing in a clinical setting, but were included in a research project based solely on prolonged QTc.11 Further details on DNA sequencing, bioinformatics, and RNA analyses are found in Data S1.
Variant Interpretation
An alternative allele frequency cutoff of 0.1% from the Exome Aggregation Consortium database (populations: all or non‐Finnish Europeans) was used for filtering variants (Figure 2). Additional databases of allele frequencies, such as gnomAD,14 SweGen,15 2000 Danes,16 and the in‐house Telemark database with ≈1000 exomes, were consulted when manually reviewing the variants that remained after filtering. Sequence variants that were synonymous (predicting no change in amino acids), intronic (outside splice sites), or in untranslated regions were discarded, unless they had previously been reported as pathogenic or likely pathogenic. Genes and variants that remained after filtering were manually reviewed in the light of available clinical and biological data to evaluate causality. The key tools used for this were Alamut Visual decision‐support software (Interactive Biosoftware), Human Gene Mutation Database,5 and ClinVar.17
Figure 2.

Flow chart of filtering and evaluation of sequence variants. Bp indicates base pair; ExAC, Exome Aggregation Consortium; LQT, long QT; QC, quality control.
Sequence variants were divided into 5 classes: class 5, pathogenic; class 4, likely pathogenic; class 3, uncertain significance; class 2, likely benign; and class 1, benign. Classification was based on guidelines from the Association for Clinical Genetic Science and the American College of Medical Genetics and Genomics.18 Previously reported pathogenic and likely pathogenic variants were not accepted as pathogenic without scrutinizing newly available evidence, such as allele frequencies and in vitro studies.
Statistical Analysis
Continuous data were described by mean±SD or median (range) and compared using the unpaired Student t test or independent‐samples Mann‐Whitney U test, as appropriate. Categorical data were described as proportions and analyzed by the χ2 test (SPSS, version 23.0; IBM, Armonk, NY). A 2‐sided P<0.05 was considered statistically significant.
Ethics
The study complies with the Declaration of Helsinki. The Norwegian Regional Committee for Medical and Health Research Ethics has approved the study (2013/1090), and informed consent was obtained from all patients. Genetic counseling was offered to all patients with pathogenic or likely pathogenic mutations. Patients with a pathogenic or likely pathogenic variant were assigned an LQTS diagnosis in an inherited disease clinic.
Results
Characteristics of Study Population at Time of First ECG With QTc ≥500 ms (ECG 1)
Demographics for the 475 (65%) of 733 patients who participated in the study are shown in Table 2. They underwent genetic testing and were on average 3 years younger than those who declined (63±14 versus 66±16 years; P=0.01). There were no significant differences in QTc, sex, hypokalemia, number of QT‐prolonging conditions, number of QT‐prolonging drugs, or pro‐QTc score between study participants and nonparticipants at the time of ECG 1 (Table 2).
Table 2.
Demographics of 733 Invited Patients at the Time of the First ECG With QTc ≥500 ms (“ECG 1”)
| Demographics | Total (n=733) | Participants (n=475) | Nonparticipants (n=258) | P Value |
|---|---|---|---|---|
| Age, y | 64±15 | 63±14 | 66±16 | 0.01 |
| Female sex | 448 (61) | 294 (62) | 154 (60) | 0.58 |
| Heart rate, bpm | 78 (39–100) | 77 (39–100) | 81 (48–100) | <0.01 |
| QRS duration, ms | 94±12 | 93±12 | 94±12 | 0.32 |
| QTc, ms | 512 (500–669) | 512 (500–669) | 515 (500–634) | 0.17 |
| Hypokalemia | 200/683 (29) | 124/441 (30) | 76/242 (31) | 0.60 |
| No. of QT‐prolonging drugs | 1 (0–5) | 1 (0–5) | 1 (0–5) | 0.34 |
| No. of QT‐prolonging conditionsa | 1 (0–4) | 1 (0–4) | 1 (0–3) | 0.29 |
| Pro‐QTc score | 2.7±1.5 | 2.7±1.6 | 2.7±1.5 | 0.95 |
Data are given as mean±SD, number/total (percentage), or median (range). Bpm indicates beats per minute; QTc, corrected QT interval.
Female sex, electrolyte disturbances, and medication not included.
A pathogenic or likely pathogenic genetic variant was identified in 31 (6.5%) of 475 patients (Table 3). The criteria applied for classification of genetic variants are shown in Table 4. These patients had a lower pro‐QTc score than those without pathogenic or likely pathogenic variants (1.7±1.0 versus 2.8±1.6; P<0.001) (Table 5). Of the 31 patients with a pathogenic or likely pathogenic variant, 12 (39%) had an additional nongenetic explanation for QTc prolongation at the time of ECG 1 (Table 1). The median serum potassium level was 3.5 mmol/L (range, 3.4–3.6 mmol/L) among those with hypokalemia. The sole acute coronary syndrome was minor, without any other ECG changes than the QTc prolongation. Three patients with pathogenic or likely pathogenic variants used potent QT‐prolonging antiarrhythmic drugs (Table S1).
Table 3.
Patients With a Pathogenic or Likely Pathogenic Variant
| No. | Gene | cDNA Change | Protein Change | Age at ECG 1, y | Sex | QTc at ECG 1, ms | QTc at ECG 2, ms | Family History | Syncope |
|---|---|---|---|---|---|---|---|---|---|
| 1 | KCNH2 | c.157G>A | p.(Gly53Ser) | 56 | Male | 535 | 496 | No | Yes |
| 2 | KCNH2 | c.2257G>T | p.(Ala753Ser) | 15 | Male | 520 | 450 | No | Yes |
| 3 | KCNH2 | c.2682_2685dup | p.(Asp896Hisfs*25) | 53 | Male | 513 | 565 | No | No |
| 4 | KCNH2 | c.2775dup | p.(Pro926Alafs*14) | 58 | Female | 517 | 511 | No | No |
| 5 | KCNQ1 | c.573_577del | p.(Arg192Cysfs*91) | 62 | Male | 504 | 492 | No | No |
| 6 | KCNQ1 | c.1588C>T | p.(Gln530*) | 39 | Female | 508 | 508 | No | Yes |
| 7 | KCNQ1 | c.1588C>T | p.(Gln530*) | 39 | Male | 509 | 413 | No | No |
| 8 | KCNQ1 | c.1588C>T | p.(Gln530*) | 43 | Female | 501 | 481 | No | No |
| 9 | KCNQ1 | c.1588C>T | p.(Gln530*) | 45 | Female | 520 | 458 | No | Yes |
| 10 | KCNQ1 | c.1588C>T | p.(Gln530*) | 51 | Male | 551 | 477 | No | No |
| 11 | KCNQ1 | c.1588C>T | p.(Gln530*) | 57 | Female | 504 | 473 | No | No |
| 12 | KCNQ1 | c.1588C>T | p.(Gln530*) | 60 | Female | 504 | 469 | No | No |
| 13 | KCNQ1 | c.1588C>T | p.(Gln530*) | 62 | Female | 510 | 467 | Yes | Yes |
| 14 | KCNQ1 | c.1588C>T | p.(Gln530*) | 62 | Female | 543 | 459 | No | No |
| 15 | KCNQ1 | c.1588C>T | p.(Gln530*) | 64 | Female | 502 | 470 | No | No |
| 16 | KCNQ1 | c.1588C>T | p.(Gln530*) | 66 | Male | 501 | 486 | No | No |
| 17 | KCNQ1 | c.1588C>T | p.(Gln530*) | 68 | Female | 538 | 482 | No | No |
| 18 | KCNQ1 | c.1588C>T | p.(Gln530*) | 70 | Male | 508 | 495 | No | No |
| 19 | KCNQ1 | c.1588C>T | p.(Gln530*) | 70 | Male | 503 | 457 | No | No |
| 20 | KCNQ1 | c.1588C>T | p.(Gln530*) | 73 | Male | 577 | NA | No | No |
| 21 | KCNQ1 | c.1588C>T | p.(Gln530*) | 77 | Female | 520 | 484 | No | No |
| 22 | KCNQ1 | c.1588C>T | p.(Gln530*) | 79 | Female | 506 | 474 | No | No |
| 23 | KCNQ1 | c.1588C>T | p.(Gln530*) | 81 | Female | 506 | 499 | No | No |
| 24 | KCNQ1 | c.1588C>T | p.(Gln530*) | 83 | Female | 515 | 459 | No | No |
| 25 | KCNQ1 | c.1588C>T | p.(Gln530*) | 86 | Female | 516 | 475 | No | No |
| 26 | KCNQ1 | c.1588C>T | p.(Gln530*) | 86 | Female | 508 | 474 | No | No |
| 27 | KCNQ1 | c.1588C>T | p.(Gln530*) | 87 | Female | 532 | 520 | No | Yes |
| 28 | KCNQ1 | c.1588C>T | p.(Gln530*) | 88 | Female | 511 | 462 | Yes | No |
| 29 | KCNQ1 | c.1591‐1G>A | p.? | 56 | Female | 502 | 496 | No | No |
| 30 | KCNQ1 | c.1760C>T | p.(Thr587Met) | 38 | Female | 504 | 493 | Yes | No |
| 31 | SCN5A | c.4931G>A | p.(Arg1644His) | 58 | Male | 513 | 481 | No | Yes |
NA indicates not available; QTc, corrected QT interval.
Table 4.
ACMG Criteria Used for Classification of Pathogenic and Likely Pathogenic Variants
| Gene | cDNA Change | Protein Change | ACMG/AMP Criteria | Classification |
|---|---|---|---|---|
| KCNH2 | c.2775dup | p.(Pro926Alafs*14) | PVS1, PS3, PM2 | Pathogenic |
| KCNH2 | c.2682_2685dup | p.(Asp896Hisfs*25) | PVS1, PM2, PP5 | Pathogenic |
| KCNH2 | c.2257G>T | p.(Ala753Ser) | PM1, PM2, PP2, PP3, PP5 | Likely pathogenic |
| KCNH2 | c.157G>A | p.(Gly53Ser) | PM1, PM2, PM5‐Sa, PP2, PP5 | Pathogenic |
| KCNQ1 | c.573_577del | p.(Arg192Cysfs*91) | PVS1, PP1, PP5 | Pathogenic |
| KCNQ1 | c.1588C>T | p.(Gln530*) | PVS1, PS4, PP1 | Pathogenic |
| KCNQ1 | c.1591‐1G>A | p.? | PVS1, PS3, PM2 | Pathogenic |
| KCNQ1 | c.1760C>T | p.(Thr587Met) | PS1, PS3, PM1, PM2, PP5 | Pathogenic |
| SCN5A | c.4931G>A | p.(Arg1644His) | PS1, PS3, PM2, PP3 | Pathogenic |
ACMG indicates American College of Medical Genetics and Genomics; AMP, Association for Molecular Pathology; PM, pathogenic moderate; PP, pathogenic supporting; PS, pathogenic strong; PVS, pathogenic very strong.
PM5 assigned strong evidence of pathogenicity because both Gly53Asp and Gly53Arg previously have been reported as pathogenic and both were trafficking deficient.
Table 5.
Demographics of 475 Genetically Tested Patients at the Time of the First ECG With QTc ≥500 ms (“ECG 1”)
| Demographics | Total (n=475) | Pathogenic or Likely Pathogenic Variant (n=31) | VUS and Negative Genetic Test Result (n=444) | P Value |
|---|---|---|---|---|
| Age, y | 63±14 | 63±17 | 63±14 | 0.86 |
| Female sex | 294 (62) | 20 (65) | 274 (62) | 0.85 |
| Heart rate, bpm | 77 (39–100) | 66 (41–97) | 77 (39–100) | <0.01 |
| QRS duration, ms | 93±12 | 88±10 | 94±12 | <0.01 |
| QTc, ms | 512 (500–669) | 511 (501–577) | 512 (500–669) | 0.50 |
| Hypokalemia | 124/441 (28) | 3/22 (14) | 121/419 (30) | <0.001 |
| No. of QT‐prolonging drugs | 1 (0–5) | 0 (0–2) | 1 (0–5) | 0.071 |
| No. of QT‐prolonging conditionsa | 1 (0–4) | 0 (0–1) | 1 (0–4) | 0.001 |
| Pro‐QTc score | 2.7±1.6 | 1.7±1.0 | 2.8±1.6 | <0.001 |
Data are given as mean±SD, number/total (percentage), or median (range). Bpm indicates beats per minute; QTc, corrected QT interval; VUS, variants of uncertain significance.
Female sex, electrolyte disturbances, and medication not included.
Among the patients without pathogenic or likely pathogenic variants, 75 (74%) of the 101 patients with acute coronary syndrome had ischemic ECG changes. The median serum potassium level was 3.4 mmol/L (range, 1.9–3.6 mmol/L) among those with hypokalemia.
A genetic VUS was identified in 41 (8.6%) of 475 patients (Table S2). There were no significant differences in QTc, sex, hypokalemia, number of QT‐prolonging conditions, number of QT‐prolonging drugs, or pro‐QTc score between these patients and patients with negative genetic test results (Table S3). All of the patients carrying a VUS had a plausible nongenetic explanation for QTc prolongation at the time of ECG 1 (Table 1, Table S4).
Awareness of QT Prolongation by Healthcare Provider
QTc prolongation was acknowledged in the medical records in 7 (23%) of the 31 patients with a pathogenic or likely pathogenic variant at the time of ECG 1. Only 1 of those had been offered a follow‐up visit and was found to have a QTc of 472 ms after discontinuation of QT‐prolonging medication. QT prolongation was acknowledged in the medical records in 59 (13%) of the 444 patients without a pathogenic or likely pathogenic variant.
QT Interval and LQTS Risk Score (Schwartz)
Among the patients with a pathogenic or likely pathogenic variant, the median QTc in ECG 1 was 511 ms (range, 501–577 ms) and the median QTc in ECG 2 was 479 ms (range, 413–565 ms) (Table 3). One patient missed ECG 2. The median difference between the QTc in ECG 1 and ECG 2 was −33 ms (range, −96 to 52 ms) (Figure 3). Of 30 patients, 4 (13%) had a QTc ≥500 ms and 15 (50%) had a QTc <480 ms on ECG 2 (Figure 4). Only 1 of the patients with a pathogenic or likely pathogenic variant had a longer QTc at the time of ECG 2 compared with ECG 1.
Figure 3.
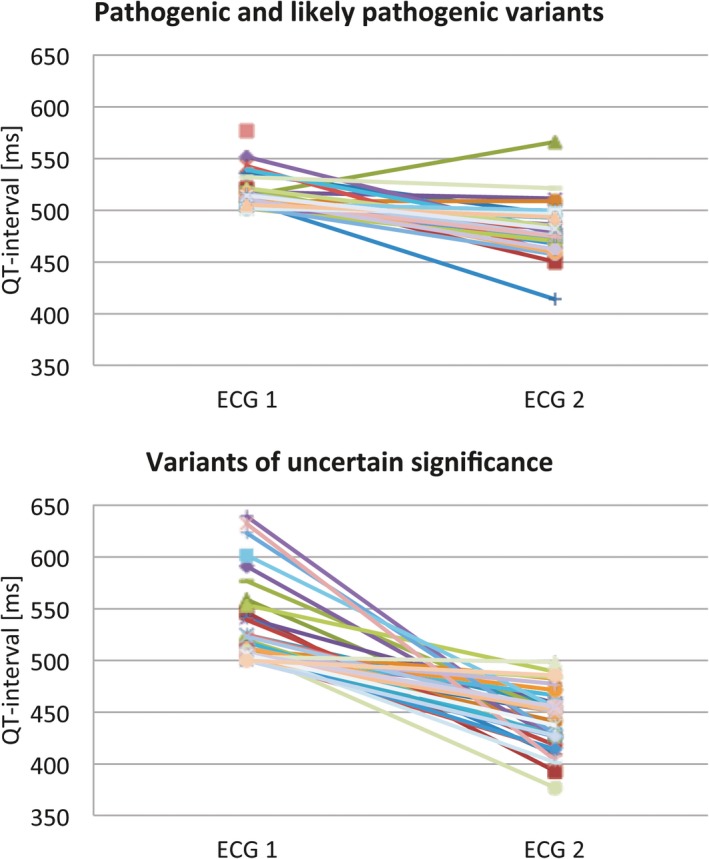
Corrected QT interval at the time of ECG 1 and ECG 2 among 31 patients with pathogenic or likely pathogenic variants and 41 patients with variants of uncertain significance.
Figure 4.
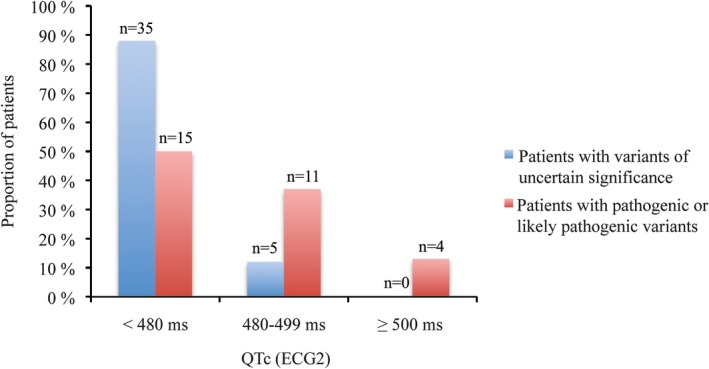
Bar chart showing the corrected QT interval (QTc) distribution at the time of ECG 2 among 30 patients with pathogenic or likely pathogenic variants (red) and 40 patients with variants of uncertain significance (blue).
Of the 31 patients with a pathogenic or likely pathogenic variant, 6 (19%) fulfilled the criteria for a clinical diagnosis of LQTS on the basis of LQTS risk score (Schwartz). One patient had a family history of sudden death in a first‐degree relative with congenital deafness who died at the age of 6 years. Another patient had a known family history of LQTS. Furthermore, 1 patient had a previously diagnosed LQTS. From the age of 6 years, 1 patient had experienced fainting spells, previously thought to be of epileptic origin, but now considered symptoms of LQTS. No patients had any documented episodes of torsade de pointes ventricular tachycardia or cardiac arrest, but 6 patients had previously had a syncope that could have been related to LQTS.
For the patients with a VUS, the median QTc in ECG 1 was 510 ms (range, 500–639 ms) and the median QTc in ECG 2 was 449 ms (range, 377–498 ms). One patient missed ECG 2. The median difference between the QTc in ECG 1 and ECG 2 was −73 ms (range, −5 to −228 ms) (Figure 3). None of the patients carrying a VUS had a QTc ≥500 ms, and 35 (88%) of 40 patients had a QTc of <480 ms on ECG 2 (Figure 4). None of the patients with VUS had an LQTS risk score (Schwartz) >3. None had a family history of sudden death in a first‐degree relative. One patient with a VUS (c.5962A>C p.[Met1988Leu] in ANK2) had a syncope that could have been related to LQTS, had documented polymorphic ventricular tachycardia, and had an implantable cardioverter‐defibrillator implanted.
Patients with a pathogenic or likely pathogenic variant had a higher median QTc in ECG 2 than the patients with VUS (479 versus 449 ms; P<0.001).
Genetic Analyses
A total of 670 sequence variants were detected in the 17 known LQTS genes. Of these, 69 were coding or splice site variants with a minor allele frequency <0.1% in the Exome Aggregation Consortium database (Figure 2). On average, the patients harbored 0.26 such rare coding variants. All pathogenic or likely pathogenic variants were heterozygous. The previously reported variant KCNQ1 c.1588C>T p.(Gln530*) was present in 23 patients. The other 8 variants were all detected in single cases. Patients heterozygous for p.(Gln530*) were older than the other patients with a pathogenic or likely pathogenic variant (67±16 versus 50±16 years; P=0.01) and more likely to be women (74% versus 38%; P=0.08) (Table S5). By excluding patients with the Gln530* variant, the 8 patients with pathogenic or likely pathogenic variants other than KCNQ1 c.1588C>T p.(Gln530*) were younger than the patients with negative genetic test results (50±16 versus 63±14 years; P<0.01).
Two novel pathogenic null variants were detected. KCNH2 c.2682_2685dup p.(Asp896Hisfs*25) is located in the 3′ end of exon 11 and within a few base pairs reach of 3 other frameshift variants previously reported in patients with LQTS.19, 20, 21 KCNQ1 c.1591‐1G>A p.(?) disrupts normal pre‐mRNA splicing (Figure S1).
Discussion
In the present study, a genetic diagnosis was obtained in 31 (6.5%) of 475 patients with at least 1 ECG with a QTc ≥500 ms. This illustrates that a QTc ≥500 ms in at least 1 ECG recording may greatly increase the likelihood of pathogenic or likely pathogenic variants in LQTS genes in hospitalized patients compared with the general population. This is a 130‐fold increase relative to the estimated population prevalence of 1:2000.1
Current guidelines from the European Society of Cardiology recommend clinical diagnosis and genetic testing in an asymptomatic individual if repetitive ECGs show a QTc ≥480 ms. An LQTS risk score (Schwartz) >3 is also clinically diagnostic.2 Genetic testing is usually reserved for patients with persistent QTc prolongation after withdrawal or resolution of the QT‐prolonging factors. We, therefore, included ECG 2 after QT‐prolonging factors had been recognized and modified when appropriate and the patients with an acute illness had recovered. If European Society of Cardiology guidelines had been followed, 47% of the patients with a pathogenic or likely pathogenic variant in the present study would probably not have been considered for the diagnosis. The Heart Rhythm Society/European Heart Rhythm Association Expert Consensus Statement recommends cutoff on repetitive ECGs to QTc >500 ms, which would have failed to diagnose 73% of the patients with a pathogenic or likely pathogenic variant in the present study.7
The awareness of QT prolongation and the possibility of LQTS was low. At Telemark Hospital Trust, the physician ordering the ECG is responsible for the interpretation. At the time of the present study, the QTc duration was indicated on the ECG, but without automated description or flagging of the QT prolongation. QT alert systems have been described previously and could increase awareness of QT prolongation.12
The previously reported KCNQ1 c.1588C>T p.(Gln530*) was identified in 23 of 31 patients with a pathogenic or likely pathogenic variant. Thus, this founder mutation inflates the prevalence of LQTS in Telemark. The Gln530* carriers were significantly older than the other patients with pathogenic or likely pathogenic variants, which could question this variant's effect on QTc interval and mortality. Several other variants previously reported to cause LQTS have recently been proved to have no effect on QTc, syncope, or overall mortality.22 This illustrates the need for reevaluation before accepting previous claims of pathogenicity. Gln530* has previously been reported as the most prevalent pathogenic KCNQ1 variant in Norway,6 and it is most common in Telemark's neighbor district, Agder. It has consistently been reported as pathogenic in several populations.17 In the gnomAD database, Gln530* is present in 7 of 55 818 non‐Finnish European individuals, but absent from all other populations. The variant is also absent from SweGen, 2000 Danes, 1000 genomes Norway, and the Telemark database, which together contain 8000 alleles.15, 16, 23 This supports that the variant is associated with a significant increase in QTc and with an increased risk of cardiac events, although this risk is reported to be lower than for KCNQ1 missense variants.24
Most studies on the risk of life‐threatening outcomes in LQTS have been performed in children and young adults, in contrast to the population in our study. Risk of adverse outcome has been assumed to be low in patients >40 years of age. However, in a previous study from the International Long QT Syndrome Registry, the risk of aborted cardiac arrest, sudden cardiac death, and implantable cardioverter‐defibrillator therapy was maintained until the age of 60 years.25 The likelihood of serious cardiac events is increased in carriers of a pathogenic variant, even when the QTc is within the normal range.3 Thus, for the patients with a pathogenic or likely pathogenic variant in our population, lifestyle modifications, evaluation of drug use, and in some cases prophylactic medical treatment are indicated.
Genetic cascade testing for detected LQTS mutations is warranted by the ability to identify relatives at risk. Many at‐risk individuals are likely to be unaware of their status in light of the disorder's incomplete penetrance, variable expressivity, and unpredictable course.6, 26 The risk of life‐threatening cardiac events is highest in young adolescence, and identification of a pathogenic variant in an LQTS gene has the potential to prevent life‐threatening cardiac events in offspring or siblings.7, 27
More patients with a pathogenic or likely pathogenic variant had a diagnostic Schwartz score, no clear QT‐prolonging factor, and persistent QT prolongation on ECG 2 compared with the patients with a VUS. Furthermore, many of the patients with a pathogenic or likely pathogenic variant had QT prolongation exceeding the values that one may expect from a specific QT‐prolonging factor, favoring these patients when considering patients for genetic testing. A role for common variants on QT interval and arrhythmia has been demonstrated, but they still have limited clinical utility and their contribution in this cohort has not been analyzed.28, 29
Of the 17 genes tested, pathogenic or likely pathogenic variants were detected only in the 3 major genes KCNQ1, KCNH2, and SCN5A. In contrast, 7 genes contained at least 1 VUS, and only 16 of 47 VUS were identified in the 3 major genes. Five patients carried 2 VUS each. One of these patients was asymptomatic but had consistently QTc >480 ms in >10 ECGs, and another patient had QTc ≥497 ms in 4 ECGs. It could be speculated that the coexistence of >1 VUS could increase QTc, because additive effects have been suggested by others.30, 31 However, the clinical characteristics of the 41 patients with VUS are comparable to the mutation‐negative group. This supports the notion that VUS are of limited clinical importance. Whether a VUS represents a benign variant, a proarrhythmic variant requiring secondary provocation, or a truly pathogenic variant remains to be determined. Until then, they should not be used for clinical decisions.
Study Limitations
All patients were recruited from Telemark County in Norway, which has 173 000 inhabitants who are relatively homogeneous from a genetic perspective. The mutation spectrum would thus likely be different in other populations. The Gln530* variant inflates the prevalence of LQTS in Telemark, and the prevalence of LQTS may be different in other populations.
Only patients still alive at the time of inclusion were included. The mutation spectrum and prevalence of pathogenic or likely pathogenic variants may be different in the deceased individuals. It is possible that patients with a history of cardiac disease (themselves or relatives) are more prone to accept genetic testing.
Only the 17 known LQTS genes have been sequenced, and not all genetic defects in these genes will be detected by next‐generation sequencing. Most intronic variants and deletions/duplications larger than ≈40 base pairs escape detection. Interpretation of the pathogenicity of genetic variation is based on current genetic and medical knowledge and is likely to change over time.
The pro‐QTc score was developed to predict mortality in hospitalized patients with QT prolongation. In the present study, the pro‐QTc score was used to assess the number of QT‐prolonging factors. The CredibleMeds QTdrugs lists were accessed in 2015, and the pro‐QTc score, therefore, does not include the most recent drugs added to the lists. Automated T‐wave morphological analyses were not performed in the present study. Future studies should assess T‐wave morphological characteristics in patients with a prolonged QT interval.
Bazett's formula overestimates QTc at high heart rates, but there were no significant differences between the groups at the time of ECG 1 using QTc Fredericia or QTc Framingham (Table S6).
Conclusion
We detected a molecular genetic diagnosis of LQTS in 31 (6.5%) of 475 patients with at least 1 ECG with a QTc ≥500 ms in a community hospital. Compared with the general population, hospitalized patients with a QTc ≥500 ms in at least 1 ECG recording had an increased likelihood for pathogenic and likely pathogenic variants in LQTS genes. If European Society of Cardiology guidelines had been followed, 47% of the patients carrying pathogenic and likely pathogenic variants in the present study would not have been diagnosed. If the Heart Rhythm Society/European Heart Rhythm Association Expert Consensus Statement had been adhered to, 73% of the patients would not have been diagnosed. We recommend increased awareness of the possibility of LQTS in patients with at least 1 ECG with a QTc ≥500 ms.
Sources of Funding
This work was supported by a grant from Telemark Hospital Trust (Skien, Norway).
Disclosures
None.
Supporting information
Data S1. Expanded Methods
Table S1. Number of Drugs From CredibleMeds QTdrugs Lists at Time of ECG 1 and ECG 2 for 31 Patients With a Pathogenic or Likely Pathogenic Variant
Table S2. Variants of Uncertain Significance
Table S3. Demographics of 41 Patients With Variants of Uncertain Significance (VUS) at Time of First ECG With QTc ≥500 ms (“ECG 1”)
Table S4. Number of Drugs From CredibleMeds QTdrugs Lists at Time of ECG 1 and ECG 2 for 41 Patients With VUS
Table S5. Demographics of 23 Patients With KCNQ1 p.(Gln530*) Compared to 8 Patients With Pathogenic or Likely Pathogenic Variants at Time of First ECG With QTc ≥500 ms (“ECG 1”)
Table S6. Uncorrected QT Duration, QTc Bazett, QTc Fredericia and QTc Framingham at Time of First ECG With QTc ≥500 ms (“ECG 1”)
Figure S1. RT‐PCR of KCNQ1 c.1591‐1G>A and SCN5A c.4501C>G.
Data S1. Expanded Methods
(J Am Heart Assoc. 2018;7:e009706 DOI: 10.1161/JAHA.118.009706.)
References
- 1. Schwartz PJ, Stramba‐Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, Mosca F, Nespoli L, Rimini A, Rosati E, Salice P, Spazzolini C. Prevalence of the congenital long‐QT syndrome. Circulation. 2009;120:1761–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Priori SG, Blomstrom‐Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez‐Madrid A, Nikolaou N, Norekval TM, Spaulding C, Van Veldhuisen DJ. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2793–2867. [DOI] [PubMed] [Google Scholar]
- 3. Goldenberg I, Horr S, Moss AJ, Lopes CM, Barsheshet A, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, Ackerman MJ, Benhorin J, Kaufman ES, Napolitano C, Platonov PG, Priori SG, Qi M, Schwartz PJ, Shimizu W, Towbin JA, Vincent GM, Wilde AA, Zhang L. Risk for life‐threatening cardiac events in patients with genotype‐confirmed long‐QT syndrome and normal‐range corrected QT intervals. J Am Coll Cardiol. 2011;57:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moss AJ, Zareba W, Hall WJ, Schwartz PJ, Crampton RS, Benhorin J, Vincent GM, Locati EH, Priori SG, Napolitano C, Medina A, Zhang L, Robinson JL, Timothy K, Towbin JA, Andrews ML. Effectiveness and limitations of beta‐blocker therapy in congenital long‐QT syndrome. Circulation. 2000;101:616–623. [DOI] [PubMed] [Google Scholar]
- 5. Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next‐generation sequencing studies. Hum Genet. 2017;136:665–677. Accessed online January 1, 2017. Downloaded version 2016‐1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berge KE, Haugaa KH, Fruh A, Anfinsen OG, Gjesdal K, Siem G, Oyen N, Greve G, Carlsson A, Rognum TO, Hallerud M, Kongsgard E, Amlie JP, Leren TP. Molecular genetic analysis of long QT syndrome in Norway indicating a high prevalence of heterozygous mutation carriers. Scand J Clin Lab Invest. 2008;68:362–368. [DOI] [PubMed] [Google Scholar]
- 7. Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze‐Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8:1308–1339. [DOI] [PubMed] [Google Scholar]
- 8. Giudicessi JR, Ackerman MJ. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl Res. 2013;161:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Online Mendelian Inheritance in Man, OMIM® . Baltimore, MD: McKusick‐Nathans Institute of Genetic Medicine, Johns Hopkins University; https://omim.org/. Accessed January 24, 2018. [Google Scholar]
- 10. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome: an update. Circulation. 1993;88:782–784. [DOI] [PubMed] [Google Scholar]
- 11. Gibbs C, Thalamus J, Heldal K, Holla OL, Haugaa KH, Hysing J. Predictors of mortality in high‐risk patients with QT prolongation in a community hospital. Europace. 2018;20:f99–f107. [DOI] [PubMed] [Google Scholar]
- 12. Haugaa KH, Bos JM, Tarrell RF, Morlan BW, Caraballo PJ, Ackerman MJ. Institution‐wide QT alert system identifies patients with a high risk of mortality. Mayo Clin Proc. 2013;88:315–325. [DOI] [PubMed] [Google Scholar]
- 13. Woosley RL, Romero KA. QTdrugs list. Oro Valley, AZ: Azcert, Inc; 2015. http://www.Crediblemeds.org. Accessed March 31, 2015.
- 14. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell‐Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce‐Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano‐Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie‐Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG; Exome Aggregation Consortium . Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ameur A, Dahlberg J, Olason P, Vezzi F, Karlsson R, Martin M, Viklund J, Kahari AK, Lundin P, Che H, Thutkawkorapin J, Eisfeldt J, Lampa S, Dahlberg M, Hagberg J, Jareborg N, Liljedahl U, Jonasson I, Johansson A, Feuk L, Lundeberg J, Syvanen AC, Lundin S, Nilsson D, Nystedt B, Magnusson PK, Gyllensten U. SweGen: a whole‐genome data resource of genetic variability in a cross‐section of the Swedish population. Eur J Hum Genet. 2017;25:1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lohmueller KE, Sparso T, Li Q, Andersson E, Korneliussen T, Albrechtsen A, Banasik K, Grarup N, Hallgrimsdottir I, Kiil K, Kilpelainen TO, Krarup NT, Pers TH, Sanchez G, Hu Y, Degiorgio M, Jorgensen T, Sandbaek A, Lauritzen T, Brunak S, Kristiansen K, Li Y, Hansen T, Wang J, Nielsen R, Pedersen O. Whole‐exome sequencing of 2,000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am J Hum Genet. 2013;93:1072–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin‐Salomon R, Rubinstein W, Maglott DR. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hedley PL, Jorgensen P, Schlamowitz S, Wangari R, Moolman‐Smook J, Brink PA, Kanters JK, Corfield VA, Christiansen M. The genetic basis of long QT and short QT syndromes: a mutation update. Hum Mutat. 2009;30:1486–1511. [DOI] [PubMed] [Google Scholar]
- 20. Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris‐Kerr C, Pollevick GD, Wilde AA, Ackerman MJ. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nishimoto O, Matsuda M, Nakamoto K, Nishiyama H, Kuraoka K, Taniyama K, Tamura R, Shimizu W, Kawamoto T. Peripartum cardiomyopathy presenting with syncope due to torsades de pointes: a case of long QT syndrome with a novel KCNH2 mutation. Intern Med. 2012;51:461–464. [DOI] [PubMed] [Google Scholar]
- 22. Ghouse J, Have CT, Weeke P, Bille Nielsen J, Ahlberg G, Balslev‐Harder M, Appel EV, Skaaby T, Olesen SP, Grarup N, Linneberg A, Pedersen O, Haunso S, Hastrup Svendsen J, Hansen T, Kanters JK, Salling Olesen M. Rare genetic variants previously associated with congenital forms of long QT syndrome have little or no effect on the QT interval. Eur Heart J. 2015;36:2523–2529. [DOI] [PubMed] [Google Scholar]
- 23. The Norwegian 1000 genomes project. http://kreftgenomikk.no/en/1000genomes. Accessed January 4, 2016.
- 24. Ruwald MH, Xu Parks X, Moss AJ, Zareba W, Baman J, McNitt S, Kanters JK, Shimizu W, Wilde AA, Jons C, Lopes CM. Stop‐codon and C‐terminal nonsense mutations are associated with a lower risk of cardiac events in patients with long QT syndrome type 1. Heart Rhythm. 2016;13:122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goldenberg I, Moss AJ, Bradley J, Polonsky S, Peterson DR, McNitt S, Zareba W, Andrews ML, Robinson JL, Ackerman MJ, Benhorin J, Kaufman ES, Locati EH, Napolitano C, Priori SG, Qi M, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Long‐QT syndrome after age 40. Circulation. 2008;117:2192–2201. [DOI] [PubMed] [Google Scholar]
- 26. Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long‐QT syndrome: clinical impact. Circulation. 1999;99:529–533. [DOI] [PubMed] [Google Scholar]
- 27. Goldenberg I, Moss AJ, Peterson DR, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, Ackerman MJ, Benhorin J, Kaufman ES, Napolitano C, Priori SG, Qi M, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long‐QT syndrome. Circulation. 2008;117:2184–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arking DE, Pulit SL, Crotti L, van der Harst P, Munroe PB, Koopmann TT, Sotoodehnia N, Rossin EJ, Morley M, Wang X, Johnson AD, Lundby A, Gudbjartsson DF, Noseworthy PA, Eijgelsheim M, Bradford Y, Tarasov KV, Dorr M, Muller‐Nurasyid M, Lahtinen AM, Nolte IM, Smith AV, Bis JC, Isaacs A, Newhouse SJ, Evans DS, Post WS, Waggott D, Lyytikainen LP, Hicks AA, Eisele L, Ellinghaus D, Hayward C, Navarro P, Ulivi S, Tanaka T, Tester DJ, Chatel S, Gustafsson S, Kumari M, Morris RW, Naluai AT, Padmanabhan S, Kluttig A, Strohmer B, Panayiotou AG, Torres M, Knoflach M, Hubacek JA, Slowikowski K, Raychaudhuri S, Kumar RD, Harris TB, Launer LJ, Shuldiner AR, Alonso A, Bader JS, Ehret G, Huang H, Kao WH, Strait JB, Macfarlane PW, Brown M, Caulfield MJ, Samani NJ, Kronenberg F, Willeit J; CARe Consortium; COGENT Consortium , Smith JG, Greiser KH, Meyer Zu Schwabedissen H, Werdan K, Carella M, Zelante L, Heckbert SR, Psaty BM, Rotter JI, Kolcic I, Polasek O, Wright AF, Griffin M, Daly MJ; DCCT/EDIC , Arnar DO, Holm H, Thorsteinsdottir U; eMERGE Consortium , Denny JC, Roden DM, Zuvich RL, Emilsson V, Plump AS, Larson MG, O'Donnell CJ, Yin X, Bobbo M, D'Adamo AP, Iorio A, Sinagra G, Carracedo A, Cummings SR, Nalls MA, Jula A, Kontula KK, Marjamaa A, Oikarinen L, Perola M, Porthan K, Erbel R, Hoffmann P, Jockel KH, Kalsch H, Nothen MM; HRGEN Consortium , den Hoed M, Loos RJ, Thelle DS, Gieger C, Meitinger T, Perz S, Peters A, Prucha H, Sinner MF, Waldenberger M, de Boer RA, Franke L, van der Vleuten PA, Beckmann BM, Martens E, Bardai A, Hofman N, Wilde AA, Behr ER, Dalageorgou C, Giudicessi JR, Medeiros‐Domingo A, Barc J, Kyndt F, Probst V, Ghidoni A, Insolia R, Hamilton RM, Scherer SW, Brandimarto J, Margulies K, Moravec CE, del Greco MF, Fuchsberger C, O'Connell JR, Lee WK, Watt GC, Campbell H, Wild SH, El Mokhtari NE, Frey N, Asselbergs FW, Mateo Leach I, Navis G, van den Berg MP, van Veldhuisen DJ, Kellis M, Krijthe BP, Franco OH, Hofman A, Kors JA, Uitterlinden AG, Witteman JC, Kedenko L, Lamina C, Oostra BA, Abecasis GR, Lakatta EG, Mulas A, Orru M, Schlessinger D, Uda M, Markus MR, Volker U, Snieder H, Spector TD, Arnlov J, Lind L, Sundstrom J, Syvanen AC, Kivimaki M, Kahonen M, Mononen N, Raitakari OT, Viikari JS, Adamkova V, Kiechl S, Brion M, Nicolaides AN, Paulweber B, Haerting J, Dominiczak AF, Nyberg F, Whincup PH, Hingorani AD, Schott JJ, Bezzina CR, Ingelsson E, Ferrucci L, Gasparini P, Wilson JF, Rudan I, Franke A, Muhleisen TW, Pramstaller PP, Lehtimaki TJ, Paterson AD, Parsa A, Liu Y, van Duijn CM, Siscovick DS, Gudnason V, Jamshidi Y, Salomaa V, Felix SB, Sanna S, Ritchie MD, Stricker BH, Stefansson K, Boyer LA, Cappola TP, Olsen JV, Lage K, Schwartz PJ, Kaab S, Chakravarti A, Ackerman MJ, Pfeufer A, de Bakker PI, Newton‐Cheh C. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat Genet. 2014;46:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi F, Guicheney P, Fressart V, Leenhardt A, Antzelevitch C, Bartkowiak S, Borggrefe M, Schimpf R, Schulze‐Bahr E, Zumhagen S, Behr ER, Bastiaenen R, Tfelt‐Hansen J, Olesen MS, Kaab S, Beckmann BM, Weeke P, Watanabe H, Endo N, Minamino T, Horie M, Ohno S, Hasegawa K, Makita N, Nogami A, Shimizu W, Aiba T, Froguel P, Balkau B, Lantieri O, Torchio M, Wiese C, Weber D, Wolswinkel R, Coronel R, Boukens BJ, Bezieau S, Charpentier E, Chatel S, Despres A, Gros F, Kyndt F, Lecointe S, Lindenbaum P, Portero V, Violleau J, Gessler M, Tan HL, Roden DM, Christoffels VM, Le Marec H, Wilde AA, Probst V, Schott JJ, Dina C, Redon R. Common variants at SCN5A‐SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45:1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoshi M, Liu H, Kaufman ES, Deschenes I. Polygenic case of long QT syndrome confirmed through functional characterization informs the interpretation of genetic screening results. HeartRhythm Case Rep. 2015;1:201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mullally J, Goldenberg I, Moss AJ, Lopes CM, Ackerman MJ, Zareba W, McNitt S, Robinson JL, Benhorin J, Kaufman ES, Towbin JA, Barsheshet A. Risk of life‐threatening cardiac events among patients with long QT syndrome and multiple mutations. Heart Rhythm. 2013;10:378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Expanded Methods
Table S1. Number of Drugs From CredibleMeds QTdrugs Lists at Time of ECG 1 and ECG 2 for 31 Patients With a Pathogenic or Likely Pathogenic Variant
Table S2. Variants of Uncertain Significance
Table S3. Demographics of 41 Patients With Variants of Uncertain Significance (VUS) at Time of First ECG With QTc ≥500 ms (“ECG 1”)
Table S4. Number of Drugs From CredibleMeds QTdrugs Lists at Time of ECG 1 and ECG 2 for 41 Patients With VUS
Table S5. Demographics of 23 Patients With KCNQ1 p.(Gln530*) Compared to 8 Patients With Pathogenic or Likely Pathogenic Variants at Time of First ECG With QTc ≥500 ms (“ECG 1”)
Table S6. Uncorrected QT Duration, QTc Bazett, QTc Fredericia and QTc Framingham at Time of First ECG With QTc ≥500 ms (“ECG 1”)
Figure S1. RT‐PCR of KCNQ1 c.1591‐1G>A and SCN5A c.4501C>G.
Data S1. Expanded Methods