

Abstract
Background
Although macrophage migration inhibitory factor (MIF) has been demonstrated to mediate cardioprotection in ischemia/reperfusion injury and antagonize fibrotic effects through its receptor, CD74, the function of the soluble CD74 receptor ectodomain (sCD74) and its interaction with circulating MIF have not been explored in cardiac disease.
Methods and Results
Cardiac fibroblasts were isolated from hearts of neonatal mice and differentiated into myofibroblasts. Co‐treatment with recombinant MIF and sCD74 induced cell death (P<0.001), which was mediated by receptor‐interacting serine/threonine‐protein kinase (RIP)1/RIP3‐dependent necroptosis (P=0.0376). This effect was specific for cardiac fibroblasts and did not affect cardiomyocytes. Gene expression analyses using microarray and RT‐qPCR technology revealed a 4‐fold upregulation of several interferon‐induced genes upon co‐treatment of myofibroblasts with sCD74 and MIF (Ifi44: P=0.011; Irg1: P=0.022; Clec4e: P=0.011). Furthermore, Western blot analysis confirmed the role of sCD74 as a modulator of MIF signaling by diminishing MIF‐mediated protein kinase B (AKT) activation (P=0.0197) and triggering p38 activation (P=0.0641). We obtained evidence that sCD74 inhibits MIF‐mediated survival pathway through the C‐X‐C chemokine receptor 4/AKT axis, enabling the induction of CD74‐dependent necroptotic processes in cardiac myofibroblasts. Preliminary clinical data revealed a lowered sCD74/MIF ratio in heart failure patients (17.47±10.09 versus 1.413±0.6244).
Conclusions
These findings suggest that treatment of cardiac myofibroblasts with sCD74 and MIF induces necroptosis, offering new insights into the mechanism of myofibroblast depletion during scar maturation. Preliminary clinical data provided first evidence about a clinical relevance of the sCD74/MIF axis in heart failure, suggesting that these proteins may be a promising target to modulate cardiac remodeling and disease progression in heart failure.
Keywords: cell death, heart failure, macrophage migration inhibitory factor, myocardial fibrosis, myofibroblast, necroptosis, soluble CD74
Subject Categories: Basic Science Research, Growth Factors/Cytokines, Fibrosis, Translational Studies, Heart Failure
Clinical Perspective
What Is New?
The effect and molecular mechanisms of the soluble CD74 receptor domain (sCD74) in cardiac fibrosis are still elusive.
Present findings demonstrated that sCD74 and macrophage migration inhibitory factor (MIF) synergistically induce necroptosis in cardiac myofibroblasts, whereas cardiomyocytes were not prone to sCD74/MIF‐induced cell death.
Preliminary clinical data revealed a lowered sCD74/MIF ratio in heart failure patients, indicating that sCD74 and MIF might affect disease progression in patients with heart failure.
What Are the Clinical Implications?
This is the first study demonstrating that cardiac myofibroblast can undergo sCD74/MIF‐regulated necroptosis, which might offer new insights into the mechanism of myofibroblast depletion during post–myocardial infarction scar maturation.
MIF and sCD74 could represent a potential therapeutic target to modulate cardiac remodeling and disease progression in heart failure.
Introduction
The healthcare system is burdened with an increasing incidence of coronary heart disease (CHD), which is the leading cause of morbidity and mortality in the Western world.1, 2 Numerous CHD patients, who experienced and survived a myocardial infarction (MI) event, consequently suffer from adverse ventricular remodeling and heart failure (HF), requiring intensive treatment strategies and hospitalization.2, 3
MI‐associated remodeling of the myocardium is required for maintaining cardiac function and integrity.4 Immediately after an ischemic insult, cardiac remodeling is initiated by a transient inflammatory response triggered through danger‐associated molecular (DAMP) patterns and cytokines released from injured cardiomyocytes, attracting immune cells into the infarcted area.5, 6 Once the wound is “cleared” and the inflammatory phase is repressed, fibroblasts are activated and recruited, followed by their proliferation and differentiation into myofibroblasts.7, 8 Myofibroblasts express and deposit large amounts of collagen to renew the extracellular matrix (ECM) compartment, therefore ensuring the integrity of the heart tissue.9, 10 However, persistence and sustained activation of myofibroblasts, especially in uninfarcted but vulnerable areas, accounts for expansion of the fibrotic core and adverse ventricular remodeling. Additionally, myofibroblasts are the major site of matrix metalloproteinases (MMPs) synthesis in the myocardium.11, 12 An increased MMP activity is observed in the myocardium of humans and animals with HF disease,13, 14, 15 underlining their key role in pathological cardiac remodeling by directly degrading ECM, which correlates with ventricular dilatation followed by a decrease in cardiac tensile strength.16 This, in turn, leads to cardiac dysfunction and ultimately to HF with adverse clinical effects on patients’ mid‐ to long‐term outcome.9, 17
The stress‐regulating chemokine‐like cytokine, macrophage migration inhibitory factor (MIF) features danger‐associated molecular pattern–like characteristics and is an important upstream regulator of the innate immune response.18, 19, 20 Although it generally promotes acute and chronic inflammatory processes, MIF has been demonstrated to also exhibit protective effects during myocardial ischemia/reperfusion injury, especially in the ischemic and early reperfusion phase. Similarly, the role of MIF in cardiac remodeling also appears to be dichotomous.21 On the one hand, MIF promotes postinfarct rupture and remodeling by promoting infiltration of immune cells as well as expression of proinflammatory and fibrosis‐related proteins.21, 22 On the other hand, MIF depletion has been reported to delay post‐MI healing and to worsen aging‐induced cardiac remodeling and fibrosis in myocardial hypertrophy.21, 23, 24
Cardioprotection by MIF is mediated through its intrinsic antioxidant capacity and by signaling through its cognate receptor CD74, a type II transmembrane glycoprotein and the surface form of class II invariant chain.25, 26, 27, 28, 29, 30, 31 In fact, The MIF/CD74/AMPK (adenosine monophosphate kinase) signaling pathway has repeatedly been demonstrated to play a pivotal protective role in acute myocardial ischemia/reperfusion injury.31, 32, 33 MIF also signals through the chemokine receptors, C‐X‐C chemokine receptor (CXCR)2 and CXCR4,34 and has been found to exhibit compartmentalized protective and detrimental effects through CXCR2 receptor in a mouse model of myocardial ischemia/reperfusion.35 Whereas the C‐X‐C motif chemokine ligand 12/CXCR4 axis also has a double‐edged role in experimental MI, the contribution of the MIF/CXCR4 ligand/receptor axis has not yet been directly studied in the heart.36
Recently, it was demonstrated that a soluble CD74 (sCD74) fragment is released from the cell surface of liver cells by ectodomain shedding to modulate MIF‐dependent activities.37 Ectodomain shedding represents an important posttranslational modification event that downregulates cell‐surface expression of various receptors and liberates biologically active fragments that often exhibit a function that is distinct from that of the membrane‐bound receptor form.38 Although the effects of the intracellular domain of CD74, released following regulated intracellular proteolysis, have been extensively studied,39, 40, 41 we are only beginning to understand the functions of the CD74 ectodomain.37, 39, 42, 43, 44
Here, we applied an in vitro model of isolated primary cardiac fibroblasts to comprehensively study the effects and mechanisms of sCD74 and MIF in myocardial fibrosis and evaluated circulating sCD74 and MIF in plasma samples of patients suffering from CHD and advanced HF.
Methods
Data, analytical methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure, because of ongoing studies.
Animals
All animal experiments were performed in accord with the local institution's Ethical Review Committee and were approved by an animal protection representative at the Institute of Animal Research of the RWTH Aachen University Hospital in accord with German Animal Protection Law §4, Section 3. All experimental procedures were approved by the Animal Care and Use Committee of the local authorities (TV11311A4, AC, LANUV NRW, Essen, Germany). Primary cultures of cardiac fibroblasts were generated from the heart of 1‐ to 7‐day‐old wild‐type (WT; C57Bl/6J) mice (Charles‐River Laboratories, Sulzfeld, Germany) or CD74 knockout (B6‐(Cd74)tm) mice.45 Mice were fed normal chow diets and housed under standardized light‐dark cycles and specific pathogen‐free conditions.
Patients
Blood samples were received from patients after informed consent, approved by the Local Ethic Committee (registration number: EK 151/09), and registered at clinical trials.gov (ClinicalTrials.gov Identifier: NCT02488876).
Reagents
All reagents were obtained from Sigma‐Aldrich (St. Louis, MO), Thermo Fisher Scientific (Schwerte, Germany), or Roth (Karlsruhe, Germany), if not stated otherwise.
Isolation and Culture of Murine Fibroblasts
Cardiac fibroblasts were isolated from hearts of 1‐ to 7‐day‐old C57BL/6J WT and/or Cd74 −/− mice. Briefly, hearts were freed from atria, valves, and vessels, washed in HBSS, and cut in 2 to 4 pieces. Heart tissue was predigested with 0.05% trypsin/HBSS solution under gentle rotation at 4°C overnight. On the next day, predigested tissue was incubated for several cycles with 250 U/mg of collagenase/HBSS solution at 37°C. The first supernatant was discarded, and the following supernatants were pooled in prechilled centrifuge tubes with growth medium consisting of low‐glucose (0.1%) DMEM, 10% horse serum, 5% FCS, 100 U/mL of penicillin/streptomycin, and 20 mmol/L of HEPES. After centrifugation (8 minutes at 500g at room temperature [RT]), the pellet was resuspended in growth medium and filtered through a prewetted 100‐μm cell strainer (BD Falcon, Durham, NC). Cardiac cells were preplated for 90 minutes to separate fast‐attaching fibroblasts from other cardiac cell types. Fibroblasts were then detached with trypsin and seeded at 100 000 cells/cm2 in cell‐culture dishes coated with 5 mg/mL of fibronectin in 0.02% gelatin and cultivated for 10 to 14 days at 37°C.
Culture of Murine Cardiomyocytes
HL1 cells were established and provided by Claycomb Laboratory (Toronto, Ontario, Canada). Because of their ability to maintain contraction and express phenotypic characteristics of cardiomyocytes, HL1 cells are a commonly used cell line for study of cardiac function. Cells were maintained in Claycomb culture medium supplemented with 10% FCS, 1% penicillin/streptomycin, 0.1 mmol/L of norepinephrine, and 2 mmol/L of l‐glutamine and routinely cultured at 37°C and 5% CO2 in a humidified incubator. The Claycomb medium was stored light‐protected. To maintain the phenotypic characteristics and the contracting ability of HL1 cells, culturing required the supplementation with 0.1 mmol/L of norepinephrine and, most important, FCS derived from the Batch 11A568, which was pretested by the Claycomb Laboratory. For experiments, 60 000 cells/cm2 were seeded on fibronectin/gelatin‐coated plates 24 hours before treatment.
Immunofluorescence Staining
Isolated primary cardiac fibroblasts were seeded at 100 000 cells/cm2 in IBIDI dishes (IBIDI, Martinsried, Germany). After 5 hours, 3 days, and 5 days, they were washed with PBS and fixed with 4% PFA for 20 minutes. Subsequently, cells were washed 3 to 5 times with PBS, followed by 2 hours of blocking (2% BSA in PBS‐T) and an overnight incubation with the primary antibody (1:200 dilution in 2% BSA in PBS‐T) in the dark at 4°C. Fibroblasts were co‐stained with vimentin, a fibroblast marker and α‐smooth muscle actin (α‐SMA), a myofibroblast marker. On the next day, cells were washed 3 to 5 times with PBS and incubated with the secondary antibody mixture (1:200 dilution in 2% BSA in PBS‐T) in the dark at RT for 2 hours (Table S1). Following 3 to 5 wash cycles with PBS, cells were covered with Fluoromount G (SouthernBiotech, Birmingham, AL), nuclei stained with 4′,6‐diamidino‐2‐phenylindole, and, finally, sealed with a cover slide. Images were recorded by Leica DM 2500 (Leica Microsystems, Wetzlar, Germany). Images were processed using DISKUS (Hilgers, Königswinter, Germany).
Experimental Setup of Cardiac Myofibroblast and Cardiomyocytes Incubations
Cardiac myofibroblasts were incubated with increasing concentrations of sCD74 (0, 0.04, 0.16, 8, 16, and 40 nmol/L; R&D Systems, Minneapolis, MN) with or without recombinant MIF (rMIF; 8 nmol/L; PeproTech. Rocky Hill, NJ). Studies in mouse cardiomyocytes (HL1) as well as signaling and mechanistic studies in myofibroblasts were then performed with the highest sCD74 concentration (40 nmol/L) in the presence versus absence of rMIF (8 nmol/L). For inhibition studies, 1 hour before sCD74/rMIF application, fibroblasts were treated with 12.6 μmol/L of CXCR4 inhibitor AMD3100, 14.2 μmol/L of CXCR2 inhibitor SB225002, or 100 μmol/L of receptor‐interacting serine/threonine‐protein kinase 1 (RIP1) inhibitor 7‐Cl‐O‐Nec‐1 (necrostatin‐1s, Nec1s; Merck Millipore, Darmstadt, Germany). The experimental setup is illustrated in Figure S1.
Survival Assays
Cell survival was assessed by trypan blue staining and counting. Cells were detached with accutase 20 to 24 hours after stimulation and mixed with the equal volume of 0.4% trypan blue solution (Bio‐Rad, Munich, Germany). The suspension was transferred to counting slides (Bio‐Rad), and total as well as living cell number was analyzed automatically by using the TC20 Automated Cell Counter (Bio‐Rad). Cell viability was calculated by the ratio of living to total cell count, and all values were normalized to cell viability of control cells.
RNA Extraction and Quantitative Real‐Time PCR
Total RNA was isolated 8 hours after stimulation using the Nucleo Spin RNA/Protein kit (Machery‐Nagel, Düren, Germany), and 1 μg of mRNA was reverse‐transcribed into cDNA using a high‐capacity cDNA reverse transcription kit (Applied Biosystems, Carlsbad, CA). mRNA quality and concentration were measured by the Infinite 200 PRO (Tecan, Männedorf, Switzerland). PCR was performed using 50 ng of cDNA and TaqMan probes (Applied Biosystems; Table S2) on a StepOne Plus Real‐Time PCR System (Applied Biosystems). Glyceraldehyde 3‐phosphate dehydrogenase was used as a housekeeping gene. Relative quantity values were calculated according to the ΔΔCt method and normalized to control.
Gene Expression Microarray
Gene expression analysis of cardiac fibroblasts for each treatment (carrier, rMIF, sCD74, and rMIF/sCD74) was carried out using the mouse Clariom S Array (Affymetrix, Santa Clara, CA) in independent triplicates according to the Minimum Information About a Microarray Experiment (MIAME) criteria. Total RNA was isolated 8 hours after stimulation using the Nucleo Spin RNA/Protein kit (Machery‐Nagel) and quantified (Nanodrop). RNA quality was assessed using the RNA 6000 Nano Assay with the 2100 Bioanalyzer (Agilent, Santa Clara, CA). Samples, each 150 ng of total RNA, for mouse Clariom S Arrays were prepared and hybridized to the arrays according to the GeneChip WT PLUS Reagent Kit (Affymetrix), according to the manufacturer's protocol. Processed samples were hybridized to mouse Clariom S Arrays at 45°C for 16 hours with 60 rpms, washed and stained on a Fluidics Station 450 (program: FS450 0007), and scanned on a GeneChip Scanner 3000 7G (both Affymetrix). Raw image data were analyzed with Affymetrix Expression Consoleä Software (Affymetrix); gene expression intensities were normalized and summarized with SST‐RMA (robust multiarray average algorithm). In order to identify genes differentially expressed between different treatments, a class comparison analysis using Affymetrix Transcriptome Analysis Console (TAC) 2.0 Software was performed. Differences were considered significant if the 2‐sided P value was <0.05. To perform pathway over‐representation analysis, data were analyzed with the software package, AltAnalyze (version 2.0.8), using KEGG (Kyoto Encyclopedia of Genes and Genomes) as the pathway database. Over‐representation parameters were a Z‐score threshold of 1.96, a Fisher's exact test P‐value threshold of 0.05, and a number of changed genes threshold of 3. Gene expression was considered as changed if transcript levels between the different treatment groups were differential with a ≥1.5‐fold change and a raw P<0.05. Microarray data from this publication have been submitted to the GEO (Gene Expression Omnibus) repository and are available under accession number GSE108999.
Western Blotting
To assess kinase activation levels, cell lysates were harvested at 0.5 and 10 hours after stimulation. Cells were lysed with 1×LDS buffer containing 50 mmol/L of DTT. Samples were sonified to shear genomic DNA, and potential cell debris were spun down at 4°C at 12 000g for 10 minutes. Afterward, supernatant was boiled at 95°C for 5 minutes and 100 000 cell equivalents/lane were loaded on a 10% SDS polyacrylamide gel (Bio‐Rad) for protein separation. Western blotting and immunodetection as well as reprobing of membranes were performed according to a previous publication.46 Briefly, for Western blotting, proteins were transferred onto a PVDF membrane (Bio‐Rad), blocked with 5% BSA or nonfat dry milk in TBS‐T, and probed with primary antibody at 4°C overnight (Table S1). On the next day, membranes were incubated with an HRP‐conjugated secondary antibody at RT for 2 hours. Blots were developed with Clarity Western ECL Substrate (Bio‐Rad) or Supersignal West Femto Maximum Sensitivity Substrate, and the resulting chemiluminescence was detected using the ChemiDoc MP System (Bio‐Rad). Band intensities were analyzed using the Image Lab software (Bio‐Rad) and normalized to unphosphorylated protein. To assess total protein levels as well as glyceraldehyde 3‐phosphate dehydrogenase or tubulin, blots were restored with stripping buffer for 15 minutes at RT, blocked again with 5% BSA or nonfat dry milk in TBS‐T, and incubated with primary antibody at 4°C overnight followed by secondary antibody incubation and detection.
ELISA
Soluble mouse tumor necrosis factor alpha (TNFα) concentrations in supernatants as well as circulating MIF levels in plasma samples were quantified by ELISA according to the manufacturer's instructions (R&D Systems). Before analysis, samples were diluted 1:10 in reaction buffer (1% BSA and PBS) for the MIF ELISA, whereas samples for the TNFα ELISA were measured undiluted.
So far, no commercial sCD74 ELISA is available. For sCD74 ELISA, the anti‐CD74 antibody (clone C‐16; Santa Cruz Biotechnology, Heidelberg, Germany) was diluted 1:800 in PBS and incubated (100 μL/well) in a 96‐well ELISA plate at 4°C overnight, followed by washing with PBS‐T. Blocking was performed with 300 μL/well of blocking buffer solution (1% BSA and PBS) at room temperature for 2 hours, followed by washing with PBS‐T. Next, plasma samples were diluted 1:10 in reaction buffer (1% BSA and PBS), added (100 μL/well) onto the plate, and incubated overnight at 4°C, followed by washing. As CD74 protein standard, we used a Chinese hamster ovary–derived Gln73‐Met232 construct with an N‐terminal HA (YPYDVPDYA) tag (R&D Systems). Anti‐CD74 detection antibody (clone LN‐2; Santa Cruz Biotechnology) was added at a 1:500 dilution in reaction buffer (100 μL/well), and the plate was incubated for 2 hours at RT, followed by washing with PBS‐T. Immunoglobulin G HRP‐linked secondary antibody (Cell Signaling Technology, Danvers, MA) then was added for 1 hour. Unbound peroxidase was removed by washing followed by application of substrate solution containing H2O2 and TMB solution (3,3′,5,5′‐tetramethylbenzidine) at a ratio of 1:1. Wells were incubated with the substrate for a maximum of 20 minutes in the dark until a change in color was visible. The colorimetric reaction was stopped by adding 1 mol/L of H2SO4. Finally, the assay was read out and quantified using a Victor Multilabel Counter at 450 nm. Ratio of sCD74/MIF was calculated by dividing the molar serum concentration ratio of circulating CD74 (19.34 kDa) by MIF (12.5 kDa).
Receptor Expression by Flow Cytometry
Cell‐surface expression of the death receptor, TNF receptor 1, Toll like receptors (TLR) 2 and TLR4 as well as MIF receptors CXCR2, CXCR4, and CD74 on myofibroblasts were analyzed with flow cytometry at different time points after stimulation (0.5, 4, and 8 hours). At indicated time points, fibroblasts were washed with glycine buffer (50 mmol/L of glycine, 150 mmol/L of NaCl in ddH2O, pH 2.8) followed by 3 sequential washing steps with PBS. Afterward, cells were detached by scraping, centrifuged (500g, 5 minutes, RT) and resuspended in an appropriate volume of prechilled FACS buffer (PBS [pH 7.2] with 0.5% BSA and 0.01% sodium azide) to obtain 4×106 cells/mL. For each preparation, 100 000 cells were stained with antibody listed in Table S3 for 20 minutes in the dark at 4°C. After incubation, cells were washed with 1 mL of FACS buffer and finally resuspended in 0.4 mL of FACS buffer to perform flow cytometry analysis using the FACSCanto II (BD Biosciences, Heidelberg, Germany). Data were analyzed using FlowJo software (Version 10.0.7; FlowJo LLC, Ashland, Oregon).
Statistical Analysis
In this exploratory analysis, after testing for normal distribution (Shapiro–Wilk test), data were statistically analyzed and graphically displayed using GraphPad PRISM (GraphPad Software Inc, La Jolla, CA). Data are represented as mean±SEM, if not stated otherwise.47 Given the exploratory‐driven character of MIF and sCD74 dose experiments on cell viability as well as signaling studies, normally distributed data were analyzed using a 2‐tailed, unpaired t test without multiple‐test adjustments in order to increase the power to detect real effects that would be otherwise compromised.48 Exploratory analysis per se should emphasize on descriptive analysis graphically or numerically.49 In contrast, after determination of promising concentrations, all other experiments were hypothesis‐driven and thus tested for significance using a 2‐tailed, unpaired t test followed by Bonferroni correction.48, 50 In all cases, P<0.05 was considered statistically significant. Besides statistical significant testing, the magnitude of the effect was evaluated by using Cohen's d as an effect size index, and statistical power calculations were performed using the free software, G*power 3.1.9.2, provided by the Heinrich‐Heine University Düsseldorf. For preliminary clinical data, the classification in small, medium, and large effects were based on Cohen's conventions d=0.2, d=0.5, and d=0.8, respectively. In contrast to clinical studies, the sample size in in vitro experiments is very limited and is commonly below n=15, requiring adaptations of the classification. Thus, only large effects were considered as meaningful effects. Assuming n=6 as an average number of biological replicates with a given type I error of α=0.05 and type II error of β=0.20, d≥1.8 were defined as a large effect size in the experimental part of our study. The calculated effect size of each experiment is listed in Table S4.
Results
Co‐Treatment With sCD74 and MIF Triggers Cell Death in Myofibroblasts but Not in Cardiomyocytes
We used an in vitro model of primary cardiac fibroblasts, which rapidly differentiate into myofibroblasts when cultured on plastic surfaces. A gain of myofibroblast‐specific markers including α‐SMA, collagen 1α1 (Col1α1), and fibronectin 1 (FN1) with an increasing cultivation period, verified the activated phenotype of cardiac fibroblasts (Figure 1A and 1B). First, we studied the influence of increasing concentrations of sCD74 and combinations of sCD74 together with rMIF on overall myofibroblast viability. Incubation with sCD74 alone reduced the viability of cardiac fibroblasts at concentrations ≥8 nmol/L to a small, but significant, degree (relative viability—control versus 40 nmol/L of sCD74: 100±1.73% versus 83.7±5.99%; P=0.008; d=1.08). Interestingly, simultaneous treatment with rMIF strongly enhanced the sCD74‐alone effect and substantially elevated the number of trypan‐blue–positive myofibroblasts in a sCD74 dose‐dependent manner, with the strongest effect observed at the highest sCD74/rMIF ratio (8 nmol/L of rMIF versus 40 nmol/L of sCD74/8 nmol/L of rMIF: 98.8±3.42% versus 47.9±4.6%; P<0.001, d=3.27; and 40 nmol/L of sCD74 versus 40 nmol/L of sCD74/8 nmol/L of rMIF: 83.7±5.99% versus 47.9±4.6%; P<0.001, d=1.86; Figure 1C). This finding suggested that MIF and the soluble CD74 ectodomain act synergistically to promote death of cardiac myofibroblasts, representing an antifibrotic property. Besides the cell‐death–inducing properties, we evaluated the de‐differentiation potential of combined sCD74/MIF treatment. However, no change in mRNA expression of typical myofibroblast‐specific markers was observed 24 hours after sCD74/MIF treatment, suggesting that sCD74/MIF is not involved in deactivation of myofibroblasts (Figure S2). Next, we subjected murine cardiomyocytes either to a solitary or combined treatment with 40 nmol/L of sCD74 and 8 nmol/L of rMIF and assessed cell survival 24 hours later by trypan blue exclusion. In contrast to myofibroblasts, cardiomyocytes were not prone to sCD74/MIF‐induced cell death (sCD74/MIF in myofibroblasts versus cardiomyocytes: 47.9±4.6% versus 93.8±4.4%; P<0.001, d=3.14), indicating a myofibroblast‐specific effect of sCD74/MIF (Figure 1D).
Figure 1.
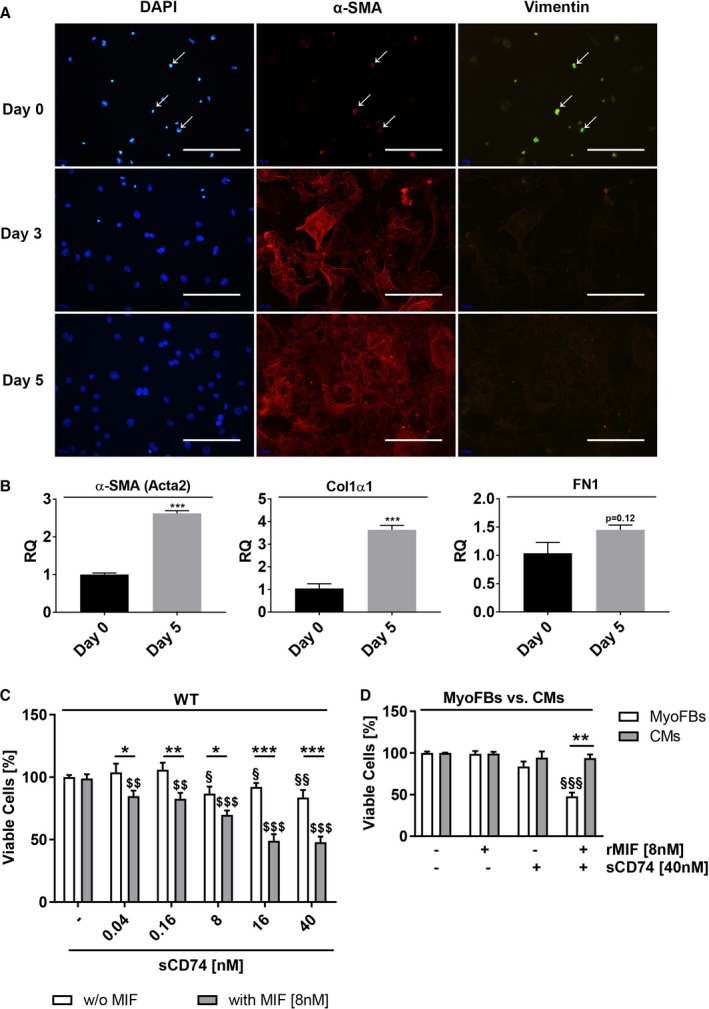
Co‐treatment with sCD74 and MIF promotes cell death in cardiac myofibroblasts. A, Primary isolated cardiac fibroblasts were co‐stained with the fibroblast marker, vimentin (green), the myofibroblast marker, α‐smooth muscle actin (α‐SMA) (red), and nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI; blue) after 0 (5 hours), 3, and 5 days in culture. Size bar, 200 μm. B, mRNA level of α‐SMA, collagen 1α1 (Col1α1), and fibronectin 1 (FN1) were determined 5 hours (day 0) and 5 days after plating by the RT‐qPCR method. Data represent mean±SEM of 3 independent experiments and were analyzed with a 2‐tailed, unpaired t test. ***P<0.001 vs day 0. C, Cardiac myofibroblasts (MyoFBs) isolated from hearts of wild‐type C57BL/6J (WT) mice were treated with increasing concentrations of sCD74 (0, 0.04, 0.16, 8, 16, and 40 nmol/L) without or with rMIF (8 nmol/L). After 24 hours, cells were stained with trypan blue and cell numbers were assessed. Data were analyzed with a 2‐tailed, unpaired t test and represent means±SEM of at least 7 independent experiments. *P<0.05; **P<0.01; ***P<0.001 without (w/o) MIF vs with MIF respectively; § P<0.05; §§ P<0.01 vs untreated control respectively; $$ P<0.01; $ P<0.001 vs MIF control respectively. D, Murine cardiomyocytes (CMs) were stimulated with 40 nmol/L of sCD74 in the absence or presence of 8 nmol/L of rMIF for 24 hours, followed by trypan blue staining. Percentage of survival of CMs was compared with MyoFBs. Data represent mean±SEM of 6 independent experiments and were analyzed with a 2‐tailed, unpaired t test with multiple correction (n=9). $$$ P<0.001 vs control of MyoFBs; **P<0.01 MyoFBs vs CMs. MIF indicates macrophage migration inhibitory factor; RQ indicates relative quantity; rMIF, recombinant MIF; sCD74, soluble CD74.
Synergistic sCD74/MIF Effect Involves Necroptotic but Not Apoptotic Cell Death Mechanisms
In order to identify mechanisms underlying the synergistic cell‐death–promoting effect of sCD74/MIF co‐treatment in myofibroblasts, we analyzed cleaved caspase‐3 levels as an indication of apoptosis execution51 using Western blot methodology. We observed no significant changes of cleaved caspase‐3 at 10 hours after stimulation (Figure 2A and 2B), which has been previously shown to be an appropriate time window for late caspase activation.52 This indicated that the effect was mediated by an apoptosis‐independent mechanism.
Figure 2.
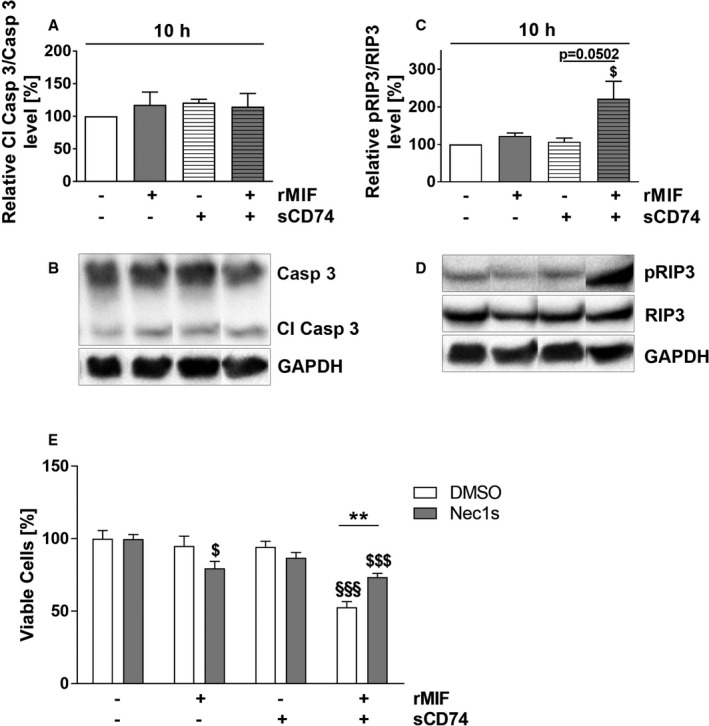
sCD74/MIF‐induced death of myofibroblasts is triggered by RIP1/RIP3‐dependent necroptosis. Cardiac wild‐type myofibroblasts were treated solitarily or simultaneously with MIF and sCD74, and lysates were taken at 10 hours. A and B, Cleaved caspase‐3 levels (Cl Casp 3) as well as (C and D) relative phosphorylation levels of receptor‐interacting serine/threonine‐protein kinase 3 (RIP3) were assessed by Western blotting and immunostaining. Instead of showing the whole blot, only relevant bands were cut out and arranged in the right order. Uncut blots are shown in Figures S3 and S4. Data represent mean±SEM of at least 3 independent experiments. Data were analyzed with a 2‐tailed, unpaired t test. $ P<0.05 vs control. E, Cardiac wild‐type fibroblasts were pretreated with dimethyl sulfoxide (DMSO) or a potent necroptosis inhibitor (Nec1s) for 1 hour followed by solitary or co‐treatment with sCD74 and rMIF. After 20 to 24 hours of incubation, cell numbers were quantified by trypan blue staining and automated counting. Data were analyzed with a 2‐tailed, unpaired t test and corrected for multiple comparison (n=9) using Bonferroni's posttest. Data represent mean±SEM of at least 8 independent experiments for inhibition studies. **P<0.01 DMSO vs Nec1s; §§§ P<0.001 vs DMSO control; $ P<0.05, $$$ P<0.001 vs Nec1s control respectively. GAPDH indicates glyceraldehyde 3‐phosphate dehydrogenase; MIF, macrophage migration inhibitory factor; pRIP3, phosphorylated RIP3; rMIF, recombinant MIF; sCD74, soluble CD74.
We therefore hypothesized that a necroptotic cell death mechanism may account for the observed effect. Necroptosis is a special subtype of programmed necrosis that is mechanistically distinct from apoptosis, but may be also triggered by members of the TNF superfamily, and thus shares some upstream events with extrinsic apoptosis cascades. Yet, necroptotic cell death cascades involve inhibition of caspase‐8 activity. The lack of proteolytic activity of caspase‐8 allows autophosphorylation of RIP1 and subsequent RIP3 and mixed lineage kinase domain‐like pseudokinase (MLKL) activation. Both phospho‐RIP3 and phospho‐MLKL assemble in so‐called necrosomes to trigger cell death.53 We assessed RIP3 activity by Western blot 10 hours after incubation with sCD74/rMIF and observed a 2‐fold increase of RIP3 phosphorylation compared with untreated control (control versus sCD74/rMIF: 100% versus 222.2±45.98%; P=0.0376, d=1.88), whereas neither rMIF nor sCD74 treatment alone affected phospho‐RIP3 levels (Figure 2C and 2D). To confirm that necroptosis contributes to cell death induction, we blocked activity of RIP1, the upstream kinase of RIP3, with the pharmacological inhibitor, 7‐Cl‐O‐Nec1 (Nec1s).54 Surprisingly, Nec1s‐pretreated myofibroblasts demonstrated a small, but significant, decrease in cell viability following MIF treatment compared with control cells (Nec1s/control versus Nec1s/MIF: 99.72±3.04% versus 79.62±4.63%; P=0.025, d=1.81). However, in accord with our hypothesis, sCD74/MIF‐induced cell death in cardiac fibroblasts was significantly rescued by inhibition of RIP1 kinase activity (DMSO versus Nec1s: 52.73±3.84% versus 73.47±2.57%; P=0.007, d=2.29), confirming that necroptosis contributes to cell death triggered by sCD74/MIF (Figure 2E).
TNFα Is Not a Mediator of sCD74/MIF‐Induced Necroptosis
It is well known that gene expression of inflammatory cytokines and soluble factors are induced by MIF and other stress stimuli. Myofibroblasts have been shown to produce and secrete TNFα,55 which mediates apoptosis‐ as well as necroptosis‐dependent cell death pathways.53 To test whether sCD74/rMIF‐triggered cell death is mediated by TNFα, we determined the mRNA level of TNFα after 8 hours as well as intra‐ and extracellular protein levels after 10 hours. Interestingly, neither mRNA nor protein levels of TNFα were influenced by the stimulation, independent of treatment (Figure S5A through S5D; whole blots of intracellular TNFα and glyceraldehyde 3‐phosphate dehydrogenase are shown in Figure S6).
To ask whether newly synthesized cytokines or other soluble factors are involved in cell death induction, WT fibroblasts (“donor cells”) were stimulated with sCD74 or rMIF either alone or in combination. After 6 and 10 hours, supernatants were transferred to unstimulated WT fibroblast (“recipient cells”). Following 20 to 24 hours of incubation, cell viability of donor and recipient cells was determined. At both time points, donor cells showed a significantly reduced survival after treatment with sCD74/rMIF compared with control, as expected (6 hours: 100±3.63% versus 69.90±5.37%; P<0.001, d=1.76; 10 hours: 100±3.13% versus 56.15±6.67%; P<0.001, d=2.99). In contrast, recipient cells showed only a slight decrease in cell viability upon stimulation with the transferred supernatant of sCD74/rMIF‐treated cells compared with control (10 hours: 102.22±3.51% versus 87.72±2.22%; P=0.032, d=1.75), which was still a significantly higher survival level than in donor cells (donor versus recipient at 10 hours: 56.15±6.67% versus 87.72±2.22%; P=0.0046, d=2.25; Figure S5E and S5F). Taken together, neither TNFα nor other newly synthesized and released factors could be identified as mediators of sCD74/MIF‐induced cell death.
Synergism of sCD74/MIF Upregulates Expression of Genes Involved in Antimicrobial Defense and Nuclear Factor Kappa B Signaling
Given that neither TNFα nor other soluble factors seem to mediate sCD74/MIF‐induced necroptosis, a microarray analysis was performed to shed light on sCD74/MIF‐dependent pathways that might explain the cell death of myofibroblasts. Following treatment with either sCD74, rMIF, or sCD74/rMIF, only a relatively low number of genes were at least 1.5‐fold differentially expressed than control (sCD74 versus control: 14 genes; rMIF versus control: 46 genes, sCD74/rMIF versus control: 55 genes; Table 1). A marginal overlap of regulated genes of rMIF alone‐ and sCD74/rMIF‐treated cells (9 genes) indicated that the combined stimulus activates a unique gene expression profile different from the individual stimuli (Figure S7A). A graphical illustration of genes that were significantly regulated by either sCD74 or rMIF treatment is shown in Figure S7B and S7C. A detailed analysis of the genes regulated by sCD74/MIF treatment demonstrated an increased expression of genes involved in antimicrobial defense mechanisms (type I interferon [IFN]‐induced genes) and nuclear factor kappa B (NF‐κB) signaling pathways (Figure 3A). An over‐representation analysis confirmed an enrichment of several genes found in pathways related to infectious diseases, NOD‐like receptor signaling and TNF signaling (Table 2). Quantitative reverse transcription PCR (RT‐qPCR) of the >2‐fold upregulated genes confirmed that IFN‐induced protein 44 (Ifi44; Figure 3B; control versus sCD74/rMIF: 1.03±1.53 versus 4.69±1.03; P=0.011, d=2.5), immunoresponsive gene 1 (Irg1; Figure 3C; control versus sCD74/rMIF: 1.00±0.32 versus 4.41±0.88; P=0.022, d=2.11), and C‐type lectin domain family 4, member e (Clec4e; Figure 3D, control versus sCD74/rMIF: 1.18±0.30 versus 4.10±0.65; P=0.011, d=2.36), as well as C‐C motif chemokine ligand (Ccl) 2 (Figure S8C; control versus sCD74/rMIF: 1.13±0.27 versus 3.18±0.58; P=0.049, d=1.83) and Ccl7 (Figure S8D; control versus sCD74/rMIF: 1.06±0.14 versus 2.70±0.22; P<0.001, d=3.65) were significantly upregulated in the presence of sCD74/MIF. Increased expression levels of 2′‐5′ oligoadenylate synthetase‐like 2 (Oasl2) and T‐cell‐specific GTPase 2 (Tgtp2) detected in the microarray was not verified by the RT‐qPCR method (Figure S8A and S8B). Taken together, our results suggest that sCD74/MIF might act as a danger‐associated molecular pattern–like signal, thereby activating the host immune response.
Table 1.
Fold Changes and P Values of Differentially Regulated Genes Following Treatment With Either sCD74/rMIF, rMIF, or sCD74
| sCD74/rMIF vs Control | rMIF vs Control | sCD74 vs Control | ||||||
|---|---|---|---|---|---|---|---|---|
| Gene Symbol | FC | P Value | Gene Symbol | FC | P Value | Gene Symbol | FC | P Value |
| Gm20917 | −2.02 | 0.0373 | Ddb2 | −1.9 | 0.0342 | Zfp616 | −2.36 | 0.0211 |
| Dsg1b | −1.9 | 0.0246 | Gm11099 | −1.82 | 0.0113 | Olfr830 | −1.67 | 0.0422 |
| Olfr77 | −1.72 | 0.0369 | Gjb6 | −1.69 | 0.0351 | Ceacam14 | −1.64 | 0.0416 |
| Akr1c12 | −1.69 | 0.0090 | Olfr74 | −1.69 | 0.0495 | Gm10735 | −1.6 | 0.0021 |
| LOC105242925 | −1.69 | 0.0263 | Ceacam13 | −1.67 | 0.0342 | Serpinb9e | −1.53 | 0.0216 |
| Afm | −1.65 | 0.0072 | Sel1 l2 | −1.66 | 0.0011 | Bpifa5 | −1.52 | 0.0254 |
| Cfap53 | −1.59 | 0.0351 | Tspan1 | −1.66 | 0.0122 | Mfap2 | −1.52 | 0.0400 |
| Pja1 | −1.58 | 0.0191 | Rs1 | −1.65 | 0.0362 | Gm13304 | −1.5 | 0.0126 |
| Ctsm | −1.56 | 0.0307 | LOC102637808 | −1.63 | 0.0205 | 1810064F22Rik | −1.5 | 0.0495 |
| Nrxn3 | −1.55 | 0.0075 | Pabpc4 l | −1.63 | 0.0420 | Kcnj10 | −1.5 | 0.0499 |
| Mmp1b | −1.55 | 0.0137 | Smr3a | −1.63 | 0.0458 | Zscan30 | 1.5 | 0.0095 |
| Vmn2r89 | −1.55 | 0.0282 | Myrip | −1.62 | 0.0180 | Olfr1184 | 1.58 | 0.0190 |
| Tnnt3 | −1.53 | 0.0181 | LOC105242925 | −1.61 | 0.0328 | Vmn2r74 | 1.71 | 0.0198 |
| Serpinb13 | −1.53 | 0.0416 | Srsx | −1.6 | 0.0289 | Gm8050 | 1.81 | 0.0283 |
| Gjb6 | −1.52 | 0.0169 | Vmn2r46 | −1.6 | 0.0309 | |||
| Hgd | −1.51 | 0.0335 | Vmn1r222 | −1.55 | 0.0145 | |||
| Nfkbia | 1.5 | 0.0077 | Gm14151 | −1.54 | 0.0074 | |||
| Scgb2b23‐ps | 1.5 | 0.0194 | Vmn1r180 | −1.53 | 0.0126 | |||
| Gpr176 | 1.5 | 0.0198 | Bcl2 l15 | −1.52 | 0.0192 | |||
| Tnip1 | 1.5 | 0.0384 | Akr1c12 | −1.5 | 0.0170 | |||
| Alg3 | 1.51 | 0.0417 | Pnmal2 | −1.5 | 0.0233 | |||
| Unc13c | 1.52 | 0.0271 | Fga | 1.5 | 0.0040 | |||
| Ripk2 | 1.52 | 0.0312 | Ets2 | 1.5 | 0.0169 | |||
| Clmn | 1.52 | 0.0490 | Serpina3 g | 1.5 | 0.0336 | |||
| Serpinf1 | 1.53 | 0.0137 | Gm5155 | 1.51 | 0.0258 | |||
| Phlda1 | 1.55 | 0.0087 | Gm8267 | 1.52 | 0.0097 | |||
| Samt2 | 1.56 | 0.0030 | 1700015F17Rik | 1.53 | 0.0028 | |||
| Ren1 | 1.59 | 0.0008 | Dnah8 | 1.53 | 0.0381 | |||
| Tdrp | 1.59 | 0.0038 | E130114P18Rik | 1.54 | 0.0150 | |||
| E130114P18Rik | 1.59 | 0.0230 | Rhox4f | 1.54 | 0.0461 | |||
| Adam18 | 1.59 | 0.0266 | Zc2hc1b | 1.55 | 0.0044 | |||
| Ptgir | 1.6 | 0.0172 | Vmn1r62 | 1.57 | 0.0262 | |||
| Tnfsf18 | 1.61 | 0.0164 | Vmn1r62 | 1.57 | 0.0262 | |||
| Tpgs1 | 1.61 | 0.0367 | Slc22a18 | 1.57 | 0.0417 | |||
| Mrpl38 | 1.63 | 0.0148 | Vmn1r228 | 1.6 | 0.0038 | |||
| Olfr1129 | 1.64 | 0.0060 | Slco3a1 | 1.6 | 0.0193 | |||
| Eif6 | 1.64 | 0.0302 | Meiob | 1.64 | 0.0315 | |||
| Slc7a2 | 1.65 | 0.0022 | Olfr1129 | 1.65 | 0.0099 | |||
| Gm21907 | 1.71 | 0.0218 | Mmp13 | 1.66 | 0.0245 | |||
| Marco | 1.73 | 0.0110 | Hoxa13 | 1.67 | 0.0468 | |||
| Cyp2j12 | 1.8 | 0.0300 | Vmn1r28 | 1.71 | 0.0257 | |||
| Isg20 | 1.81 | 0.0085 | Slc7a2 | 1.78 | 0.0325 | |||
| Iigp1 | 1.81 | 0.0318 | Ddx43 | 1.79 | 0.0217 | |||
| Ltbp2 | 1.82 | 0.0081 | Ren1 | 1.88 | 0.0059 | |||
| Pydc3 | 1.85 | 0.0472 | Ifi44 | 1.94 | 0.0419 | |||
| Birc3 | 1.87 | 0.0166 | Gm11096 | 30.61 | 0.0002 | |||
| Mapk11 | 2.06 | 0.0351 | ||||||
| Slco3a1 | 2.08 | 0.0108 | ||||||
| Ccl7 | 2.19 | 0.0005 | ||||||
| Oasl2 | 2.73 | 0.0316 | ||||||
| Ccl2 | 2.9 | 0.0191 | ||||||
| Tgtp2 | 3.12 | 0.0223 | ||||||
| Clec4e | 3.82 | 0.0234 | ||||||
| Irg1 | 4.77 | 0.0073 | ||||||
| Ifi44 | 5.22 | 0.0203 | ||||||
FC indicates fold change; rMIF, recombinant macrophage migration inhibitory factor; sCD74, soluble CD74.
Figure 3.
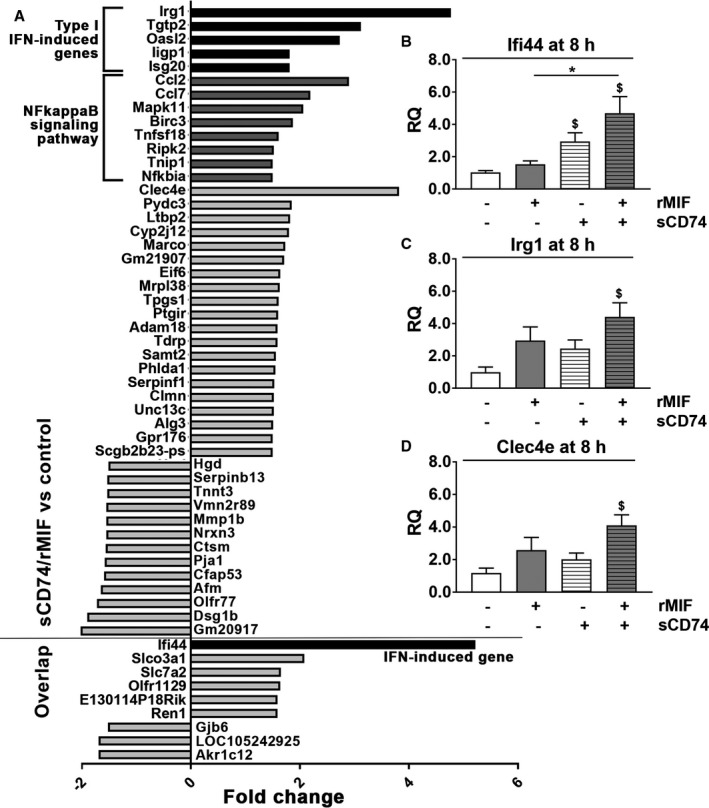
Treatment with sCD74/MIF significantly upregulates gene expression of type I interferon (IFN)‐induced genes. WT were treated with 40 nmol/L of sCD74 in the absence or presence of 8 nmol/L of rMIF. After 8 hours, mRNA was isolated. A, Microarray analysis was performed, and only genes that were at least 1.5‐fold differentially regulated upon sCD74/rMIF treatment compared with control were depicted. Independent triplicates were performed. Corresponding P values are depicted in Table 1. The 9 genes that were also significantly regulated upon MIF‐treatment were marked as overlap. Type I IFN‐induced genes and genes involved in NF‐κB signaling pathways are indicated as black and dark gray bars, respectively. Genes labeled as gray bars seem not to contribute to specialized function and pathways. B and D, RT‐qPCR was performed with the cDNA and Taqman probes specific for the type I IFN‐induced genes, interferon‐induced protein 44 (Ifi44), immunoresponsive gene 1 (Irg1), and C‐type lectin domain family 4, member e (Clec4e). GAPDH was used as a housekeeping gene. Relative quantity (RQ) values were calculated according to the ΔΔCt method and normalized to control. Data represent mean±SEM of at least 4 independent experiments and were analyzed with a 2‐tailed, unpaired t test with multiple correction (n=5). $ P<0.05 vs control; *P<0.05 vs rMIF. GAPDH indicates glyceraldehyde 3‐phosphate dehydrogenase; IFN, interferon; MIF, macrophage migration inhibitory factor; NF‐κB, nuclear factor kappa B; rMIF, recombinant MIF; sCD74, soluble CD74; WT, wild type.
Table 2.
Enrichment Analysis of sCD74/MIF‐Regulated Genes
| Database | ID | Pathway Description | C | Regulated Genes | R | P Value |
|---|---|---|---|---|---|---|
| KEGG | mmu04060 | Cytokine‐cytokine receptor interaction | 264 | Ccl2 (2.90); | 4.0 | 0.0369 |
| Ccl7 (2.19); | ||||||
| Tnfsf18 (1.61) | ||||||
| KEGG | mmu04062 | Chemokine signaling pathway | 196 | Nfkbia (1.5); | 5.4 | 0.0170 |
| Ccl2 (2.90); | ||||||
| Ccl7 (2.19) | ||||||
| KEGG | mmu05152 | Tuberculosis | 178 | Mapk11 (2.06); | 6.0 | 0.0131 |
| Ripk2 (1.52); | ||||||
| Clec4e (3.82) | ||||||
| KEGG | mmu05164 | Influenza A | 170 | Nfkbia (1.5); | 6.2 | 0.0116 |
| Mapk11 (2.06); | ||||||
| Ccl2 (2.90) | ||||||
| KEGG | mmu04722 | Neurotrophin signaling pathway | 123 | Nfkbia (1.5); | 8.6 | 0.0048 |
| Mapk11 (2.06); | ||||||
| Ripk2 (1.52) | ||||||
| KEGG | mmu05145 | Toxoplasmosis | 113 | Birc3 (1.87); | 9.4 | 0.0038 |
| Nfkbia (1.5); | ||||||
| Mapk11 (2.06) | ||||||
| KEGG | mmu05142 | Chagas disease (American trypanosomiasis) | 103 | Nfkbia (1.5); | 10.3 | 0.0029 |
| Mapk11 (2.06); | ||||||
| Ccl2 (2.90) | ||||||
| KEGG | mmu04621 | NOD‐like receptor signaling pathway | 169 | Birc3 (1.87); | 10.5 | <0.0001 |
| Nfkbia (1.5); | ||||||
| Mapk11 (2.06); | ||||||
| Ripk2 (1.52); | ||||||
| Ccl2 (2.90) | ||||||
| KEGG | mmu04668 | TNF signaling pathway | 109 | Birc3 (1.87); | 13.0 | 0.0002 |
| Nfkbia (1.5); | ||||||
| Mapk11 (2.06); | ||||||
| Ccl2 (2.90) |
Birc3 indicates baculoviral IAP repeat‐containing 3, alternative name, cellular inhibitor of apoptosis 2 (cIAP2); C, Total genes regulated in a certain pathway; Ccl, chemokine (C‐C motif) ligand; Clec4e, C‐type lectin domain family 4, member e; FC, fold change; KEGG, Kyoto Encyclopedia of Genes and Genomes; MAPK11, mitogen‐activated protein kinase 11, alternative name, p38 beta; MIF, macrophage migration inhibitory factor; sCD74, soluble CD74; Nfkbia, nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, alpha, alternative name, I‐kappa‐B‐alpha (IκBα); R, enrichment factor; Ripk2, receptor‐interacting serine‐threonine kinase 2; sCD74, soluble CD74 receptor ectodomain; Tnfsf18, tumor necrosis factor (ligand) superfamily, member 18.
Soluble CD74 Diminishes MIF‐Mediated Protein Kinase B Activation
Recently, Assis et al demonstrated that sCD74 neutralizes MIF‐dependent signaling by membrane‐bound CD74.37 Therefore, we investigated whether sCD74/MIF exhibited a different activation profile of classical MIF‐dependent kinases compared with MIF‐mediated activation of membrane CD74. As expected from previous work,56 we observed early as well as sustained protein kinase B (AKT) activation following MIF stimulation (0.5 hours: P<0.001, d=8.31; 10 hours: P=0.0485, d=1.18). This effect was significantly reduced in the presence of sCD74 (Figure 4A through 4D; whole blots of phosphorylated AKT, total AKT and tubulin are shown in Figure S9 and S10), suggesting that sCD74 inhibits MIF‐mediated AKT signaling (rMIF versus sCD74/rMIF, 0.5 hours: 198.7±8.40% versus 136.2±24.86%; P=0.0197, d=2.23; 10 hours: 157.5±30.91% versus 76.92±19.4; P=0.0422, d=1.32). Interestingly, at later time points, combined sCD74/rMIF treatment induced a 2‐fold activation of p38 compared with control (234.43±53.52%; P=0.03, d=1.88; Figure 4E through 4H; whole blots of phosphorylated p38, total p38 and glyceraldehyde 3‐phosphate dehydrogenase are shown in Figure S11 and S12). No changes were observed for other mitogen‐activated protein kinases (MAPKs) such as extracellular signal‐regulated kinase 1/2 (ERK1/2) and c‐Jun N‐terminal kinase (JNK; data not shown).
Figure 4.
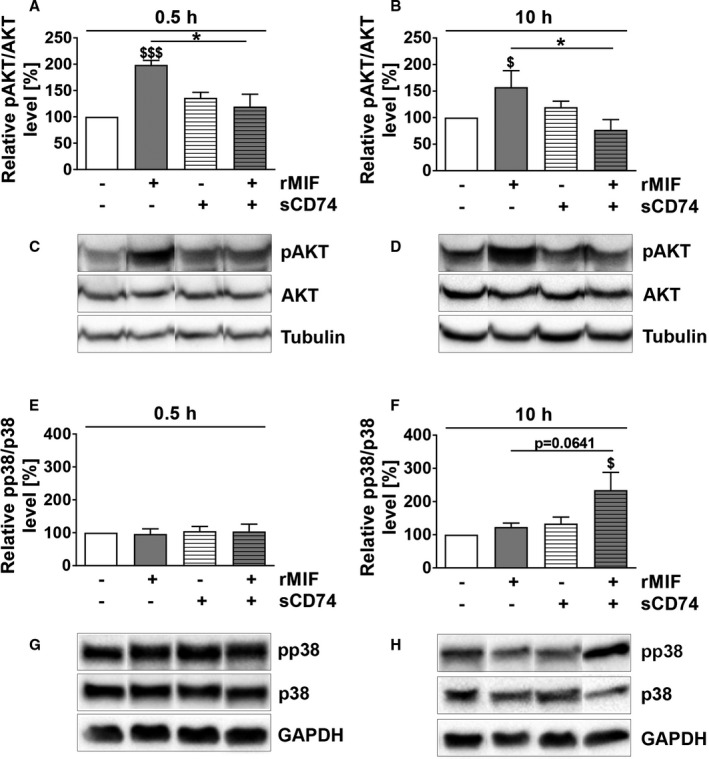
sCD74 changes the kinase activation profile of MIF. Following stimulation of WT myofibroblasts, lysates were taken after 0.5 and 10 hours. Phosphorylation and total protein levels were assessed by Western blotting, band intensities were densitometric analyzed, and relative activation levels were normalized to control. Phosphorylation levels of AKT at (A and C) 0.5 and (B and D) 10 hours as well as the mitogen‐activated protein kinase p38 at (E and G) 0.5 and (F and H) 10 hours were determined. Densitometric analysis of immunostainings as well as representative blots are shown. Instead of showing the whole blot, relevant bands were cut out and arranged in the respective order. Uncut blots are shown in Figures S9 through 12. Data represent mean±SEM of at least 4 independent experiments. Data were analyzed with a 2‐tailed, unpaired t test. $ P<0.05, $$$ P<0.001 vs control, respectively; *P<0.05 vs rMIF. AKT indicates protein kinase B; MIF, macrophage migration inhibitory factor; pAKT, phosphorylated AKT; pp38, phosphorylated p38; rMIF, recombinant MIF; sCD74, soluble CD74; WT, wild type.
Soluble CD74 Redirects the Profibrotic MIF/CXCR4 Signal into an Antifibrotic MIF/CD74 Signal
As mentioned before, the dual role of MIF in MI and cardiac remodeling has been associated with its receptors, CD74 and/or CXCR2. The role of the MIF/CXCR4 axis has not been studied in this context. To investigate whether the modulating properties of sCD74 on MIF signaling are based on a modified MIF‐mediated surface expression pattern of its receptors, we assessed the surface expression of the MIF receptors as well as that of the pattern recognition receptors, TLR2 and TLR4, and the death receptor, TNF receptor 1, in WT versus Cd74‐deficient cardiac myofibroblasts by flow cytometry analysis.
Cd74 deficiency did not affect the basal surface expression profile of CXCR2, CXCR4, TLR2, TLR4, and TNF receptor 1 (Figure S13 and S14). In accord with Schwartz et al,57 we demonstrated an MIF‐induced internalization of the CXCR4 receptor (Figure S15; 4 hours: 0.58±0.037; P=0.019, d=4.11), as well as a CD74‐dependent CXCR4 internalization (Figure S15). In contrast, a MIF‐triggered internalization of CD74 and CXCR2 was not observed (Figure S15 and S16). sCD74 did not diminish MIF‐mediated internalization of CXCR4 (Figure S15). TLR2, TLR4, and TNF receptor 1 surface expression was not affected either by sole or by co‐treatment with rMIF and sCD74 (Figure S16).
Since Heinrichs et al demonstrated, in an experimental liver disease model, that MIF‐mediated CD74 signaling reduces fibrosis; we examined Cd74 −/− myofibroblasts and asked whether endogenous CD74 contributes to the antifibrotic properties of the sCD74/rMIF mixture. Cd74 depletion rescued myofibroblasts against sCD74/MIF‐induced necroptosis observed in WT myofibroblasts (sCD74/rMIF, WT versus Cd74 −/−: 47.94±4.55 versus 80.54±6.34%; P<0.001, d=1.56; Figure 5A and 5B), indicating a contribution of endogenous CD74 to sCD74/MIF‐triggered cell death.
Figure 5.
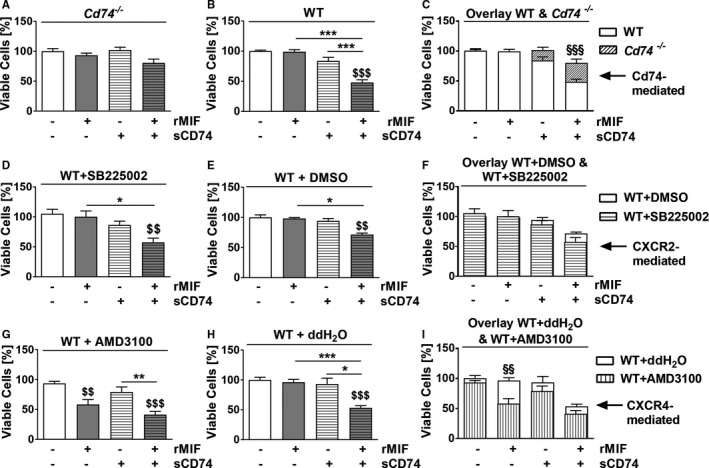
sCD74 redirects the MIF/CXCR4‐profibrotic signal into a CD74‐mediated antifibrotic signal. First, cardiac WT fibroblasts were treated with CXCR2‐inhibitor SB225002, CXCR4‐inhibitor AMD3100, or appropriate vehicle controls for 1 hour. Subsequently, (A) Cd74 −/− cells, (B) wild‐type (WT) cells, (D) SB225002‐, and (E) DMSO‐pretreated myofibroblasts as well as (G) AMD3100‐ and (H) ddH2O‐pretreated WT myofibroblasts were subjected to 40 nmol/L of sCD74 either alone or together with 8 nmol/L of MIF. Cell numbers were quantified by trypan blue staining and automated counting and normalized to untreated control. (C, F, and I) For comparison, data from Cd74 −/−, CXCR2, and CXCR4 inhibition studies were overlaid with their appropriate vehicle control. Data represent mean±SEM of at least (A) 14, (B) 12, (D) 6, (E) 6, (G) 6, and (H) 6 independent experiments. Data were analyzed with a 2‐tailed, unpaired t test and corrected for multiple comparison (A, B, D, E, G, and H: n=5; C, F, and I: n=4) using Bonferroni's posttest. $$ P<0.01, $$$ P<0.001 vs control within group respectively; *P<0.05, **P<0.01, ***P<0.001 vs MIF or sCD74 respectively; §§ P<0.01, §§§ P<0.001 comparison between WT and deficient or inhibited myofibroblasts respectively. CXCR, C‐X‐C chemokine receptor; DMSO, dimethyl sulfoxide; MIF, macrophage migration inhibitory factor; rMIF, recombinant MIF; sCD74, soluble CD74; WT, wild type.
A potential involvement of CXCR2 and CXCR4 in fibroblast survival was assessed using cardiac myofibroblasts from WT cells pretreated with either CXCR2 inhibitor SB225002 or CXCR4 inhibitor AMD3100. Survival rates of WT fibroblasts pretreated with SB225002 (sCD74/rMIF, WT+DMSO versus WT+SB225002: 71.6±2.33% versus 57.46±7.06%; P>0.05, d=1.10) and AMD3100 (sCD74/rMIF, WT+ddH2O versus WT+AMD3100: 53.3±3.58% versus 41.29±5.41%; P>0.05, d=0.96) followed by sCD74/rMIF stimulation did not change compared with sCD74/rMIF‐treated fibroblasts without inhibitor treatment (Figure 5C through 5F), suggesting that activation of the CXCR2 or CXCR4 receptor is not involved in sCD74/MIF‐induced cell death. In contrast, inhibition of CXCR4 by AMD3100 before MIF‐alone treatment resulted in significantly decreased cell viability (WT+AMD3100, control versus MIF: 93.9±53% versus 58.25±8.33%; P=0.008, d=1.95) that was comparable to sCD74/rMIF‐induced cell death (WT+AMD3100, MIF versus sCD74/rMIF: 58.25±8.33% versus 41.29±5.41%; P=0.56, d=0.86; Figure 5E and 5F). This finding suggests that sCD74 inhibits the MIF/CXCR4 axis and thus diminishes prosurvival signaling (representing a profibrotic signal) and inhibits CXCR4‐mediated counter‐regulation of the CD74 axis, resulting in a CD74‐dependent “net” pronecroptotic signal (ie, antifibrotic signal). Taken together, these results provide evidence that sCD74 reroutes the MIF/CXCR4‐profibrotic signal in myofibroblasts into a CD74‐based antifibrotic signal.
Clinical Relevance of sCD74/MIF for the Development of HF
To investigate the role of sCD74/MIF in the development of HF, we analyzed sCD74 and MIF concentrations in plasma samples of healthy volunteers, patients with CHD, and patients with advanced HF by ELISA. Compared with the healthy cohort, CHD and HF patients demonstrated a significant increase of circulating MIF (healthy: 561.7±194.6 pg/mL; CHD: 2098±231.6 pg/mL; P=0.0017, d=3.35; HF: 4729±1197 pg/mL; P=0.0139, d=2.43). Furthermore, the HF cohort had significantly elevated MIF levels compared with CHD (P=0.0456, d=1.52; Figure 6A). In contrast, levels of circulating sCD74 did not differ between cohorts (Figure 6B). Calculation of the molar ratio of sCD74/MIF revealed a 6‐ and 10‐fold decrease in CHD and HF patients, respectively, compared with healthy controls (healthy versus HF: 17.47±10.09 versus 1.41±0.62; P=0.163, d=1.12; healthy versus CHD: 17.47±10.09 versus 2.86±1.20; P=0.147, d=1.02; Figure 6C). Because of the limited cohort size, these differences did not reach significance. Effect size and statistical power calculations demonstrated that the preliminary clinical data lack statistical power (healthy versus CHD: 1‐β=0.2588; healthy versus HF: 1‐β=0.2687; CHD versus HF: 1‐β=0.1449) attributed to small sample sizes. This might explain why the statistical significant test failed to detect a meaningful or obvious effect (P>0.05) either between the diseased cohorts versus healthy subjects or between CHD versus HF patients, despite a large effect size of the sCD74/MIF ratio between healthy cohorts versus CHD and HF patients (healthy versus CHD: d=1.02 and healthy versus HF: d=1.12) and a medium effect between CHD versus HF subjects (CHD versus HF: d=0.69) were demonstrated. Thus, an adequately powered study with at least 35 patients per group is needed. Together, the findings nevertheless suggest that low sCD74 concentrations and high circulating MIF levels may affect disease progression.
Figure 6.
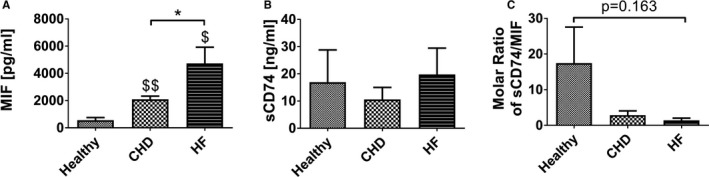
Circulating CD74 and MIF concentrations in healthy, CHD, and HF patients. Using an ELISA technique, we analyzed (A) MIF and (B) sCD74 concentrations in plasma samples of healthy volunteers (n=4), patients with coronary heart disease (CHD; n=5), and patients with advanced heart failure (HF; n=4). C, Ratio of CD74/MIF was calculated by dividing the molar serum concentration of circulating CD74 (19.4 kDa) by MIF (12.5 kDa). Data were analyzed with a 2‐tailed, unpaired t test without multiple correction and represent mean±SEM. *P<0.05 vs CHD cohort; $ P<0.05, $$ P<0.01 vs healthy cohort, respectively. MIF indicates macrophage migration inhibitory factor; sCD74, soluble CD74.
Discussion
MI is frequently associated with the development of HF because of pathological myocardial remodeling leading to stiffness of the cardiac muscle and worsening of cardiac performance.58 As key regulators of ECM turnover and the major site of collagen and MMP synthesis, myofibroblasts are presumed to be an attractive therapeutic target to minimize the expansion of fibrotic tissue.9 However, in order to develop and optimize an approach for improving recovery after MI while avoiding adverse cardiac remodeling, a detailed understanding of the complex regulation and interaction of myofibroblasts with cytokines and ECM components is mandatory.
Previous studies demonstrated that following MI, MIF is immediately released from injured cardiomyocytes, providing predominately protective properties through its receptor, CD74, and its antioxidant capacity.27, 59, 60 In contrast, a second delayed wave of MIF derived from infiltrating immune cells contributes to aggravation of cardiac function and adverse cardiac remodeling, presumably mainly through CXCR2 and CXCR4.59 The recently discovered soluble CD74 ectodomain (sCD74) fragment expands the understanding about the complex interaction within the MIF protein family and necessitates to understand its role within the MIF/receptor network. The present study identified an antifibrotic role of combined treatment with sCD74 and MIF, that is, “sCD74/MIF,” mainly by inducing programmed cell death in myofibroblasts. We are the first to demonstrate that synergism of MIF and sCD74 induces RIP‐dependent necroptosis in myofibroblasts by promoting a molecular switch from a MIF/CXCR4‐profibrotic signal into a CD74‐based antifibrotic signal (Figure 7). Given that myofibroblast activity is prominently controlled by cytokines, we hypothesized that the pleiotropic cytokine, MIF, and the soluble form of its receptor, CD74, might affect viability and activation of myofibroblasts, thereby influencing progression of fibrosis. In fact, we found that simultaneous, combined, treatment, but not individual stimulation, of myofibroblasts with recombinant MIF and sCD74 triggered a significant induction of myofibroblast death. In contrast, neither the myofibroblastic phenotype nor the survival of cardiomyocytes were negatively affected. The cell‐type–specific death effect triggered by sCD74/MIF co‐treatment is in accord with previous studies that reported that the same stimuli or pathways can induce opposing effects in cardiomyocytes and fibroblasts.61, 62 That sCD74/MIF co‐treatment does not induce de‐differentiation of myofibroblasts supports our assumption that it is unlikely that sCD74/MIF‐activated downstream pathways simultaneously affect the death and de‐differentiation of myofibroblasts. However, our assumption cannot be supported by previous literature because of a lack of studies that have interrogated the de‐differentiation of cardiac myofibroblast. Taken together, our results indicate that sCD74/MIF‐induced cell death seems to be a myofibroblast‐specific effect, suggesting that sCD74/MIF might represent a promising target to regulate the cardiac remodeling post‐MI and disease progression in patients with HF. During the healing phase, the ECM is cross‐linked and depleted from most cellular components, such as vascular cells and myofibroblasts, resulting in a mature scar.63 The understanding of the regulation of myofibroblast death during wound healing and the discovery of an “off‐switch” to rapidly remove myofibroblasts in a controlled manner would represent a therapeutic chance. However, the mechanisms that induce activation of regulated cell death in myofibroblasts are poorly understood. Although induction of apoptosis‐dependent mechanisms has been repeatedly demonstrated as a trigger of myofibroblast death,63, 64 we are the first to demonstrate an activation of a caspase‐3–independent (ie, apoptosis‐independent) death pathway in myofibroblasts. We found that myofibroblasts undergo necroptosis in a RIP1/RIP3‐dependent manner, which is consistent with previous studies demonstrating that activation of RIP1 and RIP3 are key pathway elements of necroptosis.65, 66 Thus, our findings are relevant because they extend the understanding about the regulation of myofibroblast death.
Figure 7.
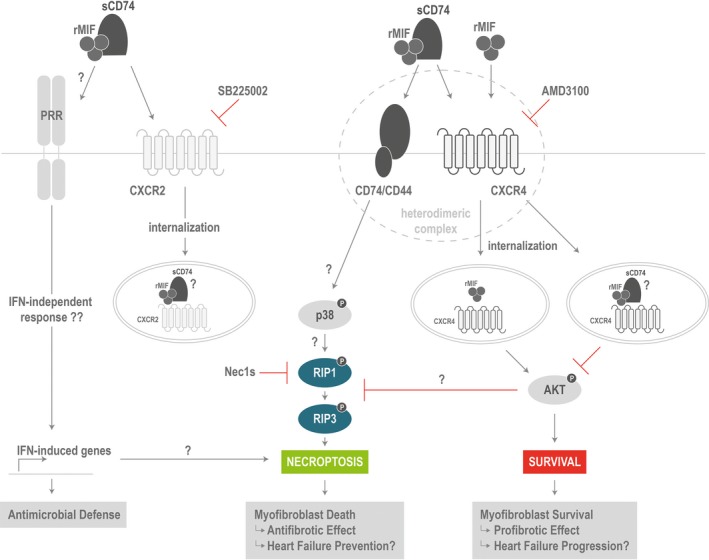
Proposed model of molecular switch between the profibrotic MIF/CXCR4 signal and antifibrotic MIF/CD74 signal. Recombinant MIF triggers CXCR4 internalization, which requires the presence of CD74. Subsequently, MIF/CXCR4 axis mediates survival by AKT activation. Although sCD74/MIF still induces CXCR4 (and CXCR2) internalization, AKT signaling is disturbed. However, the CXCR4/AKT axis seems to be important to suppress cell death. As soon as MIF‐mediated CXCR4 activation is inhibited, signaling by CD74 predominates resulting in RIP1 and RIP3 phosphorylation and, finally, necroptosis. Furthermore, sCD74/MIF seems to be recognized in a DAMP‐like manner to activate components typical for the antimicrobial defense system, such as type 1 interferon (IFN)‐induced genes. AKT indicates protein kinase B; CXCR, C‐X‐C chemokine receptor; DAMP, danger‐associated molecular pattern; MIF, macrophage migration inhibitory factor; RIP1/3, receptor‐interacting serine/threonine‐protein kinases 1 and 3; rMIF, recombinant MIF; sCD74, soluble CD74.
It has been shown that necroptosis is induced by ligand binding to TNF family death domain receptors, pattern recognition receptors, or virus sensors.53 Yet, our data suggest that neither TNFα nor other soluble factors are involved in sCD74/MIF‐induced necroptosis of cardiac myofibroblasts. Interestingly, McComb et al revealed that type I IFN (IFN‐I) signaling is a predominant mechanism of necroptosis in macrophages treated with LPS.67 In line with these results, our microarray and RT‐qPCR analysis revealed an upregulation of type 1 IFN‐regulated genes in myofibroblasts following sCD74/MIF co‐treatment, suggesting a potential contribution of type I IFN (IFN‐I) signaling to sCD74/MIF‐induced necroptosis. Furthermore, an over‐representation analysis revealed enrichment of several genes in pathways of infectious diseases such as Tuberculosis, Influenza A, Toxoplasmosis, and Chagas disease. Thus, sCD74/MIF seems to be recognized in a danger‐associated molecular pattern–like manner to activate components typical for the antimicrobial defense system.
In accord with previous studies demonstrating an attenuating effect of sCD74 on MIF‐triggered signaling,28, 37 we identified sCD74 as an inhibitor of MIF‐mediated AKT activation. MIF‐induced phosphorylation of AKT has previously been demonstrated to depend on CXCR4 and CD74.57, 68 Unlike other G‐protein‐coupled receptors that rapidly internalize following stimulation to terminate signaling,69 CXCR4 exhibits a prolonged stimulatory capacity attributed to endosomal signaling. Recruitment of signaling complexes, which can include both inhibitors or activators of signaling, to endosomes determines the signal type.70 Given that the presence of sCD74 did not inhibit MIF‐induced internalization of either CXCR2 or CXCR4, sCD74 might diminish MIF‐mediated endosomal signaling.69 This mechanistic possibility needs to be further investigated in future studies.
This study is the first to demonstrate that necroptosis is dependent on endogenous CD74. However, our study also indicates that sCD74 inhibits the MIF/CXCR4 axis and thus induces a molecular switch from MIF‐mediated prosurvival signaling through CXCR4/AKT (profibrotic) to cell death induction by CD74 (antifibrotic). Our findings are consistent with previous studies showing that CD74 mediates antifibrotic properties whereas CXCR4 promotes profibrotic effects.26, 71
Figure 7 illustrates a proposed model by which combined sCD74/MIF treatment may modulate the survival of myofibroblasts.
In addition to these mechanistic findings, we offer first clinical data from a cohort of patients suffering from CHD and advanced HF compared with healthy volunteers. These data provide first evidence that low sCD74 concentrations and high circulating MIF levels might affect disease progression. This conclusion is supported by a recent clinical study demonstrating that low levels of the circulating receptor for advanced glycation end products ectodomain were associated with increased risk of HF.72 In contrast, overexpression of a syndecan‐4 ectodomain preceding MI induction augmented the incidence of cardiac rupture and impaired heart function, apparently attributed to impaired granulation tissue formation and reduced myofibroblast numbers.73 However, a synergistic effect of a soluble receptor ectodomain moiety and its ligand triggering cell death has not been described yet. Taken together, we suggest that a high sCD74/MIF ratio in the early/acute phase of post‐MI remodeling (“reparative phase”) would reduce the healing capacity by affecting myofibroblast number and ECM production negatively. In contrast, in the late/chronic phase of post‐MI remodeling (after‐healing phase), an increased sCD74/MIF ratio would enhance myofibroblast depletion from the infarct scar, attenuating reactive fibrosis.
Limitations
We acknowledge that our study has several limitations. First, the pathological mechanisms of cardiac remodeling and HF are highly complex and involve fibrosis, inflammation, cardiomyocyte hypertrophy, and apoptosis. We mainly focused on fibrosis and used a simplistic in vitro model of cardiac myofibroblasts to investigate the biological function of combined sCD74/MIF treatment in myocardial remodeling. Nevertheless, we suggest that our findings are relevant because they extend the understanding about the regulation of myofibroblast death, in which necroptosis has never been reported before. Additionally, the cell‐type–specific induction of death in cardiac myofibroblasts by sCD74/MIF and first clinical results indicate that the sCD74/MIF molecular pair might represent a promising target to regulate the cardiac remodeling post‐MI and disease progression in patients with HF.
Second, the results of the studies have to be considered as purely hypothesis‐generating and need further confirmation. On the one hand, the sample size of some in vitro experiments should be increased especially in experiments that demonstrated large effects, but reached no statistical significance to avoid negation or underestimation of effects, which would lead to misinterpretation or wrong conclusions of underlying mechanisms. Most importantly, on the other hand, the new hypotheses have to be confirmed in comprehensive in vivo models. Yet, first clinical data support the evidence for the influence of sCD74/MIF in the disease progression of HF.
Third, we acknowledge that cardiac fibroblasts were isolated from hearts of neonatal mice. Yet, previous studies repeatedly demonstrated the reliability of using a model of neonatal fibroblasts, which provides comparable results to a model with adult fibroblasts.74, 75 Notwithstanding, this is the first study addressing the molecular mechanisms of action of sCD74 and MIF in cardiac fibrosis.
Fourth, the preliminary clinical evidence should be cautiously considered within the limits of an exploratory analysis. An additional adequately powered clinical trial with at least 35 patients per group is certainly needed to further confirm the findings. Nevertheless, the present study offers first evidence on the clinical significance of MIF and sCD74 and its synergistic action in cardiac and HF patients.
Conclusion
In summary, our study provides first evidence about an antifibrotic role of sCD74/MIF by inducing necroptosis in a myofibroblast‐specific manner. Mechanistically, we demonstrated that sCD74 inhibits the MIF‐mediated survival pathway through the CXCR4/AKT axis, enabling for activation of necroptosis in a CD74/RIP3‐dependent manner. If confirmed in further clinical cohorts, sCD74/MIF‐induced effects may represent a promising target to regulate disease progression in patients with HF.
Sources of Funding
This study was supported by the Deutsche Forschungsgemeinschaft (DFG; STO 1099/‐2 to Stoppe and BE1977/9‐1 to Bernhagen), by the Else Kröner‐Fresenius‐Stiftung (EKFS; 2014_A216 to Bernhagen), and by the Interdisciplinary Centre for Clinical Research (IZKF) at the RWTH Aachen University (SP5 to Stoppe).
Disclosures
None.
Supporting information
Table S1. Antibody List Used for Western Blotting and Immunofluorescence Staining
Table S2. Gene Expression Analysis Using TaqMan Assays From Applied Biosystems
Table S3. Antibody List Used for Flow Cytometry Analysis
Table S4. Effect Size
Figure S1. Experimental setup. Cardiac myofibroblasts were randomized into 4 groups, which were incubated either with medium (group1: control), 8 nmol/L of rMIF (group2: rMIF), 40 nmol/L of sCD74 (group3: sCD74), or 5‐fold molar excess of sCD74 to rMIF (group4: sCD74/rMIF) for 24 hours. For inhibition studies in WT myofibroblasts, cells were treated with either the inhibitor compound (AMD3100, SB225002, and Nec1s) or the appropriate solvent as control (ddH2O or DMSO). Following 1 hour of incubation, both solvent and inhibitor‐pretreated cells were stimulated either with medium, rMIF, sCD74, or sCD74/rMIF and maintained for further 20 to 24 hours.
Figure S2. Treatment with sCD74/MIF did not induce de‐differentiation of myofibroblasts to a quiescent fibroblast. Following treatment of cardiac myofibroblasts with vehicle, sCD74, rMIF or sCD74/rMIF for 24 hours, mRNA expression of (A) α‐smooth muscle actin (α‐SMA), (B) collagen 1α1 (Col1α1), and (C) fibronectin 1 (FN1) were assessed by the RT‐qPCR method. Data represent mean±SEM of 6 independent experiments and were analyzed with a 2‐tailed, unpaired t test corrected for multiple comparison (n=5). *P<0.05 vs control.
Figure S3. Representative blots of cleaved caspase‐3 and GAPDH 10 hours after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, Full‐length and cleaved caspase‐3 and (B) GAPDH were assessed 10 hours after stimulation by western blotting.
Figure S4. Representative blots of pRIP3, RIP3, and GAPDH 10 hours after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, Phosphorylation of RIP3, (B) total RIP3, and (C) GAPDH were assessed 10 hours after stimulation by western blotting.
Figure S5. TNFα could not be identified as a mediator of sCD74/MIF‐induced necroptosis. WT fibroblasts were stimulated solitarily or simultaneously with 40 nmol/L of sCD74 and 8 nmol/L of rMIF. A, mRNA levels (by RT‐PCR) as well as (B) extracellular (by ELISA) and (C and D) intracellular protein levels of TNFα (by Western blotting) were determined. Instead of showing the whole blot, relevant bands were cut out and arranged in the respective order. Uncut blots are shown in Figure S6. E and F, For the supernatant transfer experiments, WT myofibroblasts were stimulated solitarily or simultaneously with sCD74 and rMIF (donor). After supernatant was transferred to untreated WT cells (recipient), both donor and recipient cells were maintained for further 20 to 24 hours followed by trypan blue staining and automated counting. Data represent mean±SEM of at least (A) 8, (B) 6, (D) 8, (E) 10, and (F) 8 independent experiments. Data were analyzed with a 2‐tailed, unpaired t test and corrected for multiple comparison (A through D: n=5; E and F: n=7) using Bonferroni's posttest. §§§ P<0.001 vs control of donor cells; $ P<0.05 vs control of recipient cells; **P<0.01 donor vs recipient.
Figure S6. Representative blots of TNFα and GAPDH 10 hours after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, TNFα and (B) GAPDH were assessed 10 hours after stimulation by western blotting.
Figure S7. sCD74/MIF‐dependent gene expression differs largely from those induced by MIF or sCD74. A, Venn diagram of overlapping and unique effects of sCD74, rMIF, and sCD74/rMIF on gene expression. A total of 115 genes with a fold change ≥1.5 among the differentially expressed genes are represented. Venn diagram was generated using Vennplex. B, Genes with at least 1.5‐fold change following rMIF treatment compared with control were depicted. The marginal overlap between rMIF and sCD74/rMIF stimulation were marked. C, Genes with at least 1.5‐fold change following sCD74 treatment compared with control were depicted. Type I IFN‐induced genes are labeled as black bars. Genes labeled as gray bars seem not to contribute to specialized function and pathways. Independent triplicates were performed. Corresponding P values are listed in Table S1.
Figure S8. Treatment with sCD74/MIF significantly upregulates gene expression of cytokines. RT‐qPCR was performed with the cDNA and Taqman probes. Data represent mean±SEM of at least 4 independent experiments and were analyzed with a 2‐tailed, unpaired t test with multiple correction (n=5). $ P<0.05; $$$ P<0.001 vs control, respectively; **P<0.01 vs sCD74.
Figure S9. Representative blots of pAKT, AKT, and Tubulin 30 minutes after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, Phosphorylation of AKT, (B) total AKT, and (C) Tubulin were assessed 30 minutes after stimulation by western blotting.
Figure S10. Representative blots of pAKT, AKT, and Tubulin 10 hours after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, Phosphorylation of AKT, (B) total AKT, and (C) Tubulin were assessed 10 hours after stimulation by western blotting.
Figure S11. Representative blots of pp38, p38, and GAPDH 30 minutes after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, Phosphorylation of p38, (B) total p38, and (C) GAPDH were assessed 30 minutes after stimulation by western blotting.
Figure S12. Representative blots of pp38, p38, and GAPDH 10 hours after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, Phosphorylation of p38, (B) total p38, and (C) GAPDH were assessed 10 hours after stimulation by western blotting.
Figure S13. Basal surface expression levels of MIF receptors CD74, CXCR2, and CXCR4 in WT and Cd74−/− myofibroblasts. WT and Cd74−/− myofibroblasts were detached by scraping and cell‐surface expression of (A through C) CD74, (D–F) CXCR2, and (G through I) CXCR4 receptor were analyzed by flow cytometry. Median fluorescence intensity (MFI) of isotype control was subtracted from MFI of its appropriate antibody preparation. Data represent mean±SEM of at least 3 independent experiments.
Figure S14. Basal surface expression of TLR2, TLR4, and TNFRI in WT and Cd74−/− myofibroblasts. WT and Cd74−/− myofibroblasts were detached by scraping and cell‐surface expression of (A through C) TLR2, (D through F) TLR4, and (G through I) TNFRI receptor were analyzed by flow cytometry. Median fluorescence intensity (MFI) of isotype control was subtracted from MFI of its appropriate antibody preparation. Data represent mean±SEM of at least 3 independent experiments.
Figure S15. sCD74/rMIF induces rapid and prolonged chemokine receptor internalization. WT and Cd74−/− myofibroblasts were stimulated with sCD74 either alone or with MIF for (A through C) 0.5 hours, (D through F) 4 hours, and (G through I) 8 hours. Subsequently, cells were detached by scraping and cell‐surface expression of (A, D, and G) CD74, (B, E, and H) CXCR2, and (C, F, and I) CXCR4 receptor were analyzed by flow cytometry. Relative fluorescence intensity (RFI) of isotype control was subtracted from MFI of its appropriate antibody preparation. Data represent mean±SEM of at least 3 independent experiments. $ P<0.05, $$ P<0.01 vs control, respectively.
Figure S16. Increased TLR2 expression following sCD74/rMIF stimulation. WT and Cd74−/− myofibroblasts were stimulated with sCD74 either alone or with MIF for (A) 5 minutes (B through D) 0.5 hours, (E through G) 4 hours, and (H through J) 8 hours. Subsequently, cells were detached by scraping and cell‐surface expression of (A) CD74, (B, E, and H) TLR2, (C, F, and I) TLR4, and (D, G, and J ) TNFRI receptor were analyzed by flow cytometry. Relative fluorescence intensity (RFI) of isotype control was subtracted from MFI of its appropriate antibody preparation. Data represent mean±SEM of at least 3 independent experiments.
Acknowledgments
We are indebted to the laboratory staff of the Department of Thoracic & Cardiovascular Surgery, the Department of Intensive Care Medicine, and the Institute of Biochemistry and Molecular Cell Biology of RWTH Aachen University and of the Department of Vascular Biology at LMU Munich for excellent scientific and technical assistance. Performance and analysis of the microarray were supported by the Chip Facility, a core facility of the Interdisciplinary Center for Clinical Research (IZKF) Aachen within the Faculty of Medicine at RWTH Aachen University.
(J Am Heart Assoc. 2018;7:e009384 DOI: 10.1161/JAHA.118.009384.)
Contributor Information
Jürgen Bernhagen, Email: juergen.bernhagen@med.uni-muenchen.de.
Christian Stoppe, Email: christian.stoppe@gmail.com.
References
- 1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER III, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB. Executive summary: heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation. 2016;133:447–454. [DOI] [PubMed] [Google Scholar]
- 2. Roger VL. Epidemiology of myocardial infarction. Med Clin North Am. 2007;91:537–552; ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lloyd‐Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel‐Smoller S, Wong ND, Wylie‐Rosett J. Executive summary: heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation. 2010;121:948–954. [DOI] [PubMed] [Google Scholar]
- 4. Frangogiannis NG. The mechanistic basis of infarct healing. Antioxid Redox Signal. 2006;8:1907–1939. [DOI] [PubMed] [Google Scholar]
- 5. Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011;8:292–300. [DOI] [PubMed] [Google Scholar]
- 6. Lin L, Knowlton AA. Innate immunity and cardiomyocytes in ischemic heart disease. Life Sci. 2014;100:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Daskalopoulos EP, Janssen BJ, Blankesteijn WM. Myofibroblasts in the infarct area: concepts and challenges. Microsc Microanal. 2012;18:35–49. [DOI] [PubMed] [Google Scholar]
- 10. Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. [DOI] [PubMed] [Google Scholar]
- 11. Baudino TA, Carver W, Giles W, Borg TK. Cardiac fibroblasts: friend or foe? Am J Physiol Heart Circ Physiol. 2006;291:H1015–H1026. [DOI] [PubMed] [Google Scholar]
- 12. Porter KE, Turner NA, O'Regan DJ, Ball SG. Tumor necrosis factor alpha induces human atrial myofibroblast proliferation, invasion and MMP‐9 secretion: inhibition by simvastatin. Cardiovasc Res. 2004;64:507–515. [DOI] [PubMed] [Google Scholar]
- 13. Graham HK, Trafford AW. Spatial disruption and enhanced degradation of collagen with the transition from compensated ventricular hypertrophy to symptomatic congestive heart failure. Am J Physiol Heart Circ Physiol. 2007;292:H1364–H1372. [DOI] [PubMed] [Google Scholar]
- 14. Polyakova V, Hein S, Kostin S, Ziegelhoeffer T, Schaper J. Matrix metalloproteinases and their tissue inhibitors in pressure‐overloaded human myocardium during heart failure progression. J Am Coll Cardiol. 2004;44:1609–1618. [DOI] [PubMed] [Google Scholar]
- 15. Thomas CV, Coker ML, Zellner JL, Handy JR, Crumbley AJ III, Spinale FG. Increased matrix metalloproteinase activity and selective upregulation in LV myocardium from patients with end‐stage dilated cardiomyopathy. Circulation. 1998;97:1708–1715. [DOI] [PubMed] [Google Scholar]
- 16. Mujumdar VS, Smiley LM, Tyagi SC. Activation of matrix metalloproteinase dilates and decreases cardiac tensile strength. Int J Cardiol. 2001;79:277–286. [DOI] [PubMed] [Google Scholar]
- 17. Kim HE, Dalal SS, Young E, Legato MJ, Weisfeldt ML, D'Armiento J. Disruption of the myocardial extracellular matrix leads to cardiac dysfunction. J Clin Invest. 2000;106:857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Noels H, Bernhagen J, Weber C. Macrophage migration inhibitory factor: a noncanonical chemokine important in atherosclerosis. Trends Cardiovasc Med. 2009;19:76–86. [DOI] [PubMed] [Google Scholar]
- 20. Pawig L, Klasen C, Weber C, Bernhagen J, Noels H. Diversity and inter‐connections in the CXCR4 chemokine receptor/ligand family: molecular perspectives. Front Immunol. 2015;6:429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. White DA, Su Y, Kanellakis P, Kiriazis H, Morand EF, Bucala R, Dart AM, Gao XM, Du XJ. Differential roles of cardiac and leukocyte derived macrophage migration inhibitory factor in inflammatory responses and cardiac remodelling post myocardial infarction. J Mol Cell Cardiol. 2014;69:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xue YM, Deng CY, Wei W, Liu FZ, Yang H, Liu Y, Li X, Wang Z, Kuang SJ, Wu SL, Rao F. Macrophage migration inhibitory factor promotes cardiac fibroblast proliferation through the Src kinase signaling pathway. Mol Med Rep. 2018;17:3425–3431. [DOI] [PubMed] [Google Scholar]
- 23. Koga K, Kenessey A, Ojamaa K. Macrophage migration inhibitory factor antagonizes pressure overload‐induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2013;304:H282–H293. [DOI] [PubMed] [Google Scholar]
- 24. Xu X, Pang J, Chen Y, Bucala R, Zhang Y, Ren J. Macrophage migration inhibitory factor (MIF) deficiency exacerbates aging‐induced cardiac remodeling and dysfunction despite improved inflammation: role of autophagy regulation. Sci Rep. 2016;6:22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Borghese F, Clanchy FI. CD74: an emerging opportunity as a therapeutic target in cancer and autoimmune disease. Expert Opin Ther Targets. 2011;15:237–251. [DOI] [PubMed] [Google Scholar]
- 26. Heinrichs D, Berres ML, Coeuru M, Knauel M, Nellen A, Fischer P, Philippeit C, Bucala R, Trautwein C, Wasmuth HE, Bernhagen J. Protective role of macrophage migration inhibitory factor in nonalcoholic steatohepatitis. FASEB J. 2014;28:5136–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koga K, Kenessey A, Powell SR, Sison CP, Miller EJ, Ojamaa K. Macrophage migration inhibitory factor provides cardioprotection during ischemia/reperfusion by reducing oxidative stress. Antioxid Redox Signal. 2011;14:1191–1202. [DOI] [PubMed] [Google Scholar]
- 28. Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197:1467–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miller J, Hatch JA, Simonis S, Cullen SE. Identification of the glycosaminoglycan‐attachment site of mouse invariant‐chain proteoglycan core protein by site‐directed mutagenesis. Proc Natl Acad Sci USA. 1988;85:1359–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Naujokas MF, Morin M, Anderson MS, Peterson M, Miller J. The chondroitin sulfate form of invariant chain can enhance stimulation of T cell responses through interaction with CD44. Cell. 1993;74:257–268. [DOI] [PubMed] [Google Scholar]
- 31. Qi D, Hu X, Wu X, Merk M, Leng L, Bucala R, Young LH. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J Clin Invest. 2009;119:3807–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH. Macrophage migration inhibitory factor stimulates amp‐activated protein kinase in the ischaemic heart. Nature. 2008;451:578–582. [DOI] [PubMed] [Google Scholar]
- 33. Luedike P, Hendgen‐Cotta UB, Sobierajski J, Totzeck M, Reeh M, Dewor M, Lue H, Krisp C, Wolters D, Kelm M, Bernhagen J, Rassaf T. Cardioprotection through S‐nitros(yl)ation of macrophage migration inhibitory factor. Circulation. 2012;125:1880–1889. [DOI] [PubMed] [Google Scholar]
- 34. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle‐Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–596. [DOI] [PubMed] [Google Scholar]
- 35. Liehn EA, Kanzler I, Konschalla S, Kroh A, Simsekyilmaz S, Sonmez TT, Bucala R, Bernhagen J, Weber C. Compartmentalized protective and detrimental effects of endogenous macrophage migration‐inhibitory factor mediated by CXCR2 in a mouse model of myocardial ischemia/reperfusion. Arterioscler Thromb Vasc Biol. 2013;33:2180–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liehn EA, Tuchscheerer N, Kanzler I, Drechsler M, Fraemohs L, Schuh A, Koenen RR, Zander S, Soehnlein O, Hristov M, Grigorescu G, Urs AO, Leabu M, Bucur I, Merx MW, Zernecke A, Ehling J, Gremse F, Lammers T, Kiessling F, Bernhagen J, Schober A, Weber C. Double‐edged role of the cxcl12/CXCR4 axis in experimental myocardial infarction. J Am Coll Cardiol. 2011;58:2415–2423. [DOI] [PubMed] [Google Scholar]
- 37. Assis DN, Leng L, Du X, Zhang CK, Grieb G, Merk M, Garcia AB, McCrann C, Chapiro J, Meinhardt A, Mizue Y, Nikolic‐Paterson DJ, Bernhagen J, Kaplan MM, Zhao H, Boyer JL, Bucala R. The role of macrophage migration inhibitory factor in autoimmune liver disease. Hepatology. 2014;59:580–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hayashida K, Bartlett AH, Chen Y, Park PW. Molecular and cellular mechanisms of ectodomain shedding. Anat Rec (Hoboken). 2010;293:925–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Becker‐Herman S, Arie G, Medvedovsky H, Kerem A, Shachar I. CD74 is a member of the regulated intramembrane proteolysis‐processed protein family. Mol Biol Cell. 2005;16:5061–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matza D, Kerem A, Shachar I. Invariant chain, a chain of command. Trends Immunol. 2003;24:264–268. [DOI] [PubMed] [Google Scholar]
- 41. Gil‐Yarom N, Radomir L, Sever L, Kramer MP, Lewinsky H, Bornstein C, Blecher‐Gonen R, Barnett‐Itzhaki Z, Mirkin V, Friedlander G, Shvidel L, Herishanu Y, Lolis EJ, Becker‐Herman S, Amit I, Shachar I. CD74 is a novel transcription regulator. Proc Natl Acad Sci USA. 2017;114:562–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stoppe C, Rex S, Goetzenich A, Kraemer S, Emontzpohl C, Soppert J, Averdunk L, Sun Y, Rossaint R, Lue H, Huang C, Song Y, Pantouris G, Lolis E, Leng L, Schulte W, Bucala R, Weber C, Bernhagen J. Interaction of MIF family proteins in myocardial ischemia/reperfusion damage and their influence on clinical outcome of cardiac surgery patients. Antioxid Redox Signal. 2015;23:865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim BS, Stoppe C, Grieb G, Leng L, Sauler M, Assis D, Simons D, Boecker AH, Schulte W, Piecychna M, Hager S, Bernhagen J, Pallua N, Bucala R. The clinical significance of the MIF homolog d‐dopachrome tautomerase (MIF‐2) and its circulating receptor (sCD74) in burn. Burns. 2016;42:1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu G, Sun Y, Wang K, Chen Z, Wang X, Chang F, Li T, Feng P, Xia Z. Relationship between elevated soluble CD74 and severity of experimental and clinical ALI/ARDS. Sci Rep. 2016;6:30067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Elliott EA, Drake JR, Amigorena S, Elsemore J, Webster P, Mellman I, Flavell RA. The invariant chain is required for intracellular transport and function of major histocompatibility complex class II molecules. J Exp Med. 1994;179:681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mayer B, Soppert J, Kraemer S, Schemmel S, Beckers C, Bleilevens C, Rossaint R, Coburn M, Goetzenich A, Stoppe C. Argon induces protective effects in cardiomyocytes during the second window of preconditioning. Int J Mol Sci. 2016;17:E1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cumming G, Fidler F, Vaux DL. Error bars in experimental biology. J Cell Biol. 2007;177:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moye L. Statistical methods for cardiovascular researchers. Circ Res. 2016;118:439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tukey JW. Exploratory Data Analysis: Past, Present and Future. Reading, MA: Addison‐Wesley; 1993.
- 50. Glickman ME, Rao SR, Schultz MR. False discovery rate control is a recommended alternative to Bonferroni‐type adjustments in health studies. J Clin Epidemiol. 2014;67:850–857. [DOI] [PubMed] [Google Scholar]
- 51. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Johansson AC, Steen H, Ollinger K, Roberg K. Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ. 2003;10:1253–1259. [DOI] [PubMed] [Google Scholar]
- 53. Vanlangenakker N, Vanden Berghe T, Vandenabeele P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 2012;19:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W, Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P. Necrostatin‐1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Baum J, Duffy HS. Fibroblasts and myofibroblasts: what are we talking about? J Cardiovasc Pharmacol. 2011;57:376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lue H, Thiele M, Franz J, Dahl E, Speckgens S, Leng L, Fingerle‐Rowson G, Bucala R, Luscher B, Bernhagen J. Macrophage migration inhibitory factor (MIF) promotes cell survival by activation of the AKT pathway and role for CSN5/JAB1 in the control of autocrine MIF activity. Oncogene. 2007;26:5046–5059. [DOI] [PubMed] [Google Scholar]
- 57. Schwartz V, Kruttgen A, Weis J, Weber C, Ostendorf T, Lue H, Bernhagen J. Role for CD74 and CXCR4 in clathrin‐dependent endocytosis of the cytokine MIF. Eur J Cell Biol. 2012;91:435–449. [DOI] [PubMed] [Google Scholar]
- 58. Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. [DOI] [PubMed] [Google Scholar]
- 59. Dayawansa NH, Gao XM, White DA, Dart AM, Du XJ. Role of MIF in myocardial ischaemia and infarction: insight from recent clinical and experimental findings. Clin Sci (Lond). 2014;127:149–161. [DOI] [PubMed] [Google Scholar]
- 60. Kleemann R, Kapurniotu A, Frank RW, Gessner A, Mischke R, Flieger O, Juttner S, Brunner H, Bernhagen J. Disulfide analysis reveals a role for macrophage migration inhibitory factor (MIF) as thiol‐protein oxidoreductase. J Mol Biol. 1998;280:85–102. [DOI] [PubMed] [Google Scholar]
- 61. deBlois D, Orlov SN, Hamet P. Apoptosis in cardiovascular remodeling—effect of medication. Cardiovasc Drugs Ther. 2001;15:539–545. [DOI] [PubMed] [Google Scholar]
- 62. Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, Conway SJ, Graff JM, Frangogiannis NG. Opposing actions of fibroblast and cardiomyocyte Smad3 signaling in the infarcted myocardium. Circulation. 2018;137:707–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146:56–66. [PMC free article] [PubMed] [Google Scholar]
- 64. Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, Hovell C, Arthur MJ. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102:538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation‐driven assembly of the RIP1‐RIP3 complex regulates programmed necrosis and virus‐induced inflammation. Cell. 2009;137:1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase‐3 determines cellular necrotic response to TNF‐alpha. Cell. 2009;137:1100–1111. [DOI] [PubMed] [Google Scholar]
- 67. McComb S, Cessford E, Alturki NA, Joseph J, Shutinoski B, Startek JB, Gamero AM, Mossman KL, Sad S. Type‐I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci USA. 2014;111:E3206–E3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schwartz V, Lue H, Kraemer S, Korbiel J, Krohn R, Ohl K, Bucala R, Weber C, Bernhagen J. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009;583:2749–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tilton B, Ho L, Oberlin E, Loetscher P, Baleux F, Clark‐Lewis I, Thelen M. Signal transduction by CXC chemokine receptor 4. Stromal cell‐derived factor 1 stimulates prolonged protein kinase B and extracellular signal‐regulated kinase 2 activation in T lymphocytes. J Exp Med. 2000;192:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Murphy JE, Padilla BE, Hasdemir B, Cottrell GS, Bunnett NW. Endosomes: a legitimate platform for the signaling train. Proc Natl Acad Sci USA. 2009;106:17615–17622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hong F, Tuyama A, Lee TF, Loke J, Agarwal R, Cheng X, Garg A, Fiel MI, Schwartz M, Walewski J, Branch A, Schecter AD, Bansal MB. Hepatic stellate cells express functional CXCR4: role in stromal cell‐derived factor‐1alpha‐mediated stellate cell activation. Hepatology. 2009;49:2055–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lazo M, Halushka MK, Shen L, Maruthur N, Rebholz CM, Rawlings AM, Hoogeveen RC, Brinkley TE, Ballantyne CM, Astor BC, Selvin E. Soluble receptor for advanced glycation end products and the risk for incident heart failure: the atherosclerosis risk in communities study. Am Heart J. 2015;170:961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Matsui Y, Ikesue M, Danzaki K, Morimoto J, Sato M, Tanaka S, Kojima T, Tsutsui H, Uede T. Syndecan‐4 prevents cardiac rupture and dysfunction after myocardial infarction. Circ Res. 2011;108:1328–1339. [DOI] [PubMed] [Google Scholar]
- 74. Santiago JJ, Dangerfield AL, Rattan SG, Bathe KL, Cunnington RH, Raizman JE, Bedosky KM, Freed DH, Kardami E, Dixon IM. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev Dyn. 2010;239:1573–1584. [DOI] [PubMed] [Google Scholar]
- 75. Zhou Y, Richards AM, Wang P. Characterization and standardization of cultured cardiac fibroblasts for ex vivo models of heart fibrosis and heart ischemia. Tissue Eng Part C Methods. 2017;23:422–433. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Antibody List Used for Western Blotting and Immunofluorescence Staining
Table S2. Gene Expression Analysis Using TaqMan Assays From Applied Biosystems
Table S3. Antibody List Used for Flow Cytometry Analysis
Table S4. Effect Size
Figure S1. Experimental setup. Cardiac myofibroblasts were randomized into 4 groups, which were incubated either with medium (group1: control), 8 nmol/L of rMIF (group2: rMIF), 40 nmol/L of sCD74 (group3: sCD74), or 5‐fold molar excess of sCD74 to rMIF (group4: sCD74/rMIF) for 24 hours. For inhibition studies in WT myofibroblasts, cells were treated with either the inhibitor compound (AMD3100, SB225002, and Nec1s) or the appropriate solvent as control (ddH2O or DMSO). Following 1 hour of incubation, both solvent and inhibitor‐pretreated cells were stimulated either with medium, rMIF, sCD74, or sCD74/rMIF and maintained for further 20 to 24 hours.
Figure S2. Treatment with sCD74/MIF did not induce de‐differentiation of myofibroblasts to a quiescent fibroblast. Following treatment of cardiac myofibroblasts with vehicle, sCD74, rMIF or sCD74/rMIF for 24 hours, mRNA expression of (A) α‐smooth muscle actin (α‐SMA), (B) collagen 1α1 (Col1α1), and (C) fibronectin 1 (FN1) were assessed by the RT‐qPCR method. Data represent mean±SEM of 6 independent experiments and were analyzed with a 2‐tailed, unpaired t test corrected for multiple comparison (n=5). *P<0.05 vs control.
Figure S3. Representative blots of cleaved caspase‐3 and GAPDH 10 hours after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, Full‐length and cleaved caspase‐3 and (B) GAPDH were assessed 10 hours after stimulation by western blotting.
Figure S4. Representative blots of pRIP3, RIP3, and GAPDH 10 hours after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, Phosphorylation of RIP3, (B) total RIP3, and (C) GAPDH were assessed 10 hours after stimulation by western blotting.
Figure S5. TNFα could not be identified as a mediator of sCD74/MIF‐induced necroptosis. WT fibroblasts were stimulated solitarily or simultaneously with 40 nmol/L of sCD74 and 8 nmol/L of rMIF. A, mRNA levels (by RT‐PCR) as well as (B) extracellular (by ELISA) and (C and D) intracellular protein levels of TNFα (by Western blotting) were determined. Instead of showing the whole blot, relevant bands were cut out and arranged in the respective order. Uncut blots are shown in Figure S6. E and F, For the supernatant transfer experiments, WT myofibroblasts were stimulated solitarily or simultaneously with sCD74 and rMIF (donor). After supernatant was transferred to untreated WT cells (recipient), both donor and recipient cells were maintained for further 20 to 24 hours followed by trypan blue staining and automated counting. Data represent mean±SEM of at least (A) 8, (B) 6, (D) 8, (E) 10, and (F) 8 independent experiments. Data were analyzed with a 2‐tailed, unpaired t test and corrected for multiple comparison (A through D: n=5; E and F: n=7) using Bonferroni's posttest. §§§ P<0.001 vs control of donor cells; $ P<0.05 vs control of recipient cells; **P<0.01 donor vs recipient.
Figure S6. Representative blots of TNFα and GAPDH 10 hours after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, TNFα and (B) GAPDH were assessed 10 hours after stimulation by western blotting.
Figure S7. sCD74/MIF‐dependent gene expression differs largely from those induced by MIF or sCD74. A, Venn diagram of overlapping and unique effects of sCD74, rMIF, and sCD74/rMIF on gene expression. A total of 115 genes with a fold change ≥1.5 among the differentially expressed genes are represented. Venn diagram was generated using Vennplex. B, Genes with at least 1.5‐fold change following rMIF treatment compared with control were depicted. The marginal overlap between rMIF and sCD74/rMIF stimulation were marked. C, Genes with at least 1.5‐fold change following sCD74 treatment compared with control were depicted. Type I IFN‐induced genes are labeled as black bars. Genes labeled as gray bars seem not to contribute to specialized function and pathways. Independent triplicates were performed. Corresponding P values are listed in Table S1.
Figure S8. Treatment with sCD74/MIF significantly upregulates gene expression of cytokines. RT‐qPCR was performed with the cDNA and Taqman probes. Data represent mean±SEM of at least 4 independent experiments and were analyzed with a 2‐tailed, unpaired t test with multiple correction (n=5). $ P<0.05; $$$ P<0.001 vs control, respectively; **P<0.01 vs sCD74.
Figure S9. Representative blots of pAKT, AKT, and Tubulin 30 minutes after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, Phosphorylation of AKT, (B) total AKT, and (C) Tubulin were assessed 30 minutes after stimulation by western blotting.
Figure S10. Representative blots of pAKT, AKT, and Tubulin 10 hours after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, Phosphorylation of AKT, (B) total AKT, and (C) Tubulin were assessed 10 hours after stimulation by western blotting.
Figure S11. Representative blots of pp38, p38, and GAPDH 30 minutes after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, Phosphorylation of p38, (B) total p38, and (C) GAPDH were assessed 30 minutes after stimulation by western blotting.
Figure S12. Representative blots of pp38, p38, and GAPDH 10 hours after treatment with increasing concentrations of sCD74 either with or without MIF. WT myofibroblasts were stimulated with medium and 40 nmol/L of sCD74 either in the absence or presence of 8 nmol/L of rMIF. A, Phosphorylation of p38, (B) total p38, and (C) GAPDH were assessed 10 hours after stimulation by western blotting.
Figure S13. Basal surface expression levels of MIF receptors CD74, CXCR2, and CXCR4 in WT and Cd74−/− myofibroblasts. WT and Cd74−/− myofibroblasts were detached by scraping and cell‐surface expression of (A through C) CD74, (D–F) CXCR2, and (G through I) CXCR4 receptor were analyzed by flow cytometry. Median fluorescence intensity (MFI) of isotype control was subtracted from MFI of its appropriate antibody preparation. Data represent mean±SEM of at least 3 independent experiments.
Figure S14. Basal surface expression of TLR2, TLR4, and TNFRI in WT and Cd74−/− myofibroblasts. WT and Cd74−/− myofibroblasts were detached by scraping and cell‐surface expression of (A through C) TLR2, (D through F) TLR4, and (G through I) TNFRI receptor were analyzed by flow cytometry. Median fluorescence intensity (MFI) of isotype control was subtracted from MFI of its appropriate antibody preparation. Data represent mean±SEM of at least 3 independent experiments.
Figure S15. sCD74/rMIF induces rapid and prolonged chemokine receptor internalization. WT and Cd74−/− myofibroblasts were stimulated with sCD74 either alone or with MIF for (A through C) 0.5 hours, (D through F) 4 hours, and (G through I) 8 hours. Subsequently, cells were detached by scraping and cell‐surface expression of (A, D, and G) CD74, (B, E, and H) CXCR2, and (C, F, and I) CXCR4 receptor were analyzed by flow cytometry. Relative fluorescence intensity (RFI) of isotype control was subtracted from MFI of its appropriate antibody preparation. Data represent mean±SEM of at least 3 independent experiments. $ P<0.05, $$ P<0.01 vs control, respectively.
Figure S16. Increased TLR2 expression following sCD74/rMIF stimulation. WT and Cd74−/− myofibroblasts were stimulated with sCD74 either alone or with MIF for (A) 5 minutes (B through D) 0.5 hours, (E through G) 4 hours, and (H through J) 8 hours. Subsequently, cells were detached by scraping and cell‐surface expression of (A) CD74, (B, E, and H) TLR2, (C, F, and I) TLR4, and (D, G, and J ) TNFRI receptor were analyzed by flow cytometry. Relative fluorescence intensity (RFI) of isotype control was subtracted from MFI of its appropriate antibody preparation. Data represent mean±SEM of at least 3 independent experiments.