

Abstract
Background
Recent studies suggest that mavacamten (Myk461), a small myosin‐binding molecule, decreases hypercontractility in myocardium expressing hypertrophic cardiomyopathy‐causing missense mutations in myosin heavy chain. However, the predominant feature of most mutations in cardiac myosin binding protein‐C (cMyBPC) that cause hypertrophic cardiomyopathy is reduced total cMyBPC expression, and the impact of Myk461 on cMyBPC‐deficient myocardium is currently unknown.
Methods and Results
We measured the impact of Myk461 on steady‐state and dynamic cross‐bridge (XB) behavior in detergent‐skinned mouse wild‐type myocardium and myocardium lacking cMyBPC (knockout (KO)). KO myocardium exhibited hypercontractile XB behavior as indicated by significant accelerations in rates of XB detachment (krel) and recruitment (kdf) at submaximal Ca2+ activations. Incubation of KO and wild‐type myocardium with Myk461 resulted in a dose‐dependent force depression, and this impact was more pronounced at low Ca2+ activations. Interestingly, Myk461‐induced force depressions were less pronounced in KO myocardium, especially at low Ca2+ activations, which may be because of increased acto‐myosin XB formation and potential disruption of super‐relaxed XBs in KO myocardium. Additionally, Myk461 slowed krel in KO myocardium but not in wild‐type myocardium, indicating increased XB “on” time. Furthermore, the greater degree of Myk461‐induced slowing in kdf and reduction in XB recruitment magnitude in KO myocardium normalized the XB behavior back to wild‐type levels.
Conclusions
This is the first study to demonstrate that Myk461‐induced force depressions are modulated by cMyBPC expression levels in the sarcomere, and emphasizes that clinical use of Myk461 may need to be optimized based on the molecular trigger that underlies the hypertrophic cardiomyopathy phenotype.
Keywords: cardiac muscle, cardiomyopathy, contractile function, cross‐bridge behavior, Mavacamten, myocardium, steady‐state force generation
Subject Categories: Cardiomyopathy
Clinical Perspective
What Is New?
-
☐
This is the first study to show that the impact of myosin modulator mavacamten (Myk461) to reduce myocardial force generation is modulated by the expression level of cardiac myosin binding protein‐C.
-
☐
Myk461‐induced force depressions were less pronounced in myocardium lacking cardiac myosin binding protein‐C (knockout (KO)) when compared with wild‐type myocardium, especially at submaximal Ca2+ activations, suggesting a reduced impact of Myk461 on force generation.
-
☐
Myk461 treatment induced a greater degree of slowing in XB behavior in KO myocardium, which led to normalization of XB behavior to wild‐type myocardium levels.
What Are the Clinical Implications?
-
☐
Our observations suggest that Myk461 may have beneficial effects on in vivo cardiac function in cardiac myosin binding protein‐C–related hypertrophic cardiomyopathy by attenuating hyperdynamic systolic function.
-
☐
Optimized clinical utility will necessitate accounting for the additional functional effects of reduced cardiac myosin binding protein‐C expression in the sarcomere.
Introduction
Hypertrophic cardiomyopathy (HCM) is a common heritable cardiac muscle disease1 that primarily stems from mutation‐induced structural and functional defects in myofilament contractile proteins. Phenotypically, HCM is characterized by varying degrees of cardiac remodeling that ultimately leads to asymmetrical thickening of the left ventricular wall and can potentially cause sudden cardiac death.1, 2 At the whole heart level, HCM is characterized by hypercontractility, as indicated by an increased left ventricular ejection fraction3 and power output,4 and also diastolic dysfunction.5, 6 At the myofilament level, HCM‐related hypercontractility is often linked to enhanced sensitivity of the contractile apparatus to Ca2+,7, 8 and accelerated rates of cross‐bridge (XB) cycling.9
Among the myofilament proteins, mutations in β‐myosin heavy chain (β‐MHC) and cardiac myosin binding protein‐C (cMyBPC) account for ≈80% of all known cases of inherited HCM.10, 11 This observation emphasizes the importance of understanding the mechanisms that underlie cardiac hypercontractility caused by MHC and cMyBPC mutations, and how these functional defects can be reversed or normalized using genetic or pharmacological approaches.12 Missense mutations in human cardiac β‐MHC result in variable effects on contractile function including reduced13, 14 or enhanced intrinsic force generation15, 16; decreased14, 16 or enhanced15 myosin ATPase activity; and accelerated13, 15, 17 or slowed16 actin sliding velocities. Importantly, recent data show that HCM‐causing mutations in β‐MHC that cause hypercontractility weaken myosin's S1‐S2 intradomain interactions, thus effectively increasing the total number of myosin heads that can interact with actin during systole,11 thereby chronically elevating left ventricular ejection fraction.5, 18 Thus, recent studies have attempted to normalize HCM‐related hypercontractility by directly targeting the myosin motor rather than using β‐blockers and calmodulin antagonists, which may trigger the activation of unwanted signaling pathways.19, 20 Specifically, the myosin modulator mavacamten (Myk461) has been recently shown to decrease both myosin ATPase activity and the transition of XB's from their weakly to strongly bound conformations.21, 22 Notably, transgenic mice expressing missense mutations in MHC that were treated with Myk461 displayed significant reductions in systolic function indicated by reduced fractional shortening, and resulted in regression of pathological remodeling as evidenced by reduced left ventricular wall thickness, myocyte disarray and fibrosis, such that overall cardiac function was improved.21 Similarly, acute MyK461 infusion in a feline model of HCM led to reduced left ventricular outflow obstruction and improved systolic function.23
Although mutations in β‐MHC and cMyBPC are frequent causes of HCM, the molecular mechanisms by which they affect cardiac contractility may differ. This is mainly because missense mutations in β‐MHC that cause HCM result in incorporation of mutant proteins into the sarcomere, whereas the majority of mutations in cMyBPC that cause HCM often result in the production of truncated cMyBPC peptides, which are not incorporated into the sarcomere as they are likely removed by cellular quality control mechanisms such as non‐sense‐mediated mRNA decay or ubiquitin‐proteasome systems.24 The net result is decreased incorporation of full‐length cMyBPC into the sarcomere leading to haploinsufficiency.7, 8, 25, 26 Functionally, decreased incorporation of cMyBPC in human HCM samples expressing cMyBPC truncation mutations led to accelerated XB kinetics at submaximal Ca2+ activations, in part, contributing to cardiac hypercontractility and hypertrophy.27 Mouse models of reduced cMyBPC expression or cMyBPC ablation also exhibit accelerated rates of XB recruitment and XB detachment rates at submaximal Ca2+ activations,28, 29, 30 in part because reduced expression of cMyBPC radially displaces the myosin heads closer to actin, thereby effectively reducing the distance between actin and myosin and increasing the probability of acto‐myosin XB interactions.31 However, the impact of Myk461 on accelerated XB behavior induced by reduced cMyBPC expression has yet to be investigated. Considering the high prevalence of cMyBPC mutations that are predicted to result in reduced cMyBPC expression, it is of clinical importance to establish the effectiveness of Myk461 on normalization of hypercontractile function in a model of cMyBPC‐related HCM. Thus, for the first time, in this study we examined the impact of Myk461 on XB function in myocardium isolated from hearts exhibiting HCM caused by cMyBPC deficiency.32
Methods
The data, methods, and materials will be provided to other researchers for purposes of replicating the results upon request.
Ethical Approval and Animal Treatment Protocols
Experiments were conducted as per the procedures of the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85‐23, Revised 1996), and as per the Case Western Reserve University's guidelines of the Institutional Animal Care and Use Committee. Wild‐type (WT) mice expressing full‐length cMyBPC were used as controls. Mice lacking cMyBPC (knockout (KO)) in their myocardium used in this study were previously generated and well‐characterized.32 Male and female mice (SV/129 strain) aged 3 to 6 months were used in this study.
Determination of Sarcomeric Protein Expression and Phosphorylation Levels
Pro‐Q Diamond phosphoprotein stain (Life Technologies) was used to assess myofilament protein phosphorylation in the cardiac samples as described previously29, 33 (Figure 1A). Cardiac myofibrils were prepared by homogenizing frozen ventricular tissue chunks for ≈15 s using a homogenizer (PowerGen 500; Thermo Fischer Scientific) in a relaxing solution containing protease and phosphatase inhibitors (PhosSTOP and cOmplete ULTRA tablets; Roche Applied Science). Myofibrils were then detergent‐skinned for 15 minutes using 1% Triton X‐100, centrifuged at 10 000g for 5 minutes, resuspended in a fresh relaxing solution, and stored on ice until used. To assess myofilament protein phosphorylation status, WT and KO samples were solubilized by adding Laemmli buffer and heated to 90°C for 5 minutes. To determine the effects of Myk461 incubation on myofilament protein phosphorylation status, WT and KO samples were incubated with 1.0 μmol/L Myk461 for 2 minutes. Myofibrils were then separated on 4% to 20% Tris‐glycine gels, followed by Pro‐Q phosphostaining to determine phosphorylation levels (Figure 1B). Phosphorylation levels of myofilament proteins were normalized to protein expression levels, which were determined by counterstaining the gels with Coomassie blue. Densitometric scanning of stained gels was performed using Image J software available from the US National Institutes of Health, Bethesda, MD.33, 34
Figure 1.
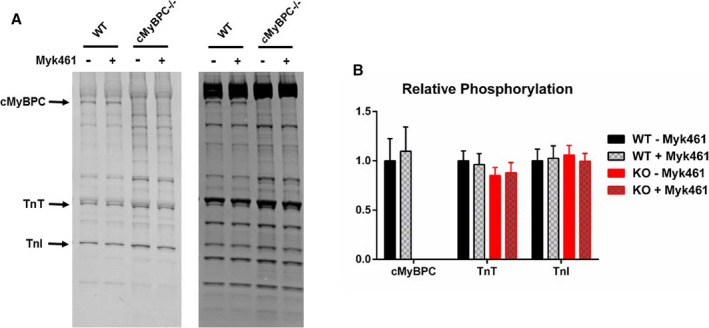
Pro‐Q analysis to assess the impact of Myk461 incubation on phosphorylation status of myofilament proteins in wild‐type (WT) and knockout (KO) myocardium. A, Representative Coomassie‐stained (right) and Pro‐Q Diamond‐stained (left) SDS gels showing the expression and phosphorylation status of myofilament proteins in WT and KO samples before and following a 2‐minute incubation with 1.0 μmol/L Myk461. B, Quantification of phosphorylation of cardiac myosin binding protein‐C (cMyBP‐C), troponin T (TnT), and troponin I (TnI) in WT and KO samples. In both groups, 6 samples were used to analyze contractile protein expression and phosphorylation. No differences in the basal phosphorylation status of myofilament proteins were observed between WT and KO samples. Furthermore, incubation with 1.0 μmol/L Myk461 did not induce any significant changes in the phosphorylation status of myofilament proteins in WT and KO samples. Linear mixed model was used for data analysis with Tukey post hoc tests for multiple comparisons between groups of data. Values are expressed as mean±SE of mean.
Preparation of Detergent‐Skinned Multicellular Myocardial Preparations and Ca2+ Solutions
Frozen ventricular tissue chunks were homogenized in a relaxing solution followed by detergent‐skinning using 1% Triton‐X 100 for 1 hour.33 Multicellular ventricular preparations measuring ≈100 μm in width and ≈400 μm in length were selected for the experiments (Figures 2 and 3). The composition of various Ca2+ solutions used for the experiments was calculated using a computer program35 and using the established stability constants.36 Ca2+ solutions contained the following (in mmol/L): 14.5 creatine phosphate, 7 EGTA, and 20 imidazole. The maximal activating solution (pCa 4.5; pCa=−log [Ca2+]free) also contained 65.45 KCl, 7.01 CaCl2, 5.27 MgCl2, and 4.81 ATP, whereas the relaxing solution (pCa 9.0) contained 72.45 KCl, 0.02 CaCl2, 5.42 MgCl2, and 4.76 ATP. The pH of the Ca2+ solutions was set to 7.0 with KOH with an ionic strength of 180 mmol/L. A range of pCa solutions (pCa's 6.3–5.7), containing varying amounts of [Ca2+]free, were then prepared by mixing appropriate volumes of pCa 9.0 and 4.5 stock solutions and experiments were conducted at ≈25°C.
Figure 2.
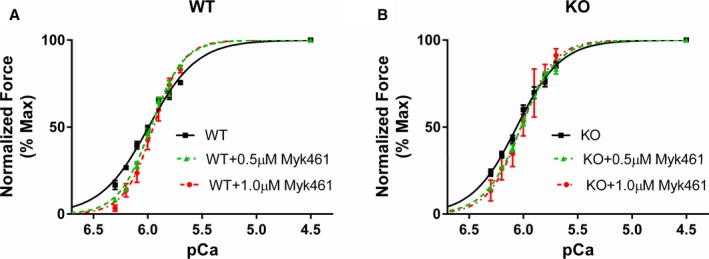
Effect of Myk461 on myofilament Ca2+ sensitivity (pCa50) in wild‐type (WT) and knockout (KO) myocardium. Force‐pCa relationships were constructed by plotting normalized forces generated against a range of pCa to assess pCa50 in WT and KO myocardial preparations.37, 38, 40 Detergent‐skinned myocardial preparations were exposed to a 2‐minute incubation with either 0.5 or 1.0 μmol/L Myk461 to assess the impact of Myk461 on force‐pCa relationships in (A) WT and (B) KO myocardium. Twelve skinned myocardial preparations (3 fibers each from 4 hearts) were used for both groups. Linear mixed model was used for data analysis with Tukey post hoc tests for multiple comparisons between groups of data. pCa50 is unaltered following incubation with Myk461.
Figure 3.
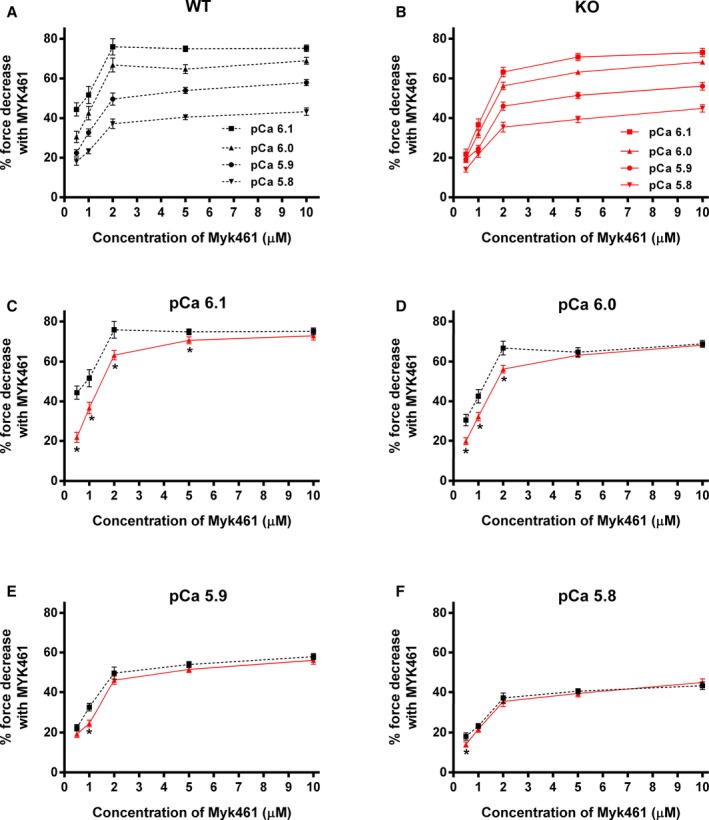
Effect of Myk461 on the magnitude of force generation at various levels of Ca2+ activation in wild‐type (WT) and knockout (KO) myocardium. Baseline forces generated by the skinned ventricular preparations were initially measured in Ca2+ solutions with pCa ranging from 6.1 to 5.8. Forces were subsequently measured on the same preparations using the same range of pCa solutions following a 2‐minute incubation with Myk461. The net decrease in force generation following Myk461 incubation at each pCa was calculated and expressed as % decrease in force from the pre‐incubation baseline force in (A) WT and (B) KO preparations as done earlier.37, 40 Incubation with Myk461 significantly decreased the myocardial force generation at all the concentrations of Myk461 tested and at all the pCa's tested in the WT and KO groups, but the Myk461‐induced force depression is attenuated as the level of Ca2+ activation increases. C through F, The effect of Myk461 in WT vs KO preparations at pCa's 6.1, 6.0, 5.9, and 5.8, respectively. Myk461‐induced force depression was less pronounced in KO myocardium when compared with WT myocardium, especially at low Ca2+ activations with concentrations of 0.5 to 2.0 μmol/L Myk461 (pCa 6.1 and 6.0; C and D). Increasing the Myk461 concentrations to 5.0 and 10.0 μmol/L did not dramatically reduce force generation beyond the effects observed with 2.0 μmol/L, suggesting that force depressions were nearly saturated at 2.0 μmol/L Myk461 at submaximal Ca2+ activations. Twenty‐four myocardial preparations (3–6 fibers from each heart) were analyzed from 5 WT and 7 KO hearts for both 0.5 and 1.0 μmol/L Myk461 incubations. Twelve myocardial preparations (3 fibers from each heart) from 4 hearts were used for both WT and KO groups for 2.0, 5.0, and 10.0 μmol/L Myk461 incubations. Linear mixed model was used for data analysis with Tukey post hoc tests for multiple comparisons between groups of data. Values are expressed as mean±SE of mean. *P<0.05 when comparing the magnitude of force depression level in KO vs force depression level in WT post Myk461 incubation.
Preparation of Myk461 Solution
Myk461 was procured from Axon Medchem (Groningen, The Netherlands) and was dissolved in dimethyl sulfoxide as per the manufacturer's instructions to prepare a stock solution. Myk461 stock solution was then added to a relaxing solution to achieve final concentrations of 0.5, 1.0, 2.0, 5.0, or 10.0 μmol/L Myk461. The impact of Myk461 on steady‐state force generation and dynamic XB behavior was measured in myocardial preparations that were exposed to a range of pCa solutions before or following a 2‐minute incubation with 0.5, 1.0, 2.0, 5.0, or 10.0 μmol/L Myk461.
Experimental Setup for Measuring Steady‐State and Dynamic Contractile Properties in Skinned Myocardial Preparations
Detergent‐skinned multicellular ventricular preparations were attached between a motor arm (312C; Aurora Scientific Inc., Aurora, Ontario, Canada) and a force transducer (403A; Aurora Scientific Inc.), as described previously.37, 38, 39, 40 Changes in the motor arm position and force transducer signals were sampled at 2000 Hz using a custom‐built sarcomere length control software.41 For all mechanical measurements, the sarcomere length of the ventricular preparations was set to 2.1 μm.33, 37, 38, 40 Force‐pCa relationships were determined by measuring the forces generated by the skinned myocardial preparations in a range of pCa solutions that yield submaximal to maximal forces. The apparent cooperativity of force generation was estimated from the steepness of Hill plot transformation of the force‐pCa relationships.29 The force‐pCa data were fit using the equation P/Po=[Ca2+]nH/(knH+[Ca2+]nH), where nH is the Hill coefficient and k is the pCa needed to elicit half‐maximal force (ie, pCa50).29
Stretch Activation Experiments to Measure Dynamic XB Parameters
The stretch activation protocol used here was described in detail in previous studies.33, 39, 42 Skinned myocardial preparations were activated with pCa that generated a range of submaximal forces (eg, ≈40%–45% of maximal force at pCa 6.1 and ≈52%–57% of maximal force at pCa 6.0). When the myocardial preparations attained a steady‐state force, they were subjected to a sudden 2% stretch of their initial muscle length (ML), held at the new ML for 8 s, and were then returned back to their initial ML. The key features of the cardiac muscle stretch activation responses have been described earlier33, 37, 39 (Figure 4A). Briefly, a sudden 2% stretch in ML evokes an instantaneous spike in the force response (P1) because of a sudden strain of elastic elements within the strongly bound XBs (Phase 1). The force then rapidly decays (Phase 2) to a minimum, because of the detachment of strained XBs into a non‐force‐bearing state, with a dynamic rate constant krel (an index of XB detachment from actin). The lowest point of Phase 2 (nadir) is shown by P2, and is an index of the magnitude of XB detachment. Following P2, there is a gradual ascent of force (Phase 3), with a dynamic rate constant kdf (an index of the rate of XB recruitment), which occurs because of stretch‐induced recruitment of XBs into the force‐bearing state.39 Stretch activation amplitudes were normalized to prestretch steady‐state Ca2+‐activated force as done before.37, 38, 39, 40 The magnitude of new steady‐state force (P3) was measured from prestretch steady‐state force to the peak force value attained in Phase 3, and P2 was measured from prestretch steady‐state force to the nadir of the force response in Phase 2, whereas Pdf was measured as the difference between P3 and P2 values.37, 38, 39, 40
Figure 4.
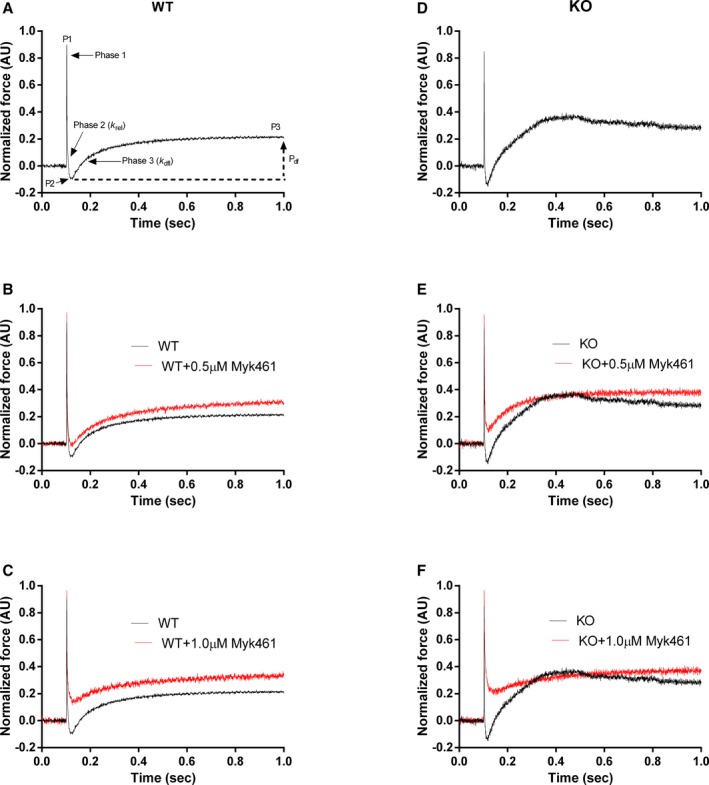
Representative stretch activation responses in wild‐type (WT) and knockout (KO) myocardium before and following Myk461 incubation. Representative force responses following a sudden 2% stretch in muscle length (ML) in isometrically contracting WT (black traces), and KO (red traces) myocardial preparations are shown. A through C, Representative WT traces before and following incubation with 0.5 or 1.0 μmol/L Myk461. D through F, Representative KO traces before and following incubation with 0.5 or 1.0 μmol/L Myk461. Incubation with Myk461 slowed krel only in the KO preparations. On the other hand, incubation with Myk461 slowed kdf in both WT and KO preparations. A, Highlights the important phases of the force transients and various stretch activation parameters that are measured from force responses to a sudden 2% stretch in ML (explained in detail in the Methods section). AU indicates arbitrary units.
krel was measured by fitting a single exponential equation to the time course of force decay using the equation: F(t)=a(−1+exp(‐krel*t)) where “a” is the amplitude of the single exponential phase and krel is the rate constant of the force decay as described earlier.39 kdf, which represents the rate of recruitment of all XBs that give rise to the delayed force transient following the sudden stretch in ML, was estimated by linear transformation of the half‐time of force redevelopment,39 ie, kdf=0.693/t1/2, where t1/2 is the time (in ms) taken from the nadir (ie, the point of force redevelopment at the end of phase 2) to the point of half maximal force in Phase 3 of the force response, where maximal force is indicated by a plateau region of Phase 3 (ie, P3)39 (Figure 4A).
Data Analysis
Data were analyzed with a linear mixed‐effect model in which multiple measurements from 1 heart were clustered and each heart was considered as an independent unit. An unstructured covariance matrix was applied to the random variable (the hearts in WT and KO groups) and not to the fixed variable (Myk461 treatments). The unstructured covariance matrix was chosen in order to account for variations within heart groups and across Myk461 treatments and because it makes no assumptions about the underlying covariances and allows the linear mixed model the most flexibility when fitting the data. R package lme4 was used to implement the linear mixed analysis43, 44 as previously done.40 Asterisks in figures and tables indicate statistical significance using a Tukey post hoc multiple comparisons test, and type‐I error was controlled by adjusting P values for multiple comparisons using a false discovery rate method.40 Values are reported as mean±SE of mean and the criterion for statistical significance was set at P<0.05.
Results
Data were analyzed using a linear mixed model and Tukey post hoc tests were used for multiple comparisons between groups of data.
Effect of Myk461 on Sarcomeric Protein Phosphorylation
Pro‐Q phospho‐staining was done before and following 1.0 μmol/L Myk461 incubation on WT myocardium and myocardium lacking cMyBPC (KO) (Figure 1A). Pro‐Q analysis shows that cardiac troponin T and cardiac troponin I phosphorylation levels were unaltered with Myk461 incubation in both WT and KO samples. Furthermore, Myk461 incubation did not alter cMyBPC phosphorylation status in WT samples (Figure 1B). Phosphorylation levels of cardiac troponin T and cardiac troponin I were not significantly different between WT and KO myocardial samples as previously reported29, 37 (Figure 1B). As expected, cMyBPC was absent in KO samples (Figure 1A).
Effect of Myk461 on Myofilament Ca2+ Sensitivity (pCa50) and Cooperativity of Force Development (nH)
Myk461 incubations did not affect the responsiveness of myofilaments to Ca2+ (pCa50) in both WT and KO preparations (Table 1). This is likely because Myk461‐induced depression of force generation was most prominent at low Ca2+ levels in both WT and KO groups (Figure 2), but the impact was gradually reduced with increased Ca2+ levels. Under basal conditions, KO myocardium showed a slight increase in pCa50 when compared with WT myocardium, a trend that was maintained after Myk461 incubation (Table 1).
Table 1.
Steady‐State Parameters Measured Before and Following Myk461 Incubation in WT and KO Myocardium
| Group | pCa50 | nH | Fmax (mN/mm2) | Fmin (mN/mm2) |
|---|---|---|---|---|
| WT | ||||
| Pre‐Myk461 | 5.99±0.02 | 2.01±0.18 | 12.52±1.63 | 1.00±0.09 |
| 0.5 μmol/L Myk461 | 5.97±0.02 | 3.04±0.09a | 11.10±1.41 | 0.82±0.09 |
| 1.0 μmol/L Myk461 | 5.95±0.01 | 3.24±0.07a | 9.42±0.97 | 0.78±0.09 |
| KO | ||||
| Pre‐Myk461 | 6.05±0.02b | 2.24±0.21 | 12.21±1.29 | 1.13±0.17 |
| 0.5 μmol/L Myk461 | 6.02±0.01b | 2.66±0.12 | 10.41±1.28 | 1.29±0.15 |
| 1.0 μmol/L Myk461 | 6.01±0.02b | 2.91±0.25a | 11.10±1.70 | 1.38±0.18b |
Values are expressed as mean±SE of mean. Twelve skinned myocardial preparations (3 fibers each from 4 hearts) were used for both groups. Linear mixed model was used for data analysis with Tukey post hoc tests for multiple comparisons between groups of data. Fmax indicates Ca2+‐activated maximal force measured at pCa 4.5; Fmin, Ca2+‐independent force measured at pCa 9.0; KO, knockout; nH, cooperativity of force production; pCa50, myofilament Ca2+ sensitivity; WT, wild‐type.
Significantly different vs the corresponding pre‐Myk461 group (P<0.05).
Significantly different vs the corresponding WT group (P<0.05).
Furthermore, cooperativity of force development (nH) increased post‐Myk461 incubations in WT group, but increased only following 1.0 μmol/L Myk461 incubation in KO myocardium (Table 1), presumably because of the smaller Myk461‐induced force depressions in KO myocardium. Taken together, our data suggest a differential impact of Myk461 on nH in WT and KO myocardium.
Effect of Myk461 on Ca2+‐Activated Force Generation
A different set of experiments were conducted to specifically assess the impact of Myk461 on force generation at pCa's 6.1 to 5.8 and 4.5. Force generation was first measured in skinned myocardial preparations in solutions containing increasing amounts of Ca2+ (pCa's 6.1, 6.0, 5.9, and 5.8 that produce ≈40% to 75% of maximal force, which was tested using pCa 4.5). This was followed by a 2‐minute incubation of those myocardial preparations in 0.5, 1.0, 2.0, 5.0, or 10.0 μmol/L Myk461 and measurement of Ca2+‐activated force generation in the same pCa solutions, as done in our earlier studies.37, 40 Our results show a significant decrease in force generation (expressed as % decrease in force from the corresponding baseline force considered as 100% force that can be generated at a given Ca2+ activation) in both WT and KO preparations following Myk461 incubations (Table 2; Figure 3). Furthermore, our results also show that Myk461‐induced force decreases were more pronounced at low Ca2+ activation levels and were progressively diminished at high Ca2+ activation levels, a trend that was observed in both WT and KO preparations (Figure 3A and 3B). Importantly, however, Myk461‐induced force decreases were less pronounced in KO myocardium when compared with WT myocardium, suggesting that Myk461 was not as effective in decreasing force generation in myocardium lacking cMyBPC, especially at low Ca2+ levels (at pCa's 6.1 and 6.0) (Figure 3C and 3D). On the other hand, Myk461‐induced force decreases were nearly equivalent in both WT and KO groups at high Ca2+ levels (at pCa's 5.9 and 5.8) (Figure 3E and 3F). Our results also show that increasing the Myk461 concentrations to 5.0 or 10.0 μmol/L did not induce dramatic force depressions beyond the effects observed with 2.0 μmol/L, suggesting that force depressions are nearly saturated with 2.0 μmol/L Myk461, especially at submaximal Ca2+ activations.
Table 2.
Percentage Decreases in Force Generation in WT and KO Myocardial Preparations Following Myk461 Incubations at Different Levels of Ca2+ Activation
| Group | pCa 6.1 | pCa 6.0 | pCa 5.9 | pCa 5.8 | pCa 4.5 |
|---|---|---|---|---|---|
| WT | |||||
| 0.5 μmol/L Myk461 | 44.4±3.40 | 30.47±2.89 | 22.44±1.67 | 18.06±1.87 | 14.09±1.74 |
| 1.0 μmol/L Myk461 | 51.69±4.36 | 42.51±3.43 | 32.70±1.87 | 23.28±1.27 | 14.40±1.18 |
| 2.0 μmol/L Myk461 | 75.97±4.14 | 66.86±3.49 | 49.64±3.08 | 37.19±2.44 | 23.93±1.17 |
| 5.0 μmol/L Myk461 | 74.97±1.40 | 64.79±2.29 | 53.92±1.49 | 40.47±1.27 | 30.91±1.40 |
| 10.0 μmol/L Myk461 | 75.23±1.68 | 68.93±1.74 | 57.90±1.56 | 43.21±1.95 | 35.11±1.26 |
| KO | |||||
| 0.5 μmol/L Myk461 | 21.77±2.55a | 19.81±1.75a | 19.22±1.73 | 14.02±1.28a | 10.81±1.37a |
| 1.0 μmol/L Myk461 | 36.69±2.95a | 32.18±2.15a | 24.57±1.75a | 21.72±1.58 | 15.14±1.73 |
| 2.0 μmol/L Myk461 | 63.32±2.35a | 56.18±1.93a | 45.98±2.09 | 35.43±2.50 | 19.71±1.03a |
| 5.0 μmol/L Myk461 | 70.78±1.76a | 63.20±0.96 | 51.43±1.60 | 39.33±1.67 | 29.59±1.36 |
| 10.0 μmol/L Myk461 | 73.05±2.17 | 68.35±1.18 | 56.08±2.02 | 44.88±1.82 | 33.36±1.69 |
The impact of Myk461 on force generation was measured by incubating myocardial preparations in various concentrations of Myk461. Myk461 incubations caused significant force depression at all concentrations and pCa that was tested when compared with forces generated before Myk461 incubations. However, the magnitudes of Myk461‐induced force decreases were less pronounced with increasing Ca2+ activations, a trend that was observed in both WT and KO groups. Interestingly, Myk461‐induced force decreases were less pronounced in the KO group, especially at low Ca2+ activations and especially at concentrations of 0.5 to 2.0 μmol/L, suggesting a differential impact of Myk461 on force generation in the myocardium that lacks cardiac myosin binding protein‐C. Twenty‐four skinned myocardial preparations (3–6 fibers each from 5 hearts) were used in the WT group for 0.5 and 1.0 μmol/L Myk461 incubations. Twenty‐four skinned myocardial preparations (3–6 fibers each from 7 hearts) were used in the KO group for 0.5 and 1.0 μmol/L Myk461 incubations. Twelve skinned myocardial preparations (3 fibers each from 4 hearts) were used for both groups for 2.0, 5.0, and 1.0 μmol/L Myk461 incubation. Linear mixed model was used for data analysis with Tukey post hoc tests for multiple comparisons between groups of data. Values are expressed as mean±SE of mean. KO indicates knockout; WT, wild‐type.
Significantly different vs the corresponding WT group (P<0.05).
Myk461 incubation did not alter the Ca2+‐independent force, Fmin measured at pCa 9.0 and maximal Ca2+‐activated force, Fmax measured at pCa 4.5 in either group (Table 1). However, Fmin was significantly greater in KO myocardium when compared with WT myocardium after 1.0 μmol/L Myk461 incubation, suggesting a differential effect of Myk461 on Fmin in WT and KO myocardium.
Effect of Myk461 on Dynamic Stretch Activation Parameters
Stretch activation parameters were assessed following 0.5 or 1.0 μmol/L Myk461 incubations at pCa's 6.1 and 6.0 (Figures 4 and 5; Tables 3 and 4). The impact of Myk461 on XB detachment rate was assessed by measuring krel. Before Myk461 incubation, KO myocardium displayed accelerated krel (≈107%) compared with WT myocardium (Tables 3 and 4).29, 37 At pCa 6.1, Myk461 incubation did not alter krel in WT myocardium, but slowed krel by ≈36% and ≈51% in KO myocardium at 0.5 and 1.0 μmol/L Myk461, respectively, such that krel was no longer different between WT and KO myocardium following Myk461 incubations (Table 3). Likewise, krel was unaffected in WT myocardium at higher Ca2+ activation (pCa 6.0), but was slowed by ≈27% and ≈35% in KO myocardium at 0.5 and 1.0 μmol/L Myk461, respectively (Table 4). However, unlike results obtained at pCa 6.1, krel in KO myocardium was normalized to WT myocardium levels only following 1.0 μmol/L Myk461 incubation.
Figure 5.
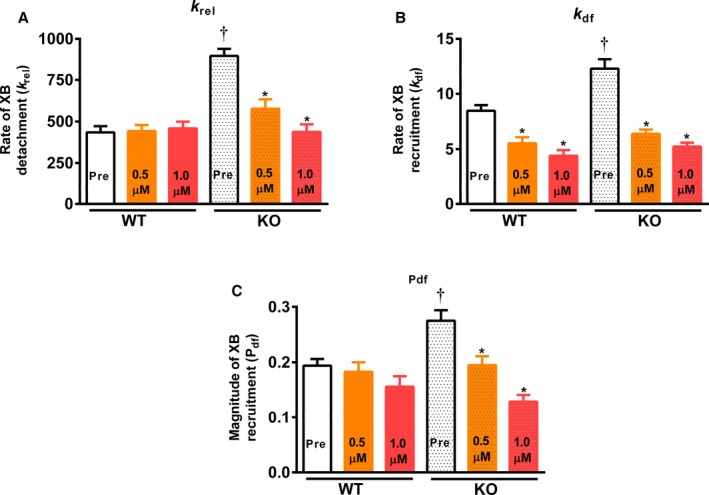
Effect of Myk461 on rate of cross‐bridge (XB) detachment (krel), rate of XB recruitment (kdf), and magnitude of XB recruitment (Pdf) in wild‐type (WT) and knockout (KO) myocardium. Shown are the effects of Myk461 in myocardial preparations at pCa 6.1. A, krel before and following Myk461 incubations in WT and KO myocardium. B, kdf before and following Myk461 incubations in WT and KO myocardium. C, Pdf before and following Myk461 incubation in WT and KO myocardium. Values are expressed as mean±SE of mean. Twelve skinned myocardial preparations (3 fibers each from 4 hearts) were used for both groups. Linear mixed model was used for data analysis with Tukey post hoc tests for multiple comparisons between groups of data. *Different vs the corresponding pre‐Myk461 group (P<0.05); †Different vs the corresponding WT group (P<0.05).
Table 3.
Dynamic Stretch‐Activation Parameters Measured Before and Following Myk461 Incubation in WT and KO Myocardium at pCa 6.1
| Group | krel (s−1) | kdf (s−1) | P1 | P2 | P3 | |
|---|---|---|---|---|---|---|
| WT | ||||||
| Pre‐Myk461 | 434.45±37.30 | 8.46±0.52 | 0.521±0.017 | −0.050±0.013 | 0.144±0.008 | 0.194±0.012 |
| 0.5 μmol/L Myk461 | 442.91±35.82 | 5.53±0.55a | 0.556±0.025 | 0.040±0.017a | 0.223±0.013a | 0.183±0.017 |
| 1.0 μmol/L Myk461 | 459.02±40.84 | 4.41±0.44a | 0.597±0.030 | 0.123±0.022a | 0.278±0.029a | 0.156±0.019 |
| KO | ||||||
| Pre‐Myk461 | 898.05±42.65b | 12.31±0.85b | 0.473±0.044 | −0.057±0.014 | 0.217±0.012b | 0.275±0.019b |
| 0.5 μmol/L Myk461 | 576.77±57.39a | 6.40±0.39a | 0.590±0.022a | 0.038±0.023a | 0.233±0.009 | 0.195±0.016a |
| 1.0 μmol/L Myk461 | 437.60±45.40a | 5.25±0.35a | 0.637±0.021a | 0.139±0.014a | 0.268±0.007a | 0.129±0.012a |
kdf, rate of XB recruitment; krel, rate of XB detachment; KO, knockout; P1, XB stiffness; P2, magnitude of XB detachment; P3, the new steady‐state force attained following a 2% stretch in muscle length; Pdf, magnitude of XB recruitment; WT, wild‐type; XB, cross‐bridge. Stretch activation amplitudes were normalized to prestretch steady‐state Ca2+‐activated force, which refers to the activation level (≈40%–45% of maximal Ca2+‐activated force) as described in the Methods section. Values are expressed as mean±SE of mean. Twelve skinned myocardial preparations (3 fibers each from 4 hearts) were used for both groups. Linear mixed model was used for data analysis with Tukey post hoc tests for multiple comparisons between groups of data.
Significantly different vs the corresponding pre‐Myk461 group (P<0.05).
Significantly different vs the corresponding WT group (P<0.05).
Table 4.
Dynamic Stretch‐Activation Parameters Measured Before and Following Myk461 Incubation in WT and KO Myocardium at pCa 6.0
| Group | krel (s−1) | kdf (s−1) | P1 | P2 | P3 | |
|---|---|---|---|---|---|---|
| WT | ||||||
| Pre‐Myk461 | 376.37±27.86 | 11.54±0.51 | 0.541±0.019 | −0.017±0.010 | 0.130±0.009 | 0.149±0.012 |
| 0.5 μmol/L Myk461 | 410.39±32.74 | 7.43±0.58a | 0.551±0.023 | 0.014±0.010 | 0.184±0.014a | 0.171±0.016 |
| 1.0 μmol/L Myk461 | 424.23±42.04 | 6.00±0.84a | 0.567±0.021 | 0.050±0.010a | 0.217±0.012a | 0.167±0.016 |
| KO | ||||||
| Pre‐Myk461 | 792.67±53.56b | 18.37±1.22b | 0.526±0.021 | −0.028±0.016 | 0.178±0.008b | 0.206±0.013 |
| 0.5 μmol/L Myk461 | 578.47±65.90a,b | 9.00±0.42a | 0.573±0.021 | 0.021±0.024 | 0.209±0.010 | 0.189±0.018 |
| 1.0 μmol/L Myk461 | 513.60±52.89a | 8.14±0.49a | 0.611±0.019 | 0.062±0.023a | 0.235±0.010a | 0.173±0.017 |
Stretch activation amplitudes were normalized to prestretch steady‐state Ca2+‐activated force, which refers to the activation level (≈52%–57% of maximal Ca2+‐activated force) as described in the Methods section. Values are expressed as mean±SE of mean. Twelve skinned myocardial preparations (3 fibers each from 4 hearts) were used for both groups. Linear mixed model was used for data analysis with Tukey post hoc tests for multiple comparisons between groups of data. kdf indicates rate of XB recruitment; krel, rate of XB detachment; KO, knockout; P1, XB stiffness; P2, magnitude of XB detachment; P3, the new steady‐state force attained following a 2% stretch in muscle length; Pdf, magnitude of XB recruitment; WT, wild‐type.
Significantly different vs the corresponding pre‐Myk461 group (P<0.05).
Significantly different vs the corresponding WT group (P<0.05).
Measurement of XB stiffness (P1) revealed that at pCa 6.1, Myk461 incubations increased P1 in KO but not in the WT group, suggesting a differential impact of Myk461 on P1. However, at pCa 6.0, P1 was unaffected following Myk461 incubations in WT myocardium but showed an increasing trend (P=0.07) following 1.0 μmol/L Myk461 incubation in KO myocardium (Tables 3 and 4). It is unlikely that Myk461‐induced P1 changes are simply a result of changes in absolute magnitude of tension developed but most likely indicate a direct effect of Myk461 on the binding properties of individual XBs as it has been previously shown that changes in Ca2+ activation state per se do not affect P1.45 It is possible that the structural conformation of myosin heads and stability of XBs and response to increased strain may be different in myocardium lacking cMyBPC, and that Myk461 binding may increase the stability of these XBs, resulting in increases in P1 in KO myocardium. Myk461 incubations also resulted in more positive P2 values, indicating that the magnitude of strain‐induced XB detachment was decreased in both WT and KO groups at pCa 6.1 (Table 3); however, this trend was only observed after 1.0 μmol/L Myk461 incubation in both groups at a higher pCa of 6.0 (Table 4).
The impact of Myk461 on the rate of XB recruitment was assessed by measuring kdf. Before Myk461 incubation, KO myocardium displayed accelerated kdf (by ≈45% and ≈59%) compared with WT myocardium (Tables 3 and 4),29, 37 at pCa's 6.1 and 6.0, respectively. Myk461 incubations slowed kdf in both WT and KO groups at both pCa's tested, such that basal differences in kdf between WT and KO were abolished following Myk461 incubations (Tables 3 and 4), indicative of normalized kdf in KO myocardium.
Before Myk461 incubation, P3 was significantly greater in KO myocardium when compared with WT myocardium, but no differences in P3 were observed following Myk461 incubations at either pCa, suggesting normalization of P3. At pCa 6.1, P3 was significantly greater in WT myocardium after 0.5 and 1.0 μmol/L Myk461 incubations. However, this trend was only observed following 1.0 μmol/L Myk461 incubation in KO myocardium. At pCa 6.0, P3 was increased in the WT group following Myk461 incubations but P3 increased only at 1.0 μmol/L Myk461 incubation in the KO myocardium. The magnitude of XB recruitment (Pdf) was greater in KO myocardium compared with WT myocardium before Myk461 incubation (≈42% at pCa 6.1; Table 3). Interestingly, following Myk461 incubations, no significant differences were observed in Pdf between the WT and KO groups at either pCa and Myk461 concentration tested (Tables 3 and 4). Specifically, at pCa 6.1, 0.5 and 1.0 μmol/L Myk461 incubations resulted in ≈29% and ≈53% decreases in Pdf, respectively, in KO myocardium, but Pdf was unaffected in WT myocardium. On the other hand, at pCa 6.0, Pdf was unaffected by Myk461 incubations in both WT and KO myocardium, suggesting a Ca2+‐dependence of Myk461 on the magnitude of XB recruitment.
Discussion
Previous studies have shown that the myosin modulator mavacamten (Myk461) has a potential role in attenuating cardiac hypercontractility caused by β‐MHC mutations by decreasing myosin ATPase activity and XB transitions to force‐generating states.21, 22 However, the effects of Myk461 on cardiac hypercontractility resulting from decreased cMyBPC expression have not been investigated until now. Here we examined the impact of Myk461 on both steady‐state force generation and dynamic XB behavior in myocardial preparations lacking cMyBPC. Our results indicate that Myk461 modulates both the rates of XB recruitment and XB detachment, and are the first to demonstrate that Myk461‐induced force depression and slowing of XB kinetics differ in a myocardium lacking cMyBPC.
Myk461 Differentially Impacts Steady‐State Force Generation in KO Myocardium
A recent study has shown that pCa50 of in vitro motility assays is enhanced in regions of the sarcomere containing cMyBPC because the N‐terminal domains of cMyBPC may bind the thin filaments and shift the tropomyosin from its closed to blocked state leading to Ca2+ sensitization of thin filaments.46 This mechanism would be beneficial in normalizing the intrinsic nonuniformity in Ca2+ gradient that occurs as Ca2+ diffuses from the sarcomeric ends towards the central regions of the sarcomere.46 In contrast, here using skinned myocardium with an intact sarcomere lattice, we observed a small but significant increase in Ca2+ sensitivity (pCa50) in myocardium lacking cMyBPC (Table 1). This result can be explained by the cMyBPC ablation‐induced displacement of XBs towards thin filaments and possibly reduced repression of XB movement, such that acto‐myosin interactions are increased, thereby resulting in enhanced XB‐mediated XB recruitment and force generation at submaximal Ca2+‐activations.31, 47 Previous findings show that Myk461 inhibits myosin ATPase activity, decreases force generation, and prevents adverse cardiac remodeling in hypercontractile mouse hearts expressing HCM‐causing MHC missense mutations.21 It has been shown that myocardial samples isolated from HCM patients expressing cMyBPC mutations often display reduced total cMyBPC expression7, 25 and accelerated XB kinetics27; therefore, we sought to investigate the effects of Myk461 on the contractile function of myocardium lacking cMyBPC. In this context, recent data show that Myk461 stabilizes an autoinhibited state of 2‐headed myosin structure48 and also strengthens a folded‐back sequestered super‐relaxed state of myosin heads onto the thick filament backbone49—an effect that contributes to Myk461‐induced force depression by stabilizing the “off” state of myosin heads and preventing acto‐myosin formation.
New findings from our study show that Myk461‐induced reductions in force generation are dependent on the level of thin filament Ca2+ activation, in that force depression was more pronounced at low Ca2+ activations and is blunted with increased Ca2+ activations, irrespective of whether cMyBPC is present in the sarcomere (Figure 3A and 3B). It has been proposed that Myk461 binds directly to myosin and prolongs the overall duration of the ATPase cycle by decreasing the number of XBs transitioning from the weakly bound to the strongly bound state and the rate of phosphate release, which together reduces the total number of XBs that are in a force‐generating state at a given time.21, 22 Thus, in our study, it is apparent that Myk461 reduces force generation more at low Ca2+ levels than at high Ca2+ levels in detergent‐skinned mouse myocardium. This finding differs from a prior study,21 which reports a large reduction (≈70%) in force generation at maximal Ca2+ activation without altering force generation at submaximal Ca2+ activations in detergent‐skinned rat myocardium. While the reasons for this discrepancy are not entirely clear, it is possible that differences in experimental conditions such as temperature could have played a role in the discrepancies we observed in the effects on maximal force. However, our previous studies with myosin modulators such as the myosin activator omecamtiv mecarbil37, 40 have also shown that force enhancement occurs predominantly at submaximal Ca2+ activations, whereas at maximal Ca2+ activation the force enhancement is minimal. The data presented here are consistent with our previous findings and show that Myk461 predominantly depresses force at low Ca2+ activations, likely because at low Ca2+ activations the dominant mechanism underlying force propagation along the thin filaments is XB‐mediated cooperative XB recruitment,50 which influences XB transitions from their weakly to strongly bound states. Presumably, Myk461 directly influences this phase of XB cycle and prevents the formation of strongly bound XBs,22 thereby reducing the magnitude and rate of myofilament force generation. However, as Ca2+ levels rise within the sarcomere, most of the regulatory units will be switched to the “on” state and thus at high Ca2+ levels force generation is less dependent on XB‐mediated cooperative XB recruitment as Ca2+ binding to thin filaments directly opens the actin molecules for myosin binding,50 attenuating the impact of Myk461 on force generation.
Consistent with the idea that Myk461 mainly reduces force generation at low Ca2+‐activation, in this study we observed a minimal impact of Myk461 on myofilament Ca2+ sensitivity (assessed by pCa50) (Table 1; Figure 2). Importantly, it is also interesting to note that the magnitudes of Myk461‐induced force depression are less pronounced in the KO myocardium when compared with WT myocardium especially at low Ca2+ activations, but are nearly equivalent in both groups at high Ca2+ activations (Figure 3C to 3F). This diminished impact of Myk461 on force depression in the KO myocardium is likely because of the fact that cMyBPC normally tethers to myosin heads and prevents binding of myosin heads to actin,10, 51, 52 while the absence of cMyBPC relieves this physical constraint on myosin heads, allowing the myosin heads to move away from the thick filament backbone and towards actin binding sites, thus enhancing acto‐myosin XB formation.31 On the other hand, Myk461 has not been shown to prevent the binding of myosin heads to actin per se, but rather, acts to reduce the number of XBs transitioning from a weakly to a strongly bound state.22 Thus, depletion of cMyBPC in the thick filament increases the population of XBs that are binding to actin and transitioning to force‐generating states, such that in the KO myocardium greater concentrations of Myk461 are required to achieve the same degree of force depression in WT myocardium, as we observed here (Table 2). An additional mechanism underlying the diminished impact of Myk461 on force generation in KO myocardium may be related to the fact cMyBPC normally sequesters XBs in a nonactivated state by maintaining the super‐relaxed state of myosin heads53; therefore, cMyBPC ablation disrupts the super‐relaxed state, allowing an increased number of myosin heads to bind actin. It is possible that at an equivalent dose of Myk461, there is a greater stabilization of the super‐relaxed state of myosin heads in WT myocardium compared with KO myocardium, which supposedly contains a smaller population of myosin heads in the super‐relaxed state, thereby resulting in the observed differential impact of Myk461 on force generation. Apart from reducing force generation, Myk461 has been reported to decrease ATPase activity in cardiac preparations.21 Thus, based on our observation that the impact of Myk461 on force generation is less pronounced in KO myocardium at low Ca2+ activations, we expect that the Myk461‐induced reduction in steady‐state ATPase activity may be less pronounced in KO when compared with WT myocardium, especially at low Ca2+ activations.
Myk461 Differentially Impacts the XB Behavior in KO Myocardium
We performed stretch activation experiments to probe the impact of Myk461 on XB cycling kinetics by measuring both the rates of recruitment (kdf) and XB detachment (krel) in WT and KO myocardium. Consistent with the finding that Myk461 slows the rate of phosphate release and the transition of XBs from a weakly bound to a strongly bound state,22 our data show that Myk461 incubation significantly slows kdf in both WT and KO myocardium (Tables 3 and 4; Figure 5). Furthermore, recent data show that the actin association rate of ADP‐bound myosin heads is significantly reduced in the presence of Myk461,22 likely contributing to the slowed kdf observed post Myk461 incubations in WT and KO preparations (Tables 3 and 4). Importantly, basal kdf differences between the WT and KO groups, observed at low Ca2+ activations, were no longer evident after Myk461 incubation, suggesting a greater degree of Myk461‐induced slowing in kdf in KO myocardium. As a consequence, Myk461 incubation also resulted in a decreased magnitude of XB recruitment (Pdf) in the KO myocardium, which is indicative of reduced stretch‐mediated recruitment of additional XBs to a force‐producing state (Table 3).
In agreement with previous studies,29, 54 KO myocardium displayed accelerated XB detachment under basal conditions when compared with WT myocardium. This effect is likely because of the fact that the presence of cMyBPC imparts an elastic and viscous load on the myosin heads, which acts to slow force relaxation and that absence of cMyBPC leads to a more compliant XB and possibly a greater degree of radial compliance of the sarcomeric lattice55 in the KO myocardium, resulting in an abbreviated XB “on” time. Interestingly, our data also show that Myk461 incubation significantly slowed krel in the KO myocardium but not in the WT myocardium, effectively normalizing the basal differences in krel between KO and WT groups (Table 3; Figure 5), suggesting a slowing in the strain‐induced XB detachment from actin in the KO myocardium. Our observation of unaltered krel in WT myocardium agrees with results from solution‐based transient kinetic studies, which show that Myk461 does not affect the ADP release rate from actomyosin.22, 48 However, our result that Myk461 slows XB detachment in KO indicates a differential impact of Myk461 on XB detachment, suggesting that the impact of Myk461 may vary depending on the presence or absence of cMyBPC in an intact sarcomeric lattice system that is influenced by strain‐dependent mechanisms, a phenomenon that is difficult to replicate using solution‐based transient kinetic studies. Furthermore, the impact of Myk461 on myosin head kinetics varies depending on the presence of a 2‐headed cardiac heavy meromyosin or a single‐headed S1,48 suggesting that the structural complexity of the experimental preparations may be an important determinant in interpreting the effects of Myk461 on cardiac muscle contractile function.
Although the precise mechanism by which Myk461 differentially slows krel in KO myocardium is unclear, there is evidence to show that the inhibitory effects of Myk461 on ATPase activity are attenuated in HCM‐causing mutant myosins displaying more accelerated XB behavior.22 Thus, the differential impact of Myk461 on krel observed here is likely related to inherent differences in XB behavior between KO and WT myocardium. It is known that cMyBPC plays a dominant role in strain‐dependent XB detachment from actin and that XBs are destabilized in the absence of cMyBPC, thus resulting in faster rates of force decay.30, 37, 54 Perhaps Myk461 counters this effect by altering myosin head conformation in a manner that increases XB stability and/or slows XB turnover in KO myocardium, partly explaining the reduced force deficits induced by Myk461 in KO myocardium—a hypothesis that requires further validation using molecular studies specifically geared towards elucidating the contributions of cMyBPC on Myk461‐mediated modulation of XB behavior. Nevertheless, the Myk461‐induced slowing in krel in the KO myocardium would be expected to reduce overall ATP turnover rate and tension cost as XB detachment was shown to correlate well with tension cost,56, 57 which may improve systolic function in vivo and reduce pathological hypertrophy and remodeling in models of hypertrophy induced by reductions in cMyBPC.30, 32 Notably, a slowed krel and increased XB “on” time following Myk461 incubation may underlie our observation that Myk461‐induced force generation reductions were not as pronounced in the KO myocardium (Table 2; Figure 3).
Study Limitations
A limitation of this study is that the effects of MyK461 were investigated in cMyBPC ablated myocardium, which is an extreme case of cMyBPC deficiency as the majority of human cMyBPC truncation mutations are heterozygous and express at least 50% of normal full‐length cMyBPC protein in the sarcomere.8, 58 Although far less common, there is clinical evidence showing that homozygous truncating mutations in cMyBPC exist and result in severe infantile HCM with poor prognosis and early death,59, 60, 61 likely because cMyBPC expression is either absent, or significantly lower than heterozygous mutation carriers. Our results therefore may be relevant in elucidating the clinical impact of Myk461 in conditions of severe cMyBPC deficiency, and provide insights of the role of cMyBPC in modulating the effects of Myk461 at the sarcomere level.
Potential In Vivo Impact of Myk461 on Hearts Expressing cMyBPC Deficiency
Recent findings show that Myk461 administration in mice expressing α‐MHC HCM‐mutations results in reduced hypercontractility evidenced by decreased fractional shortening and a concomitant regression of pathological ventricular remodeling as supported by reductions in ventricular wall thickness, myocyte disarray, and fibrosis.21 In addition to its beneficial effects in the murine models of MHC‐mediated HCM, Myk461 has also been shown to have utility improving cardiac contractile function in feline models of HCM.23 Here we show that Myk461 has salutary effects in reducing hypercontractility in myocardium lacking cMyBPC by decreasing the magnitude of force generation and the tension cost of force generation, which at the whole organ level would be expected to reduce systolic pressure generation, improve contractile efficiency, and reduce myocardial oxygen demands. Thus, development of Myk461‐based therapies may have utility in treating HCM patients expressing cMyBPC mutations, which are known to reduce the total amount of cMyBPC7 and induce severe cardiac dysfunction and death, especially at a young age.59, 60, 61 Our results define the mechanistic impact of Myk461 in hypercontractile myocardium lacking cMyBPC, and importantly, demonstrate that in KO myocardium Myk461‐induced force reductions were less pronounced than in WT myocardium. Furthermore, in conjunction with significantly slowed rates of XB recruitment and transitions to strongly bound states, Myk461 also induced significant reductions in the rate of XB detachment in KO myocardium compared with WT myocardium, which would be expected to increase XB “on” time, contributing to the observed attenuated force depressions. Taken together, our findings suggest that the impact of Myk461 to decrease force generation may be modulated by the level of cMyBPC expression, highlighting the potential need for optimization of clinical use of Myk461 based on the molecular trigger (ie, mutations in MHC, cMyBPC, or thin filament proteins) that underlies the HCM phenotype.
Author Contributions
Mamidi and Stelzer contributed to the conception and design of the experiments. Mamidi, Li, Doh, and Stelzer participated in performing the experiments, data acquisition, data analysis, data interpretation, drafting, and revising the manuscript. Verma participated in data analysis, data interpretation, drafting, and revising the manuscript. All authors approved the final version of the manuscript.
Sources of Funding
This work was supported by the National Institutes of Health (HL‐114770 to Stelzer; T32‐HL007567 and GM007250 to Li), American Heart Association (16POST30730000 to Mamidi), and National Institutes of Health (P30 EY011373) grants.
Disclosures
None.
(J Am Heart Assoc. 2018;7:e009627 DOI: 10.1161/JAHA.118.009627.)
References
- 1. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the cardia study. Coronary artery risk development in (young) adults. Circulation. 1995;92:785–789. [DOI] [PubMed] [Google Scholar]
- 2. van der Velden J, Ho CY, Tardiff JC, Olivotto I, Knollmann BC, Carrier L. Research priorities in sarcomeric cardiomyopathies. Cardiovasc Res. 2015;105:449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ho CY, Carlsen C, Thune JJ, Havndrup O, Bundgaard H, Farrohi F, Rivero J, Cirino AL, Andersen PS, Christiansen M, Maron BJ, Orav EJ, Kober L. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Spudich JA. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J. 2014;106:1236–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ho CY, Sweitzer NK, McDonough B, Maron BJ, Casey SA, Seidman JG, Seidman CE, Solomon SD. Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation. 2002;105:2992–2997. [DOI] [PubMed] [Google Scholar]
- 6. Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775–785. [DOI] [PubMed] [Google Scholar]
- 7. Jacques AM, Copeland O, Messer AE, Gallon CE, King K, McKenna WJ, Tsang VT, Marston SB. Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. J Mol Cell Cardiol. 2008;45:209–216. [DOI] [PubMed] [Google Scholar]
- 8. van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ, van der Velden J. Cardiac myosin‐binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. [DOI] [PubMed] [Google Scholar]
- 9. Witjas‐Paalberends ER, Ferrara C, Scellini B, Piroddi N, Montag J, Tesi C, Stienen GJ, Michels M, Ho CY, Kraft T, Poggesi C, van der Velden J. Faster cross‐bridge detachment and increased tension cost in human hypertrophic cardiomyopathy with the R403Q MYH7 mutation. J Physiol. 2014;592:3257–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein‐C in health and disease. J Mol Cell Cardiol. 2010;48:866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nag S, Trivedi DV, Sarkar SS, Adhikari AS, Sunitha MS, Sutton S, Ruppel KM, Spudich JA. The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat Struct Mol Biol. 2017;24:525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mamidi R, Li J, Gresham KS, Stelzer JE. Cardiac myosin binding protein‐C: a novel sarcomeric target for gene therapy. Pflugers Arch. 2014;466:225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kawana M, Sarkar SS, Sutton S, Ruppel KM, Spudich JA. Biophysical properties of human beta‐cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Sci Adv. 2017;3:e1601959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nag S, Sommese RF, Ujfalusi Z, Combs A, Langer S, Sutton S, Leinwand LA, Geeves MA, Ruppel KM, Spudich JA. Contractility parameters of human beta‐cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Sci Adv. 2015;1:e1500511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adhikari AS, Kooiker KB, Sarkar SS, Liu C, Bernstein D, Spudich JA, Ruppel KM. Early‐onset hypertrophic cardiomyopathy mutations significantly increase the velocity, force, and actin‐activated ATPase activity of human beta‐cardiac myosin. Cell Rep. 2016;17:2857–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sommese RF, Sung J, Nag S, Sutton S, Deacon JC, Choe E, Leinwand LA, Ruppel K, Spudich JA. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human beta‐cardiac myosin motor function. Proc Natl Acad Sci USA. 2013;110:12607–12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Palmiter KA, Tyska MJ, Haeberle JR, Alpert NR, Fananapazir L, Warshaw DM. R403Q and L908V mutant beta‐cardiac myosin from patients with familial hypertrophic cardiomyopathy exhibit enhanced mechanical performance at the single molecule level. J Muscle Res Cell Motil. 2000;21:609–620. [DOI] [PubMed] [Google Scholar]
- 18. Garfinkel AC, Seidman JG, Seidman CE. Genetic pathogenesis of hypertrophic and dilated cardiomyopathy. Heart Fail Clin. 2018;14:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Malik FI, Morgan BP. Cardiac myosin activation part 1: from concept to clinic. J Mol Cell Cardiol. 2011;51:454–461. [DOI] [PubMed] [Google Scholar]
- 20. Teerlink JR. A novel approach to improve cardiac performance: cardiac myosin activators. Heart Fail Rev. 2009;14:289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Green EM, Wakimoto H, Anderson RL, Evanchik MJ, Gorham JM, Harrison BC, Henze M, Kawas R, Oslob JD, Rodriguez HM, Song Y, Wan W, Leinwand LA, Spudich JA, McDowell RS, Seidman JG, Seidman CE. A small‐molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351:617–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kawas RF, Anderson RL, Ingle SRB, Song Y, Sran AS, Rodriguez HM. A small‐molecule modulator of cardiac myosin acts on multiple stages of the myosin chemomechanical cycle. J Biol Chem. 2017;292:16571–16577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stern JA, Markova S, Ueda Y, Kim JB, Pascoe PJ, Evanchik MJ, Green EM, Harris SP. A small molecule inhibitor of sarcomere contractility acutely relieves left ventricular outflow tract obstruction in feline hypertrophic cardiomyopathy. PLoS One. 2016;11:e0168407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carrier L, Schlossarek S, Willis MS, Eschenhagen T. The ubiquitin‐proteasome system and nonsense‐mediated mRNA decay in hypertrophic cardiomyopathy. Cardiovasc Res. 2010;85:330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marston S, Copeland O, Gehmlich K, Schlossarek S, Carrier L. How do MYBPC3 mutations cause hypertrophic cardiomyopathy? J Muscle Res Cell Motil. 2012;33:75–80. [DOI] [PubMed] [Google Scholar]
- 26. Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–222. [DOI] [PubMed] [Google Scholar]
- 27. Hoskins AC, Jacques A, Bardswell SC, McKenna WJ, Tsang V, dos Remedios CG, Ehler E, Adams K, Jalilzadeh S, Avkiran M, Watkins H, Redwood C, Marston SB, Kentish JC. Normal passive viscoelasticity but abnormal myofibrillar force generation in human hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2010;49:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng Y, Wan X, McElfresh TA, Chen X, Gresham KS, Rosenbaum DS, Chandler MP, Stelzer JE. Impaired contractile function due to decreased cardiac myosin binding protein C content in the sarcomere. Am J Physiol Heart Circ Physiol. 2013;305:H52–H65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mamidi R, Gresham KS, Stelzer JE. Length‐dependent changes in contractile dynamics are blunted due to cardiac myosin binding protein‐C ablation. Front Physiol. 2014;5:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Merkulov S, Chen X, Chandler MP, Stelzer JE. In vivo cardiac myosin binding protein c gene transfer rescues myofilament contractile dysfunction in cardiac myosin binding protein C null mice. Circ Heart Fail. 2012;5:635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Colson BA, Bekyarova T, Fitzsimons DP, Irving TC, Moss RL. Radial displacement of myosin cross‐bridges in mouse myocardium due to ablation of myosin binding protein‐C. J Mol Biol. 2007;367:36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein‐C knockout mice. Circ Res. 2002;90:594–601. [DOI] [PubMed] [Google Scholar]
- 33. Gresham KS, Mamidi R, Stelzer JE. The contribution of cardiac myosin binding protein‐C Ser282 phosphorylation to the rate of force generation and in vivo cardiac contractility. J Physiol. 2014;592:3747–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gresham KS, Mamidi R, Li J, Kwak H, Stelzer JE. Sarcomeric protein modification during adrenergic stress enhances cross‐bridge kinetics and cardiac output. J Appl Physiol (1985). 2017;122:520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 1988;157:378–417. [DOI] [PubMed] [Google Scholar]
- 36. Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. J Gen Physiol. 1982;80:279–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mamidi R, Gresham KS, Li A, Dos Remedios CG, Stelzer JE. Molecular effects of the myosin activator omecamtiv mecarbil on contractile properties of skinned myocardium lacking cardiac myosin binding protein‐C. J Mol Cell Cardiol. 2015;85:262–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mamidi R, Gresham KS, Li J, Stelzer JE. Cardiac myosin binding protein‐C Ser(302) phosphorylation regulates cardiac beta‐adrenergic reserve. Sci Adv. 2017;3:e1602445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mamidi R, Gresham KS, Verma S, Stelzer JE. Cardiac myosin binding protein‐C phosphorylation modulates myofilament length‐dependent activation. Front Physiol. 2016;7:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mamidi R, Li J, Gresham KS, Verma S, Doh CY, Li A, Lal S, Dos Remedios CG, Stelzer JE. Dose‐dependent effects of the myosin activator omecamtiv mecarbil on cross‐bridge behavior and force generation in failing human myocardium. Circulation Heart Failure. 2017;10:e004257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Campbell KS, Moss RL. Slcontrol: PC‐based data acquisition and analysis for muscle mechanics. Am J Physiol Heart Circ Physiol. 2003;285:H2857–H2864. [DOI] [PubMed] [Google Scholar]
- 42. Ford SJ, Chandra M, Mamidi R, Dong W, Campbell KB. Model representation of the nonlinear step response in cardiac muscle. J Gen Physiol. 2010;136:159–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bates D, Martin M, Ben B, Steve W. Fitting linear‐mixed effects model using Ime4. J Stat Softw. 2015;67:1–48. [Google Scholar]
- 44. Core TR . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2017. [Google Scholar]
- 45. Stelzer JE, Larsson L, Fitzsimons DP, Moss RL. Activation dependence of stretch activation in mouse skinned myocardium: implications for ventricular function. J Gen Physiol. 2006;127:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Previs MJ, Prosser BL, Mun JY, Previs SB, Gulick J, Lee K, Robbins J, Craig R, Lederer WJ, Warshaw DM. Myosin‐binding protein C corrects an intrinsic inhomogeneity in cardiac excitation‐contraction coupling. Sci Adv. 2015;1:e1400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moss RL, Fitzsimons DP, Ralphe JC. Cardiac MyBP‐C regulates the rate and force of contraction in mammalian myocardium. Circ Res. 2015;116:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rohde AJ, Thomas DD, Muretta MJ. Mavacamten stabilizes the auto‐inhibited state of two‐headed cardiac myosin. PNAS. 2018;115:E7486–7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Anderson RL, Trivedi DV, Sarkar SS, Henze M, Ma W, Gong H, Rogers CS, Wong F, Morck M, Seidman JG, Ruppel K, Irving TC, Cooke R, Green EM, Spudich JA. Mavacamten stabilizes a folded‐back sequestered super‐relaxed state of β‐cardiac myosin. BioRxiv. 2018. Avaialble at: https://www.biorxiv.org/content/biorxiv/early/2018/03/10/266783.full.pdf. Accessed August 24, 2018. [Google Scholar]
- 50. Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. [DOI] [PubMed] [Google Scholar]
- 51. Gruen M, Prinz H, Gautel M. Capk‐phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin‐S2 in an on‐off fashion. FEBS Lett. 1999;453:254–259. [DOI] [PubMed] [Google Scholar]
- 52. Trivedi DV, Adhikari AS, Sarkar SS, Ruppel KM, Spudich JA. Hypertrophic cardiomyopathy and the myosin mesa: viewing an old disease in a new light. Biophys Rev. 2018;10:27–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McNamara JW, Li A, Smith NJ, Lal S, Graham RM, Kooiker KB, van Dijk SJ, Remedios CG, Harris SP, Cooke R. Ablation of cardiac myosin binding protein‐C disrupts the super‐relaxed state of myosin in murine cardiomyocytes. J Mol Cell Cardiol. 2016;94:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stelzer JE, Dunning SB, Moss RL. Ablation of cardiac myosin‐binding protein‐C accelerates stretch activation in murine skinned myocardium. Circ Res. 2006;98:1212–1218. [DOI] [PubMed] [Google Scholar]
- 55. Palmer BM, Sadayappan S, Wang Y, Weith AE, Previs MJ, Bekyarova T, Irving TC, Robbins J, Maughan DW. Roles for cardiac MyBP‐C in maintaining myofilament lattice rigidity and prolonging myosin cross‐bridge lifetime. Biophys J. 2011;101:1661–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brenner B. Effect of Ca2+ on cross‐bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc Natl Acad Sci USA. 1988;85:3265–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. de Tombe PP, Stienen GJ. Protein kinase a does not alter economy of force maintenance in skinned rat cardiac trabeculae. Circ Res. 1995;76:734–741. [DOI] [PubMed] [Google Scholar]
- 58. van Dijk SJ, Paalberends ER, Najafi A, Michels M, Sadayappan S, Carrier L, Boontje NM, Kuster DW, van Slegtenhorst M, Dooijes D, dos Remedios C, ten Cate FJ, Stienen GJ, van der Velden J. Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ Heart Fail. 2012;5:36–46. [DOI] [PubMed] [Google Scholar]
- 59. Lekanne Deprez RH, Muurling‐Vlietman JJ, Hruda J, Baars MJ, Wijnaendts LC, Stolte‐Dijkstra I, Alders M, van Hagen JM. Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene. J Med Genet. 2006;43:829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ortiz MF, Rodriguez‐Garcia MI, Hermida‐Prieto M, Fernandez X, Veira E, Barriales‐Villa R, Castro‐Beiras A, Monserrat L. A homozygous MYBPC3 gene mutation associated with a severe phenotype and a high risk of sudden death in a family with hypertrophic cardiomyopathy. Rev Esp Cardiol. 2009;62:572–575. [DOI] [PubMed] [Google Scholar]
- 61. Zahka K, Kalidas K, Simpson MA, Cross H, Keller BB, Galambos C, Gurtz K, Patton MA, Crosby AH. Homozygous mutation of MYBPC3 associated with severe infantile hypertrophic cardiomyopathy at high frequency among the Amish. Heart. 2008;94:1326–1330. [DOI] [PubMed] [Google Scholar]