

Abstract
Background
Glycosphingolipid accumulation in Fabry cells generates a proinflammatory response that may influence disease evolution and responsiveness to enzyme replacement therapy. This study evaluated incidence, mechanism, and impact of myocarditis in Fabry disease cardiomyopathy (FDCM).
Methods and Results
Myocarditis, defined as CD3+ T lymphocytes >7/mm2 associated with necrosis of glycolipid‐laden myocardiocytes, was retrospectively evaluated in endomyocardial biopsies from 78 patients with FDCM: 13 with maximal wall thickness (MWT) <11 mm (group 1), 17 with MWT 11 to 15 mm (group 2), 30 with MWT 16 to 20 mm (group 3), and 18 with MWT >20 mm (group 4). Myocarditis was investigated by polymerase chain reaction for cardiotropic viruses, by serum antiheart and antimyosin antibodies, and by cardiac magnetic resonance. Myocarditis was recognized at histology in 48 of 78 patients with FDCM (38% of group 1, 41% of group 2, 66% of group 3, and 72% of group 4). Myocarditis was characterized by positive antiheart and antimyosin antibodies and negative polymerase chain reaction for viral genomes. CD3+ cells/mm2 correlated with myocyte necrosis, antimyosin autoantibody titer, and MWT (P<0.001,r=0.79; P<0.001, r=0.84; P<0.001, r=0.61, respectively). Cardiac magnetic resonance showed myocardial edema in 24 of 78 patients (31%): 0% of group 1, 23% of group 2, 37% of group 3, and 50% of group 4.
Conclusions
Myocarditis is detectable at histology in up to 56% of patients with FDCM. It is immune mediated and correlates with disease severity. It can be disclosed by antiheart/antimyosin autoantibodies and in the advanced phase by cardiac magnetic resonance. It may contribute to progression of FDCM and resistance to enzyme replacement therapy.
Keywords: cardiomyopathy, Fabry disease, heart failure, myocarditis
Subject Categories: Cardiomyopathy, Inflammatory Heart Disease, Heart Failure
Clinical Perspective
What Is New?
Pathophysiology of Fabry disease is still elusive.
This study demonstrates that the highly immunogenic globotriaosylceramide released by Fabry myocytes through a constitutional secretory pathway or cell necrosis activates immune‐mediated myocardial inflammation that occurs with disease progression and may limit the impact of enzyme replacement therapy.
What Are the Clinical Implications?
In patients with Fabry disease cardiomyopathy in whom an overlapping inflammatory process is shown by T2‐mapping signals at cardiac magnetic resonance and/or histology, further studies may be warranted to determine whether immunosuppressive therapy may improve the disease and perhaps the response to enzyme replacement therapy.
Introduction
Fabry disease (FD) is an X‐linked inborn error of glycosphingolipid catabolism caused by deleterious mutations in the GLA (α‐galactosidase A) gene encoding the lysosomal hydrolase GAL.1, 2 The marked deficiency or absence of GAL activity results in the systemic accumulation of globotriaosylceramide and related glycosphingolipids within the lysosomes, particularly in microvascular endothelial cells, vascular smooth muscle cells, renal tubular cells, podocytes, and cardiomyocytes.3, 4, 5, 6, 7
FD cardiomyopathy (FDCM) is a major determinant of patient survival, and its management represents a main therapeutic challenge. Indeed, the impact of enzyme replacement therapy (ERT) on FDCM is still controversial,8, 9, 10, 11, 12 and although there is agreement that early ERT administration, particularly in prehypertrophic FDCM, prevents progression of the disease, the advanced form is believed to be irreversible.
Mechanisms of resistance to ERT are still unclear, although expansion of interstitial space and myocardial fibrosis appear to be implicated.
Glycosphingolipid accumulation in Fabry cardiomyocytes promotes a proinflammatory response that may influence disease progression and responsiveness to ERT. In this report, myocarditis is assessed, characterized, and tested for possible infectious and/or autoimmune origin in a large FDCM series. Inflammatory damage at histology is compared with cardiac magnetic resonance (CMR) features.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Patient Population
From January 2000 to May 2016 in our department, 78 patients (51 male, 27 female; mean age 45.5±9.7 years) received a diagnosis of FDCM. Among them, 60 had classic FD mutations, whereas 18 had a late‐onset cardiac variant due to N215S mutation. The diagnosis was based on detection of a pathogenic mutation of GLA and the identification of low‐level GLA enzyme activity in the peripheral leukocytes of male participants and on an abnormal endomyocardial biopsy (EMB) showing accumulation of glycosphingolipids in the cardiac cells. Of the diagnosed patients, 13 were in a prehypertrophic phase (maximal wall thickness [MWT] <11 mm), 17 had mild hypertrophy (MWT 11–15 mm), 30 had moderate left ventricular (LV) hypertrophy (MWT 16–20 mm), and 18 had advanced disease (MWT >20 mm). MWT was defined as the greatest thickness of any segment of the LV wall. All clinical and pathological data were obtained at the time of diagnosis when patients were not yet on ERT. The study was approved by the ethics committee of our institution, and the participants gave written informed consent.
Cardiac Studies
Extensive clinical examination, including FD systemic manifestations, noninvasive (resting ECG, Holter monitoring, echocardiography with tissue Doppler analysis, CMR) and invasive cardiac studies, was performed in all patients, as described.13
Invasive cardiac exams were performed after patient written informed consent and approval by the ethics committee of our institution and included cardiac catheterization, coronary and LV angiography, and biventricular or LV EMB. The presence of slow coronary flow, defined as the delayed opacification of the distal vasculature in angiographically normal or nearly normal coronary arteries, was determined and defined in accordance with TIMI (Thrombolysis in Myocardial Infarction) frame counting.14
CMR Image Acquisition
All CMR exams were performed on a 1.5‐T scanner (Magnetom Avanto; Siemens Medical Systems) using body and phase‐array coils.
The CMR protocol included the following elements:
Cine balanced steady‐state free precession images were acquired during breath holds in the short‐axis, 2‐chamber, and 4‐chamber planes.
Black blood T2‐weighted short τ inversion recovery (T2w‐STIR) images for myocardial edema detection used a triple‐inversion recovery preparation module; the body coil was adapted for the measurement of a relative myocardial‐to‐skeletal muscle signal intensity (SI) ratio because of its more homogeneous signal (repetition time, 2 R‐to‐R intervals (the interval between two consecutive normal ECG waveforms); echo time, 75 ms; flip angle, 180°; inversion time, 170 ms; slice thickness, 8 mm; no interslice gap; field of view, 340–400 mm; matrix, 256×256; voxel size, 2.3×1.3×8 mm). The T2w‐STIR images were acquired on short‐axis planes covering the entire left ventricle during 6 to 8 consecutive breath holds.
ECG‐gated single‐shot modified Look‐Locker inversion recovery sequence with the original 3(3)3(3)5 scheme and motion‐correction postprocessing algorithm (Siemens WIP package no. 448) for native T1 mapping was acquired on 2 basal and midventricular short‐axis planes.
Late gadolinium–enhanced (LGE) imaging was performed 15 minutes after bolus injection of 0.2 mmol/kg of body weight of gadoterate meglumine (gadolinium‐DOTA; Dotarem), using a segmented phase‐sensitive inversion recovery gradient echo pulse sequence, setting the acquisition window to mid–end diastole and paying extreme care in setting the inversion time to null for the signal of normal myocardium (values ranging from 250 to 300 ms) Sequence parameters were as follows: repetition time, 9.6/4.4; matrix, 256×208; flip angle, 25°; slice thickness, 8.0 mm; slice spacing, 2.0 mm.
Gadolinium injection was avoided in 6 patients because of severe chronic renal failure (estimated glomerular filtration rate <30 mL/min per 1.73 m2).
CMR Image Analysis
Image analysis was performed using dedicated software (Cvi42, v5.3.0; Circle Cardiovascular Imaging). End‐diastolic MWT, LV myocardial mass, ventricular volumes (indexed to body surface area), and ejection fraction were measured in short‐axis cine balanced steady‐state free precession images.
Myocardial edema was assessed in T2w‐STIR images by comparing the LV myocardial SI against skeletal muscle included in the same slice, as recommended in the “Lake Louise Criteria.”15 The T2 SI ratio was calculated as T2 SI ratio=SI myocardium/SI skeletal muscle. Myocardial edema was diagnosed when the global or focal T2 SI ratio was >1.9.
Native T1 maps were automatically elaborated by applying a nonlinear curve fitting, using the 3‐parameter signal method. Global T1 value was measured as the mean of T1 values calculated in all 3 planes, avoiding LGE areas and leaving adequate margins of separation from tissue interfaces with adipose epicardial tissue and ventricular cavity. In addition, the T1 value of a 1‐cm2 region of interest within the midventricular septum was calculated as a more reproducible measure to minimize in‐plane and through‐plane partial volume effects.16
LGE areas were defined as a myocardial zone of SI >5 SD above the remote myocardium, as reported elsewhere.17
Invasive and EMB Studies
Cardiac catheterization with LV or biventricular ventriculography and coronary angiography was obtained in all patients. EMB was performed in the septal–apical region of the left or both ventricles approached using a 7F (501‐613A Cordis) long sheath introduced from a femoral access. The site of the biopsy was identified on a radiographic view using flashing of contrast medium. Three to 6 right ventricular biopsies or 3 to 6 LV biopsies (from 2 to 5 mm3) were drawn from each patient. Myocardial samples were processed for routine histological and histochemical analysis and for transmission electron microscopy. Two frozen endomyocardial samples per patient were processed for real‐time polymerase chain reaction screening of the most common cardiotropic viruses in the presence of myocardial inflammation at histology.
Histology and Electron Microscopy
For histological analysis, the EMB samples were fixed in 10% buffered formalin and paraffin embedded. Sections 5‐µm thick were stained with hematoxylin and eosin, Masson trichrome, and Miller's elastic van Gieson. Histochemistry with periodic acid–Schiff and Sudan black stains was obtained in frozen sections to evaluate the presence of intracellular glycolipid material.
For the phenotypic characterization of the inflammatory infiltrates, immunohistochemistry for CD3, CD43, CD45RO, and CD68 was performed. Briefly, myocardial sections were incubated with mouse monoclonal primary antibodies and a peroxidase/antiperoxidase complex, followed by labeling with the chromogen diaminobenzidine. The presence of an inflammatory infiltrate ≥14 leukocytes/mm2 including up to 4 monocytes/mm2, with the presence of CD3+ T lymphocytes ≥7 cells/mm2 associated with evidence of degeneration and/or necrosis of the adjacent cardiomyocytes, was considered diagnostic for myocarditis. The number of CD3+ cells was manually counted using a tally counter on a high‐power field (×400) scanning the entire slide. The area of tissue samples was measured using a computerized system (Imaging Software/NIS‐Elements AR 4.30; Nikon Instruments). The number of CD3+ cells was expressed as the number of cells/mm2. Morphometric evaluation was performed by a pathologist blinded to clinical data.
For transmission electron microscopy, additional samples were fixed in 2% glutaraldehyde in 0.1 mol/L phosphate buffer (pH 7.3), post fixed in osmium tetroxide, and processed following a standard schedule for embedding in Epon resin. Ultrathin sections were stained with uranyl acetate and lead hydroxyde. A Philips CM‐10 transmission electron microscope was used for observation and photographic analysis.
Molecular Biology Studies
Real‐time polymerase chain reaction was performed on 2 frozen EMB tissue samples to search for the most common DNA (adenovirus, cytomegalovirus, parvovirus B19, Epstein–Barr virus, human herpes virus 6, and herpes simplex virus 1 and 2) and RNA (enterovirus, influenza virus A and B, hepatitis C virus) for cardiotropic viruses in patients with histological evidence of myocarditis.18
Antiheart Autoantibodies
Patient sera were tested for the presence of circulating cardiac autoantibodies using a standard indirect immunofluorescence technique with a substrate of 4‐μm‐thick unfixed cryostat sections of human atrium and intercostal skeletal muscle of blood group O patients.18, 19
Briefly, patient sera were diluted 1/10 in pH 7.2 phosphate‐buffered saline solution, and fluorescein isothiocyanate–labeled rabbit antihuman immunoglobulin was used at 1:100 dilution. The intensity of immunofluorescence of the positive control (known positive serum) at 1/40 dilution was used as the cutoff point for positivity. Omission of the patient serum and known negative serum were included in every assay (negative controls).
ELISA Test for Antimyosin Antibodies
Patient sera were screened for the presence of antimyosin antibodies, detected by a human myosin ELISA kit. As controls, we used a pool of normal sera from participants matched with patients for age and sex. Standards and undiluted samples were added to the micro‐ELISA plate wells and combined with the specific antibody. The optical density proportional to the concentration of human myosin was measured spectrophotometrically at a wavelength of 450 nm±2 nm. The detection range of the human myosin ELISA kit is 1.56 to 100 ng/mL. The concentration in the samples was calculated comparing the optical density of the samples with the standard curve using the software interface GraphExpert Professional 1.4.0 (Hyams Development) and expressed as ng/mL.
Statistical Analysis
Descriptive analysis was conducted using median and interquartile range for numeric variables and proportions for qualitative variables. Statistical analysis was performed with a nonparametric Kruskal–Wallis test to evaluate differences within groups. Multiple comparisons of the median values were performed with the Dunn post test. Significance was set as P<0.05. The χ2 test was used for comparison of categorical variables. The correlations between CD3 grading and human myosin antibodies, MWT, age, and enzyme activity were explored with the Spearman ρ test. Data analysis was performed using GraphPad Prism v6.04 for Windows (GraphPad Software).
Results
Figure 1 shows the main clinical, histological, and immunological characteristics of 78 patients with FDCM enrolled in the study and divided in 4 groups depending on the extent of LV MWT at CMR. Age, sex, genetic mutation, enzymatic activity, cardiac symptoms, and extracardiac manifestations are reported in Table.
Figure 1.
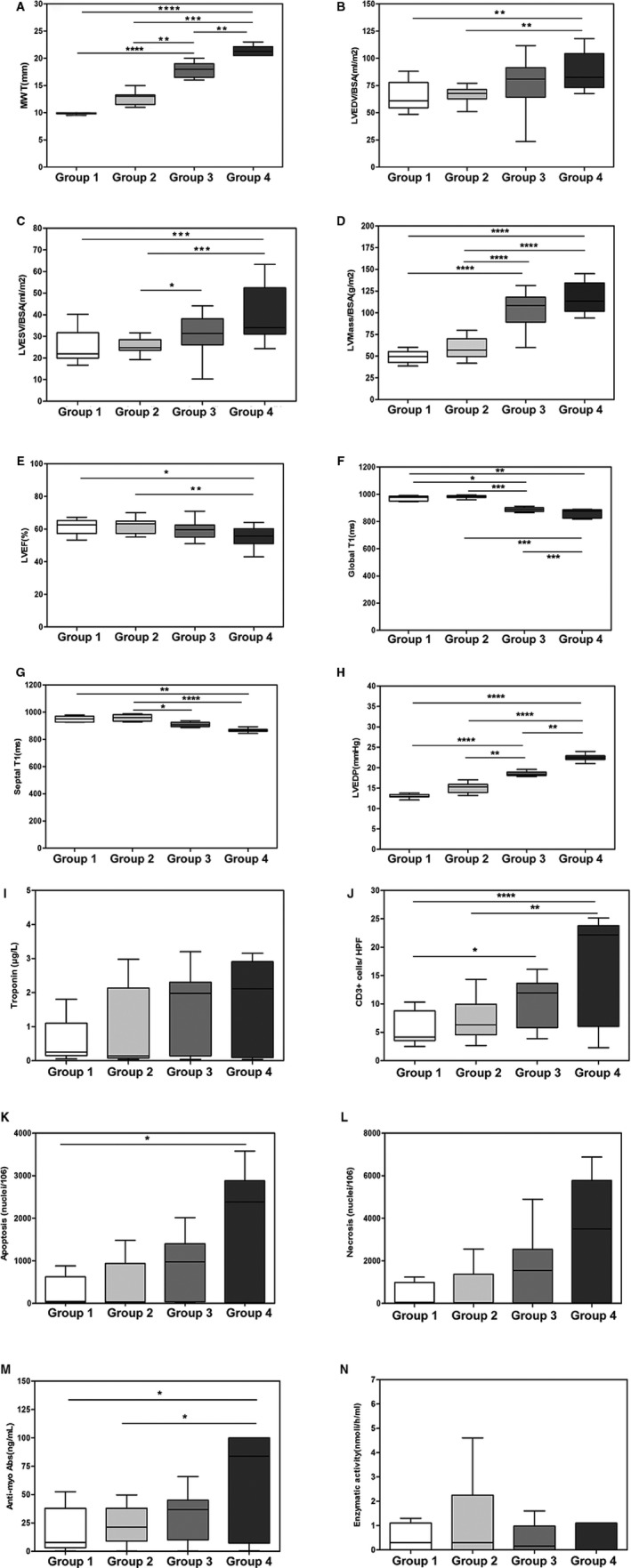
Box plot distribution by group for definition of relevant variables. A, Maximal wall thickness (MWT; mm). B, Left ventricular end‐diastolic volume/body surface area (LVEDV/BSA; mL/m2). C, Left ventricular end‐systolic volume/body surface area (LVESV/BSA; mL/m2). D, Left ventricular mass/body surface area (LV mass/BSA; g/m2). E, Left ventricular ejection fraction (LVEF; %). F, Global T1 (ms). G, Septal T1 (ms). H, Left ventricular end‐diastolic pressure (LVEDP; mm Hg). I, Troponin I (μg/L). L, CD3+ cells/high‐power field (HPF). J, Apoptosis (nuclei/106). K, Necrosis (nuclei/106). L, Antimyosin autoantibodies (ng/mL). M, Enzymatic activity (nmol/h/mL). Comparison among groups was performed using the Dunn test. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
Table 1.
Gene Mutation, Enzymatic Activity, and Clinical Manifestations of the 4 Groups of Patients With Fabry Disease
| Patient | Age y/Sex | Cardiac Symptoms | Extracardiac Manifestations | Enzymatic Activity nmol/h/mL (n.v >3) | Genotype |
|---|---|---|---|---|---|
| Group 1 | |||||
| 1 | 36/F | P, A | K | 4.6 | R220X |
| 2 | 20/M | P | Ap, Ak | 0 | R220X |
| 3 | 28/M | P | ··· | 0.5 | N215S |
| 4 | 35/M | P | Ap, Ak, K, E | 0 | Q279K |
| 5 | 39/F | P, A | ··· | 0.3 | N215S |
| 6 | 29/M | P | CV, Ap | 0.1 | G328R |
| 7 | 22/M | A | Ap, Ak, CV, K | 0 | c946delG |
| 8 | 25/M | P | Ap, Ak, CV, E | 0 | Q279K |
| 9 | 39/F | A, D | Ap, CV | 1.3 | C378Y |
| 10 | 44/F | P | CV | 0.9 | R220X |
| 11 | 31/M | P | ··· | 0.2 | N215S |
| 12 | 26/M | A | ··· | 0.3 | N215S |
| 13 | 37/F | A | CV, Ap | 3.5 | R227Q |
| Group 2 | |||||
| 1 | 38/M | D, P | K, Ap, CNS, E | 0 | G328R |
| 2 | 43/M | A, P | K, Ap, CNS | 0 | G238R |
| 3 | 46/F | P | CV, K | 2.5 | C223Y |
| 4 | 35/F | P | ··· | 6.3 | N215S |
| 5 | 38/F | A, P | ··· | 1.0 | N215S |
| 6 | 39/F | P | CV, Ap | 3.2 | R227Q |
| 7 | 60/F | D, P | CV, Ap | 0.5 | C223Y |
| 8 | 53/F | D, P | CV, Ap | 2.0 | R227Q |
| 9 | 36/F | A, D, P | K, Ap, CNS | 0 | C946delG |
| 10 | 28/M | P | CV, Ak, Ap | 0 | Q279K |
| 11 | 52/F | D, P | K, CV | 1.4 | C378Y |
| 12 | 42/M | D, P | K, Ap, E | 0 | R220X |
| 13 | 48/M | A | ··· | 0.2 | N215S |
| 14 | 38/M | A | K, CV, Ak | 0.1 | Q279K |
| 15 | 52/F | A | K, Ap, CNS | 4.6 | C378Y |
| 16 | 42/M | A, D | K, Ap, Ak, CV | 0 | R220X |
| 17 | 48/M | A, D | ··· | 0.3 | N215S |
| Group 3 | |||||
| 1 | 50/M | A, D, P | K, E, CV | 0 | R220X |
| 2 | 53/M | D | ··· | 0.9 | N215S |
| 3 | 45/M | D, P | CNS, E | 0 | Q279K |
| 4 | 59/F | D, P | ··· | 2.8 | N215S |
| 5 | 44/M | A, D, P | K, Ap, Ak, E | 0 | G328R |
| 6 | 58/F | D, P | CV | 1.2 | C223Y |
| 7 | 50/M | D | ··· | 0.3 | N215S |
| 8 | 41/M | A, D | Ap, E, CV | 0 | C378Y |
| 9 | 52/M | D, P | ··· | 0.5 | N215S |
| 10 | 39/M | D, P | ··· | 0.1 | N215S |
| 11 | 49/F | D, P | K, Ap | 5.6 | R227Q |
| 12 | 44/F | D | Ap, CV | 3.5 | C378Y |
| 13 | 60/M | A, D, P | K, CNS, Ap, E, CV | 0 | C946delG |
| 14 | 51/M | D, P | K, Ap, Ak, E, CV | 0 | Q279K |
| 15 | 53/M | D, P | K, Ap, Ak, E, CV | 0 | Q279K |
| 16 | 54/M | A, D, P | K, CNS, Ap, CV | 0.1 | R220X |
| 17 | 52/M | D, P | ··· | 0.2 | N215S |
| 18 | 59/F | A, D, P | K, CNS, Ap, Ak, E | 0.5 | C946delG |
| 19 | 39/M | P | K, Ap, Ak, E | 0 | Q279K |
| 20 | 46/M | A, P | K, Ap, CV | 0 | C378Y |
| 21 | 55/F | A, P | Ap, CNS, | 1.2 | R220X |
| 22 | 43/M | D | ··· | 0 | N215S |
| 23 | 44/M | D | K, Ak, E | 0 | C378Y |
| 24 | 44/M | D | K, CNS, CV, E | 0.2 | R220X |
| 25 | 47/M | D, P | ··· | 0.2 | N215S |
| 26 | 49/F | A, D, P | K, CNS, CV | 0.6 | C946delG |
| 27 | 39/M | A, P | K, Ap, Ak, E, CV | 0 | Q279K |
| 28 | 46/F | A, P | E, CV | 0 | C378Y |
| 29 | 55/F | A, P | E, CV | 1.6 | R220X |
| 30 | 63/M | A, D, P | ··· | 1.3 | N215S |
| Group 4 | |||||
| 1 | 55/M | A, D, P | K, Ak, CV, E | 0 | pR227X |
| 2 | 52/M | A, D, P | K, CNS, Ap, E | 0 | C427delACTT |
| 3 | 53/M | D, P | K, CNS, Ak, Ap, E | 0 | C946delG |
| 4 | 48/M | D, P | ··· | 0.2 | N215S |
| 5 | 53/M | A, D, P | K, Ap, E | 0 | Y216C |
| 6 | 57/F | A, D, P | K, Ap, E | 4.1 | Q279K |
| 7 | 62/M | D, P | K, Ak, Ap, E | 0.2 | R227Q |
| 8 | 50/F | A, D, P | K, CV, E, Ak, Ap, CNS | 0 | C946delG |
| 9 | 56/M | D, P | CV, Ak, Ap | 0 | Q279k |
| 10 | 47/M | A, D, P | K, Ak, Ap, CV, E, CNS, E | 0 | C946delG |
| 11 | 51/M | A, D, P | K, Ap, CV | 0 | Q279K |
| 12 | 60/F | D, P | K, Ap, Ak, CV | 0 | C946delG |
| 13 | 69/F | A, D, P | K, CV | 3.8 | Q279K |
| 14 | 48/M | A, D, P | K, Ap, Ak, CNS, CV, E | 0 | C946delG |
| 15 | 52/M | D, P | K, Ap, E | 0 | pR227X |
| 16 | 54/F | D, P | ··· | 0 | N215S |
| 17 | 53/F | D, P | K, E | 5.1 | R22XQ |
| 18 | 52/M | D, P | K, Ap, Ak, E | 4.8 | C378Y |
A indicates angina; Ak, angiokeratoma; Ap, acroparesthesia; CNS, central nervous system (transient ischemic attack and/or stroke and/or progressive white matter lesions and/or dolichoectasia of the basilar artery); CV, cornea verticillata; D, dyspnea; E, ear; F, female; K, kidney involvement (ie, pathological microalbuminuria, and/or proteinuria and/or estimated glomerular filtration rate reduction); M, male; P, palpitation.
Participant age increased among groups. Patients in groups 1 and 2 were younger (median ages of 31 and 42 years, respectively) than those in groups 3 and 4 (median ages of 50 and 53 years, respectively); differences were significant for group 1 versus groups 3 and 4 and for group 2 versus group 4 (P<0.05; Table).
Cardiac Studies
Group 1 patients presented normal ECG voltages and normal LV wall thickness and motion at 2‐dimensional echocardiogram. They were biopsied because of persistent chest pain with abnormal ST‐segment deflection at stress ECG (5 patients) or palpitations and Lown class >3 ventricular ectopic beats at ECG/Holter (8 patients) associated with a pathogenic mutation of GLA.
In group 2, increased ECG voltages (Sokolov–Lyon criteria) were observed in 10 patients, and major complaints were palpitation and chest pain; ventricular ectopic beats with Lown class ≥3 were documented in 6 patients.
Group 3 patients complained of dyspnea on effort, and 14 in the group also reported anginal pain. LV hypertrophy was observed at ECG in 24 patients, and left bundle‐branch block was observed in 6 patients. Lown class ≥4 ventricular arrhythmias requiring antiarrhythmics were relieved in 4 patients.
Group 4 patients were always symptomatic, showing New York Heart Association class II–IV and often chest pain. Occurrence of pain was usually associated with increase of serum troponin I. ECG was systematically abnormal with elevated QRS voltages, suggesting LV hypertrophy and ventricular repolarization abnormalities in 14 patients, left bundle‐branch block in 4 patients, and atrial fibrillation in 5 patients. Ventricular arrhythmias at ECG/Holter required antiarrhythmic drugs for 3 patients (nonsustained ventricular tachycardia) and implantable converter–defibrillator implantation for 2 patients (because of sustained ventricular tachycardia). Pacemaker implantation was needed for 2 patients.
Cardiac Magnetic Resonance
CMR features are reported in Figure 1. CMR images are reported in Figures 2, 3, 4 through 5 (panels A–C) representing groups 1 through 4, respectively.
Figure 2.
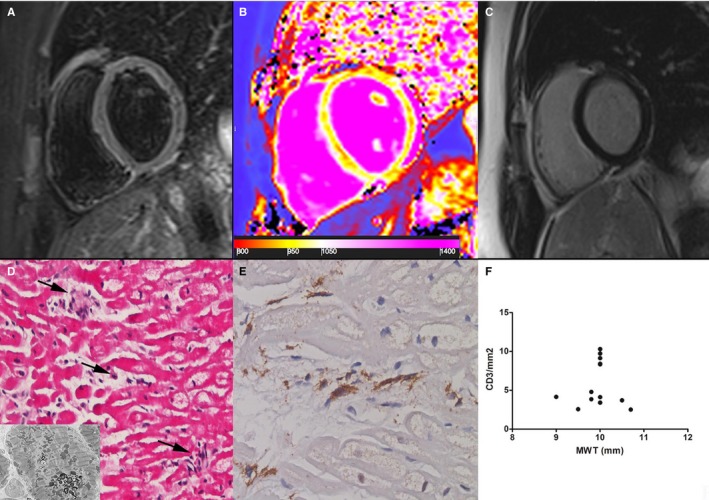
Group 1 patient. A 22‐year‐old woman with prehypertrophic Fabry disease cardiomyopathy (maximal wall thickness [MWT]: 8 mm) showing normal myocardium in a T2‐weighted short τ inversion recovery image (A) and a late gadolinium–enhanced image (C). Analysis of T1 mapping (B) showed a reduced septal T1 value (923±30 ms, n.v. 960–990 ms), reflecting initial tissue lipid accumulation. D, At histology (hematoxylin and eosin, ×200), cardiomyocytes are intermittently enlarged because of cytoplasm vacuoles that, at electron microscopy, consist of myelin bodies (insert) and are focally (arrows) surrounded by CD3+ infiltrates (E) with cell necrosis. F, Correlation between MWT and CD3 count (P=ns) in all group 1 patients (n=13).
Figure 3.
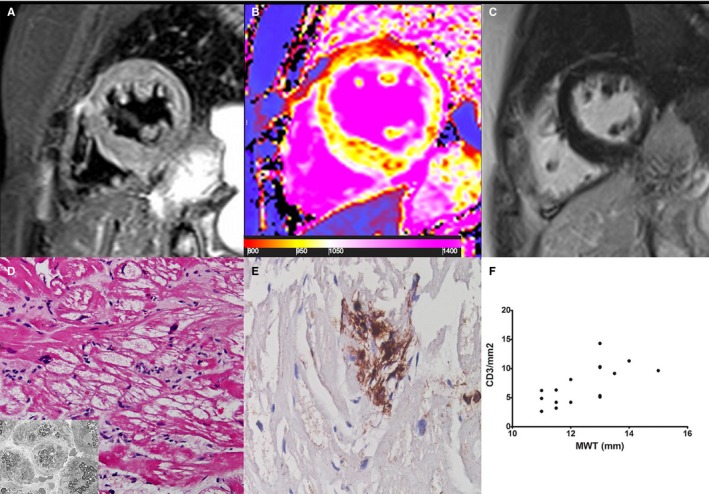
Group 2 patient. A 61‐year‐old woman with Fabry disease cardiomyopathy and mild hypertrophy (maximal wall thickness [MWT]: 14 mm). A T2‐weighted short τ inversion recovery image (A) and a late gadolinium–enhanced image (C) do not show clear areas of myocardial edema or enhancement (T2 ratio: 1.7). Native T1 map (B) shows some patchy areas of focal reduction of T1 (red spots within septum and ventricular wall) with a global mild decrease in myocardial T1 value (946±39 ms, n.v. 960–990 ms) as an expression of tissue accumulation of sphingolipidis. D, At histology (hematoxylin and eosin, ×200), cardiomyocytes are intermittently hypertrophied and vacuolated. Vacuoles, consisting of myelin bodies (insert), are focally damaged by CD3+ inflammatory cells (E). F, Correlation between MWT and CD3 count (P<0.001, ρ=0.70) in all group 2 patients (n=17).
Figure 4.
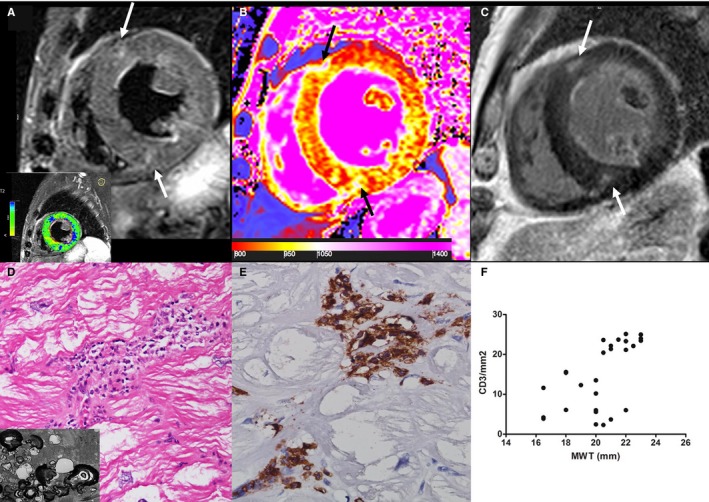
Group 3 patient. A 65‐year‐old man with Fabry disease cardiomyopathy and moderate cardiac hypertrophy (maximal wall thickness [MWT]: 19 mm). On a T2‐weighted short τ inversion recovery image (A), some mesocardial foci (arrows) of myocardial edema are detectable at anterior and inferior interventricular junctions, matching enhanced areas (arrows) on a late gadolinium–enhanced image (C); analysis of T2‐weighted myocardium‐over‐skeletal muscle signal intensity ratio reveals myocardial edema in the inferolateral wall (blue myocardium in the lower box is T2 ratio ≥2.0). B, A diffuse marked decrease of T1 value (septum: 834±57 ms) is represented by a predominant red color in myocardium with areas of increase signal (1093–1170 ms) of fibrous replacement at interventricular insertions (black arrows). D, At histology (hematoxylin and eosin, ×200), severely hypertrophied and vacuolated cardiomyocytes are surrounded by CD3+ inflammatory cells (D and E). Insert in D shows vacuoles consisting of glycolipid bodies. F, Correlation between MWT and CD3 count (P<0.001, ρ=0.62) in all group 3 patients (n=30).
Figure 5.
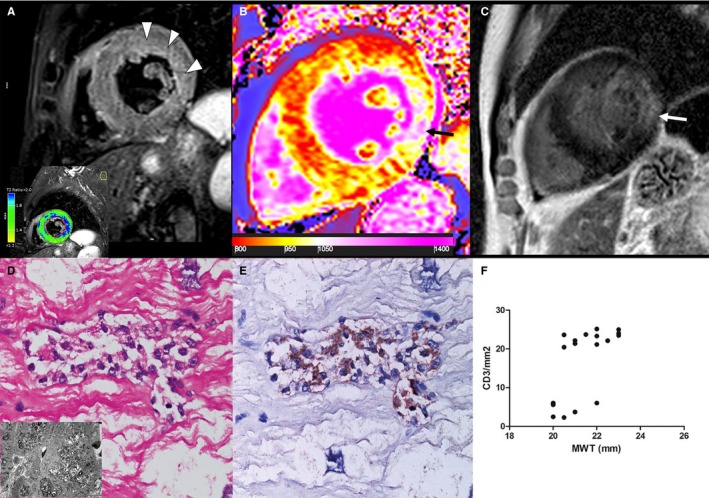
Group 4 patient. A 65‐year‐old man with Fabry disease cardiomyopathy (FDCM) and severe hypertrophy (maximal wall thickness [MWT]: 22 mm). A, T2‐weighted short τ inversion recovery shows a diffuse hyperintense signal of subendocardial midwall layers of lateral wall (arrowheads), as detected by the comparison with skeletal muscle signal (blue color, T2 ratio ≥2.0), reflecting myocardial edema. Native T1 map (B) and a late gadolinium–enhanced (LGE) image (C) demonstrate a severe decrease of myocardial T1 (septum: 867±42 ms), reflecting lipid accumulation and an LGE area at inferolateral wall (arrows), as a distinctive cardiac magnetic resonance hallmark of FDCM, corresponding to fibrous replacement (nT1: 1131±64 ms). D, Inflammatory infiltrate (hematoxylin and eosin, ×200), mainly of CD3+lymphocytes (E), was associated with necrosis of adjacent glycolipid‐laden myocytes. Insert in D shows vacuoles consisting of myelin bodies. F, Correlation between MWT and CD3 count (P<0.001, ρ=0.67) in all group 4 patients (n=18).
The analysis of T2w‐STIR images showed evidence of myocardial edema in 24 of 78 patients (31%) that increased with the degree of LV hypertrophy. No significant differences were found in the global myocardium‐to‐skeletal muscle T2 SI ratio values among 4 groups, ranging from 1.43 (group 1) to 1.78 (group 4).
In 87% (21/24) of patients, myocardial edema areas in T2w‐STIR images were evident in hypertrophic myocardium (myocardial segments with MWT >11 mm); 17 of 24 patients (70%) had patchy midwall distribution in the basal anteroseptal wall and 7 of 24 had involvement of the anterolateral wall. LGE was documented in 53 patients (68%). In 46 of 53 patients (87%), LGE was located in the inferolateral wall with subendocardial midwall distribution and was not associated with segments in which MWT was measured, which was mainly the basal septum. LGE increased with increasing severity by group and was detected in 23% of group 1 patients, 53% of group 2 patients, 77% of group 3 patients, and 100% of group 4 patients (P<0.05 versus group 1).
T1 measurements were available in only 34 of 78 patients (44%) because the sequence package became available at our institution in 2013. Both global and septal mean T1 values were higher in pre‐ and mild hypertrophic groups compared with the groups with moderate and advanced disease (P<0.05), reflecting the greater tissue accumulation of glycosphingolipids in patterns with MWT >14 mm.
Invasive Cardiac Studies
Cardiac catheterization showed an increased LV or biventricular end‐diastolic pressure (>12 mm Hg) in 7 of 13 patients in group 1 and in all patients in groups 2 through 4. LV end‐diastolic pressure was increasingly raised from group 1 to group 4. LV ejection fraction was reduced (<50%) in 6 patients in group 4. Coronary angiography showed epicardial coronary stenosis in 4 patients, requiring percutaneous transluminal coronary angioplasty and stenting in 3 and coronary artery bypass grafting in 1. Coronary flow was usually slow in patients in groups 3 and 4, in 4 patients in group 1, and in 7 patients in group 2 (TIMI frame count, mean±SD: 45.3±7.3 for left anterior descending artery, 43.6±8.2 for circumflex artery, and 42.2±8.7 for right coronary artery).
EMB was devoid of major complications. Two patients reported transient headache with visual disturbances, likely from brain microembolism following LV biopsy.
Histology and Electron Microscopy
In all patients, cardiomyocytes showed perinuclear and/or cytoplasmic vacuoles stained positively with periodic acid–Schiff and Sudan black and consisting, at electron microscopy, of myelin bodies (Figures 2, 3, 4 through 5, panel D).
Interstitial and replacement fibrosis was absent or mild in patients in groups 1 and 2, whereas it was pronounced in patients in groups 3 and 4. Inflammatory infiltrates consisting mainly of CD3+ T lymphocytes >7/mm2 focally associated with necrosis of adjacent glycolipid‐laden myocytes were observed in 38% of group 1, 41% of group 2, 66% of group 3, and 72% of group 4 (Figures 2, 3, 4 through 5, panel E). The number of CD3+ cells was not correlated with enzyme activity, whereas it was weakly correlated with age (P<0.05, Spearman ρ=0.3292) and mainly with the extent of LV MWT at CMR (P<0.001, Spearman ρ=0.6133) globally and in groups 2 through 4 separately (Figures 2, 3, 4 through 5, panel F). In group 4 patients, myocarditis was associated with focal inflammatory infiltration of small coronary vessels (mostly capillaries and venules; Figure 6).
Figure 6.
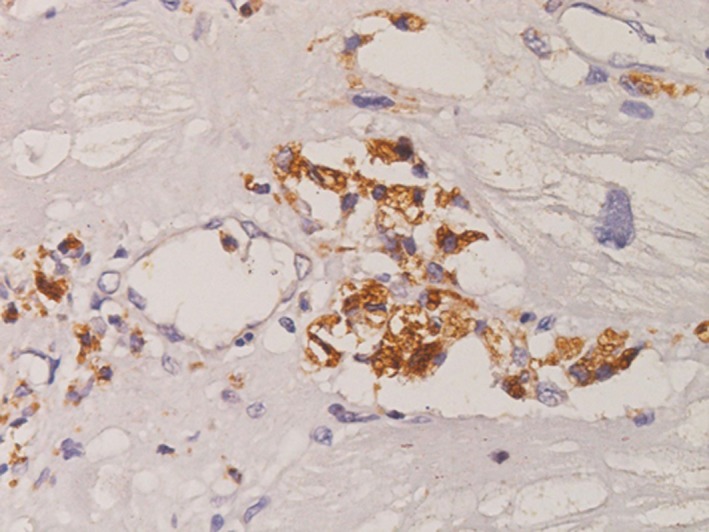
CD3+ T lymphocytes infiltrating myocardial capillaries in advanced (group 4) Fabry disease cardiomyopathy (×200 magnification, immunoperoxidase for T lymphocyte CD3 antigen).
Molecular Studies
No viral genome was detectable in real‐time polymerase chain reaction for any patients in groups 1 through 4.
Circulating Autoantibodies
Antiheart and antimyosin autoantibodies were detectable in all patients with myocarditis. Antiheart autoantibodies showed a partially organ‐specific (or cross‐reactive 1) pattern in all patients (increasing with severity; 38% of group 1, 41% of group 2, 67% of group 3, 72% of group 4), with a strongly positive fine striational pattern on human heart tissue and a weak positive fine striational pattern on human skeletal muscle19 (Figure 7). Antimyosin autoantibodies were documented in 38% of patients in prehypertrophic FDCM (group 1), suggesting an easy and sensitive marker for early assessment of myocarditis, particularly following the negative results of CMR. In patients with myocarditis, serum concentration of antimyosin autoantibodies correlated with the number of CD3+ cells/mm2 (P<0.001, Spearman ρ=0.8476).
Figure 7.
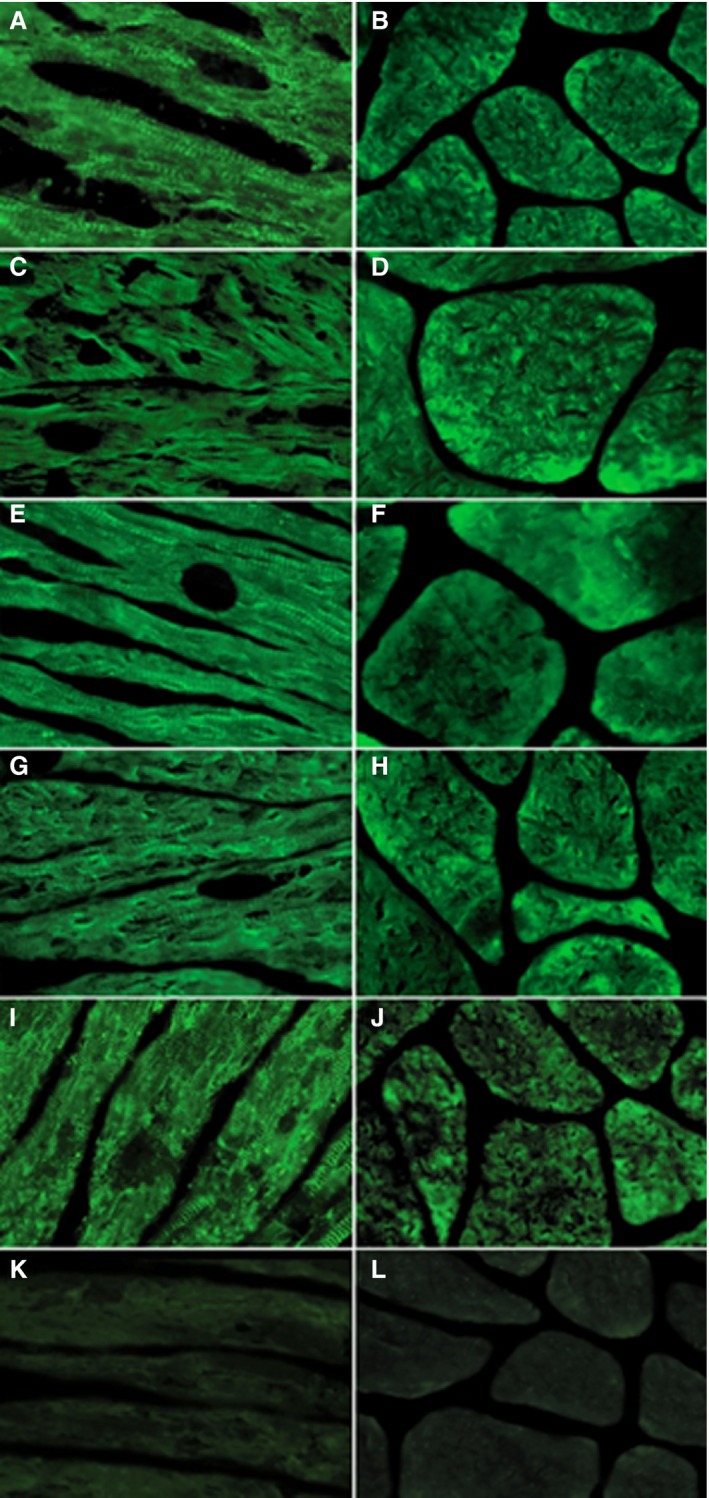
Antiheart autoantibodies in Fabry disease patients by indirect immunofluorescence. A, C, E and G, Strongly positive fine striational pattern of positivity on human heart tissue in sera from groups 1, 2, 3, and 4, respectively (×400). B, D, F and H, Weakly positive fine striational pattern on human skeletal muscle in sera from groups 1, 2, 3, and 4, respectively (×400), suggesting a partially organ‐specific (or cross‐reactive 1) pattern. I, Positive antiheart autoantibodies control serum on heart tissue (×400). J, Positive antiheart autoantibodies control serum on human skeletal muscle (×400). K, Negative antiheart autoantibodies control serum on human heart tissue (×400). L, Negative control serum on human skeletal muscle (×400).
Discussion
Accumulation of glycosphingolipids in Fabry cells is known to generate a proinflammatory response by the human immune system. In particular, self‐glycosphingolipids, released in the systemic circulation by a constitutional secretory pathway, are recognized as antigens and are exposed by the CD1d molecule on dendritic antigen‐presenting cells, which, under the action of TLR4 (Toll‐like receptor 4), activate the invariant natural killer T cells to a humoral (including probably anti–glycospingolipid antibodies) and cellular immune response.20 What is poorly known is the incidence of tissue inflammation in the affected organs and the impact it may have on disease progression and responsiveness to ERT. The heart is a major target of FD, and its involvement strongly influences life expectancy that was reduced, before ERT introduction, to 58.2 years for men and to 75.4 years for women.21 The impact of ERT on FDCM is still controversial because some studies show benefits from ERT administration in terms of relief,8, 9, 10 and others deny benefits, reporting an unaffected progressive trend.11, 12 Most experts agree on evidence showing that early ERT administration, particularly in prehypertrophic FDCM (MWT <11 mm), prevents disease progression, whereas the advanced form (MWT >15 mm) appears to be irreversible.12 The cause of FDCM resistance to ERT is unknown, although the expansion of myocardial interstitium and the advent of fibrosis are believed to play roles. In this regard, myocarditis may occur with interstitial widening through inflammatory cell infiltration, edema, and cell necrosis and may also be a source of myocardial fibrosis through the activation of Smad and TGF‐β1 (transforming growth factor β1). Interstitial expansion may then impair ERT pharmacokynetics, particularly through glycosphingolipids that may block and inactivate the infused enzyme with a basic pH, compromising its entrance into cardiomyocytes and lysosomal targeting.
Our study reports, for the first time, a high histological incidence (56%) of myocarditis in a large series of patients with FDCM undergoing EMB. The diagnosis followed the indications of Dallas criteria implemented by immunohistochemical characterization of inflammatory infiltrates.18 Notably, inflammatory cells were mostly CD3+ T lymphocytes >7/mm2, and their increasing number correlated with degree of MWT, severity of cell necrosis, and serum antimyosin antibody titer. Specifically, myocarditis was detected in 38% of FDCM patients in the prehypertrophic phase, 41% with MWT of 11 to 15 mm, 66% with MWT between 16 and 20 mm, and 72% with MWT >20 mm. This progressive inflammatory trend can be explained by the increasing amount of glycosphingolipids likely released by larger myocardiocytes activating a parallel immune response and proportional immune‐mediated myocardial damage. Release from damaged myocardiocytes of segregated antigens such as myosin may contribute to enhancement and perpetuation of myocardial inflammation. Indeed, in our study, myocarditis was associated with negative polymerase chain reaction for viral genomes and positive antiheart and antimyosin, suggesting an immune‐mediated mechanism of damage. Furthermore, the severity of myocarditis strictly correlated with the serum concentration of antimyosin antibodies, which appeared to be a sensitive marker revealing the early phase of inflammation. In this regard, the consistent incidence (38%) of myocarditis in prehypertrophic FDCM patients suggests that presenting symptoms like chest pain and cardiac arrhythmias, often associated with increased serum troponin I, can be caused by an immunological response to glycosphyngolipid exposition rather than to FD itself. Finally, the partially organ‐specific pattern of autoantibodies detected in our patients suggests that, in addition to cardiomyocyte antigens like myosin, other non–organ‐specific autoantigens, such as GB3, can be involved with a widespread autoimmune inflammatory reaction.
The presence of myocarditis in FDCM had a negative impact on the disease being associated with disturbing symptoms like chest pain and palpitations, occurrence of arrhythmias at ECG, and detrimental biological features such as elevation of troponin I and increased cell necrosis. At CMR, myocardial inflammation was detected as a focal increase of myocardial signal on T2w‐STIR images, reflecting tissue edema, in 31% of cases. Incidence of myocardial edema at T2w‐STIR imaging was low in patients with MWT ≤15 mm but appreciable, reaching 50%, in those with MWT >20 mm (ie, 0% in group 1, 29% in group 2, 43% in group 3, and 56% in group 4; P<0.05 versus group 1).
The limited sensitivity of T2‐weighted CMR imaging has been already reported in other clinical settings16 and explained by the low‐resolution power of the technique and the variable contribution of cell necrosis to the expansion of interstitial space.
In addition, it is conceivable that the inflammation mechanism in FDCM may also involve the skeletal muscle22; therefore, the resulting reduction of T2 myocardium‐to‐skeletal muscle SI ratio, which is commonly used to assess myocardial edema on CMR, may be an additional reason for underestimation. Conversely, the importance of CMR in detecting myocardial inflammation in FDCM has been raised recently.23 Our report correlating CMR findings with histology and immunological profile confirms this observation, particularly for patients with MWT >15 mm, and adds a new topic for recently published guidelines on hypertrophic cardiomyopathy.24
Study Limitations
A limitation of this research was the small sample size; however, given the rarity of the disease, the study benefited from the possibility of analyzing cardiac biopsies from these patients. We acknowledge that the differences in age among the 4 groups might affect some of the comparisons of clinical CMR and histopathological measures, independent of LV wall thickness. Nevertheless, it is inherent to the progressive nature of the disease that clinical and histological severity increases with age. A proper analysis that could account for age differences was not performed, given the small number of observations.
Perspectives
Clinicopathological evidence of myocarditis potentially contributing to disease progression and ERT resistance may change the therapeutic approach to FDCM. Immunosuppressive therapy (ie, a combination of prednisone with azathioprine17) or immunomodulating therapy (ie, immunoadsorption25) can be associated with ERT in those FDCM patients with advanced disease (groups 3 and 4) and CMR and/or histological evidence of myocarditis and refractory to the conventional management. The impact of immunosuppression can be relieved with improvement of major symptoms like chest pain, dyspnea, and cardiac arrhythmias and with the decrease or disappearance of serum antimyosin and antiheart antibodies.
CMR can be a valuable tool for evaluation of inflammatory response to immunosuppression. Reduction of LV mass and improvement in diastolic and systolic dysfunction associated with edema reabsorption would suggest efficacy of immunosuppression and a major pathogenetic role of myocarditis in FDCM.
Finally, inflammation is believed to affect other organs with FD, including kidney,26 vessels,27 and brain.28 Control of systemic inflammation in FD may result in the improvement of patient prognosis and quality of life.
Conclusions
Myocarditis is detectable at histology in up to 56% of patients with FDCM. It is immune mediated and correlates with disease severity. It can be disclosed by serum antiheart or antimyosin autoantibodies and, in the advanced stage, by CMR. It may contribute to disease progression and ERT resistance.
Sources of Funding
The study was supported by Shire International GmbH project number IIR‐ITA‐00169, “Myocardial Inflammation in Fabry Disease and the Mechanism of Therapeutical Responsiveness and/or Resistance to Enzyme Replacement Therapy” and Telethon Foundation project number GGP17055.
Disclosures
None.
(J Am Heart Assoc. 2018;7:e009052 DOI: 10.1161/JAHA.118.009052.)
References
- 1. Desnick RJ, Ioannou YA, Eng CM. Alpha‐galactosidase a deficiency: Fabry disease In: Scriver CR, Beaudet Al, Sly WS, Valle D, eds. The metabolic and molecular bases of inherited disease. New York, NY: McGraw‐Hall; 2001:3733–3774. [Google Scholar]
- 2. Desnick RJ. Fabry's disease (αGalactosidase A Deficiency): an X‐linked Nephropathy In: Lifton R, Somlo S, Giebisch, G, Seldin D, eds. Genetic Diseases of the Kidney. San Diego, CA: Elsevier Academic Press; 2009:597–616. [Google Scholar]
- 3. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:1–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thurberg BL, Fallon JT, Mitchell R, Aretz T, Gordon RE, O'Callaghan MW. Cardiac microvascular pathology in Fabry disease: evaluation of endomyocardial biopsies before and after enzyme replacement therapy. Circulation. 2009;119:2561–2567. [DOI] [PubMed] [Google Scholar]
- 5. Najafian B, Savarstad E, Bostad L, Gubler MC, Tondel C, Whitley C, Mauer M. Progressive podocyte injury and globotriasylceramide (GL‐3) accumulation in young patients with Fabry disease. Kidney Int. 2011;79:663–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chimenti C, Morgante E, Tanzilli G, Mangieri E, Critelli G, Russo MA, Frustaci A. Angina in Fabry disease reflects coronary small vessel disease. Circ Heart Fail. 2008;1:161–169. [DOI] [PubMed] [Google Scholar]
- 7. von Scheidt W, Eng CM, Fitzmaurice TF, Erdmann E, Hubner G, Olsen EG, Christomanou H, Kandolf R, Bishop DF, Desnick RJ. An atypical variant of Fabry's disease with manifestations confined to the myocardium. N Engl J Med. 1991;324:395–399. [DOI] [PubMed] [Google Scholar]
- 8. Shiffmann R, Murray GJ, Treco D, Daniel P, Sellos‐Moura M, Quirk JM, Zirzow GC, Borowski M, Loveday K, Anderson T, Gillespie F, Oliver KL, Jeffries NO, Doo E, Liang TJ, Kreps C, Gunter K, Frei K, Crutchfield K, Selden RF, Brady RO. Infusion of alphagalactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci USA. 2000;97:365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schiffmann R, Floeter MK, Dambrosia JM, Gupta S, Moore DF, Sharabi Y, Khurana RK, Brady RO. Enzyme replacement therapy improves peripheral nerve and sweat function in Fabry disease. Muscle Nerve. 2003;28:703–710. [DOI] [PubMed] [Google Scholar]
- 10. Tondel C, Bostad L, LarsenK K. Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol. 2013;24:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rombach SM, Smid BE, Bouwman MG, Linthorst GE, Dijkgraaf MG, Hollak CE. Longterm enzyme replacement therapy for Fabry disease: effectiveness on Kidney, heart and brain. Orphanet J Rare Dis. 2013;8:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weidemann F, Niemann M, Stork S, Breunig F, Beer M, Sommer C, Hermann S, Ertl G, Wanner C. Long‐term outcome of enzyme replacement therapy in advanced Fabry disease:evidence for disease progression towards serious complications. J Intern Med. 2013;274:331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pieroni M, Chimenti C, De Cobelli F, Morgante E, Gaudio C, Russo MA, Frustaci A. Fabry cardiomyopathy: echocardiographic detection of endomyocardial glycosphingolipids compartmentalization. J Am Coll Cardiol. 2006;47:1663–1671. [DOI] [PubMed] [Google Scholar]
- 14. Gibson CM, Cannon CP, Daley WL, Dodge JT, Alexander B, Marble SJ, McCabe CH, Raymond L, Fortin T, Poole WK ,Braunwald E; for the TIMI 4 Study Group . TIMI Frame Count: a quantitative method of assessing coronary artery flow. Circulation. 1996;93:879–888. [DOI] [PubMed] [Google Scholar]
- 15. Friedrich MG, Sechtem U, Schulz‐Menger J, Holmvang G, Alakija P, Cooper LT, White JA, Abdel‐Aty H, Gutberlet M, Prasad S, Aletras A, Laissy JP, Paterson I, Filipchuk NG, Kumar A, Pauschinger M, Liu P. Cardiovascular magnetic resonance in myocarditis: a JACC White Paper. J Am Coll Cardiol. 2009;53:1475–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu S, Han J, Nacif MS, Jones J, Kawel N, Kellman P, Sibley CT, Bluemke DA. Diffuse myocardial fibrosis evaluation using cardiac magnetic resonance T1 mapping: sample size considerations for clinical trials. J Cardiovasc Magn Reson. 2012;14:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moon JC, Messroghli DR, Kellman P, Piechnik SK, Robson MD, Ugander M, Gatehouse PD, Arai AE, Friedrich MG, Neubauer S, Schulz‐Menger J, Schelbert EB; Society for Cardiovascular Magnetic Resonance Imaging; Cardiovascular Magnetic Resonance Working Group of the European Society of Cardiology . Myocardial T1 mapping and extracellular volume quantification: a Society for Cardiovascular Magnetic Resonance (SCMR) and CMR Working Group of the European Society of Cardiology consensus statement. J Cardiovasc Magn Reson. 2013;15:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frustaci A, Chimenti C, Calabrese F, Pieroni M, Thiene G, Maseri A. Immunosuppressive therapy for active lymphocytic myocarditis: virological and immunologic profile of responders versus nonresponders. Circulation. 2003;107:857–863. [DOI] [PubMed] [Google Scholar]
- 19. Caforio AL, Goldman JH, Baig MK, Keeling PJ, Bottazzo GF, McKenna WJ. Organspecific cardiac autoantibodies in dilated cardiomyopathy—an update. Eur Heart J. 1995;16 suppl O:68–70. [DOI] [PubMed] [Google Scholar]
- 20. Mauhin W, Lidove O, Masat E, Mingozzi F, Mariampillai K, Ziza JM, Benveniste O. Innate and adaptive immune response in Fabry disease. J Inherit Metab Dis. 2015;22:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry registry. Genet Med. 2009;11:790–796. [DOI] [PubMed] [Google Scholar]
- 22. Chimenti C, Padua L, Pazzaglia C, Morgante E, Centurion C, Antuzzi D, Russo MA, Frustaci A. Cardiac and skeletal myopathy in Fabry disease: a clinicopathologic correlative study. Hum Pathol. 2012;43:1444–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nordin S, Kozor R, Bulluck H, Castelletti S, Rosmini S, Abdel‐Gadir A, Baig S, Mehta A, Hughes D, Moon JC. Cardiac Fabry disease with late gadolinium enhancement is a chronic inflammatory cardiomyopathy. J Am Coll Cardiol. 2016;68:1707–1708. [DOI] [PubMed] [Google Scholar]
- 24. Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2733–2779. [DOI] [PubMed] [Google Scholar]
- 25. Felix SB, Beug D, Dörr M. Immunoadsorption therapy in dilated cardiomyopathy. Expert Rev Cardiovasc Ther. 2015. Feb;13:145–152. [DOI] [PubMed] [Google Scholar]
- 26. Barbey F, Lidove O, Schwarting A. Fabry nephropathy: 5 years of enzyme replacement therapy‐ a short review. NDT Plus. 2008;1:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rombach SM, Twickler TB, Aerts JM, Linthorst GE, Wijburg FA, Hollak CE. Vasculopathy in patients with Fabry disease: current controversies and research directions. Mol Genet Metab. 2010;99:99–108. [DOI] [PubMed] [Google Scholar]
- 28. De Francesco PN, Mucci JM, Ceci R, Fossati CA, Rozenfeld PA. Fabry disease peripheral blood immune cells release inflammatory cytokines: role of globotriaosylceramide. Mol Genet Metab. 2013;109:93–99. [DOI] [PubMed] [Google Scholar]