

Abstract
Background
Soluble urokinase plasminogen activator receptor (suPAR) is a proinflammatory biomarker associated with immune activation and fibrinolysis inhibition. Plasminogen activator inhibitor (PAI‐1) is associated with excessive fibrin accumulation, thrombus formation, and atherosclerosis. The relationship between cross‐coronary suPAR and PAI‐1 production and endothelial dysfunction remains unknown.
Methods and Results
Seventy‐nine patients (age 53±10 years, 75% women) with angina and normal coronary arteries or mild coronary artery disease (<40% stenosis) on angiogram underwent acetylcholine assessment of epicardial endothelial dysfunction (mid–left anterior descending coronary artery diameter decrease >20% after acetylcholine) and mircovascular endothelial dysfunction (coronary blood flow change <50% after acetylcholine). Simultaneous left main and coronary sinus suPAR and PAI‐1 levels were measured in each patient before acetylcholine administration, and cross‐coronary suPAR and PAI‐1 production rates were calculated. Patients’ characteristics, except for age (51±10 versus 57±9, P=0.02), and resting coronary hemodynamics were not significantly different between patients with (26%) versus without (74%) epicardial endothelial dysfunction. Patients’ characteristics and resting coronary hemodynamics were not significantly different between those with (62%) and those without (38%) mircovascular endothelial dysfunction. Patients with mircovascular endothelial dysfunction demonstrated local coronary suPAR production versus suPAR extraction in patients with normal microvascular function (median 25.8 [interquartile range 121.6, −23.7] versus −12.7 [52.0, −74.8] ng/min, P=0.03). Patients with epicardial endothelial dysfunction had higher median coronary PAI‐1 production rates compared with those with normal epicardial endothelial function (1224.7 [12 940.7, −1915.4] versus −187.4 [4444.7, −4535.8] ng/min, P=0.03).
Conclusions
suPAR is released in coronary circulation of patients with mircovascular endothelial dysfunction and extracted in those with normal microvascular function. Cross‐coronary PAI‐1 release is higher in humans with epicardial endothelial dysfunction.
Keywords: coronary circulation, endothelial dysfunction, epicardial, microvascular dysfunction, plasminogen activator, soluble urokinase plasminogen activator receptor
Subject Categories: Coronary Artery Disease, Coronary Circulation, Biomarkers, Endothelium/Vascular Type/Nitric Oxide
Clinical Perspective
What Is New?
Humans with early atherosclerosis and endothelial dysfunction have higher local cross‐coronary soluble urokinase plasminogen activator receptor and plasminogen activator inhibitor‐1 production.
Increased local coronary soluble urokinase plasminogen activator receptor production in the coronary circulation of patients with microvascular endothelial dysfunction implicates immune and fibrinolytic pathways in the development of coronary endothelial dysfunction and increased cardiac events.
Increased local coronary production of plasminogen activator inhibitor in patients with epicardial endothelial dysfunction suggests that impaired or ineffective local fibrinolysis in response to possible subclinical coronary thrombosis occurs concomitantly with endothelial dysfunction in the development of coronary artery disease.
What Are the Clinical Implications?
Findings of this study may allow for identification of patients with impaired coronary vasomotion and increased risk of coronary artery disease progression who would benefit from initiation of early aggressive medical management.
Introduction
Coronary endothelial dysfunction is the earliest clinically detectable form of atherosclerosis,1, 2, 3, 4 ultimately associated with coronary plaque progression,5 presence of rupture‐prone vulnerable plaque,6 and increased risk of adverse cardiovascular events.7, 8, 9, 10 Endothelial dysfunction is associated with abnormal coronary vasoreactivity of epicardial vessels and/or intramyocardial microvasculature and is characterized by imbalance between vasodilator and vasoconstrictor responses to endothelial‐dependent vasodilating agents such as acetylcholine.11, 12, 13
Plasminogen activator inhibitor (PAI‐1), a potent inhibitor of fibrinolysis, is associated with excessive fibrin accumulation, thrombus formation, and atherosclerosis.14 Soluble urokinase plasminogen activator receptor (suPAR) is a proinflammatory biomarker associated with immune activation and fibrinolysis inhibition. Elevated systemic plasma suPAR and PAI‐1 levels have been associated with increased risk of major adverse cardiovascular outcomes in advanced coronary artery disease (CAD).15, 16 However, whether elevated suPAR and PAI‐1 levels are also associated with the early phase of atherosclerosis, as represented by endothelial dysfunction, remains unknown. To date, it also remains unclear whether suPAR is directly involved in the pathogenesis of atherosclerosis or is merely a biomarker of adverse risk. Therefore, we assessed the relationship between local coronary suPAR and PAI‐1 production in early atherosclerosis, measured by epicardial endothelial dysfunction (EED) and microvascular endothelial dysfunction (MED). We hypothesized that coronary endothelial dysfunction is associated with local production of PAI‐1 and suPAR in humans.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request. Data sharing is subject to limitations of the informed consent and Mayo Institutional Review Board approval.
Patient Population
Patients who underwent a clinically indicated coronary angiography for angina symptoms at our institution were screened for inclusion in this study. The study was approved by the Mayo Institutional Review Board, and written informed consent was obtained. The study population consisted of 79 consecutive patients with angina who were found to have angiographically normal coronary arteries or mild epicardial CAD (<40% stenosis). Those patients subsequently underwent coronary vasomotor testing during the coronary catheterization procedure described below. Exclusion criteria included acute coronary syndrome presentation, myocardial infarction, or cerebrovascular accident within the past 6 months, previous percutaneous coronary intervention, use of radiographic contrast agents within 12 hours before catheterization, advanced chronic kidney disease, cardiomyopathy (ejection fraction <45%), active malignancy, local or systemic infectious disease within 4 weeks before catheterization, inflammatory diseases, and pregnancy. All patients fasted for at least 8 hours and withheld all prescription medications that could affect coronary vasoreactivity for at least 48 hours before the study procedure.
Study Protocol
Systemic and Local Coronary Blood Sampling
Routine preprocedural laboratory values including complete blood count, electrolyte panel, fasting lipid profile, and systemic C‐reactive protein (CRP) were obtained per institutional protocol. Serum CRP concentrations were measured on a Hitachi 912 automated chemistry analyzer using a high‐sensitivity polystyrene particle–enhanced immunoturbidimetric assay from DiaSorin (Stillwater, MN). Immediately following right femoral access, simultaneous left main coronary artery and coronary sinus blood samples were obtained before endothelial function assessment and stored at −80°C until assay. Left main and coronary sinus suPAR and PAI‐1 levels were measured, and trans–left anterior descending coronary artery (LAD) biomarker gradients were calculated as described below. The suPAR concentrations were measured using Human suPAR ELISA Kit (MyBioSource, San Diego, CA; MBS2516189). PAI‐1 concentrations were measured using MILLIPLEX Human Adipokine Magnetic Bead Panel 1 (EMD Millipore Corporation, Burlington, MA; HADK1MAG‐61K).
Measurement of Local Coronary suPAR and PAI‐1 Production
The gradients of suPAR and PAI‐1 across the LAD circulation were calculated by subtracting the left main arterial concentration from the coronary sinus concentration. Net production of each biomarker in the LAD territory was then calculated, using a previously well‐validated method, as the gradient times the coronary blood flow (CBF).17, 18, 19, 20, 21 For measurement of CBF in the LAD, a 6F or 7F guiding catheter was placed in the left main coronary artery, and a Doppler guidewire (Flowire, Volcano Therapeutics Inc, Rancho Cordova, CA) was positioned in the midportion of the LAD for blood velocity measurements. Velocity signals were instantaneously obtained from the Doppler wire by an online fast Fourier transform, and average peak velocity was determined. Coronary artery diameter was measured by an independent investigator in the segment 5 mm distal to the tip of the Doppler wire offline with a quantitative coronary angiography program (Medis Corp, Leiden, the Netherlands) as previously described.22 CBF was calculated from the Doppler‐derived time velocity integral and vessel diameter, where CBF=π×(coronary artery diameter/2)2×(average peak velocity/2).23
Invasive Physiologic Assessment
Endothelium‐independent coronary flow reserve (CFR) in response to intracoronary adenosine administration and endothelium‐dependent coronary vasoreactivity in response to intracoronary acetylcholine infusion were performed. In brief, intracoronary bolus injections of incremental doses (18‐48 μg) of adenosine were administered until maximal hyperemia was achieved or the largest dose was given to evaluate CFR. CFR was calculated as the average hyperemic velocity postadenosine/average baseline velocity. Subsequently, to assess endothelium‐dependent vasoreactivity, acetylcholine at increasing concentrations (10−6, 10−5, and 10−4 mol/L) was selectively infused into the LAD for 3 minutes at each concentration.9, 24 Endothelium‐independent microvascular dysfunction was defined as CFR <2.5 at maximal hyperemia after adenosine infusion.9, 25, 26, 27 Coronary vasoreactivity was assessed as both epicardial endothelial function and endothelium‐dependent microvascular function in response to acetylcholine. EED was defined as coronary artery vasoconstriction,9, 24, 28, 29 with percentage change (∆) in coronary artery diameter (D) <−20%, in response to intracoronary acetylcholine infusion.30 MED was defined as <50% ∆CBF in response to acetylcholine infusion.24, 31, 32, 33, 34
Statistical Analysis
Continuous variables are described as mean±SD if normally distributed or median and interquartile range (IQR) if nonnormally distributed, and categorical variables as proportions. Student t test and chi‐squared test were used for comparison of means or proportions for continuous and categorical variables, respectively. Nonnormally distributed variables were compared using the Mann‐Whitney test and were log transformed as required. The associations between those variables were investigated using univariate and multivariate linear regression analyses after adjustment for traditional cardiovascular risk factors, including age, sex, race, diabetes mellitus, hypertension, dyslipidemia, and smoking. A 2‐sided P<0.05 was considered statistically significant. The statistical analyses were performed using SPSS 22 (IBM SPSS Statistics, Chicago, IL).
Results
Mean age was 53±10 years, 59 (75%) patients were women, 39 (49%) had hypertension, 49 (62%) had hyperlipidemia, 26 (33%) were current smokers, and 2 (3%) had diabetes mellitus. A majority, 71 (89.9%), of study patients were white, 2 (2.5%) were black, 1 (1.3%) was Hispanic, 1 (1.3%) was Native American, and 4 (5.1%) were other. Among all patients, 22 (28%) had endothelium‐independent microvascular dysfunction with CFR <2.5 after adenosine, 49 (62%) patients had MED with ∆CBF <50% after acetylcholine, and 22 (26%) patients had EED with %∆D <−20% after intracoronary acetylcholine infusion. Baseline demographic, laboratory, and clinical characteristics, as well as resting CBF, were not significantly different between patients with and those without MED (Table 1). Medication profiles were also similar between the 2 groups except for higher frequency of β‐blocker usage in patients with MED (84% versus 16%, P=0.01). Baseline demographic, laboratory, and clinical characteristics were not different, except age (57±9 versus 51±10 years, P=0.02), which was higher in patients with than in those without EED (Table 1). Resting CBF was not significantly different between the 2 groups. Similarly, medication profiles were similar in both groups of patients with and without EED. Median local coronary suPAR production rate was 14.9 (IQR 86.9; −38.0) ng/min, and median PAI‐1 production rate was 126.3 (IQR 5012.8; −2518.7) ng/min.
Table 1.
Demographic, Laboratory, and Clinical Characteristics of Study Patients Stratified by Presence or Absence of Microvascular Endothelial Dysfunction and by Presence or Absence of Epicardial Endothelial Dysfunction
| All Patients (n=79) | Normal Microvascular Endothelial Function 30 (38%) | Microvascular Endothelial Dysfunction 49 (62%) | Normal Epicardial Endothelial Function 58 (74%) | Epicardial Endothelial Dysfunction 20 (26%) | |
|---|---|---|---|---|---|
| Age, y | 53±10 | 51±10 | 54±11 | 51±10 | 57±9a |
| Sex (women) | 59 (75) | 23 (39) | 36 (61) | 44 (76) | 14 (24) |
| Body mass index, kg/m2 | 30±7 | 30±7 | 31±7 | 31±7 | 29±5 |
| Baseline mean arterial pressure, mm Hg | 99±14 | 100±10 | 99±16 | 101±13 | 95±15 |
| Baseline heart rate, beats per minute | 73±17 | 71±14 | 74±19 | 75±17 | 67±15 |
| Total cholesterol, mg/dL | 189±45 | 198±45 | 184±45 | 186±45 | 198±46 |
| Triglycerides, mg/dL | 130±79 | 151±102 | 117±59 | 135±82 | 117±73 |
| High‐density lipoprotein, mg/dL | 60±19 | 58±19 | 61±19 | 57±18 | 67±21 |
| Low‐density lipoprotein, mg/dL | 104±39 | 112±36 | 100±40 | 103±37 | 109±44 |
| Serum CRP, mg/L | 3.96±5.58 | 4.40±7.43 | 3.68±4.02 | 4.12±6.01 | 3.36±4.51 |
| Diabetes mellitus | 2 (3) | 1 (50) | 1 (50) | 2 (100) | 0 |
| Hypertension | 39 (50) | 15 (38) | 24 (62) | 29 (74) | 9 (26) |
| Hyperlipidemia | 49 (64) | 20 (41) | 29 (59) | 36 (73) | 13 (27) |
| Tobacco smoking | 26 (35) | 10 (38) | 16 (62) | 17 (65) | 9 (35) |
Numbers are mean±SD or proportions with n (%). CRP indicates C‐reactive protein.
P<0.05 vs normal epicardial endothelial function.
Patients with MED demonstrated a median local coronary suPAR production of 25.8 (IQR 121.6, −23.7) ng/min versus local suPAR extraction, observed in patients with normal microvascular function, of −12.7 (IQR 52.0, −74.8) ng/min; P=0.03 (Figure 1). Moreover, as compared with all other patients, patients with isolated MED also had significantly higher local median coronary suPAR production (30.9 [122.2, −16.2] versus −9.6 [48.7, −74.8] ng/min; P=0.026). There was no significant difference between arterial systemic suPAR or coronary sinus suPAR levels between patients with versus those without MED (Table 2). Furthermore, there was no significant difference in median systemic CRP level (P=0.11) or local coronary PAI production (P=0.93) between patients with and those without MED. We did not observe a significant univariate linear relationship between local coronary suPAR production and %∆CBF (r=0.12, P=0.28), nor was suPAR an independent predictor of %∆CFB in a multivariate regression analysis after adjusting for age, sex, hypertension, diabetes melltus, hyperlipidemia, body mass index, and smoking status.
Figure 1.
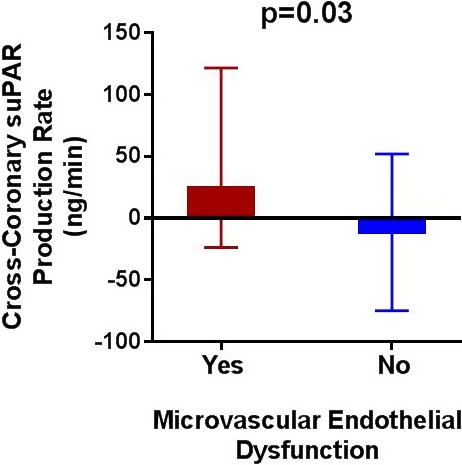
Cross‐coronary suPAR production rate in patients with vs those without microvascular endothelial dysfunction. Bars represent medians and interquartile ranges. suPAR indicates soluble urokinase plasminogen activator receptor.
Table 2.
suPAR and PAI‐1 Left Main and Coronary Sinus Concentrations as Well as Local Coronary Production Rates
| All Patients (n=79) | Normal Microvascular Endothelial Function 30 (38%) | Microvascular Endothelial Dysfunction 49 (62%) | Normal Epicardial Endothelial Function 58 (74%) | Epicardial Endothelial Dysfunction 20 (26%) | |
|---|---|---|---|---|---|
| Left main suPAR concentration, ng/mL | 9.7 (7.7, 11.1) | 9.6 (7.1, 11.2) | 9.7 (7.7, 11.3) | 9.7 (7.6, 11.5) | 9.9 (7.9, 10.6) |
| Coronary sinus suPAR concentration, ng/mL | 9.9 (8.0, 12.1) | 9.0 (7.7, 11.0) | 10.2 (8.2, 12.7) | 9.6 (7.9, 12.3) | 10.0 (8.3, 11.2) |
| Local LAD suPAR production rate, ng/min | 14.9 (86.9, −38.0) | −12.7 (52.0, −74.8) | 25.8 (121.6, −23.7)a | 12.8 (87.9, −50.2) | 11.6 (100.1, −36.3) |
| Left main PAI‐1 concentration, ng/mL | 166.1 (85.9, 281.5) | 125.0 (77.6, 214.2) | 198.0 (93.9, 300.3) | 166.1 (85.9, 275.3) | 156.3 (85.2, 302.2) |
| Coronary sinus PAI‐1 concentration, ng/mL | 167.3 (84.0, 337.0) | 154.3 (52.0, 369.2) | 168.4 (100.8, 321.3) | 154.3 (69.0, 280.3) | 169.0 (103.7, 393.1) |
| Local LAD PAI‐1 production rate, ng/min | 126.3 (5012.8, −2518.7) | 118.4 (4152.5, −1767.3) | 222.1 (6253.1, −3912.8) | −187.4 (4444.7, −4535.8) | 1224.7 (12 940.7, −1915.4)b |
Numbers are medians (IQR). IQR indicates interquartile range; LAD, left anterior descending coronary artery; PAI‐1, plasminogen activator receptor‐1; suPAR, soluble urokinase plasminogen activator receptor.
P<0.05 vs normal microvascular endothelial function.
P<0.05 vs normal epicardial endothelial function.
Patients with EED had significantly higher median local coronary PAI‐1 production as compared with local coronary PAI‐1 extraction in those patients with normal epicardial endothelial function (1224.7 [IQR 12 940.7; −1915.4] versus −187.4 [IQR 4444.7; −4535.8] ng/min, P=0.03) (Figure 2). There was no significant difference in arterial systemic PAI‐1 or coronary sinus PAI‐1 levels between patients with and those without EED (Table 2). Similarly, there was no significant difference in median systemic CRP level (P=0.79) or local coronary suPAR production (P=0.80) between patients with and those patients without EED. There was no significant linear relationship between PAI‐1 production rate and %∆D. Age (P=0.01) and current smoking (P=0.02) were the only independent predictors of EED when age, sex, hypertension, diabetes mellitus, hyperlipidemia, body mass index, smoking, and PAI‐1 production rate were accounted for.
Figure 2.
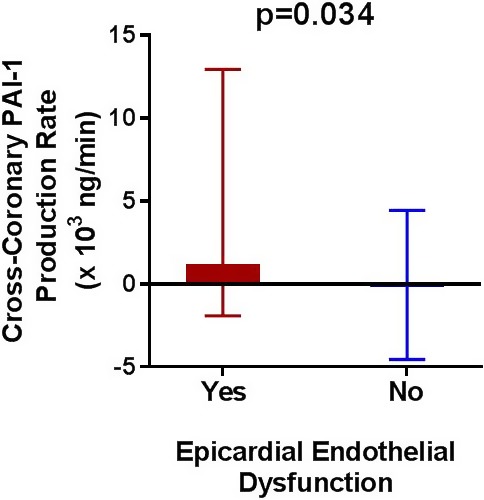
Cross‐coronary PAI‐1 production rate in patients with vs those without epicardial endothelial dysfunction. Bars represent medians and interquartile ranges. PAI‐1 indicates plasminogen activator receptor‐1.
Finally, median local coronary suPAR production (P=0.80), median local coronary PAI‐1 production (P=0.79), and median systemic CRP level (P=0.58) were not significantly different between patients with (22 [28%]) and those patients without (57 [72%]) endothelium‐independent microvascular dysfunction.
Discussion
This study shows that humans with early atherosclerosis and endothelial dysfunction have higher local cross‐coronary suPAR and PAI‐1 production. Increased local coronary suPAR production in the coronary circulation of patients with MED implicates immune and fibrinolytic pathways in the development of coronary endothelial dysfunction and increased cardiac events. Increased local coronary production of PAI‐1 in patients with EED suggests that impaired or ineffective local fibrinolysis in response to possible subclinical coronary thrombosis occurs concomitantly with endothelial dysfunction in the development of CAD.
Local Coronary suPAR Production and Endothelial Dysfunction
The study shows, for the first time, that suPAR is locally released in the coronary circulation of patients with MED and extracted from the coronary arteries of those with normal microvascular function. The association between elevated suPAR levels and non–endothelium‐dependent microvascular dysfunction has been previously reported.35 However, this is the first study to link local coronary suPAR production to endothelial dysfunction at the microvascular level and to suggest that cross‐coronary suPAR production may be a useful biomarker of early atherosclerosis and MED.
suPAR has been extensively studied as a biomarker of inflammation and immune activation. Coronary microvascular dysfunction9, 36, 37 and elevated levels of systemic suPAR15, 16, 38 have both been associated with increased major adverse coronary outcomes. Elevated levels of suPAR have also been observed in advanced atherosclerotic disease in both the carotid39, 40 and renal arteries,41 which in turn has been associated with major adverse coronary outcomes. This study links endothelial dysfunction, and therefore early atherosclerosis, with cross‐coronary suPAR production and potentially with a higher risk of adverse cardiac events.
At the molecular level, Mustjoki et al demonstrated that both unstimulated blood mononuclear and endothelial cells can release suPAR. However, suPAR release is markedly enhanced when either mononuclear cells or thrombocytes were cultured with endothelial cells. Importantly, thrombocytes cultured in the absence of endothelium demonstrated no suPAR release, whereas coculture of the above‐mentioned cell types without cell‐cell contacts failed to enhance suPAR release, both suggestive of an important role for the endothelial vascular cell layer in suPAR release.42 Other molecular studies have shown that only 10% to 20% of activated urokinase plasminogen activator receptor is secreted as suPAR from the endothelial apical surface into the blood stream while the remaining activated urokinase plasminogen activator receptor on the basolateral side of the endothelial cells is secreted into the vessel wall where activated urokinase plasminogen activator/urokinase plasminogen activator receptor system is associated with macrophage foam cell formation, extracellular matrix degradation, and smooth muscle cell migration, all of which are associated with atherosclerotic plaque progression.43
Local coronary suPAR production in patients with MED and its extraction in those with normal microvascular function demonstrate that suPAR could possibly be a novel biomarker of early atherosclerosis and implicates inflammatory, immune, and fibrinolytic pathways in the development of coronary endothelial dysfunction and increased cardiac events.
The findings of this study are in line with our prior data linking local cross‐coronary lipoprotein‐associated phospholipase A2 production, a marker of inflammation, and increased oxidative stress secondary to altered anti‐inflammatory function of the endothelium with early atherosclerosis in humans.19, 20 Taken all together, these data provide another layer of evidence demonstrating the cardinal role of inflammation in the development of endothelial dysfunction and highlighting the potential important role of new anti‐inflammatory therapy, such as canakinumab,44 in early coronary artery disease.
Local Coronary PAI‐1 Production and Endothelial Dysfunction
This study shows that patients with EED and no obstructive coronary artery disease have higher local coronary PAI‐1 production as compared with those with normal epicardial endothelial dysfunction. PAI‐1, the main inhibitor of plasminogen activators, is a major inhibitor of fibrinolysis. It is produced and released from various cell types including adipocytes, platelets, macrophages, smooth muscle cells, and endothelial cells.45
Inhibition of fibrinolysis due to elevated PAI‐1 levels has been associated with atherosclerotic plaque development, accelerated coronary atherosclerosis, thrombus formation, and an increased risk of recurrent myocardial infarctions.14, 41, 46, 47, 48, 49 Moreover, elevated levels of PAI‐1 have been documented in metabolic syndrome,50 diabetes mellitus,51 and obesity, all of which have been closely associated with endothelial dysfunction.
PAI‐1 is not only a marker of ineffective fibrinolysis but is also a marker of abnormal vascular health. Adly et al52 showed that children and adolescents with diabetes mellitus type 1 who had microvascular complications including diabetic nephropathy, retinopathy, and neuropathy had significantly higher systemic PAI‐1 levels. They also showed a significant positive linear relationship between PAI‐1 level and carotid intima media thickness, a surrogate of early atherosclerosis. Results of our study are in line with those findings where higher local coronary PAI‐1 production is associated with epicardial endothelial dysfunction and possibly increased subclinical coronary thrombosis.
Indeed, the differential association of studied biomarkers with MED (for suPAR) and EED (for PAI‐1) may possibly be related to either different mechanisms underlying endothelial cell injury and/or different subsequent responses to endothelial cell injury at the epicardial and microvascular levels. Based on our data, 1 hypothesis would be that endothelial dysfunction at the microvascular level may be driven more by vascular inflammation and accumulation of inflammatory cells and/or fibrin deposits in the microcirculation, as reflected by suPAR production in patients with MED versus coronary suPAR extraction in those patients with normal microvascular function. A second hypothesis would be that significantly higher coronary PAI‐1 production rate in patients with EED may not be directly related to the mechanism of endothelial cell injury at the epicardial level but rather be a reflection of increased subclinical thrombus formation associated with epicardial endothelial injury in the coronary circulation of “vulnerable patients,” those with a thrombotic vascular milieu. A third hypothesis would be that PAI‐1 may contribute to thrombotic events in the epicardial vessels during acute coronary syndromes. Therefore, basic mechanistic studies should be undertaken to further elucidate the cause versus effect role of these novel biomarkers, given such a role's significant implications for therapeutic and/or diagnostic value of these biomarkers in the future.
Study Limitations
Several limitations pertaining to this study should be noted. First, this is a small cross‐sectional pilot study that only demonstrates an association between local coronary suPAR and PAI‐1 biomarkers’ production and coronary endothelial dysfunction at the epicardial and microvascular levels. The detailed molecular mechanisms and possible causality among the local production of those biomarkers, immune and inflammatory pathways, early atherosclerosis, and coronary endothelial dysfunction all warrant further investigation. Second, a significantly higher number of patients with MED were on β‐blockers as compared with those without MED. However, the fact that all patients were off vasoactive medications for 48 hours before the procedure negates any meaningful effect of this difference between the 2 groups. Finally, we do not have longitudinal follow‐up data on our study patients to correlate cross‐coronary biomarker production and endothelial dysfunction with hard cardiovascular outcomes. Prospective outcome studies are warranted to further evaluate this association.
Conclusion
The current study supports a potential role for suPAR and PAI‐1 as biomarkers and potential mechanisms of endothelial dysfunction and reflects on the possible mechanisms associated with its development in humans. This study shows for the first time that humans with early atherosclerosis have higher local cross‐coronary suPAR and PAI‐1 production. Increased local coronary suPAR production in the coronary circulation of patients with MED implicates immune and fibrinolytic pathways in the development of coronary endothelial dysfunction and an increased number of cardiac events. Increased local coronary production of PAI‐1 in patients with EED suggests that impaired or ineffective local fibrinolysis occurs concomitantly with endothelial dysfunction in the development of CAD. Those findings could potentially have clinical implications, allowing for identification of patients with impaired coronary vasomotion and increased risk of CAD progression, who would benefit from initiation of early aggressive medical management. Furthermore, larger prospective clinical trials are warranted to investigate the magnitude of change in coronary suPAR and PAI‐1 production rates, in conjunction with coronary endothelial function improvement, in response to lifestyle modifications such as weight loss, optimal traditional medical therapy, and novel pharmacological agents in patients with early CAD.
Disclosures
None.
(J Am Heart Assoc. 2018;7:e009881 DOI: 10.1161/JAHA.118.009881.)
References
- 1. Gimbrone MA Jr, Garcia‐Cardena G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc Pathol. 2013;22:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gimbrone MA Jr, Garcia‐Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stary HC. Natural history and histological classification of atherosclerotic lesions: an update. Arterioscler Thromb Vasc Biol. 2000;20:1177–1178. [DOI] [PubMed] [Google Scholar]
- 4. Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. [DOI] [PubMed] [Google Scholar]
- 5. Yoon MH, Reriani M, Mario G, Rihal C, Gulati R, Lennon R, Tilford JM, Lerman LO, Lerman A. Long‐term endothelin receptor antagonism attenuates coronary plaque progression in patients with early atherosclerosis. Int J Cardiol. 2013;168:1316–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lavi S, Bae JH, Rihal CS, Prasad A, Barsness GW, Lennon RJ, Holmes DR Jr, Lerman A. Segmental coronary endothelial dysfunction in patients with minimal atherosclerosis is associated with necrotic core plaques. Heart. 2009;95:1525–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lerman A, Zeiher AM. Endothelial function: cardiac events. Circulation. 2005;111:363–368. [DOI] [PubMed] [Google Scholar]
- 8. Reriani MK, Flammer AJ, Jama A, Lerman LO, Lerman A. Novel functional risk factors for the prediction of cardiovascular events in vulnerable patients following acute coronary syndrome. Circ J. 2012;76:778–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suwaidi JA, Hamasaki S, Higano ST, Nishimura RA, Holmes DR Jr, Lerman A. Long‐term follow‐up of patients with mild coronary artery disease and endothelial dysfunction. Circulation. 2000;101:948–954. [DOI] [PubMed] [Google Scholar]
- 10. Targonski PV, Bonetti PO, Pumper GM, Higano ST, Holmes DR Jr, Lerman A. Coronary endothelial dysfunction is associated with an increased risk of cerebrovascular events. Circulation. 2003;107:2805–2809. [DOI] [PubMed] [Google Scholar]
- 11. Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003;23:168–175. [DOI] [PubMed] [Google Scholar]
- 12. Reddy KG, Nair RN, Sheehan HM, Hodgson JM. Evidence that selective endothelial dysfunction may occur in the absence of angiographic or ultrasound atherosclerosis in patients with risk factors for atherosclerosis. J Am Coll Cardiol. 1994;23:833–843. [DOI] [PubMed] [Google Scholar]
- 13. Vita JA, Treasure CB, Nabel EG, McLenachan JM, Fish RD, Yeung AC, Vekshtein VI, Selwyn AP, Ganz P. Coronary vasomotor response to acetylcholine relates to risk factors for coronary artery disease. Circulation. 1990;81:491–497. [DOI] [PubMed] [Google Scholar]
- 14. Schafer K, Muller K, Hecke A, Mounier E, Goebel J, Loskutoff DJ, Konstantinides S. Enhanced thrombosis in atherosclerosis‐prone mice is associated with increased arterial expression of plasminogen activator inhibitor‐1. Arterioscler Thromb Vasc Biol. 2003;23:2097–2103. [DOI] [PubMed] [Google Scholar]
- 15. Eapen DJ, Manocha P, Ghasemzadeh N, Patel RS, Al Kassem H, Hammadah M, Veledar E, Le NA, Pielak T, Thorball CW, Velegraki A, Kremastinos DT, Lerakis S, Sperling L, Quyyumi AA. Soluble urokinase plasminogen activator receptor level is an independent predictor of the presence and severity of coronary artery disease and of future adverse events. J Am Heart Assoc. 2014;3:e001118 DOI: 10.1161/JAHA.114.001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lyngbaek S, Andersson C, Marott JL, Moller DV, Christiansen M, Iversen KK, Clemmensen P, Eugen‐Olsen J, Hansen PR, Jeppesen JL. Soluble urokinase plasminogen activator receptor for risk prediction in patients admitted with acute chest pain. Clin Chem. 2013;59:1621–1629. [DOI] [PubMed] [Google Scholar]
- 17. Gossl M, Modder UI, Gulati R, Rihal CS, Prasad A, Loeffler D, Lerman LO, Khosla S, Lerman A. Coronary endothelial dysfunction in humans is associated with coronary retention of osteogenic endothelial progenitor cells. Eur Heart J. 2010;31:2909–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kalra PR, Clague JR, Bolger AP, Anker SD, Poole‐Wilson PA, Struthers AD, Coats AJ. Myocardial production of C‐type natriuretic peptide in chronic heart failure. Circulation. 2003;107:571–573. [DOI] [PubMed] [Google Scholar]
- 19. Lavi S, McConnell JP, Rihal CS, Prasad A, Mathew V, Lerman LO, Lerman A. Local production of lipoprotein‐associated phospholipase A2 and lysophosphatidylcholine in the coronary circulation: association with early coronary atherosclerosis and endothelial dysfunction in humans. Circulation. 2007;115:2715–2721. [DOI] [PubMed] [Google Scholar]
- 20. Lavi S, Yang EH, Prasad A, Mathew V, Barsness GW, Rihal CS, Lerman LO, Lerman A. The interaction between coronary endothelial dysfunction, local oxidative stress, and endogenous nitric oxide in humans. Hypertension. 2008;51:127–133. [DOI] [PubMed] [Google Scholar]
- 21. Traverse JH, Nesmelov YE, Crampton M, Lindstrom P, Thomas DD, Bache RJ. Measurement of myocardial free radical production during exercise using EPR spectroscopy. Am J Physiol Heart Circ Physiol. 2006;290:H2453–H2458. [DOI] [PubMed] [Google Scholar]
- 22. Al Suwaidi J, Higano ST, Holmes DR Jr, Rihal CS, Lerman A. Measuring maximal percent area stenosis poststent placement with intracoronary Doppler and the continuity equation and correlation with intracoronary ultrasound and angiography. Am J Cardiol. 1999;84:650–654. [DOI] [PubMed] [Google Scholar]
- 23. Doucette JW, Corl PD, Payne HM, Flynn AE, Goto M, Nassi M, Segal J. Validation of a Doppler guide wire for intravascular measurement of coronary artery flow velocity. Circulation. 1992;85:1899–1911. [DOI] [PubMed] [Google Scholar]
- 24. Hasdai D, Gibbons RJ, Holmes DR Jr, Higano ST, Lerman A. Coronary endothelial dysfunction in humans is associated with myocardial perfusion defects. Circulation. 1997;96:3390–3395. [DOI] [PubMed] [Google Scholar]
- 25. Geltman EM, Henes CG, Senneff MJ, Sobel BE, Bergmann SR. Increased myocardial perfusion at rest and diminished perfusion reserve in patients with angina and angiographically normal coronary arteries. J Am Coll Cardiol. 1990;16:586–595. [DOI] [PubMed] [Google Scholar]
- 26. Marinescu MA, Loffler AI, Ouellette M, Smith L, Kramer CM, Bourque JM. Coronary microvascular dysfunction, microvascular angina, and treatment strategies. JACC Cardiovasc Imaging. 2015;8:210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Serruys PW, di Mario C, Piek J, Schroeder E, Vrints C, Probst P, de Bruyne B, Hanet C, Fleck E, Haude M, Verna E, Voudris V, Geschwind H, Emanuelsson H, Muhlberger V, Danzi G, Peels HO, Ford AJ Jr, Boersma E. Prognostic value of intracoronary flow velocity and diameter stenosis in assessing the short‐ and long‐term outcomes of coronary balloon angioplasty: the DEBATE Study (Doppler Endpoints Balloon Angioplasty Trial Europe). Circulation. 1997;96:3369–3377. [DOI] [PubMed] [Google Scholar]
- 28. Hasdai D, Cannan CR, Mathew V, Holmes DR Jr, Lerman A. Evaluation of patients with minimally obstructive coronary artery disease and angina. Int J Cardiol. 1996;53:203–208. [DOI] [PubMed] [Google Scholar]
- 29. Hasdai D, Holmes DR Jr, Higano ST, Burnett JC Jr, Lerman A. Prevalence of coronary blood flow reserve abnormalities among patients with nonobstructive coronary artery disease and chest pain. Mayo Clin Proc. 1998;73:1133–1140. [DOI] [PubMed] [Google Scholar]
- 30. Ludmer PL, Selwyn AP, Shook TL, Wayne RR, Mudge GH, Alexander RW, Ganz P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315:1046–1051. [DOI] [PubMed] [Google Scholar]
- 31. Drexler H, Zeiher AM, Meinzer K, Just H. Correction of endothelial dysfunction in coronary microcirculation of hypercholesterolaemic patients by L‐arginine. Lancet. 1991;338:1546–1550. [DOI] [PubMed] [Google Scholar]
- 32. Egashira K, Inou T, Hirooka Y, Yamada A, Maruoka Y, Kai H, Sugimachi M, Suzuki S, Takeshita A. Impaired coronary blood flow response to acetylcholine in patients with coronary risk factors and proximal atherosclerotic lesions. J Clin Invest. 1993;91:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Egashira K, Inou T, Hirooka Y, Yamada A, Urabe Y, Takeshita A. Evidence of impaired endothelium‐dependent coronary vasodilatation in patients with angina pectoris and normal coronary angiograms. N Engl J Med. 1993;328:1659–1664. [DOI] [PubMed] [Google Scholar]
- 34. Quyyumi AA, Cannon RO III, Panza JA, Diodati JG, Epstein SE. Endothelial dysfunction in patients with chest pain and normal coronary arteries. Circulation. 1992;86:1864–1871. [DOI] [PubMed] [Google Scholar]
- 35. Mekonnen G, Corban MT, Hung OY, Eshtehardi P, Eapen DJ, Al‐Kassem H, Rasoul‐Arzrumly E, Gogas BD, McDaniel MC, Pielak T, Thorball CW, Sperling L, Quyyumi AA, Samady H. Plasma soluble urokinase‐type plasminogen activator receptor level is independently associated with coronary microvascular function in patients with non‐obstructive coronary artery disease. Atherosclerosis. 2015;239:55–60. [DOI] [PubMed] [Google Scholar]
- 36. Gulati M, Cooper‐DeHoff RM, McClure C, Johnson BD, Shaw LJ, Handberg EM, Zineh I, Kelsey SF, Arnsdorf MF, Black HR, Pepine CJ, Merz CN. Adverse cardiovascular outcomes in women with nonobstructive coronary artery disease: a report from the Women's Ischemia Syndrome Evaluation Study and the St James Women Take Heart Project. Arch Intern Med. 2009;169:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnson BD, Shaw LJ, Buchthal SD, Bairey Merz CN, Kim HW, Scott KN, Doyle M, Olson MB, Pepine CJ, den Hollander J, Sharaf B, Rogers WJ, Mankad S, Forder JR, Kelsey SF, Pohost GM. Prognosis in women with myocardial ischemia in the absence of obstructive coronary disease: results from the National Institutes of Health‐National Heart, Lung, and Blood Institute‐Sponsored Women's Ischemia Syndrome Evaluation (WISE). Circulation. 2004;109:2993–2999. [DOI] [PubMed] [Google Scholar]
- 38. Persson M, Ostling G, Smith G, Hamrefors V, Melander O, Hedblad B, Engstrom G. Soluble urokinase plasminogen activator receptor: a risk factor for carotid plaque, stroke, and coronary artery disease. Stroke. 2014;45:18–23. [DOI] [PubMed] [Google Scholar]
- 39. Pawlak K, Mysliwiec M, Pawlak D. The urokinase‐type plasminogen activator/its soluble receptor system is independently related to carotid atherosclerosis and associated with CC‐chemokines in uraemic patients. Thromb Res. 2008;122:328–335. [DOI] [PubMed] [Google Scholar]
- 40. Pawlak K, Pawlak D, Mysliwiec M. Urokinase‐type plasminogen activator and metalloproteinase‐2 are independently related to the carotid atherosclerosis in haemodialysis patients. Thromb Res. 2008;121:543–548. [DOI] [PubMed] [Google Scholar]
- 41. Park MY, Herrmann SM, Saad A, Eirin A, Tang H, Lerman A, Textor SC, Lerman LO. Biomarkers of kidney injury and klotho in patients with atherosclerotic renovascular disease. Clin J Am Soc Nephrol. 2015;10:443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mustjoki S, Sidenius N, Vaheri A. Enhanced release of soluble urokinase receptor by endothelial cells in contact with peripheral blood cells. FEBS Lett. 2000;486:237–242. [DOI] [PubMed] [Google Scholar]
- 43. Fuhrman B. The urokinase system in the pathogenesis of atherosclerosis. Atherosclerosis. 2012;222:8–14. [DOI] [PubMed] [Google Scholar]
- 44. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida‐Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 45. Tsantes AE, Nikolopoulos GK, Bagos PG, Bonovas S, Kopterides P, Vaiopoulos G. The effect of the plasminogen activator inhibitor‐1 4G/5G polymorphism on the thrombotic risk. Thromb Res. 2008;122:736–742. [DOI] [PubMed] [Google Scholar]
- 46. Hamsten A, de Faire U, Walldius G, Dahlen G, Szamosi A, Landou C, Blomback M, Wiman B. Plasminogen activator inhibitor in plasma: risk factor for recurrent myocardial infarction. Lancet. 1987;2:3–9. [DOI] [PubMed] [Google Scholar]
- 47. Hamsten A, Wiman B, de Faire U, Blomback M. Increased plasma levels of a rapid inhibitor of tissue plasminogen activator in young survivors of myocardial infarction. N Engl J Med. 1985;313:1557–1563. [DOI] [PubMed] [Google Scholar]
- 48. Schneiderman J, Sawdey MS, Keeton MR, Bordin GM, Bernstein EF, Dilley RB, Loskutoff DJ. Increased type 1 plasminogen activator inhibitor gene expression in atherosclerotic human arteries. Proc Natl Acad Sci USA. 1992;89:6998–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thogersen AM, Jansson JH, Boman K, Nilsson TK, Weinehall L, Huhtasaari F, Hallmans G. High plasminogen activator inhibitor and tissue plasminogen activator levels in plasma precede a first acute myocardial infarction in both men and women: evidence for the fibrinolytic system as an independent primary risk factor. Circulation. 1998;98:2241–2247. [DOI] [PubMed] [Google Scholar]
- 50. Alessi MC, Juhan‐Vague I. PAI‐1 and the metabolic syndrome: links, causes, and consequences. Arterioscler Thromb Vasc Biol. 2006;26:2200–2207. [DOI] [PubMed] [Google Scholar]
- 51. Pandolfi A, Cetrullo D, Polishuck R, Alberta MM, Calafiore A, Pellegrini G, Vitacolonna E, Capani F, Consoli A. Plasminogen activator inhibitor type 1 is increased in the arterial wall of type II diabetic subjects. Arterioscler Thromb Vasc Biol. 2001;21:1378–1382. [DOI] [PubMed] [Google Scholar]
- 52. Adly AA, Elbarbary NS, Ismail EA, Hassan SR. Plasminogen activator inhibitor‐1 (PAI‐1) in children and adolescents with type 1 diabetes mellitus: relation to diabetic micro‐vascular complications and carotid intima media thickness. J Diabetes Complications. 2014;28:340–347. [DOI] [PubMed] [Google Scholar]