

Abstract
Background
The proximal aorta normally functions as a critical shock absorber that protects small downstream vessels from damage by pressure and flow pulsatility generated by the heart during systole. This shock absorber function is impaired with age because of aortic stiffening.
Methods and Results
We examined the contribution of common genetic variation to aortic stiffness in humans by interrogating results from the AortaGen Consortium genome‐wide association study of carotid‐femoral pulse wave velocity. Common genetic variation in the N‐WASP (WASL) locus is associated with carotid‐femoral pulse wave velocity (rs600420, P=0.0051). Thus, we tested the hypothesis that decoy proteins designed to disrupt the interaction of cytoskeletal proteins such as N‐WASP with its binding partners in the vascular smooth muscle cytoskeleton could decrease ex vivo stiffness of aortas from a mouse model of aging. A synthetic decoy peptide construct of N‐WASP significantly reduced activated stiffness in ex vivo aortas of aged mice. Two other cytoskeletal constructs targeted to VASP and talin‐vinculin interfaces similarly decreased aging‐induced ex vivo active stiffness by on‐target specific actions. Furthermore, packaging these decoy peptides into microbubbles enables the peptides to be ultrasound‐targeted to the wall of the proximal aorta to attenuate ex vivo active stiffness.
Conclusions
We conclude that decoy peptides targeted to vascular smooth muscle cytoskeletal protein‐protein interfaces and microbubble packaged can decrease aortic stiffness ex vivo. Our results provide proof of concept at the ex vivo level that decoy peptides targeted to cytoskeletal protein‐protein interfaces may lead to substantive dynamic modulation of aortic stiffness.
Keywords: aging, aortic stiffness, cytoskeletal dynamics, Genome Wide Association Study, vascular smooth muscle
Subject Categories: Contractile function, Cell Biology/Structural Biology, Cell Signalling/Signal Transduction, Smooth Muscle Proliferation and Differentiation, Vascular Biology
Clinical Perspective
What Is New?
Aortic stiffness can be decreased in a mouse model of aging by using decoy peptides that compete with cytoskeletal protein‐protein interfaces.
The decoy peptides can be packaged onto microbubbles for ultrasound‐mediated targeting to the aorta.
Genetic studies suggest that the same cytoskeletal pathways may be involved in aortic stiffness in humans.
What Are the Clinical Implications?
Aortic stiffness increases with age and is an early and independent biomarker for cognitive decline, myocardial infarction, and renal disease.
Although there is currently no approved therapy to reduce aortic stiffness, our results indicate that cytoskeletal peptides may represent novel therapeutic targets.
Introduction
Vascular aging is associated with impaired endothelial function, low‐grade inflammation, and markedly increased aortic stiffness.1, 2, 3, 4 Recent clinical studies document that increased aortic stiffness, as measured by pulse wave velocity, is an early and independent biomarker for negative cardiovascular outcomes such as myocardial infarction, cognitive decline, and renal disease.2, 5, 6, 7, 8, 9 Greater aortic stiffness increases transmission of pressure and flow pulsatility associated with cardiac contraction into delicate microvessels, particularly in high‐flow organs such as the brain and kidney.2, 3, 5, 7, 9, 10 Aortic stiffening (arteriosclerosis) is distinct from atherosclerosis11 and is thought to cause secondary morphological changes in microvessels that contribute to the pathogenesis of hypertension and cardiovascular disease.10, 12, 13
Aging is associated with fragmentation of elastin and increased amounts and cross‐linking of collagen, all of which increase the passive stiffness of the extracellular matrix.14, 15 However, it has also been proposed that aging of the vascular smooth muscle cell (VSMC) can adversely modulate the fractional engagement of collagen, leading to a dynamic increase in stiffness.16, 17, 18, 19, 20, 21, 22 In fact, recent studies in a mouse model, where viable smooth muscle preparations can be readily obtained and activated with vasoactive agents to measure active stiffness, have demonstrated that close to half of the total stiffness of the aortic wall is attributable to the active stiffness of the VSMC,16 with the remaining fraction due to the extracellular matrix. Furthermore, aging has recently been shown to further increase the active stiffness of the VSMCs23 in a mouse model.
In addition to the passive stiffness of the matrix, there are at least 2 dynamic components that contribute to the material stiffness of the VSMC: first, the attachment of cycling crossbridges in the contractile filaments, and, second, the regulated transmission of force and stiffness through a nonmuscle actin cytoskeleton connected to focal adhesion (FA) complexes. The stiffness and plasticity of this nonmuscle actin cytoskeleton are regulated by proteins that control branched and linear actin polymerization such as N‐WASP and VASP, respectively.20, 24, 25, 26 The FA complexes include transmembrane integrins that connect with the extracellular matrix of the aortic wall. Thus, it has become clear in recent years that the contractile filaments are not directly connected to the integrins.20
The nonmuscle actin cytoskeleton and FAs to which it is attached have been shown to display plasticity in the presence of agonists26 and biomechanical stimuli27, 28 by exhibiting stimulus‐induced increases in actin polymerization and endosomal‐dependent remodeling of a subset of FA proteins.29 Plasticity of the cortical cytoskeleton of VSMCs may contribute to the function of the healthy, compliant proximal aorta, acting as a tunable “shock absorber” that adapts in order to limit transmission of excessive pulsatile energy into the delicate downstream microvessels. This plasticity of the cortical cytoskeleton of the aorta in young mice has been shown to utilize a Src‐dependent signaling pathway that promotes tyrosine phosphorylation of FA proteins.30 We have found that attenuated activity of this pathway with aging is associated with stiffening, measured ex vivo in a mouse model.16
In the present study we asked whether human single nucleotide polymorphism analysis might support a link between VSM cytoskeletal malfunction and aortic stiffness, and we tested the hypothesis that specific cytoskeletal protein‐protein interfaces that no longer remodel in the aged aorta could be competed with by decoy peptides to reduce increases in aortic stiffness of proximal aortas taken from aged mice. Because a simultaneous decrease of the stiffness of all smooth muscle tissues would be disadvantageous, we furthermore provide proof of concept for a microbubble‐mediated delivery of cytoskeletal decoy peptides to the aortic smooth muscle that can be ultrasound targeted and therefore shows promise as a novel potential therapeutic approach to reverse cardiovascular aging.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure. This is due to the cost of replicating the materials and limitations on the sharing of the human data.
Experimental Animals and Aortic Sample Preparation
All procedures were performed in accordance to protocols approved by the Institutional Animal Care and Use Committee of Boston University (Permit Number: A3316‐01) and used in compliance with federal, state, and local laws. C57BL6/J mice were euthanized by isoflurane inhalation. Following euthanasia, aortas were quickly excised from young (3‐4 months) or aged (24‐29 months) mice, placed directly in ice‐cold modified Krebs solution ([mmol/L]: 154 NaCl, 5.4 KCl, 1.2 MgSO4, 10 MOPS, 5.5 glucose, and 1.6 CaCl2; pH=7.4), and cleaned of excess perivascular fat. Axial rings that were 4 to 5 mm in length were cut from the proximal end of the thoracic aorta for biomechanical measurements and bath loading of peptides.
Human Studies
The AortaGen Consortium included 20 634 participants with a broad age distribution from 9 community‐based cohort studies that completed genome‐wide genotyping and had measured carotid‐femoral pulse wave velocity (CFPWV). Each study received institutional review board approval of its consent procedures, examination and surveillance components, data security measures, and DNA collection and use for genetic research. All participants in each study gave written informed consent for participation in the study and the conduct of genetic research. Details of study cohort, CFPWV measurement protocols, and inclusion and exclusion criteria are provided in the original publication.31
Expression and Purification of VASP EVH1‐TAT Recombinant Protein
VASP EVH1‐TAT recombinant protein was expressed in a pTYB1 vector (New England Biolabs, Ipswich, MA) and purified on an affinity based chitin resin column as published previously.26 Briefly, the EVH1‐TAT construct was cloned in a pTYB1 vector (New England Biolabs). This vector consists of a chitin‐binding domain (for affinity purification) and an intein tag (for self‐cleavage after purification). The construct was expressed in BL21 (DE3) cells (Invitrogen, Carlsbad, CA), which were grown in terrific broth medium at 37°C until an optical density at 600 nm reached a value of 0.8. The recombinant protein expression was induced by addition of 0.5 mmol/L Isopropyl β‐D‐thiogalactoside (Invitrogen), and cultivation was continued at 20°C for 16 hours. Cells were harvested by centrifugation, suspended in a column buffer (20 mmol/L Na‐HEPES, pH8.5, 500 mmol/L NaCl, 1 mmol/L EDTA, 0.1% Triton X‐100, and 20 μmol/L protease inhibitor), and lysed by sonication. The soluble lysate was then loaded on an affinity‐based chitin resin column. VASP EVH1‐TAT recombinant protein was purified by standard affinity chromatography procedure through a chitin column (New England Biolabs, Ipswich, MA) and concentrated by Amicon®Ultra‐15 (Millipore, Burlington, MA). The protein aliquots were flash frozen in liquid nitrogen and stored at −80°C until further use.
WASP Peptide Sequences
The human and mouse CA domain sequence used is TSGIVGALMEVMQKRSKAIHSSDEDEDEDDEEDFEDDDEWED. In parallel, as a control, we also synthesized a scrambled peptide, identical except that the cytoskeletal sequence has the amino acids arranged in a random order: for the CA domain the scrambled sequence used is DEMLEQEKGDESGIDSGDKDMEEVADSWEVEDHSETDIADDE.
Synthesis of Cell‐Permeant Fluorescently Tagged Decoy Peptides
Peptide sequences were synthesized by solid‐state peptide synthesis using fluorenylmethoxycarbonyl chemistry to protect the amino groups. They were synthetically coupled to a TAT protein transduction domain comprising residues YGRKKRRQRRR. To add fluorescence labels to the peptides, the side chain of the first lysine in the TAT sequence was protected with the acid‐labile 4‐methytrityl, which was selectively removed from the completed peptide sequence using 5 to 8 consecutive 2‐minute treatments with 2% trifluoroacetic acid. The deprotected lysine side chain was then coupled to fluorenylmethoxycarbonyl aminohexanoic acid as a spacer, which, following removal of the fluorenylmethoxycarbonyl, was labeled with isothiocyanate derivatives of the fluorophores.
Measurement of Aortic Geometry
Axial length, diameter, and wall thickness were quantitated to determine cross‐sectional area, used to calculate vessel stress and stiffness. Axial length and slack diameter were measured during dissection. For wall thickness measurements, ≈1‐mm length rings are cut just proximal and distal to the tissue used for biomechanics. For visualization, the 1‐mm rings were incubated in nuclear stain (NucBlue, Life Technologies, Carlsbad, CA) for 30 minutes and then imaged (×20) with an Eclipse TE2000‐E fluorescence microscope (Nikon Instruments, Tokyo, Japan) via NIS‐Elements software. Fifteen to 20 measurements were recorded for each ring to calculate the average wall thickness.
Biomechanics
Tissue stiffness in kilopascals was determined at baseline and in the presence of the agonist phenylephrine (10−5 mol/L) at the optimal length for force production with small‐amplitude (1% of tissue length), high‐frequency (40 Hz) sinusoidal stretches. Stiffness is defined as stress divided by the strain in response to the sinusoidal oscillations, (ΔF/cross‐sectional area)/(ΔL/L0), where ΔF is the amplitude of the force response to the cyclic stretches, ΔL is the amplitude of the cyclic length changes, and L0 is the optimal length. Stress was calculated as ΔF/cross‐sectional area. This method does not result in a phase lag of the force response and does not cause detachment of actomyosin crossbridges.32
Aortic rings were mounted onto 2 wire triangles (0.01 inch diameter). Once mounted, the vessels were incubated in baths (50 mL) containing oxygenated (95% O2+5% CO2) modified Krebs solution. The lower triangle was fixed, and the upper triangle was attached to a lever arm connected to a force transducer system (model 300C Dual‐Mode Lever Arm System by Aurora Scientific, Aurora, ON, Canada) and monitored using chart software (AD Instruments, Dunedin, New Zealand). Rings were stretched uniaxially in the circumferential direction to the optimal length L0 (1.8×slack length). Steady‐state force, F, was measured after equilibration for 30 minutes at that length. Each aortic ring was tested for viability by contracting with a depolarizing solution (Modified Krebs solution in which 51 mmol/L NaCl is replaced by 51 mmol/L KCl).
Differential Centrifugation for Tissue Fractionation
Aortic tissue was dissected and cleaned. Aortic strips were quick frozen and placed in Precellys tube (Bertin Technologies, Montigny‐Le‐Bretonneux, France) containing ice‐cold 0.1 mL buffer I containing 20 mmol/L Tris HCl, pH 7.5, 50 mmol/L NaCl, 250 mmol/L sucrose, 10 mmol/L dithiothreitol, 3 mmol/L EGTA, 5 mmol/L MgCl2, 1 mmol/L ATP, and protease inhibitor cocktail. The tissue was homogenized with a Precellys 24 unit (Bertin Technologies, Montigny‐Le‐Bretonneux, France) at 4°C. The homogenized tissue was centrifuged at 100 000g for 1 hour at 4°C. The supernatant (cytosolic fraction) was collected, and the pellet was dissolved in equal volumes of buffer II containing 20 mmol/L Tris HCl, pH 7.5, 250 mmol/L sucrose, 10 mmol/L dithiothreitol, 3 mmol/L EGTA, 5 mmol/L MgCl2, 0.5% Triton X‐100, 1 mmol/L ATP, and protease inhibitor cocktail. The resuspended pellet was shaken for 1 hour at 4°C and centrifuged at 100 000g for 1 hour. This supernatant was collected as the membrane fraction. The pellet was resuspended in buffer III (20 mmol/L Tris.HCl, pH 7.5, 250 mmol/L sucrose, 10 mmol/L dithiothreitol, 3 mmol/L EGTA, 5 mmol/L MgCl2, 0.5% Triton X‐100, 1.2% SDS, 1 mmol/L ATP and protease inhibitor cocktail), shaken for 1 hour at 4°C and briefly centrifuged. The final supernatant was collected as the cytoskeletal fraction. Equal volumes of each fraction were loaded on SDS‐PAGE for further analysis of vinculin/metavinculin levels by Western blotting with a monoclonal antivinculin antibody (catalog # V9131, Sigma, St. Louis, MO) at 1:1000 dilution and IRDye 800CW goat antimouse secondary antibody (catalog # 925‐32210, LI‐COR Biosciences, Lincoln, NE) at 1:10 000 dilution. Vinculin/metavinculin levels were determined by scanning band densitometry on the raw images obtained using an Odyssey IR imaging system (LI‐COR). Data are presented as a percentage of total vinculin/metavinculin.
Aortic Cell Culture
A7r5 aortic smooth muscle cells (ATCC, Manassas, VA) were cultured on coverslips in DMEM high glucose with 10% fetal calf serum, 1% glutamine, 50 units/mL penicillin, and 50 μg/mL streptomycin. Before experimentation, cells were serum starved for 24 hours to drive them to a differentiated state.33, 34 To confirm permeation of the peptides into cells, cultured A7r5 cells were either unloaded or loaded for 30 minutes at 37°C with 250 μmol/L N‐WASP CA domain or 100 μmol/L TLN‐VCL fluorescently labeled constructs. For focal adhesion assays, 50 μmol/L TLN‐VCL was used.
Filamentous actin was stained with Alexa Fluor 488 phalloidin (1:3000, Invitrogen, Carlsbad, CA). Cells were examined with an Eclipse TE2000‐E fluorescence microscope (Nikon Instruments) using a Nikon Plan Apochromat oil‐immersion objective and a charge‐coupled device camera (CoolSNAP HQ2, Photometrics, Tucson, AZ). NIS‐Element Advanced Research software (Nikon Instruments) was used to capture images for removal of out‐of‐focus fluorescence blur in tissues by deconvolution of Z‐sections (Richardson‐Lucy algorithm, constrained iterative–maximum likelihood estimation algorithm). Images from control and construct‐loaded samples were obtained at identical microscope and software settings (look‐up tables, offsets, and gains).
Focal Adhesion Integrity Assay
To analyze the focal adhesion size and number, A7r5 cells were grown on glass coverslips. Cells were serum‐starved for 24 hours followed by treatment with 50 μmol/L scrambled TLN‐VCL or TLN‐VCL construct for 30 minutes at 37°C. The cells were rinsed in PBS and fixed in 4% paraformaldehyde. Cells were then permeabilized and blocked concurrently with 0.1% Triton X‐100 in 1% BSA and 10% normal goat serum for 1 hour at room temperature. Labeling was accomplished with antivinculin antibody (1:200, V9131, Sigma, St. Louis, MO). For the secondary antibody, we used Alexa Fluor 488 conjugated goat antimouse IgG (H+L) (1:400, A11029 Invitrogen). F‐actin was stained with Alexa Fluor 568 Phalloidin (1:400, A12380, Invitrogen). Coverslips were mounted on glass slides with fluorsave mounting medium with DAPI (ab104139, Abcam, Cambridge, UK).
The cells were examined with an Eclipse TE2000‐E fluorescence microscope (Nikon, Tokyo, Japan) equipped with a Nikon Plan Apochromat 60× (NA 1.4) oil‐immersion objective and a charge‐coupled device camera (CoolSNAP HQ2, Photometrics, Tucson, AZ). Images were acquired by NIS‐Element Advance Research software. Only peripheral focal adhesions (≈5‐10 μm from cell edge) were measured using NIS‐Element AR Software. Focal adhesions were measured for 4 to 7 cells per condition (control, scrambled, TLN‐VCL) in 5 independent experiments each. Focal adhesion numbers were counted manually.
Microbubble Peptide Complex Production
The microbubbles were made from a lipid solution and octafluoropropane gas via mechanical agitation. The lipids distearoylphosphocholine, distearoylphosphoethanolamine‐PEG2000 (Avanti Polar Lipids, Inc, Alabaster, AL), and distearoylphosphoethanolamine‐PEG3400 linked with peptide‐FITC were mixed at a molar ratio of 8:1:1 and a final lipid concentration of 5 μmol/mL in chloroform. The lipid solution was dried under vacuum overnight, and the resultant thin lipid film was rehydrated in a solution of 10% glycerol, 10% propylene glycol, and 80% deionized water. The lipid suspension was sonicated for 1 minute and degassed at room temperature. After saturation of the lipid solution with octafluoropropane gas (Fluoromed, Round Rock, TX), the solution was shaken vigorously, producing polydispersed lipid‐coated microbubbles.
Bath Loading of Peptides Into Ex Vivo Aortic Tissue and Tissue Imaging
From lyophilized powder, loading solutions were prepared by dilution to final working concentrations (100 μmol/L for TLN‐VBS and EVH1, 250 μmol/L for N‐WASP) in modified Krebs solution and incubated with aortic rings for 30 minutes at 37°C in 1‐mL loading trays containing a disk of polydimethylsiloxane gel at the bottom of the well. Each aortic ring was threaded with a triangular wire clasp parallel to a piece of straight wire and then transferred to the loading tray. The tissue was stretched with small surgical pins to the physiologic strain (180% of slack length) by pulling the triangular clasp and wire apart to the precise length as measured with a reticule in a dissecting microscope. The tray was positioned atop a heating bath at 37°C. Successful peptide loading was confirmed at the end of each experiment by cutting a 1‐mm‐length ring, staining nuclei for orientation with NucBlue (Life Technologies, Carlsbad, CA), and examining with an Eclipse TE2000‐E fluorescence microscope (Nikon Instruments, Tokyo, Japan) as described in the above section using a Nikon Plan Apochromat oil‐immersion objective and a charge‐coupled device camera (CoolSNAP HQ2, Photometrics, Tucson, AZ). NIS‐Element Advanced Research software (Nikon Instruments) was used to capture images and for removal of out‐of‐focus fluorescence blur in tissues by deconvolution of Z‐sections. Tissue images shown are deconvolved images. In all cases images from peptide‐loaded or control samples were obtained at identical microscope and software settings (look‐up tables, offsets, and gains).
Actin Polymerization Assay
Aortic tissue strips (≈8 mg wet weight) were placed in 0.5‐mL soft tissue homogenizing tubes (Precellys, Bertin Technologies, Montigny‐Le‐Bretonneux, France) containing prewarmed lysis and F‐actin stabilization buffer (50 mmol/L PIPES pH 6.9, 50 mmol/L NaCl, 5 mmol/L MgCl2, 5 mmol/L EGTA, 5% [v/v] Glycerol, 0.1% Nonidet P40, 0.1% Triton X‐100, 0.1% Tween 20, 0.1% 2‐mercapto‐ethanol, 0.001% Antifoam C) with 1 mmol/L ATP and protease inhibitor cocktail (Cytoskeleton, Inc, Denver, CO). The tissue was quickly homogenized in a tissue homogenizer (Precellys 24, Bertin Technologies, Montigny‐Le‐Bretonneux, France), and 100 μL of lysates was incubated at 37°C for 20 minutes and then spun at 150 000g for 60 minutes at 37°C in an Optima TLX ultracentrifuge (Beckman Coulter, Brea, CA). The supernatant fraction (G‐actin) was removed gently to avoid disturbing the F‐actin pellet. Then, 100 μL of ice‐cold F‐actin depolymerization buffer (8 mol/L urea) was added to each pellet. The pellet was dissolved by pipetting up and down several times every 15 minutes for 1 hour. To remove cell debris, this solution was spun at 2300g for 5 minutes (4°C), and the supernatant was collected to give the final F‐actin fraction. The samples were stored at −80°C until further separation by SDS‐PAGE and immunoblotting with an antiactin primary antibody (Cytoskeleton) and a secondary IRDye 680–labeled goat antirabbit (LI‐COR, Lincoln, NE) and visualized on an Odyssey infrared imaging system (LI‐COR [Lincoln, NE]) for densitometric analysis with Odyssey 2.1 software. All analyses were performed on raw data.
Conjugation to Lipid for Microbubbles
Before the microbubble formation, the peptides were conjugated directly to lipid using a thiol‐maleimide reaction. In general, distearoylphosphoethanolamine‐PEG3400‐maleimide (1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐[maleimide(polyethylene glycol)‐2000], ammonium salt) (Nanocs, PG2‐DSML‐3k) was dispersed via sonication at a concentration of 0.04 mmol/L in PBS, pH 7.4 (or ultrapure water, pH 7.0) to create micelles. Micelle formation was verified via dynamic light scattering. Separately, the peptide was dissolved in modified Krebs solution (pH 7.4) containing 5 mmol/L EDTA. The lipid and peptide solutions were mixed in a 1:1.25 molar ratio and stirred, protected from light, for 24 hours at room temperature to ensure development of the thiol‐ester bond. Free peptide was separated from the protein‐lipid conjugate using centrifugal filtration. In brief, the peptide solution along with 3 mL ultrapure water was added to the filter (MWCO 10 kDa, Amicon, EMD Millipore, Billerica, MA), and spun for 10 minutes at 4000g. This process was repeated 3 times to wash the conjugate and to exchange the buffer. The peptide‐lipid was then lyophilized and resuspended in chloroform (1 mg/mL) for use in the microbubbles. Conjugation was confirmed via fluorescent microscopy of subsequently produced microbubbles.
Microbubble‐Peptide Complex Introduction Into Aortic Segments
A suspension of microbubbles loaded with peptide at a concentration of 0.2 μmol/mL was injected into the lumen of an excised murine aorta. The ends of the aorta were sealed, and the vessel was placed at the focus of a 1‐MHz focused transducer (Panametrics A392S, Waltham, MA) in a water bath. The focused transducer transmitted 200‐cycle ultrasound pulses with a frequency of 1‐MHz and acoustic pressure of 978 kPa at a pulse repetition frequency of 1 Hz for 30 seconds to fragment microbubbles, release the peptide, and reversibly disrupt the endothelium.
Statistics
We evaluated the contribution of common genetic variation at the N‐WASP locus (gene symbol WASL) to aortic stiffness in humans by interrogating results of the CFPWV genome‐wide association study performed by the AortaGen Consortium.31 The standardized phenotype in the AortaGen analysis was 1/CFPWV, which was adjusted for age,2 height, and weight and then transformed to standardized Z‐values (mean=0 and SD=1). However, because the phenotype was inverse CFPWV, a positive B corresponds to lower CFPWV (less stiff aorta), and a negative B corresponds to higher CFPWV (stiffer aorta). We selected single nucleotide polymorphisms that fell within 20 kb of the WASL gene and were nonredundant at an R 2>0.30. These selection criteria identified 9 single nucleotide polymorphisms in the WASL locus. Therefore, we prospectively set a gene‐wise Bonferroni‐adjusted threshold for significance at P<0.05/9=0.0056.
Values are presented as mean±SEM. Analysis was carried out using the GraphPad Prism (7.0) software (La Jolla, CA). For actin polymerization and ex vivo biomechanics comparisons, groups were compared using a Student t test (2‐tailed) for parametric data. Significance was assumed at P<0.05 except as noted above and indicated in the figures with an asterisk.
Results
Human Single Nucleotide Polymorphism Analysis Supports a Link Between Vascular Smooth Muscle Cytoskeletal Function and Aortic Stiffness
Results are detailed in Table. We found evidence for association at rs600420 (P=0.0051). Local linkage disequilibrium for rs600420 is plotted in Figure 1, prepared by using SNAP,35 which demonstrates evidence for a region of linkage disequilibrium that spans the WASL gene.
Table 1.
Association Results for Carotid‐Femoral Pulse Wave Velocity From the AortaGen Consortium
| SNP ID | Allele1 | Allele2 | Effect Allele | Effect Allele Frequency | Location on Chr 7 | B | SE | P Value |
|---|---|---|---|---|---|---|---|---|
| rs12706544 | A | C | C | 0.23 | 123092680 | −0.032 | 0.013 | 0.014 |
| rs1019219 | T | C | C | 0.08 | 123106873 | −0.014 | 0.020 | 0.497 |
| rs1073682 | G | C | C | 0.99 | 123110320 | −0.019 | 0.061 | 0.753 |
| rs6971373 | A | G | G | 0.01 | 123121498 | 0.016 | 0.063 | 0.795 |
| rs6975039 | T | C | C | 0.93 | 123134504 | −0.015 | 0.022 | 0.486 |
| rs12706551 | T | G | G | 0.96 | 123166013 | 0.059 | 0.031 | 0.060 |
| rs2069269 | G | C | C | 0.92 | 123193642 | −0.022 | 0.020 | 0.280 |
| rs623201 | T | C | C | 0.23 | 123194511 | −0.014 | 0.013 | 0.299 |
| rs600420 | A | G | G | 0.60 | 123195286 | −0.031 | 0.011 | 0.005 |
Carotid‐femoral pulse wave velocity was inverted and standardized before analysis. B indicates difference in standardized inverse carotid‐femoral pulse wave velocity per dose of the coded allele; Chr, chromosome; SNP, single nucleotide polymorphism.
Figure 1.
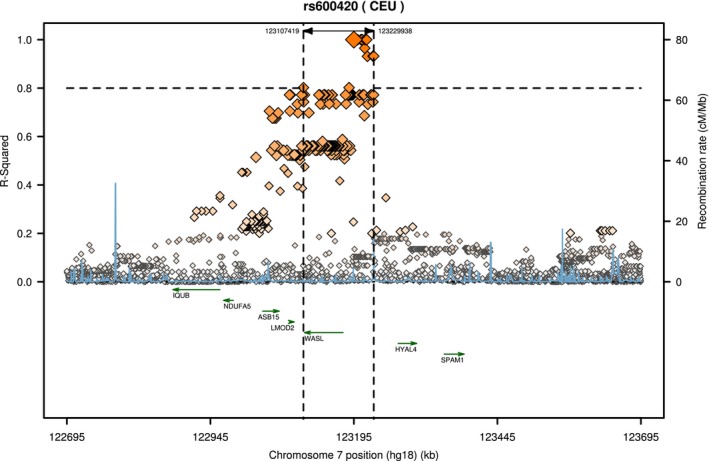
Regional linkage disequilibrium plot for rs600420. The span of single nucleotide polymorphisms with an R 2>0.80 is indicated by the vertical dashed lines. The WASL gene lies within this region of linkage disequilibrium. Figure was prepared by using the Scalable Nucleotide Alignment Program (SNAP).35
Previous studies on animal models have shown an involvement of the N‐WASP protein from the WASL gene in the regulation of actin polymerization and contractility in smooth muscle of mammalian airways,36 but effects on aortic stiffness have not been determined. Thus, N‐WASP was an attractive candidate molecule to pursue with a decoy peptide strategy to determine effects on stiffness.
Strategy of Decoy Peptide Design
The CA domain of N‐WASP is a subset of the C‐terminal VCA domain by which N‐WASP activates the Arp 2/3 complex, inducing actin polymerization at branches from the side of parent actin filaments.36, 37 The CA domain binds to G‐actin, and because it lacks the V domain, acts as a dominant negative inhibitor of N‐WASP‐mediated actin polymerization in airway smooth muscle,36 but its effects in vascular smooth muscle are unknown. In order to test the novel idea that the CA peptide and other decoy cytoskeletal peptides might function as a useful approach to inhibit aortic stiffness and vascular contractility, we synthesized a series of constructs using the generic design illustrated in Figure 2A to incorporate the TAT cell permeation tag38, 39 and a fluorescent tag together with the peptide. A scrambled CA sequence was used as a control.
Figure 2.
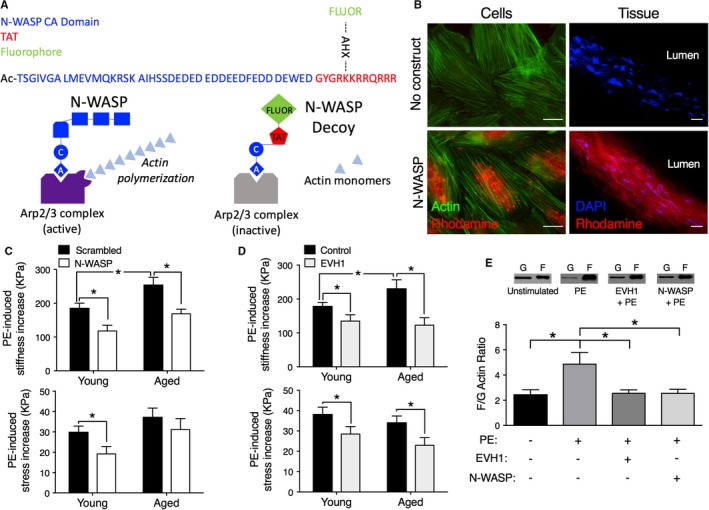
Decoy constructs targeted to actin polymerization molecules decrease aortic stiffness. A, Top, Design of prototype for cell‐permeant decoy peptides. Blue type, protein‐specific domain sequence, acetylated on the N‐terminus to maintain stability. N‐WASP CA dominant negative peptide sequence shown. Red type, sequence derived from the TAT known to be a cell permeation sequence38, 39 added to C‐terminus. Green type, a fluorescent dye (FITC or rhodamine) added to an ε‐aminohexanoic acid (AXH) bridge as a side chain off a lysine of the TAT sequence in order to confirm intracellular loading. The AHX bridge allows the fluorophore to be added without adverse effects on peptide stability. Bottom, Diagram illustrating the method of inhibition by the decoy peptide. The CA domain inhibits the binding of N‐WASP to the Arp2/3 complex and prevents its activation. B, Decoy constructs permeate into aortic cells (left) and tissue (right). Left panels, No added construct (top) or rhodamine‐labeled (red) N‐WASP CA peptide‐loaded cells (bottom). Costained with phalloidin (green) for actin. Incorporated peptide is especially noticeable in the perinuclear region, the thickest part of the cell. Right panels, No added construct (top) or rhodamine‐labeled N‐WASP CA peptide‐loaded into ex vivo aortic tissue (bottom). Tissue costained with DAPI to locate nuclei. Images are obtained at identical microscope and software settings (look‐up tables, offsets, and gains). Scale bars: 20 μm. C, Top panel, Aortic tissues from aged mice show significantly increased active, PE‐induced stiffness compared with those from young mice (black bars). The introduction of the N‐WASP CA domain peptide (white vs black bars) significantly decreases active stiffness (top panel) in both young (3‐month‐old, n=6) and old (24‐29 months, n=6) mice. The peptide decreases PE‐induced stress significantly only in young mice (bottom panel). Asterisks indicate significant differences (P<0.05) from scrambled peptides. D, Top panel, Aortic tissues from aged mice show significantly increased active, PE‐induced stiffness compared with those from young mice (black bars). Decoy construct targeted to EVH1 decreases PE‐mediated aortic stiffness (top panel) and stress (bottom panel) (tan vs black bars) ex vivo in young (n=12) and aged (n=7) mice. Asterisks indicate significant differences P<0.05 from control peptides. E, Inhibition of PE‐induced actin polymerization (n=7, young adult, 3‐ to 4‐month‐old mice) by the N‐WASP CA domain peptide (n=8) and EVH1 construct (n=8), compared with baseline values (n=7), demonstrate on‐target effects. PE indicates phenylephrine.
Decoy Peptides Can Be Introduced Into Vascular Smooth Muscle Cells of the Blood Vessel Wall When Coupled to a Cell Permeation Sequence
The left‐hand panels of Figure 2B show images of A7r5 aortic cells that were subjected to serum starvation and labeled with phalloidin (green) for actin. The cells were serum starved to promote a differentiated phenotype. The top image shows control cells to which no construct was added. The lower image shows similar cells but to which the rhodamine‐labeled N‐WASP CA decoy peptide was added, demonstrating incorporation into the cells.
The right‐hand panels show aortic tissue preparations stained with DAPI (blue) to locate nuclei for orientation. No construct was added to the top tissue. The bottom image is of another aortic preparation, taken with identical microscope acquisition and software settings, stained with DAPI but also loaded with a rhodamine (red)‐labeled peptide for 30 minutes, which is clearly detectable in the smooth muscle cells of the wall. Thus, we are able to load decoy peptides successfully into aortic smooth muscle tissue by virtue of the cell permeation tag alone.
N‐WASP CA Domain Introduced Ex Vivo Into Young and Old Mouse Aortas Decreases Aortic Stiffness
As we have previously reported,23 ex vivo mouse aorta preparations display a significantly higher level of α agonist–activated stiffness when taken from aged animals compared with young animals (Figure 2C top graph, black bars, right versus left). Furthermore, when aortas from both young and aged mice were bath loaded with the N‐WASP CA domain decoy peptide, active stiffening of the aortas in the presence of phenylephrine was reduced (Figure 2C top graph, black bar versus white bar). The decoy peptide also inhibited the contractile stress response to phenylephrine in aortic preparations from young mice (lower panel). The decreased stress response is expected because the cortical cytoskeleton, which transmits forces generated by the contractile filaments to the integrins and the vessel wall, has been targeted by this construct. But, interestingly, stress was not significantly changed in aged mice, suggesting a difference in mechanisms of force generation or transmission with age. No change in unstimulated, ie, baseline stiffness or stress was seen with the peptide, consistent with these parameters being set constant by passive extracellular matrix responses at these levels of tissue stretch.
A Decoy Construct From the EVH1 Domain of Ena‐VASP Family Proteins, Targeted to a Different Mechanism of Actin Polymerization, Is Also Effective in Decreasing Ex Vivo Aortic Stiffness of Aortas From Young and Old Mice
We were next interested in whether a decoy peptide strategy is more broadly applicable. Actin polymerization can occur in 2 major ways in cytoskeletal structures: (1) by the activation of the Arp 2/3‐N‐WASP complex, which adds branched actin, or (2) by linear extension of the barbed end of actin filaments by the activity of the ena‐VASP family of cytoskeletal proteins.40 The latter has been shown by our group to occur in vascular smooth muscle in a regulated manner.26 Hence, we determined whether regulation of linear actin polymerization could be targeted to decrease ex vivo aortic stiffness. The N‐terminal EVH1 domain of ena‐VASP family members localizes VASP to zyxin in vascular FAs, facilitating linear actin polymerization at those sites.41, 42 Because of the length of the EVH1 domain, we synthesized the analogous decoy construct by making EVH1 domain, attached to the TAT cell permeation tag, as a recombinant protein. This protein was bath loaded into aortic rings ex vivo and, as shown in Figure 2D, was effective in decreasing both stiffness (top panel) and stress (bottom panel) compared with control loading in young and aged mice. The control used was either a mutant (Phe78Ser) EVH1 domain, which has no effect on contractility,26 or sham loading with no peptide. No significant difference (P<0.05) was seen between the use of these 2 controls on either stiffness or stress so their values are merged in Figure 2D.
We further confirmed that the effects of these 2 types of decoy constructs, designed to interfere with actin polymerization mechanisms, altered biomechanics by on‐target, direct effects on actin polymerization (Figure 2E). The phenylephrine‐induced increase in actin polymerization as shown by the actin filamentous/globular actin ratio was significantly decreased by both the WASP CA domain and EVH1 constructs, essentially to unstimulated levels.
A Decoy Peptide Based on a Protein‐Protein Interface Between FA Proteins is Also Effective in Decreasing Ex Vivo Aortic Stiffness in Aortas From Young and Old Mice
Given that FA remodeling in smooth muscle has been shown to occur in response to vasoactive stimuli,29, 43 and that Src‐dependent pathways of FA remodeling are lost with aging,16 we asked whether protein‐protein interfaces in the FA complex could be targeted with a similar cell‐permeant decoy peptide approach. Based on the widely known importance of interactions of vinculin with talin in the FA,44, 45 we designed a peptide (TLN‐VCL) mimicking a vinculin binding site on talin. There are several talin‐vinculin binding sites, and although several of these sequences had no effect on smooth muscle cell function, the sequence GRPLLQAAKGLAGAVSELLRSAQPA was effective. A scrambled version of this peptide, LLRRSQAALGAAAEVLPQALSGPGK, was used as a control. The sequence was substituted for the N‐WASP sequence in the generic construct described in Figure 2A. We demonstrate, in Figure 3A, the ability of the construct to incorporate into smooth muscle cells in both serum‐starved aortic cells (Figure 3A, left column, bottom panel compared with unloaded cells, top panel) and tissue (Figure 3A, right column, bottom panel compared to unloaded tissue, top panel). The construct is also effective in decreasing tissue stiffness (Figure 3B, top) and stress (Figure 3B, bottom) after bath loading at 100 μmol/L in proximal aortas from both young and aged animals.
Figure 3.
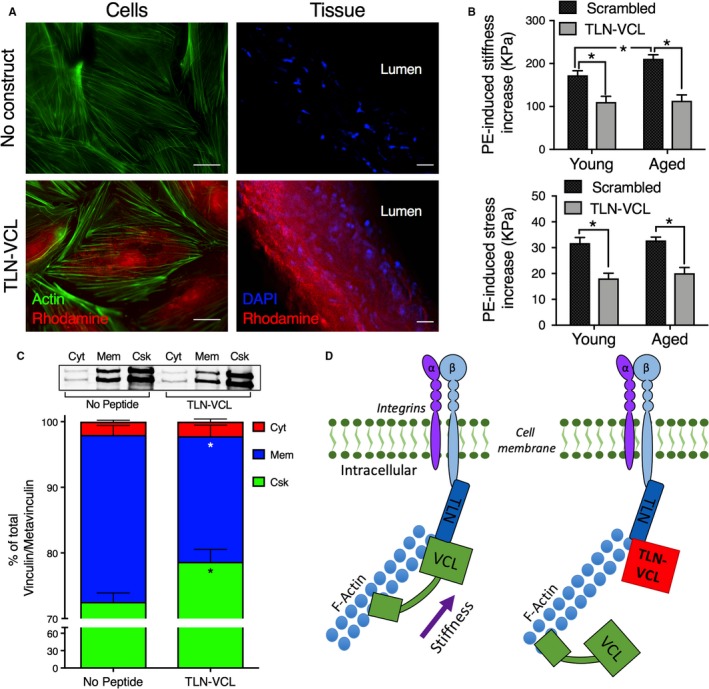
Decoy construct targeted to focal adhesion protein‐protein interfaces decrease aortic stiffness through an on‐target cytoskeletal mechanism. A, Immunofluorescent images demonstrate that the decoy talin‐vinculin (TLN‐VCL) construct permeates into aortic cells and tissue ex vivo. Left panels, Rhodamine‐labeled (red) TLN‐VCL construct loaded cells (bottom) or not loaded (top). All cells costained with phalloidin (green) for actin. Right panels, Rhodamine‐labeled (red) TLN‐VCL construct loaded into ex vivo aortic tissue (bottom) or not loaded (top). Both tissues costained with DAPI (blue) for nuclei. Scale bars: 20 μm. All images were obtained with identical microscope settings. B, Targeting of the talin‐vinculin binding interface in FAs decreases PE‐induced stiffness (top) and stress (bottom) in young (n=12, left panels) and aged (n=7, right panels) mice. Asterisks indicate significant differences (P<0.05) between TLN‐VCL peptide and scrambled peptide or between ages. C, Fractionation of aortic tissue by differential ultracentrifugation demonstrates that TLN‐VCL disrupts cytoskeletal integrity. Top, Representative Western blots depicting metavinculin (top band) and vinculin (bottom band) from cytosolic (Cyt), membrane (Mem) and cytoskeletal (Csk) fractions from control (no peptide), sham loaded, tissues (left, n=9) and TLN‐VCL–treated (right, n=9) aortic tissue. Bottom, vinculin/metavinculin in each fraction expressed as a percentage of total vinculin/metavinculin. D, Model of mechanism by which TLN‐VCL decoy construct reduces ex vivo aortic stiffness. Left, Talin binds integrins at plasmalemma. Vinculin binds talin to facilitate transmission of force and stiffness from the actin cytoskeleton through the integrins to the extracellular matrix. Right, TLN‐VCL decoy peptide reduces vinculin binding to talin at the membrane, attenuating force and stiffness transmission to the extracellular matrix.
We next sought to confirm that the TLN‐VCL peptide disrupts cytoskeletal/contractile complexes by an on‐target effect of interfering with vinculin binding to talin. To do this, we used a focal adhesion redistribution assay as described in Methods. Aortic tissue was fractionated into cytosolic, membrane, and cytoskeletonal fractions by differential ultracentrifugation, allowing the determination of the subcellular location of vinculin in the 3 fractions. In addition to vinculin itself, smooth muscle also expresses the slightly larger metavinculin, which is a muscle‐specific splice variant of vinculin.46 We analyzed the amounts of both vinculin and metavinculin in each fraction by immunoblot. In TLN‐VCL peptide–treated tissue, the amount of vinculin/metavinculin was significantly decreased in the membrane fraction compared with control tissue not exposed to peptide. In parallel, the vinculin/metavinculin in the cytoskeletal fraction of peptide‐treated tissue was increased as compared with control tissue (Figure 3C). Altogether, these data are consistent with the concept that the TLN‐VCL peptide decreases stress and stiffness by disrupting cytoskeletal protein‐protein interfaces (Figure 3D).
To further confirm that the TLN‐VCL peptides altered focal adhesion integrity, we used a model system, the serum‐starved A7r5 cells. Although this cultured cell line is noncontractile, these A7r5 cells express vascular smooth muscle differentiation markers when serum starved. When these cells were treated with the same concentration of TLN‐VCL peptide as shown above to decrease vessel stiffness, we noticed that focal adhesions were very difficult to visualize. Thus, we decreased the concentration of the TLN‐VCL peptide to half of that used for the biomechanics experiments, ie, to 50 μmol/L. At this lower concentration, we quantitated the size and number of peripheral focal adhesions in cells treated with either control (scrambled peptide) or TLN‐VCL peptides.
Typical cell images for cells exposed to no peptide (control), scrambled TLN‐VCL peptide, and TLN‐VCL peptide are shown in Figure 4A. In general, both control cells and cells treated with the scrambled peptide show numerous peripheral focal adhesions, but the TLN‐VCL–treated cells show sparse, small focal adhesions. We quantitated these findings by measuring the area of individual FAs (Figure 4B) and the number of peripheral FAs per cell (Figure 4C). As can be seen in Figure 4B, no significant difference occurred between the control and scrambled peptide–treated cells in FA size, but the cells treated with TLN‐VCL peptide displayed a significant decrease in FA size. Similarly, as shown in Figure 4C, no difference was detectable between the control and scrambled peptide–treated cells with respect to the number of peripheral FAs. However, the FA number was significantly decreased in the cells treated with the TLN‐VCL peptide. Thus, the TLN‐VCL peptide directly alters FA properties.
Figure 4.
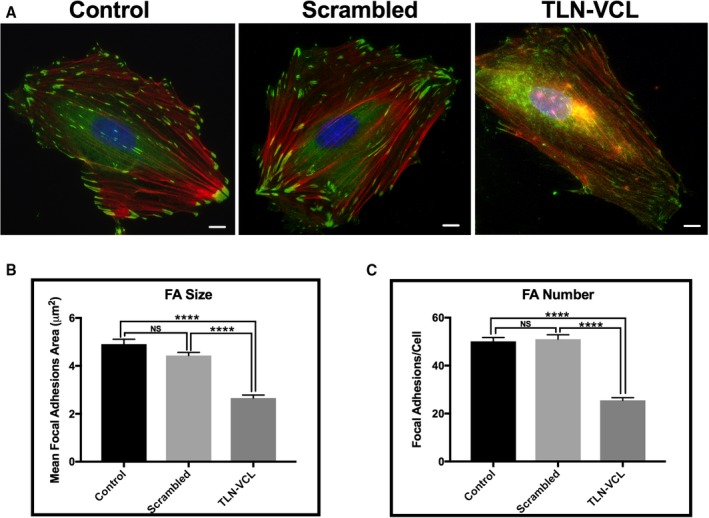
Talin‐vinculin (TLN‐VCL) decoy construct decreases focal adhesion size and number in A7r5 cells. A, Immunofluorescence microscopy of representative serum‐starved A7r5 cells treated with no peptide (control), rhodamine‐labeled scrambled peptide, or rhodamine‐labeled TLN‐VCL peptide. Cultured A7r5 cells were treated for 30 minutes with either scrambled or TLN‐VCL peptides followed by immunolabeling with vinculin to image focal adhesions (green) and phalloidin to image actin filaments (red). Nuclei were stained with DAPI (blue). Scale bars: 10 μm. B, Focal adhesion area is significantly decreased in cells treated with TLN‐VCL peptide as compared with cells treated with scrambled or no peptide. C, Treatment with the TLN‐VCL peptide significantly reduced focal adhesion numbers in A7r5 cells compared with scrambled peptide–treated cells or no peptide. No significant difference was seen in focal adhesion size and number in control cells compared to scrambled peptide–treated cells (n=65 cells). ****P<0.0001, 1‐way ANOVA, Tukey multiple comparisons test. FA indicates focal adhesion.
Coupling of Peptides to Microbubbles Allows Ultrasound‐Mediated Targeting Deep Into the Aortic Wall
The question arises as to how these peptides could be introduced into the aortic media in a living organism. One possibility is to use microbubbles47 to sequester the peptides after injection into the bloodstream and then to use ultrasound to create targeted release into only the proximal aorta. As is shown in Figure 5B, a FITC‐labeled TLN‐VCL peptide was conjugated to lipid‐coated microbubbles through a stable thioether linker. The linkage sequesters the peptide, but the peptide can be released inside the aorta by fragmenting the microbubbles with ultrasound pulses transmitted by a 1.1‐MHz focused transducer (Figure 5A). Stresses generated during bubble oscillation and destruction can permeabilize the endothelium reversibly48 and allow the peptide to cross the intima and reach smooth muscle cells in the media of the aorta. In Figure 5C, lower panel, the tissue has been ultrasound‐loaded, and the green peptide is seen appearing in cells at least half way across the thickness of the vessel wall. A summary of biomechanics measurements made on these tissues is shown in Figure 5D, where loading of a scrambled TLN‐VCL sequence peptide attached to microbubbles has no effect (black bar). Additionally, the active peptide, when attached to microbubbles in the absence of ultrasound has no effect (gray bar). The latter result confirms that the microbubbles do effectively sequester the peptide. In contrast, ultrasound‐released microbubble‐attached active peptides significantly decrease vessel stiffness (green bar). This, therefore, illustrates a potential approach by which these decoy peptides could be targeted to the aorta to reverse increased aortic stiffness.
Figure 5.
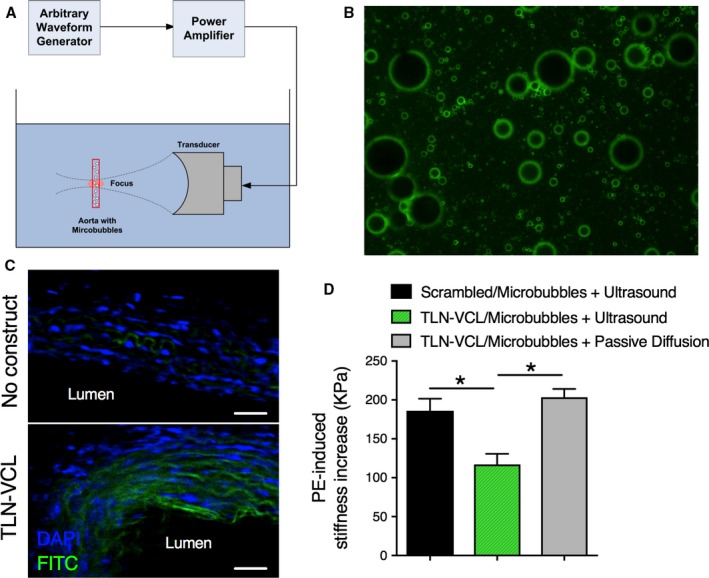
Coupling of peptides to microbubbles allows ultrasound‐mediated targeting into the aortic wall. A, Ultrasound apparatus used to release decoy peptide and load it into aortic tissues from young animals by bursting the microbubbles. B, Microbubbles coated with phospholipids conjugated to green FITC‐labeled peptides. C, Top, An unloaded ring with blue DAPI labeling of nuclei. Bottom, Green peptide loaded into cells by ultrasound‐mediated destruction of the peptide‐coated microbubbles. Both panels were obtained at identical microscope settings. D, Ultrasound‐mediated release of TLN‐VCL peptide decreases PE‐induced stiffness (green, n=6). No effect on stiffness was observed after ultrasound‐mediated release of scrambled peptide (black, n=5) or by passive diffusion of TLN‐VCL peptide conjugated to microbubbles (grey, n=3). *Difference between the bracketed bars is significant at a level of P<0.05.
Thus, we can successfully target vascular smooth muscle cytoskeletal function in 3 different ways: by interfering with the talin‐vinculin binding interface, the VASP EVH1 domain, or the N‐WASP CA domain.
Discussion
We have previously reported that increased aortic stiffness with aging, as measured in vivo by pulse wave velocity, occurs in C57BL/6J mice.23 We have also observed an age‐related increase in ex vivo activated smooth muscle stiffness of the mouse.16 Indeed, there is now a significant amount of evidence from multiple laboratories to suggest that the smooth muscle cell contributes significantly to the development of age‐related increases in active aortic stiffness.16, 17, 18, 19, 20, 21, 22, 49, 50 Our findings point to the FA complex and the attached, nonmuscle actin cytoskeleton as key sources for the increase in smooth muscle cell stiffness.
The data presented here are consistent with a model in which an age‐related impairment of FA signaling leads to impaired plasticity of VSMCs. We have previously demonstrated that Src tyrosine kinase is essential in the regulation of VSMC stiffness through its effects on a program of FA and actin cytoskeletal remodeling pathways. The tyrosine phosphorylation of the FA proteins CAS pY‐165, FAK pY‐925, and paxillin pY‐118 causes FA remodeling,20, 50, 51 which we previously found to occur through an endocytic recycling pathway, allowing cyclic remodeling of the FAs. The mechanisms that trigger the growth or degradation of individual FAs in the intact aorta are controversial, but the previously described concept of “slip” and “catch” bond formation at high versus low forces, respectively,52 may apply. Perhaps during the high forces of systole, some focal adhesion proteins break loose from the FA and enter into endosomal recycling pathways, allowing compliance and expansion of the aorta, but during lower, diastolic, forces, the FA surface rebuilds, promoting the return to the diastolic aortic diameter. This process may be typical of the young compliant aorta, but it appears to be lost in the aged, stiffened aorta.
Therefore, the FA remodeling pathway is a rational target for efforts designed to reduce aortic stiffness. Indeed, we show here that the targeting of the talin‐vinculin interface with a synthetic decoy peptide directly decreases FA size and number in serum‐starved A7r5 vascular smooth muscle cells and attenuates ex vivo stiffness, providing further evidence that the focal adhesion complex is a key modulator of vascular stiffness. Similarly, in the presence of peptides designed to interfere with actin polymerization, the actin peptides decrease actin polymerization as measured by the filamentous/globular actin ratio and decrease ex vivo stiffness. In future studies it will be of interest to see if this approach is more broadly effective in additional situations involving integrin signaling, which could be relevant not only to vascular muscle function but also to endothelial function, where changes in endothelial stiffness or adhesion of leukocytes induced by inflammatory cytokines have been demonstrated.53
Additionally, we demonstrated an association between increased vascular stiffness and common genetic variation of the gene encoding N‐WASP by interrogating results from the AortaGen Consortium genome‐wide association study of CFPWV. The AortaGen Consortium focused on CFPWV because it is the gold standard for clinical assessment of aortic stiffness.54 CFPWV has been shown to predict risk for cardiovascular disease and to reclassify risk after accounting for standard cardiovascular disease risk factors.2, 55
Because N‐WASP has previously been demonstrated to modulate smooth muscle contraction through actin polymerization/depolymerization pathways, we considered it as a candidate to influence vascular stiffness. Indeed, a decoy peptide targeted to the N‐WASP CA domain is effective in decreasing ex vivo aortic stiffness by directly inhibiting actin polymerization. In addition, by inhibiting VASP function using an EVH1 recombinant protein construct, we demonstrated that ex vivo stiffness could also be attenuated by influencing this separate actin polymerization pathway in vascular smooth muscle. Therefore, in the current study, we have shown, for the first time, that aortic stiffness can be modulated by targeting these 2 separate types of interfaces, FA protein‐protein and actin‐actin protein interfaces. We propose that the connections between the FAs and the nonmuscle actin cytoskeleton allow force and stiffness generated by the contractile filaments to be transmitted to the extracellular matrix. We found both strategies to be effective in decreasing ex vivo stiffness following VSMC activation. In vivo, this activation of the VSMC is likely caused by circulating catecholamines, inflammatory angiotensin receptor stimulation, or other circulating vasoactive agents.
We have shown here that the permeation tag and design of these peptides allow passive diffusion of the peptides into the vascular smooth muscle cells. With aging, additional barriers may arise from increased thickness and stiffness of the adventitia, but because luminal delivery is planned, we do not anticipate this to cause significant difficulties.
Importantly, we also report that by coupling the peptides with microbubbles and triggering release via ultrasound, we can generate focused ultrasound‐targeted microbubble delivery of the peptides into the media of the aortic wall, where the cell permeation moiety of the peptides can carry them into the intracellular space. It is possible that the endothelial cells may produce some age‐related changes in their barrier function, but the cavitation forces caused by microbubble rupture should be able to overcome any unexpected age‐related increases in the endothelial barrier.
There are limitations to our study that should be considered. Alteration of aortic stiffness with decoy peptides might cause other sorts of dysfunction. If the ultrasound‐mediated peptide release is targeted exclusively to the proximal aorta, adverse effects on the resistance vessels would be avoided. However, the decoy peptides decrease local vascular tone. Increased smooth muscle tone is known to increase damping of high‐frequency energy content, which raises concerns that too much of a decrease in stress and stiffness could promote thoracic aortic aneurysm. An inverse relationship between higher vessel wall stiffness and lower risk for aneurysm has been reported56 in patients with diabetes mellitus, where there is a decreased incidence of thoracic aortic aneurysms and dissection, which is thought to be due to protective cross‐linking of structural proteins. Additional study may be warranted to test the hypothesis that excessive reduction in aortic stiffness could have the negative side effect of increasing risk for aneurysm development. In vivo aging studies, first in animal models and eventually in human patients, will be needed to test all of these possibilities. Finally, cohorts included in the AortaGen Consortium meta‐analysis comprised exclusively white participants of European descent. Thus, our findings may not be generalizable to other racial or ethnic groups.
Conclusions
We have shown here that, first, perturbation of the cytoskeleton of the vascular smooth muscle cell can modulate aortic stiffness; second, it is possible to modulate this process with microbubble‐loaded decoy peptide constructs targeted to vascular smooth muscle cytoskeletal protein‐protein interfaces; and, third, genetic studies suggest that similar pathways may be involved in aortic stiffness in humans.
Appendix
The AortaGen Consortium
Gary F. Mitchell, MD, Cardiovascular Engineering, Inc, Norwood, MA; Germaine C. Verwoert, MSc, Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands; Department of Internal Medicine, Erasmus Medical Center, Rotterdam, The Netherlands; Member of the Netherlands Consortium on Healthy Aging (NCHA), Leiden, The Netherlands; Kirill V. Tarasov, MD, PhD, Laboratory of Cardiovascular Science, Intramural Research Program, National Institute on Aging, NIH, Baltimore, MD; Aaron Isaacs, PhD, Genetic Epidemiology Unit, Department of Epidemiology, Erasmus University Medical Center, Rotterdam, The Netherlands; Centre for Medical Systems Biology, Leiden, the Netherlands; Albert V. Smith, PhD, Icelandic Heart Association, Kopavogur, Iceland; University of Iceland, Reykjavik, Iceland; Yasmin, BSc, MA, PhD, Clinical Pharmacology Unit, Addenbrooke's Hospital, University of Cambridge, Cambridge, UK; Ernst R. Rietzschel, MD, PhD, Department of Cardiovascular Diseases, Ghent University Hospital, Ghent, Belgium; Department of Public Health, Ghent University, Ghent, Belgium; Toshiko Tanaka, PhD, Clinical Research Branch, Intramural Research Program, National Institute on Aging, NIH, Baltimore, MD; Yongmei Liu, MD, PhD, Division of Public Health Sciences, Department of Epidemiology and Prevention, Wake Forest University School of Medicine, Winston‐Salem, NC; Afshin Parsa, MD, MPH, University of Maryland School of Medicine, Baltimore, MD; Samer S. Najjar, MD, Laboratory of Cardiovascular Science, Intramural Research Program, National Institute on Aging, NIH, Baltimore, MD; Division of Cardiology, Washington Hospital Center, Washington, DC; Kevin M. O'Shaughnessy, MA, BM, DPhil, FRCP, Clinical Pharmacology Unit, Addenbrooke's Hospital, University of Cambridge, Cambridge, UK; Sigurdur Sigurdsson, MS, Icelandic Heart Association, Kopavogur, Iceland; Marc L. De Buyzere, MSc, Heart Centre, Ghent University Hospital, Ghent, Belgium; Martin G. Larson, ScD, Department of Mathematics and Statistics, Boston University, Boston, MA; NHLBI's Framingham Study, Framingham, MA; Mark P. S. Sie, MD, PhD, Genetic Epidemiology Unit, Department of Epidemiology, Erasmus University Medical Center, Rotterdam, The Netherlands; Jeanette S. Andrews, MS, Division of Public Health Sciences, Department of Biostatistical Sciences, Wake Forest University School of Medicine, Winston‐Salem, NC; Wendy S. Post, MD, MS, The Johns Hopkins Hospital, Baltimore, MD; Francesco U. S. Mattace‐Raso, MD, PhD, Department of Internal Medicine, Erasmus Medical Center, Rotterdam, The Netherlands; Member of the Netherlands Consortium on Healthy Aging (NCHA), Leiden, The Netherlands; Carmel M. McEniery, BSc, PhD, Clinical Pharmacology Unit, Addenbrooke's Hospital, University of Cambridge, Cambridge, UK; Gudny Eiriksdottir, MSc, Icelandic Heart Association, Kopavogur, Iceland; Patrick Segers, PhD, Biofluid, Tissue and Solid Mechanics for Medical Applications (bioMMeda), Institute Biomedical Technology, Ghent University, Belgium; Ramachandran S. Vasan, MD, NHLBI's Framingham Study, Framingham, MA; Evans Department of Medicine, Whitaker Cardiovascular Institute, and Preventive Medicine and Cardiology Sections, Boston University School of Medicine, Boston, MA; Marie Josee E. van Rijn, MD, PhD, Genetic Epidemiology Unit, Department of Epidemiology, Erasmus University Medical Center, Rotterdam, The Netherlands; Timothy D. Howard, PhD, Center for Human Genomics, Wake Forest University Health Sciences, Winston‐Salem, NC; Patrick F. McArdle, PhD, University of Maryland School of Medicine, Baltimore, MD; Abbas Dehghan, MD, DSc, Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands; Member of the Netherlands Consortium on Healthy Aging (NCHA), Leiden, The Netherlands; Elizabeth Jewell, BA, Department of Biostatistics, Center for Statistical Genetics, University of Michigan, Ann Arbor, MI; Stephen J. Newhouse, MSc, PhD, Clinical Pharmacology Unit, Addenbrooke's Hospital, University of Cambridge, Cambridge, UK; Sofie Bekaert, PhD, Department of Molecular Biotechnology, Faculty of Bioscience Engineering, Ghent University, Ghent, Belgium; Naomi M. Hamburg, MD, Evans Department of Medicine, Whitaker Cardiovascular Institute, and Preventive Medicine and Cardiology Sections, Boston University School of Medicine, Boston, MA; Anne B. Newman, MD, MPH, Department of Epidemiology, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA; Albert Hofman, MD, PhD, Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands; Angelo Scuteri, MD, PhD, Laboratory of Cardiovascular Science, Intramural Research Program, National Institute on Aging, NIH, Baltimore, MD; Dirk De Bacquer, PhD, Department of Public Health, Ghent University, Ghent, Belgium; Mohammad Arfan Ikram, MD, Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands; Member of the Netherlands Consortium on Healthy Aging (NCHA), Leiden, The Netherlands; Bruce Psaty, MD, PhD, Cardiovascular Health Study, Departments of Epidemiology, Medicine and Health Services, University of Washington, Seattle, WA; Center for HealthStudies, Group Health, Seattle, WA; Christian Fuchsberger, PhD, Department of Biostatistics, Center for Statistical Genetics, University of Michigan, Ann Arbor, MI; Matthias Olden, PhD, Department of Internal Medicine II, Department of Epidemiology and Preventive Medicine, University Medical Center Regensburg, Regensburg, Germany; Louise V. Wain, PhD, Department of Health Sciences, University of Leicester, Leicester, UK; Paul Elliott, MB, PhD, MRC‐HPA Centre for Environment and Health, School of Public Health, Imperial College London, St Mary's Campus, London, UK; Nicholas L. Smith, PhD, Department of Epidemiology, University of Washington, Seattle, WA; Seattle Epidemiologic Research and Information Center of the Department of Veterans Affairs Office of Research and Development, Seattle, WA; Group Health Research Institute, Group Health, Seattle, WA; Janine F. Felix, MD, PhD, Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands; Member of the Netherlands Consortium on Healthy Aging (NCHA), Leiden, The Netherlands; German Cancer Research Center (DKFZ), Division of Clinical Epidemiology and Aging Research, Heidelberg, Germany; Jeanette Erdmann, PhD, Medizinische Klinik II, Universität zu Lübeck, Lübeck, Germany; Joseph A. Vita, MD, Evans Department of Medicine, Whitaker Cardiovascular Institute, and Preventive Medicine and Cardiology Sections, Boston University School of Medicine, Boston, MA; Kim Sutton‐Tyrrell, PhD, Department of Epidemiology, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA; Eric J. G. Sijbrands, MD, PhD, Department of Internal Medicine, Erasmus Medical Center, Rotterdam, The Netherlands; Serena Sanna, PhD, Istituto di Neurogenetica e Neurofarmacologia, Consiglio Nazionale delle Ricerche, Cittadella Universitaria di Monseratto, Monserrato, Cagliari, Italy; Lenore J. Launer, MS, PhD, Intramural Research Program, Laboratory of Epidemiology, Demography and Biometry, National Institute on Aging, Bethesda, MD; Tim De Meyer, PhD, Department of Molecular Biotechnology, Faculty of Bioscience Engineering, Ghent University, Ghent, Belgium; Andrew D. Johnson, MD, NHLBI's Framingham Study, Framingham, MA; Center for Population Studies, National Heart, Lung, and Blood Institute, Bethesda, MD; Anna F. C. Schut, MD, PhD, Genetic Epidemiology Unit, Department of Epidemiology, Erasmus University Medical Center, Rotterdam, The Netherlands; David M. Herrington, MD, MHS, Department of Cardiology, Wake Forest University School of Medicine, Winston‐Salem, NC; Fernando Rivadeneira, MD, PhD, Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands; Department of Internal Medicine, Erasmus Medical Center, Rotterdam, The Netherlands; Member of the Netherlands Consortium on Healthy Aging (NCHA), Leiden, The Netherlands; Manuela Uda, PhD, Istituto di Neurogenetica e Neurofarmacologia, Consiglio Nazionale delle Ricerche, Cittadella Universitaria di Monseratto, Monserrato, Cagliari, Italy; Ian B. Wilkinson, MA, BM, FRCP, Clinical Pharmacology Unit, Addenbrooke's Hospital, University of Cambridge, Cambridge, UK; Thor Aspelund, PhD, Icelandic Heart Association, Kopavogur, Iceland; University of Iceland, Reykjavik, Iceland; Thierry C. Gillebert, MD, PhD, Department of Cardiovascular Diseases, Ghent University Hospital, Ghent, Belgium; Luc Van Bortel, MD, PhD, Heymans Institute of Pharmacology, Ghent University, Ghent, Belgium; Emelia J. Benjamin, MD, MSc, NHLBI's Framingham Study, Framingham, MA; Evans Department of Medicine, Whitaker Cardiovascular Institute, and Preventive Medicine and Cardiology Sections, Boston University School of Medicine, Boston, MA; Ben A. Oostra, PhD, Genetic Epidemiology Unit, Department of Epidemiology, Erasmus University Medical Center, Rotterdam, The Netherlands; Centre for Medical Systems Biology, Leiden, the Netherlands; Jingzhong Ding, MD, PhD, Sticht Center on Aging, Wake Forest University School of Medicine, Winston‐Salem, NC; Quince Gibson, MBA, University of Maryland School of Medicine, Baltimore, MD; André G. Uitterlinden, PhD, Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands; Department of Internal Medicine, Erasmus Medical Center, Rotterdam, The Netherlands; Member of the Netherlands Consortium on Healthy Aging (NCHA), Leiden, The Netherlands; Gonçalo R. Abecasis, PhD, Department of Biostatistics, Center for Statistical Genetics, University of Michigan, Ann Arbor, MI; John R. Cockcroft, BSc, MB, ChB, FRCP, Department of Cardiology, Wales Heart Research Institute, Cardiff University, Cardiff, UK; Vilmundur Gudnason, MD, PhD, Icelandic Heart Association, Kopavogur, Iceland; University of Iceland, Reykjavik, Iceland; Guy G. De Backer, MD, PhD, Department of Public Health, Ghent University, Ghent, Belgium; Luigi Ferrucci, MD, Clinical Research Branch, Intramural Research Program, National Institute on Aging, NIH, Baltimore, MD; Tamara B. Harris, MD, MS, Intramural Research Program, Laboratory of Epidemiology, Demography and Biometry, National Institute on Aging, Bethesda, MD; Alan R. Shuldiner, MD, University of Maryland School of Medicine, Baltimore, MD; Geriatric Research and Education Clinical Center, Veterans Administration Medical Center, Baltimore, MD; Cornelia M. van Duijn, PhD, Genetic Epidemiology Unit, Department of Epidemiology, Erasmus University Medical Center, Rotterdam, The Netherlands; Centre for Medical Systems Biology, Leiden, the Netherlands; Daniel Levy, MD, NHLBI's Framingham Study, Framingham, MA; National Heart, Lung, and Blood Institute, Bethesda, MD; Edward G. Lakatta, MD, Laboratory of Cardiovascular Science, Intramural Research Program, National Institute on Aging, NIH, Baltimore, MD; Jacqueline C. M. Witteman, PhD, Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands; Member of the Netherlands Consortium on Healthy Aging (NCHA), Leiden, The Netherlands.
Author Contributions
All authors contributed to the drafting of the paper and planning of experiments. Nicholson and Singh made equal primary contributions. Verwoert, Mitchell, Li, and Leavis contributed human data analysis, or unique methods or reagents. Experimental data were collected by Nicholson, Singh, Saphirstein, Gao, Li, and Chiu, under guidance from Morgan and Porter.
Sources of Funding
These studies were funded in part by the National Institute of Aging grants AG050599 and AG053274, the National Heart, Lung and Blood Institute's Framingham Heart Study (Contracts No. N01‐HC‐25195 and HHSN268201500001I), by HL080124, HL107385, and HL126136 and by the Aortic Stiffness Affinity Research Collaborative of the Evans Center at Boston University.
Disclosures
Dr Mitchell is the owner of Cardiovascular Engineering, Inc, a company that develops and manufactures devices to measure vascular stiffness and serves as a consultant to and receives grants and honoraria from Novartis, Merck, Servier, and Philips. The remaining authors have no disclosures to report.
Acknowledgments
The authors thank Dr Francesca Seta for critical input on the article.
(J Am Heart Assoc. 2018;7:e008926 DOI: 10.1161/JAHA.118.008926.)
Contributor Information
Kathleen G. Morgan, Email: kmorgan@bu.edu.
AortaGen Consortium:
Kirill V. Tarasov, Aaron Isaacs, Albert V. Smith, Yasmin, Ernst R. Rietzschel, Toshiko Tanaka, Yongmei Liu, Afshin Parsa, Samer S. Najjar, Kevin M. O'Shaughnessy, Sigurdur Sigurdsson, Marc L. De Buyzere, Martin G. Larson, Mark P. S. Sie, Jeanette S. Andrews, Wendy S. Post, Francesco U. S. Mattace‐Raso, Carmel M. McEniery, Gudny Eiriksdottir, Patrick Segers, Ramachandran S. Vasan, Marie Josee E. van Rijn, Timothy D. Howard, Patrick F. McArdle, Abbas Dehghan, Elizabeth Jewell, Stephen J. Newhouse, Sofie Bekaert, Naomi M. Hamburg, Anne B. Newman, Albert Hofman, Angelo Scuteri, Dirk De Bacquer, Mohammad Arfan Ikram, Bruce Psaty, Christian Fuchsberger, Matthias Olden, Louise V. Wain, Paul Elliott, Nicholas L. Smith, Janine F. Felix, Jeanette Erdmann, Joseph A. Vita, Kim Sutton‐Tyrrell, Eric J. G. Sijbrands, Serena Sanna, Lenore J. Launer, Tim De Meyer, Andrew D. Johnson, Anna F. C. Schut, David M. Herrington, Fernando Rivadeneira, Manuela Uda, Ian B. Wilkinson, Thor Aspelund, Thierry C. Gillebert, Luc Van Bortel, Emelia J. Benjamin, Ben A. Oostra, Jingzhong Ding, Quince Gibson, André G. Uitterlinden, Gonçalo R. Abecasis, John R. Cockcroft, Vilmundur Gudnason, Guy G. De Backer, Luigi Ferrucci, Tamara B. Harris, Alan R. Shuldiner, Cornelia M. van Duijn, Daniel Levy, Edward G. Lakatta, and Jacqueline C. M. Witteman
References
- 1. Mitchell GF, Guo CY, Benjamin EJ, Larson MG, Keyes MJ, Vita JA, Vasan RS, Levy D. Cross‐sectional correlates of increased aortic stiffness in the community: the Framingham Heart Study. Circulation. 2007;115:2628–2636. [DOI] [PubMed] [Google Scholar]
- 2. Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, Vita JA, Levy D, Benjamin EJ. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation. 2010;121:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci. 2010;65:1028–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nilsson PM, Boutouyrie P, Cunha P, Kotsis V, Narkiewicz K, Parati G, Rietzschel E, Scuteri A, Laurent S. Early vascular ageing in translation: from laboratory investigations to clinical applications in cardiovascular prevention. J Hypertens. 2013;31:1517–1526. [DOI] [PubMed] [Google Scholar]
- 5. Mitchell GF. Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end‐organ damage. J Appl Physiol. 2008;105:1652–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mitchell GF. Arterial stiffness and wave reflection: biomarkers of cardiovascular risk. Artery Res. 2009;3:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mitchell GF, van Buchem MA, Sigurdsson S, Gotal JD, Jonsdottir MK, Kjartansson O, Garcia M, Aspelund T, Harris TB, Gudnason V, Launer LJ. Arterial stiffness, pressure and flow pulsatility and brain structure and function: the Age, Gene/Environment Susceptibility—Reykjavik study. Brain. 2011;134:3398–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cooper LL, Woodard T, Sigurdsson S, van Buchem MA, Torjesen AA, Inker LA, Aspelund T, Eiriksdottir G, Harris TB, Gudnason V, Launer LJ, Mitchell GF. Cerebrovascular damage mediates relations between aortic stiffness and memory. Hypertension. 2016;67:176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Woodard T, Sigurdsson S, Gotal JD, Torjesen AA, Inker LA, Aspelund T, Eiriksdottir G, Gudnason V, Harris TB, Launer LJ, Levey AS, Mitchell GF. Mediation analysis of aortic stiffness and renal microvascular function. J Am Soc Nephrol. 2015;26:1181–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaess BM, Rong J, Larson MG, Hamburg NM, Vita JA, Levy D, Benjamin EJ, Vasan RS, Mitchell GF. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA. 2012;308:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elias MF, Robbins MA, Budge MM, Abhayaratna WP, Dore GA, Elias PK. Arterial pulse wave velocity and cognition with advancing age. Hypertension. 2009;53:668–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Najjar SS, Scuteri A, Shetty V, Wright JG, Muller DC, Fleg JL, Spurgeon HP, Ferrucci L, Lakatta EG. Pulse wave velocity is an independent predictor of the longitudinal increase in systolic blood pressure and of incident hypertension in the Baltimore Longitudinal Study of Aging. J Am Coll Cardiol. 2008;51:1377–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dernellis J, Panaretou M. Aortic stiffness is an independent predictor of progression to hypertension in nonhypertensive subjects. Hypertension. 2005;45:426–431. [DOI] [PubMed] [Google Scholar]
- 14. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–146. [DOI] [PubMed] [Google Scholar]
- 15. Janic M, Lunder M, Sabovic M. Arterial stiffness and cardiovascular therapy. Biomed Res Int. 2014;2014:621437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao YZ, Saphirstein RJ, Yamin R, Suki B, Morgan KG. Aging impairs smooth muscle‐mediated regulation of aortic stiffness: a defect in shock absorption function? Am J Physiol Heart Circ Physiol. 2014;307:H1252–H1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lacolley P, Regnault V, Segers P, Laurent S. Vascular smooth muscle cells and arterial stiffening: relevance in development, aging, and disease. Physiol Rev. 2017;97:1555–1617. [DOI] [PubMed] [Google Scholar]
- 18. Qiu H, Depre C, Ghosh K, Resuello RG, Natividad FF, Rossi F, Peppas A, Shen YT, Vatner DE, Vatner SF. Mechanism of gender‐specific differences in aortic stiffness with aging in nonhuman primates. Circulation. 2007;116:669–676. [DOI] [PubMed] [Google Scholar]
- 19. Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, Resuello RR, Natividad FF, Hunter WC, Genin GM, Elson EL, Vatner DE, Meininger GA, Vatner SF. Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res. 2010;107:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saphirstein RJ, Gao YZ, Jensen MH, Gallant CM, Vetterkind S, Moore JR, Morgan KG. The focal adhesion: a regulated component of aortic stiffness. PLoS One. 2013;8:e62461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sehgel NL, Sun Z, Hong Z, Hunter WC, Hill MA, Vatner DE, Vatner SF, Meininger GA. Augmented vascular smooth muscle cell stiffness and adhesion when hypertension is superimposed on aging. Hypertension. 2015;65:370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sehgel NL, Vatner SF, Meininger GA. “Smooth muscle cell stiffness syndrome”—revisiting the structural basis of arterial stiffness. Front Physiol. 2015;6:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nicholson CJ, Seta F, Lee S, Morgan KG. MicroRNA‐203 mimics age‐related aortic smooth muscle dysfunction of cytoskeletal pathways. J Cell Mol Med. 2017;21:81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burianek LE, Soderling SH. Under lock and key: spatiotemporal regulation of WASP family proteins coordinates separate dynamic cellular processes. Semin Cell Dev Biol. 2013;24:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang W, Huang Y, Wu Y, Gunst SJ. A novel role for RhoA GTPase in the regulation of airway smooth muscle contraction. Can J Physiol Pharmacol. 2015;93:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim HR, Graceffa P, Ferron F, Gallant C, Boczkowska M, Dominguez R, Morgan KG. Actin polymerization in differentiated vascular smooth muscle cells requires vasodilator‐stimulated phosphoprotein. Am J Physiol Cell Physiol. 2010;298:C559–C571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cipolla MJ, Osol G. Vascular smooth muscle actin cytoskeleton in cerebral artery forced dilatation. Stroke. 1998;29:1223–1228. [DOI] [PubMed] [Google Scholar]
- 28. Moreno‐Dominguez A, El‐Yazbi AF, Zhu HL, Colinas O, Zhong XZ, Walsh EJ, Cole DM, Kargacin GJ, Walsh MP, Cole WC. Cytoskeletal reorganization evoked by Rho‐associated kinase‐ and protein kinase C‐catalyzed phosphorylation of cofilin and heat shock protein 27, respectively, contributes to myogenic constriction of rat cerebral arteries. J Biol Chem. 2014;289:20939–20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Poythress RH, Gallant C, Vetterkind S, Morgan KG. Vasoconstrictor‐induced endocytic recycling regulates focal adhesion protein localization and function in vascular smooth muscle. Am J Physiol Cell Physiol. 2013;305:C215–C227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ohanian J, Pieri M, Ohanian V. Non‐receptor tyrosine kinases and the actin cytoskeleton in contractile vascular smooth muscle. J Physiol. 2015;593:3807–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mitchell GF, Verwoert GC, Tarasov KV, Isaacs A, Smith AV, Yasmin, Rietzschel ER, Tanaka T, Liu Y, Parsa A, Najjar SS, O'Shaughnessy KM, Sigurdsson S, De Buyzere ML, Larson MG, Sie MP, Andrews JS, Post WS, Mattace‐Raso FU, McEniery CM, Eiriksdottir G, Segers P, Vasan RS, van Rijn MJ, Howard TD, McArdle PF, Dehghan A, Jewell ES, Newhouse SJ, Bekaert S, Hamburg NM, Newman AB, Hofman A, Scuteri A, De Bacquer D, Ikram MA, Psaty BM, Fuchsberger C, Olden M, Wain LV, Elliott P, Smith NL, Felix JF, Erdmann J, Vita JA, Sutton‐Tyrrell K, Sijbrands EJ, Sanna S, Launer LJ, De Meyer T, Johnson AD, Schut AF, Herrington DM, Rivadeneira F, Uda M, Wilkinson IB, Aspelund T, Gillebert TC, Van Bortel L, Benjamin EJ, Oostra BA, Ding J, Gibson Q, Uitterlinden AG, Abecasis GR, Cockcroft JR, Gudnason V, De Backer GG, Ferrucci L, Harris TB, Shuldiner AR, van Duijn CM, Levy D, Lakatta EG, Witteman JC. Common genetic variation in the 3′‐BCL11B gene desert is associated with carotid‐femoral pulse wave velocity and excess cardiovascular disease risk: the AortaGen Consortium. Circ Cardiovasc Genet. 2012;5:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brozovich FV, Morgan KG. Stimulus‐specific changes in mechanical properties of vascular smooth muscle. Am J Physiol. 1989;257:H1573–H1580. [DOI] [PubMed] [Google Scholar]
- 33. Kimes BW, Brandt BL. Characterization of two putative smooth muscle cell lines from rat thoracic aorta. Exp Cell Res. 1976;98:349–366. [DOI] [PubMed] [Google Scholar]
- 34. Firulli AB, Han D, Kelly‐Roloff L, Koteliansky VE, Schwartz SM, Olson EN, Miano JM. A comparative molecular analysis of four rat smooth muscle cell lines. In Vitro Cell Dev Biol Anim. 1998;34:217–226. [DOI] [PubMed] [Google Scholar]
- 35. Johnson AD, Handsaker RE, Pulit SL, Nizzari MM, O'Donnell CJ, de Bakker PI. SNAP: a web‐based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang W, Wu Y, Du L, Tang DD, Gunst SJ. Activation of the Arp2/3 complex by N‐WASp is required for actin polymerization and contraction in smooth muscle. Am J Physiol Cell Physiol. 2005;288:C1145–C1160. [DOI] [PubMed] [Google Scholar]
- 37. Machesky LM, Mullins RD, Higgs HN, Kaiser DA, Blanchoin L, May RC, Hall ME, Pollard TD. Scar, a WASp‐related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc Natl Acad Sci USA. 1999;96:3739–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Violini S, Sharma V, Prior JL, Dyszlewski M, Piwnica‐Worms D. Evidence for a plasma membrane‐mediated permeability barrier to Tat basic domain in well‐differentiated epithelial cells: lack of correlation with heparan sulfate. Biochemistry. 2002;41:12652–12661. [DOI] [PubMed] [Google Scholar]
- 39. Ciobanasu C, Siebrasse JP, Kubitscheck U. Cell‐penetrating HIV1 TAT peptides can generate pores in model membranes. Biophys J. 2010;99:153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P. Mena, a relative of VASP and drosophila enabled, is implicated in the control of microfilament dynamics. Cell. 1996;87:227–239. [DOI] [PubMed] [Google Scholar]
- 41. Reinhard M, Jouvenal K, Tripier D, Walter U. Identification, purification, and characterization of a zyxin‐related protein that binds the focal adhesion and microfilament protein VASP (vasodilator‐stimulated phosphoprotein). Proc Natl Acad Sci USA. 1995;92:7956–7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, Gertler FB. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. [DOI] [PubMed] [Google Scholar]
- 43. Tang DD, Wu MF, Opazo Saez AM, Gunst SJ. The focal adhesion protein paxillin regulates contraction in canine tracheal smooth muscle. J Physiol. 2002;542:501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gilmore AP, Burridge K. Regulation of vinculin binding to talin and actin by phosphatidyl‐inositol‐4‐5‐bisphosphate. Nature. 1996;381:531–535. [DOI] [PubMed] [Google Scholar]
- 45. Gingras AR, Ziegler WH, Frank R, Barsukov IL, Roberts GC, Critchley DR, Emsley J. Mapping and consensus sequence identification for multiple vinculin binding sites within the talin rod. J Biol Chem. 2005;280:37217–37224. [DOI] [PubMed] [Google Scholar]
- 46. Witt S, Zieseniss A, Fock U, Jockusch BM, Illenberger S. Comparative biochemical analysis suggests that vinculin and metavinculin cooperate in muscular adhesion sites. J Biol Chem. 2004;279:31533–31543. [DOI] [PubMed] [Google Scholar]
- 47. Fisher NG, Christiansen JP, Klibanov A, Taylor RP, Kaul S, Lindner JR. Influence of microbubble surface charge on capillary transit and myocardial contrast enhancement. J Am Coll Cardiol. 2002;40:811–819. [DOI] [PubMed] [Google Scholar]
- 48. Aryal M, Arvanitis CD, Alexander PM, McDannold N. Ultrasound‐mediated blood‐brain barrier disruption for targeted drug delivery in the central nervous system. Adv Drug Deliv Rev. 2014;72:94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sehgel NL, Zhu Y, Sun Z, Trzeciakowski JP, Hong Z, Hunter WC, Vatner DE, Meininger GA, Vatner SF. Increased vascular smooth muscle cell stiffness: a novel mechanism for aortic stiffness in hypertension. Am J Physiol Heart Circ Physiol. 2013;305:H1281–H1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saphirstein RJ, Gao YZ, Lin QQ, Morgan KG. Cortical actin regulation modulates vascular contractility and compliance in veins. J Physiol. 2015;593:3929–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Min J, Reznichenko M, Poythress RH, Gallant CM, Vetterkind S, Li Y, Morgan KG. Src modulates contractile vascular smooth muscle function via regulation of focal adhesions. J Cell Physiol. 2012;227:3585–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barsegov V, Thirumalai D. Dynamics of unbinding of cell adhesion molecules: transition from catch to slip bonds. Proc Natl Acad Sci USA. 2005;102:1835–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huveneers S, Daemen MJ, Hordijk PL. Between Rho(k) and a hard place: the relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease. Circ Res. 2015;116:895–908. [DOI] [PubMed] [Google Scholar]
- 54. Townsend RR, Wilkinson IB, Schiffrin EL, Avolio AP, Chirinos JA, Cockcroft JR, Heffernan KS, Lakatta EG, McEniery CM, Mitchell GF, Najjar SS, Nichols WW, Urbina EM, Weber T; American Heart Association Council on Hypertension . Recommendations for improving and standardizing vascular research on arterial stiffness: a scientific statement from the American Heart Association. Hypertension. 2015;66:698–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ben‐Shlomo Y, Spears M, Boustred C, May M, Anderson SG, Benjamin EJ, Boutouyrie P, Cameron J, Chen CH, Cruickshank JK, Hwang SJ, Lakatta EG, Laurent S, Maldonado J, Mitchell GF, Najjar SS, Newman AB, Ohishi M, Pannier B, Pereira T, Vasan RS, Shokawa T, Sutton‐Tyrell K, Verbeke F, Wang KL, Webb DJ, Willum Hansen T, Zoungas S, McEniery CM, Cockcroft JR, Wilkinson IB. Aortic pulse wave velocity improves cardiovascular event prediction: an individual participant meta‐analysis of prospective observational data from 17,635 subjects. J Am Coll Cardiol. 2014;63:636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Prakash SK, Pedroza C, Khalil YA, Milewicz DM. Diabetes and reduced risk for thoracic aortic aneurysms and dissections: a nationwide case‐control study. J Am Heart Assoc. 2012;1:e000323 DOI: 10.1161/JAHA.111.000323. [DOI] [PMC free article] [PubMed] [Google Scholar]