

Abstract
Background
Exosomes are membranous vesicles generated by almost all cells. Recent studies demonstrated that mesenchymal stem cell–derived exosomes possessed many effects, including antiapoptosis, anti‐inflammatory effects, stimulation of angiogenesis, anticardiac remodeling, and recovery of cardiac function on cardiovascular diseases. However, targeting of exosomes to recipient cells precisely in vivo still remains a problem. Ligand fragments or homing peptides discovered by phage display and in vivo biopanning methods fused to the enriched molecules on the external part of exosomes have been exploited to improve the ability of exosomes to target specific tissues or organs carrying cognate receptors. Herein, we briefly elucidated how to improve targeting ability of exosomes to ischemic myocardium.
Methods and Results
We used technology of molecular cloning and lentivirus packaging to engineer exosomal enriched membrane protein (Lamp2b) fused with ischemic myocardium‐targeting peptide CSTSMLKAC (IMTP). In vitro results showed that IMTP‐exosomes could be internalized by hypoxia‐injured H9C2 cells more efficiently than blank‐exosomes. Compared with blank‐exosomes, IMTP‐exosomes were observed to be increasingly accumulated in ischemic heart area (P<0.05). Meanwhile, attenuated inflammation and apoptosis, reduced fibrosis, enhanced vasculogenesis, and cardiac function were detected by mesenchymal stem cell–derived IMTP‐exosome treatment in ischemic heart area.
Conclusions
Our research concludes that exosomes engineered by IMTP can specially target ischemic myocardium, and mesenchymal stem cell–derived IMTP‐exosomes exert enhanced therapeutic effects on acute myocardial infarction.
Keywords: acute myocardial infarction, cardiac regeneration, exosomes, ischemic myocardium‐targeting peptide, mesenchymal stem cell
Subject Categories: Translational Studies, Myocardial Regeneration, Ischemia, Stem Cells, Cell Therapy
Clinical Perspective
What Is New?
Exosomes engineered with the ischemic myocardium‐targeting peptide CSTSMLKAC motif can preferentially target ischemic myocardium, and mesenchymal stem cell–derived ischemic myocardium‐targeting peptide CSTSMLKAC–exosomes play important roles in the treatment of acute myocardial infarction.
What Are the Clinical Implications?
Mesenchymal stem cell–derived exosomes targeting to ischemic myocardium could be supplied as a novel carrier to transport therapeutic molecules, such as drugs or microRNAs, into ischemic heart tissues.
Introduction
Treatment strategies for acute myocardial infarction (AMI), such as traditional medicine, percutaneous coronary intervention therapy, and coronary artery bypass grafting, are insufficient to stave off cardiomyocyte losses, prevent the remodeling of the left ventricle (LV), and terminate the progress of heart failure. Regenerative medicine has been developed in recent years, such as mesenchymal stem cell (MSC) transplantation to repair myocardial infarction (MI) and restore heart function in both animal experiments1, 2, 3, 4 and patients.5, 6, 7, 8, 9, 10 There is mounting evidence that MSCs help repair or regenerate damaged tissues, primarily by means of secreting paracrine factors,11 including antiapoptotic factors,12 proangiogenic factors,13 and exosomes,14 rather than via the differentiation into cardiomyocytes.15, 16, 17
Exosomes are nano‐sized membrane vesicles, with the diameter ranging from 30 to 150 nm. As the natural carrier, they transfer signal molecules, such as protein, DNA, mRNAs, and microRNA,18 from original cells to recipient cells to facilitate cell‐to‐cell communication.19 Compared with conventional carriers, such as liposomes, nanospheres, micelles, microemulsion, and different kinds of conjugates, exosomes afford all the desirable advantages, such as low toxicity, low immunogenicity, high stability in circulation, biocompatibility, and biological barrier permeability,20 which makes them promising carriers for efficient drug or therapeutic gene delivery. Nevertheless, both the conventional carriers and exosomes are apt to be trapped in nonspecific organs, especially in the lung and the liver, leading to insufficiency in targeting myocardial ischemia area.21 Therefore, attempts to modify exosomes as effective carriers directly targeting ischemic myocardium have been considered. One method that has been harnessed is to restructure transmembrane proteins of exosomes to fuse with ligands or homing peptides, which confers exosomes’ targeting capability to tissues or organs carrying the corresponding receptors. However, only a few studies on tumor or nervous system exploited engineered exosomes for targeted disease therapy.22, 23 Recently, a new peptide sequence, CSTSMLKAC, that can preferentially target to ischemic region of the heart has been discovered via in vivo phage display technique.13 In our study, we clearly elucidated that fusion with ischemic myocardium‐targeting peptide CSTSMLKAC (IMTP) could enhance the specificity and efficiency of exosomes directly targeting ischemic myocardium.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure on request from the Institute for Cardiovascular Science, Soochow University (Suzhou, China).
Animals
One commonly used mouse strain, C57BL/6, was selected (sex unlimited) from the Experimental Animal Center of Soochow University. All animal procedures in this protocol were conducted in accordance with the Guidelines for the Care and Use of Research Animals, established by Soochow University.
BMSC Isolation and Culture Condition
A new improved approach of isolating BMSCs was adopted in our study. Humeri, tibiae, and femurs were severed from 2‐ or 3‐week‐old C57BL/6 mice. The bones were placed in a sterile culture dish containing appropriate DMEM/F‐12 (Gibco, United States) added with 0.1% penicillin/streptomycin and 2% fetal bovine serum (Hyclone, United States). Bone marrow cells were flushed thoroughly with a 26‐gauge syringe needle inserted into the marrow cavity. Afterwards, the diaphyses of humeri, tibiae, and femurs were chopped into ≈3‐mm3 sclerites and digested with collagenase II for 1 to 2 hours at 37°C in a shaker with a rotating speed of 40 g. During digestion, bone marrow cells were filtered using a 70‐μm strainer filter and centrifuged at 200 g for 5 minutes. The cell pellet was resuspended in C57BL/6 mBMSC complete culture medium (Cyagen Biosciences Inc, United States) and seeded in culture dishes. After digestion, bone chips were washed 3 times with basic DMEM/F‐12 and seeded into the culture dishes cocultured with the bone marrow cells at 37°C containing 5% CO2. After being seeded for 3 days, mBMSCs attached to the bottom of culture dishes, whereas hematopoietic cells were still suspended in the medium. The nonadherent cells were eliminated through the exchange of the cell medium at 72 hours and every 2 to 3 days thereafter. The mBMSCs from passage 3 to passage 8 were applied to in vivo and in vitro experiments.
Construction of Lamp2b With IMTP Plasmid and Transfection
Clonal expansion of Lamp2b was conducted using cDNA extracted from mouse skeletal muscle cells. Lamp2b+IMTP gene sequence was synthesized and purified by the GenePharma (China). Polymerase chain reaction (PCR) can be applied to expand the cDNA sequences. After being digested by BamHI (ThermoFisher Scientific, United States) and EcorI (ThermoFisher Scientific) restriction enzymes, the Lamp2b+IMTP and the lentivirus‐based vector pCDH‐CMV‐MCS‐EF1‐copGFP were run on a gel for purification. Then, the Lamp2b+IMTP gene sequence was joined to the vector via T4 DNA ligase (ThermoFisher Scientific) at 4°C for >12 hours. Then, transformation was conducted to drive the plasmid vector into the competent cells that would clone it. Next, bacteria were coated on agarose plates with ampicillin and incubated overnight at 37°C. An individual colony was singled out from the transformed plates, added into liquid Luria‐Bertani media in numbered tubes, and incubated in a shaker incubator at the shaking speed of 230 rpm/min overnight at 37°C. After centrifugation, the plasmid was extracted and sequenced to confirm the correct insertion.
Lentivirus packaging was conducted using 293T cells. The day before transfection, cells were trypsinized and counted. The cells were plated at a density of 2×106 cells in each culture dish so that they were 90% to 95% confluent on the day of transfection. A total of 8 μg of Lamp2b+IMTP plasmid DNA, 6 μg of psPAX, and 4 μg of pMD were diluted in 500 μL Opti‐MEM (Gibco). Lipofectamine 2000 transfection reagent (Invitrogen, United States) was diluted in Opti‐MEM to a total volume of 500 μL and was incubated for 5 minutes at room temperature. They were gently mixed together and incubated for 20 minutes at room temperature. The mixture was added to each 293T culture dish, and the cells were incubated at 37°C for 72 hours before harvesting lentivirus suspension. PEG 8000 (Sigma, Japan) was used to concentrate the lentivirus. After determination of virus titer, appropriate lentivirus was applied to infect BMSCs.
Purification of Exosomes
BMSCs were cultivated in DMEM/F‐12, added with 10% fetal bovine serum, which was previously centrifuged at 110 000g for 10 hours to remove exosomes already in bovine serum, and serum‐free medium (Cyagen, China) was used as the control exosome group. The BMSC culture supernatant containing exosomes was harvested 48 hours after transfection by centrifugation at 500g for 5 minutes. Exosomes were extracted by using Total Exosome Isolation reagent (Invitrogen), strictly according to the instructions. PBS was used to resuspend the purified exosomes. Exosomes were kept either at −80°C for long‐term preservation or at −20°C for short‐term preservation.
Identification of Exosomes by Western Blot and Transmission Electron Microscopy
Purified exosomes from BMSCs were identified using TSG101 (Abcam, United Kingdom), CD63 (Abcam), and CD9 (Abcam) by Western blot analysis. Modified exosomes were detected by Lamp2 (Abcam) by Western blot analysis. Purified exosomes from BMSCs were resuspended in PBS and fixed with 3% glutaraldehyde solution for half an hour at room temperature. Exosomes (20 µL) were added to a copper grid and dyed with 1% phosphotungstanic acid for 5 minutes at room temperature. The dried grid was examined using a Zeiss Libra 120 (Zeiss, Germany) electron microscope at 120 kV.
Characterization of Exosomes Using Nanoparticle Tracking Analysis
Nanoparticle tracking analysis (NTA) was conducted with a NanoSight LM10‐HSB instrument (A&P Instrument Co, United Kingdom) to automatically track and size purified exosomes simultaneously in real time. The determination of capturing and analyzing parameters was manually set (refer to the NanoSight Technical note). A high‐resolution particle‐size distribution was detected and processed with the NTA 2.2 Analytical Software Suite.
Internalization of Exosomes by H9C2
To investigate whether IMTP‐exosomes were able to target more efficiently and selectively to ischemic myocardium, IMTP‐exosomes, blank‐exosomes, and control‐exosomes were labeled with DiI (1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethylindocarbocyanine perchlorate) and cocultured with the rat cardiomyoblast cell line H9C2, which induced hypoxia by culturing in DMEM‐HG (Hyclone) containing 3% fetal bovine serum and incubating in a hypoxic incubator containing 94% N2, 5% CO2, and 1% O2 for >24 hours. After 2 hours of coculturing, the percentage of H9C2 internalized exosomes was confirmed by flow cytometry.
To verify whether IMTP‐exosomes fused with H9C2 cells, we labeled the exosomes with DiI (Thermo Fisher Scientific) and labeled hypoxia pretreated H9C2 cells with DiO (3,3?‐dioctadecyloxacarbocyanine perchlorate) (Thermo Fisher Scientific). Then, the exosomes were cocultured with the H9C2 cells for 30 or 60 minutes at 37°C. Images of colocalized exosomes and the H9C2 membrane were observed under fluorescent microscopy (Olympus, Japan).
Exosome Labeling
Near‐infrared fluorescent dye, 1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethyl indotricarbocyanine iodide (DiR), was purchased from Invitrogen and used to label exosomes. The dye powder was dissolved in dimethyl sulfoxide to prepare stock solution in accordance with specification. Purified exosomes were incubated with 50 mmol/L DiR for 30 minutes at 37°C. After being washed in 20 mL of PBS, excessive dye was eliminated by ultracentrifugation at 100 000g for 90 minutes. Then, the labeled exosomes were resuspended in PBS before use.
Mouse MI Model and Exosome Injection
MI was performed in female C57BL/6 mice (aged 8 weeks; weight, 20–25 g), according to the previous report.24 Briefly, animals were anesthetized with 4% chloral hydrate through intraperitoneal injection for one time and underwent tracheal intubation. Operations were performed by left thoracotomy and ligating the left anterior descending coronary artery with a 6‐0 nondestructive suture. Successful MI model was confirmed by hemodynamic parameters recorded by electrocardiography. The chest was carefully closed with a 3‐0 sterile suture after the lungs were fully inflated, and incisions were cleaned and disinfected.
DiR‐labeled exosomes derived from BMSCs were intravenously injected via the tail vein (4×109 particles/50 μg/100 μL PBS/mouse) immediately after coronary artery ligation. Internal fluorescent signals were monitored using the In Vivo Imaging System (IVIS) at 0.5, 2, 24, 48, and 72 hours after the injection to acquire serial fluorescence images. Wavelength was set for excitation at 745 nm and emission at 780 nm. The animals were imaged at 5 time points while supine. At the last time point, heart, liver, spleen, lung, and kidney from each mouse were harvested for ex vivo imaging. Fluorescence signal intensity was analyzed using the IVIS Spectrum in vivo imaging system and Living Image Software (PerkinElmer, United States) to determine tissue distribution of DiR‐labeled exosomes.
Cardiac Function
Transthoracic echocardiography was performed to assess cardiac function in response to MI and/or exosome therapy. Four time points, at postoperative day and day 3, day 14, and day 28 after the MI model was established, were selected to monitor echocardiography. Anesthesia was induced in the mice with 5% isoflurane and maintained with 1.5% isoflurane. The precordial region was dehaired and examined with VevoStrain software integrated with the Vevo2100 application (Visual Sonics Inc, Canada) furnished with a 400‐MHz transducer. Measurements were designed to be applied on parasternal long‐axis view and short‐axis view of the heart, achieved in B mode at the level of the papillary muscles. From these images, the following calculations, including ejection fraction, fractional shortening, LV end‐diastolic diameter, and LV end‐systolic diameter, were completed.
Inflammatory Response and Ventricular Remodeling
Hematoxylin and eosin staining, immunofluorescence staining of macrophages, and reverse transcription–quantitative PCR (RT‐qPCR) were applied to evaluate inflammatory response. Terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling assay, Masson trichrome staining, and immunohistochemistry were used to measure the degree of ventricular remodeling.
All mice were euthanized at either day 3 or day 28 after experimental infarction. Hearts were harvested and simply trimmed to dissociate out the upper part of ligation. Then, tissues were immersed in 4% paraformaldehyde for 48 hours at 4°C. Cryosections (6 μm thick) and paraffin‐embedded sections (5 μm thick) were prepared, according to the standard protocol mentioned in Cryosectioning Tissues25 and Cutting Sections of Paraffin‐Embedded Tissues,26 respectively.
Hematoxylin and eosin staining (Solarbio, China), Masson trichrome staining (Solarbio), and terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling assay (Roche, Sweden) were conducted according to reagent specification. High‐magnification light micrographs were captured using light microscopy. Image Pro Plus 6.0 was used to determine the collagen volume fraction. The number of apoptotic cells was manually counted in 10 randomly selected areas at a magnification of ×200.
Macrophages were polarized into the M1 or M2 populations. M1 macrophages (classically activated macrophages) were proinflammatory and characterized by the expression of tumor necrosis factor (TNF)‐α, whereas M2 macrophages (alternatively activated macrophages) were associated with anti‐inflammatory and expressed CD206. Immunofluorescence staining for TNF‐α, CD206, and CD68 was conducted. In brief, cryosections were blocked with 3% BSA for half an hour at room temperature. After incubation with first antibodies TNF‐α (1:100; Abcam), CD206 (1:100; Abcam), and CD68 (1:100; Abcam) overnight at 4°C, sections were then incubated with appropriate fluorescence‐labeled second antibodies (1:500; Beyotime, China) in a moist cassette at room temperature for 1 hour. All sections were stained with 4′,6‐diamidino‐2‐phenylindole (Invitrogen) and mounted with medium. A light microscope at ×200 magnification was used to detect fluorescence. The images were analyzed using Image Pro Plus 6.0.
The mRNA expression for the interleukin‐6, TNF‐α, and interleukin‐1β was analyzed using RT‐qPCR. Briefly, after MI, mice randomly received intravenous injection of blank‐exosomes or IMTP‐exosomes (50 μg/100 μL PBS/mouse). Three days after surgery, mice were euthanized and hearts were harvested. Total RNA of the hearts was extracted and reversely transcribed into cDNA by PrimeScript RT Reagen Kit (Takara, Japan). Reactions were conducted using the SYBR Premix Ex TaqRox plus (Takara, Japan) in a StepOnePlus qPCR equipment (Thermo Fisher Scientific), as follows: 95°C for 30 seconds, 40 cycles of 95°C for 5 seconds and 60°C for 30 seconds, and finally a melting curve of 95°C for 15 seconds, 60°C for 1 minute, and 95°C for 15 seconds. PCR amplification of cDNA was repeated 3 times. GAPDH was used as housekeeping gene. The mean value of each sample was expressed as the cycle threshold. Gene expression was determined as the difference in cycle threshold between target gene and GAPDH.
At 28 days after MI, each mouse received intracardial injection of BS1‐lectin (Vector, Germany) at 250 μg. Ten minutes later, anesthetized mice were lavaged with saline, followed by 10 mL of 4% paraformaldehyde through intracardiac injection. For immunohistochemistry, myocardial sections (5 μm thick) were deparaffinized and blocked with 3% BSA for half an hour at room temperature. After being incubated with anti–BS1‐lectin goat polyclonal antibody (1:100; Life Technologies, United States) and anti–α‐smooth muscle actin mouse polyclonal antibody (1:100; Life Technologies) overnight, all slides were incubated with appropriate fluorescence‐labeled second antibodies (1:500) at room temperature for 1 hour. All sections were mounted and detected using a light microscope at ×200 magnification. The images were analyzed using Image Pro Plus 6.0.
Statistical Analysis
Quantitative data were exhibited as mean±SEM. Nonparametric data were analyzed by 2‐tailed Mann‐Whitney U test. Comparisons between multiple groups were specified for significance by Kruskal‐Wallis test, with Dunn's posttest to compare all pairs of groups. The data reflected changes over time and were analyzed with 2‐way ANOVA with repeated measures. P<0.05 was defined as statistically significant.
Results
Flowchart for Reconstruction of IMTP‐Exosomes
Processes of production, purification, and systematic injection of targeted exosomes for MI therapy are exhibited in the schematic diagram (Figure 1A). Molecular cloning and lentivirus packaging techniques were used to introduce the IMTP identified by in vivo phage display fused with Lamp2b on the external surface of exosomes. Lamp2b was cloned with cDNA extracted from mouse skeletal muscle cells, and BamHI and EcorI restriction sites were inserted after the signal peptide sequence, together with glycine linkers using appropriate primers. IMTP was cloned into Lamp2b between linkers after the signal peptide. The full gene sequence was then cloned downstream of CMV (cytomegalovirus) promoter with BamHI and EcorI restriction sites into the vector plasmid (Figure 1B).
Figure 1.
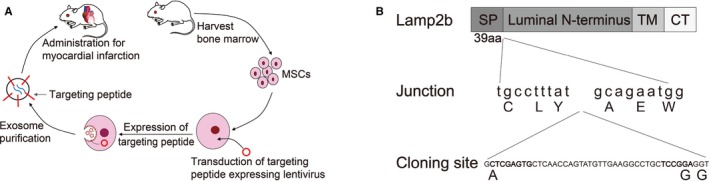
Reconstruction of exosomes exhibited with ischemic myocardium‐targeting peptide CSTSMLKAC fused with Lamp2b. A, Schematic diagram of production, purification, and systematic injection of targeted exosome for myocardial infarction therapy. B, Schematic diagram of readministration of Lamp2b protein. CT indicates C terminus; MSC, mesenchymal stem cell; SP, signal peptide; TM, transmembrane domain.
Isolation, Identification, and Successful Modification of Mouse MSCs
Cell morphological features of mouse MSCs were observed using inverted phase contrast microscopy (Figure 2A). At 72 hours after initial culture, fibroblastoid cells moved out from the collagenase II digested humerus, tibia, and femur sclerites. After 1 more passage, more cells migrated out and grew around the bone chips. A spindle‐ or spiral‐shaped fibroblastic colony formed. A morphologically homogeneous population of fibroblast‐like cells was developed in the third passage. Cell population trended toward stability and homogenized after being subcultured up to the third passage. These cells were homogeneously positive for mesenchymal markers CD44 (72.83%) and CD105 (31.82%) and progenitor cell marker Sca‐1 (stem cell antigen‐1) (93.79%) but negative for hematopoietic markers CD45 (0.64%) and CD11b (0.5%) and endothelial cell marker CD31 (0.38%) (Figure 2B). Results obtained showed that the cultured cells possessed the characteristic phenotype of BMSCs, and BMSCs generated by the improved protocol had less hematopoietic cell contamination by comparison with traditional bone marrow culture method. The day before transfection, BMSCs were trypsinized and counted. The cells were plated at a density of 2×106 cells in each culture dish so that they were ≈70% confluent on the day of transfection. After 48 hours, obvious green fluorescence signals were detected in the cells using inverted phase contrast microscopy, which indicated that BMSCs were successfully infected with lentivirus. The infection efficiency of both Lamp2b+IMTP gene overexpression lentivirus and empty vector lentivirus achieved 80% to 90% (Figure 2C). To identify whether the IMTP was fused with Lamp2b, agarose gel electrophoresis of RT‐qPCR products and Western blot of cell proteins were performed and revealed that the IMTP was successfully cloned into Lamp2b (Figure 2D and 2E).
Figure 2.
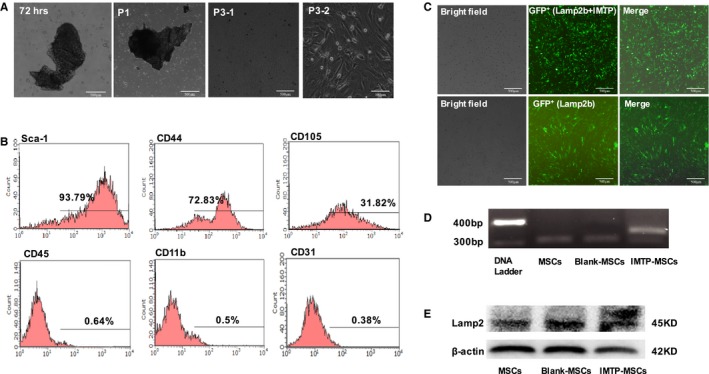
Isolation, identification, and modification of mouse mesenchymal stem cells (mMSCs). A, Cell morphological observation of mMSCs using inverted phase contrast microscopy. Bars: 500 μm (72 hours, passage [P] 1 and P3‐1) and 100 μm (P3‐2). B, Identification of mMSCs with Sca‐1, CD44, CD11b, CD31, CD105, and CD45. C, Infection efficiency of lentivirus in BMSCs. The infection efficiency of both Lamp2b+ischemic myocardium‐targeting peptide CSTSMLKAC (IMTP) gene overexpression lentivirus (top panel) and empty vector lentivirus (bottom panel) achieved 80% to 90%. D, Agarose gel electrophoresis of reverse transcription–quantitative polymerase chain reaction products in normal mMSCs and mMSCs that were transfected with either plasmid encoding Lamp2b or plasmid encoding Lamp2b fused with IMTP (Lamp2b+IMTP). GAPDH was used as internal reference and was detected at similar levels in all groups. E, Western blot analysis of proteins in normal mMSCs and mMSCs that were transfected with either plasmid encoding Lamp2b or plasmid encoding Lamp2b fused with IMTP (Lamp2b+IMTP). β‐Actin was used as control and was detected at similar levels in all groups. GFP indicates green fluorescent protein.
Identification of Exosomes
Total exosomes were isolated from untransfected and transfected BMSC culture supernatant using Total Exosomes Isolation Reagent in accordance with the reagent specification. The purified exosomes were identified according to protein expression, morphological features, and size and by Western blot analysis, transmission electron microscopy, and NTA, respectively. We obtained 400 to 600 μg (3.2–4.8×1010 particles) of exosomes per 107 cells. Western blot analysis confirmed that mouse MSC exosomes expressed typical exosomal protein TSG101, CD63, and CD9 without contamination of cellular protein (Figure 3A). The exosomes were morphologically homogeneous, with size ranging from 30 to 150 nm and peaking at 134 nm, as examined by NTA. Reconstruction did not affect the physical properties of the IMTP‐exosomes, according to the transmission electron microscope images (Figure 3B) and the NTA results (Figure 3C).
Figure 3.
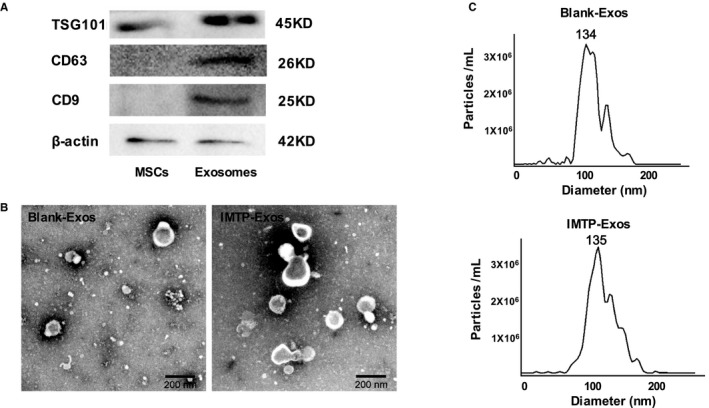
A, Western blot analysis of mouse mesenchymal stem cells (MSCs) and exosomes by TSG101, CD63, and CD9. B, Transmission electron microscope images of control (blank‐exosomes [Exos]) and ischemic myocardium‐targeting peptide CSTSMLKAC (IMTP)–positive exosomes (IMTP‐Exos). Bar=200 nm. C, Size distribution of blank‐Exos and IMTP‐Exos on the basis of nanoparticle tracking analysis measurements peaking at 134 and 135 nm, respectively.
In Vitro Targeting of IMTP‐Exosomes
To investigate whether IMTP‐exosomes were able to target H9C2 cells subjected to hypoxic injury more efficiently, IMTP‐exosomes or blank‐exosomes were labeled with DiI and cocultured with hypoxia preconditioned H9C2 cells. Flow cytometry illustrated that IMTP‐exosomes bound to injured H9C2 cells more efficiently than blank‐exosomes and control‐exosomes (43.96±1.21% versus 38.66±0.86% and 38.82±0.52%, respectively) (Figure 4A), indicating that the IMTP augmented the binding ability of exosomes to injured H9C2 cells. To verify whether IMTP‐exosomes fused with injured H9C2 cells, we labeled the exosomes with DiI (red) and H9C2 cell membranes with DiO (green). Then, we cocultured them for 30 or 60 minutes at 37°C. Fluorescent microscopy analysis observed relatively low levels of interaction in control‐exosome– and blank‐exosome–treated cells up to 60 minutes (Figure 4B and 4C). By contrast, merging of red and green fluorescence on the cell surface appeared within 30 minutes and increased over time (up to 60 minutes) in the IMTP‐exosome–treated group (Figure 4D); this approved the targeting ability of IMTP‐exosomes to injured H9C2 cells.
Figure 4.
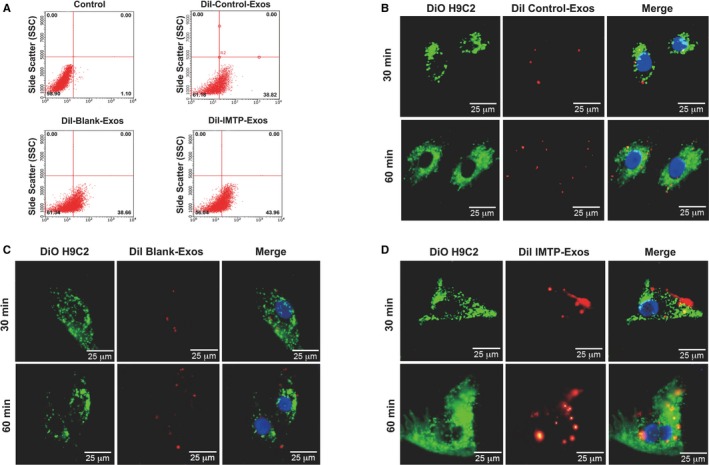
Binding of ischemic myocardium‐targeting peptide CSTSMLKAC–exosomes (Exos) to a rat cardiomyoblast cell line H9C2 in vitro. A, Flow cytometric analysis of exosomes labeled with DiI binding to H9C2 cells. The percentages represent the proportion of H9C2 cells that internalized DiI‐labeled exosomes (DiI‐positive H9C2 cells). B through D, Confocal microscopy images of colocalization of DiI‐labeled exosomes (red) and the DiO‐labeled H9C2 cell membrane (green) at different time points. Cell nuclei were dyed with 4′,6‐diamidino‐2‐phenylindole (blue). Bar=25 μm.
To detect the antiapoptosis function of IMTP‐exosomes, H9C2 cells were induced by hypoxia and treated with different exosomes. Although more IMTP‐exosomes were internalized by injured H9C2 cells than control‐exosomes and blank‐exosomes, antiapoptosis function of IMTP‐exosomes did not achieve the desired effect (Figure 5A and 5B).
Figure 5.
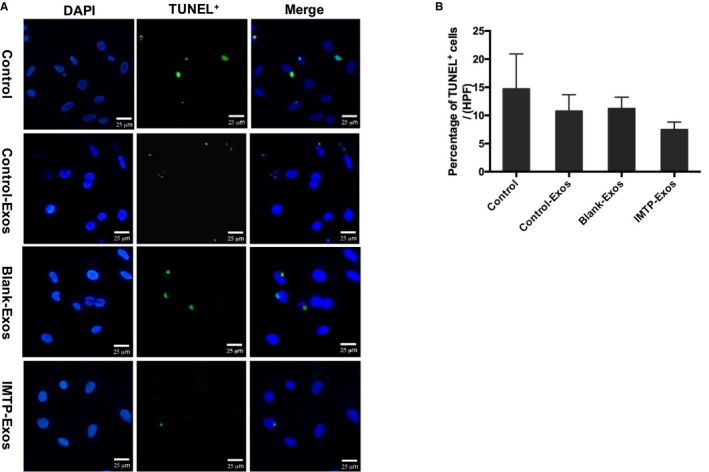
Measurements of H9C2 apoptosis by terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling (TUNEL) assays. A, Representative images of TUNEL staining (green) in the hypoxic injured H9C2 cells in different groups. Bar=25 μm. Many TUNEL‐positive nuclei were observed in the H9C2 cells from the control group. In contrast, in the control‐exosomes (Exos), blank‐Exos, and ischemic myocardium‐targeting peptide CSTSMLKAC (IMTP)–Exos treated groups, TUNEL‐positive nuclei were much less abundant, especially in the IMTP‐Exos treated group. B, Statistical analysis of TUNEL‐positive cells in each group using Image‐Pro Plus 6.0 software. At ×200 magnification, 5 regions in the peri‐infarct areas on each slide were randomly examined. Data were presented as mean±SEM. The same results with the fluorescence images were achieved; however, no statistical differences were found between the groups. DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
Biodistribution and Delivery Efficiency of Modified Exosomes
To track exosomes and their targeting ability in vivo, near‐infrared fluorescence tracer DiR‐labeled blank‐exosomes and IMTP‐exosomes were injected systematically into the mice experiencing MI. Imaging was performed with an IVIS Spectrum using an excitation spectrum at 750 nm and an emission spectrum at 782 nm. Image processing and the region of interest calculation were performed using Living Image Software. As depicted in Figure 6A, mice were optically imaged at 0.5, 2, 12, 48, and 72 hours after injection of exosomes. The strongest signal was observed in the mouse tail immediately after injection, and a gradually enhanced fluorescence was also detected in the abdomen and chest. Stable and clear fluorescent signal was achieved after 12 hours, peaked at 48 hours, and continued to 72 hours. After 72 hours, the signal began to degrade (data not shown). To further figure out the targeting ability of blank‐exosomes and IMTP‐exosomes, animals were euthanized and organs were separated at the time point of 72 hours. We detected the signals of the liver, spleen, lung, and kidney at the same time (Figure 6B); almost no marked between‐group difference was achieved in terms of passive exosome transfer. Data showed that the signal detected in the MI region in the IMTP‐exosome treatment group was significantly stronger than that of the blank‐exosome treatment group (P<0.05) (Figure 6C and 6D).
Figure 6.
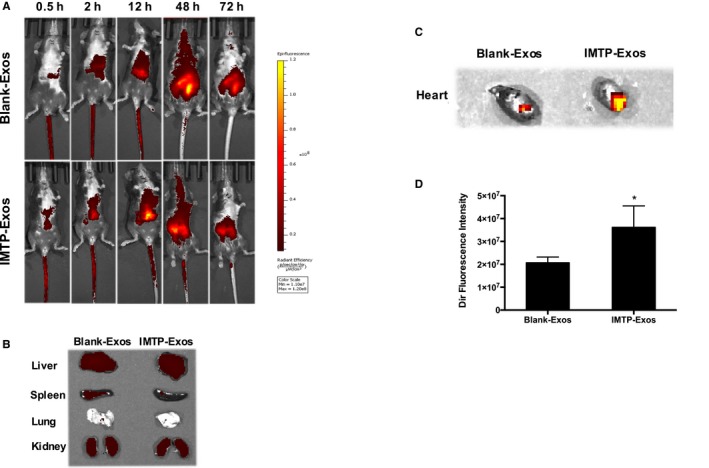
In vivo tracking of 1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethyl indotricarbocyanine iodide (DiR)–labeled ischemic myocardium‐targeting peptide CSTSMLKAC–exosomes (IMTP‐Exos). A, Mice underwent myocardial infarction (MI) surgery and received DiR‐labeled blank‐Exos or IMTP‐Exos treatment through tail intravenous injection. Fluorescence signals were detected up to 72 hours after injection. B, Ex vivo fluorescence imaging of major organs from mice 72 hours after intravenous injection with DiR‐labeled blank‐Exos or IMTP‐Exos. C, Signal accumulation in the MI region in the IMTP‐Exos group and the blank‐Exos group. D, The intensity of fluorescence signals from the ischemic myocardium quantified using an IVIS. Data are shown as mean±SEM. n=9 mice. *P<0.05.
IMTP‐Exosome Treatment Significantly Inhibited Inflammatory Response
We again verified the targeting ability of IMTP‐exosomes to ischemic myocardium through intravenous injection of DiI‐labeled exosomes. As depicted in Figure 7A, more DiI‐labeled IMTP‐exosomes were accumulated in the MI region contrasted with blank‐exosomes. Quantification results of fluorescence signals in ischemic myocardium confirmed that signal in the IMTP‐exosome group was significantly higher than in the blank‐exosome group (Figure 7B).
Figure 7.
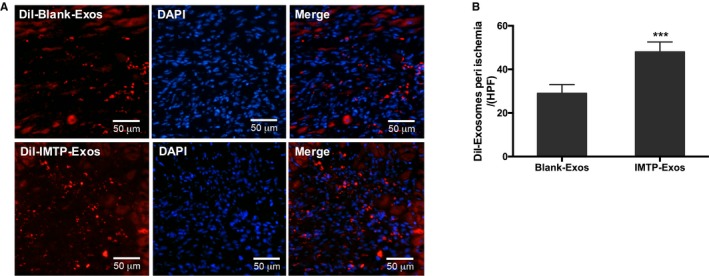
Recruitment of exosomes in the myocardial infarction region. A, Representative images of recruitment of DiI‐labeled exosomes in ischemic myocardium. n=3 mice. Bar=50 μm. B, Quantification of fluorescence signals in ischemic myocardium. n=3 mice. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; Exos, exosomes; IMTP, ischemic myocardium‐targeting peptide CSTSMLKAC. ***P<0.001.
To detect the effect of exosomes on inflammation within the ischemic area, the mRNA levels of the proinflammatory factors, including interleukin‐6, TNF‐α, and interleukin‐1β, were analyzed using RT‐qPCR. The mRNA levels of interleukin‐6, TNF‐α, and interleukin‐1β in the mice treated with blank‐exosomes were statistically higher than those treated with IMTP‐exosomes (P<0.0001) (Figure 8A). Hematoxylin and eosin staining confirmed the condition of inflammatory cellular infiltration. In the PBS group, cardiomyocytes showed intracellular edema, swollen and arranged irregularly. Inflammatory cells in the PBS group were remarkably elevated compared with the other 2 groups, and the IMTP‐exosome group exhibited the slightest inflammation (Figure 8B).
Figure 8.
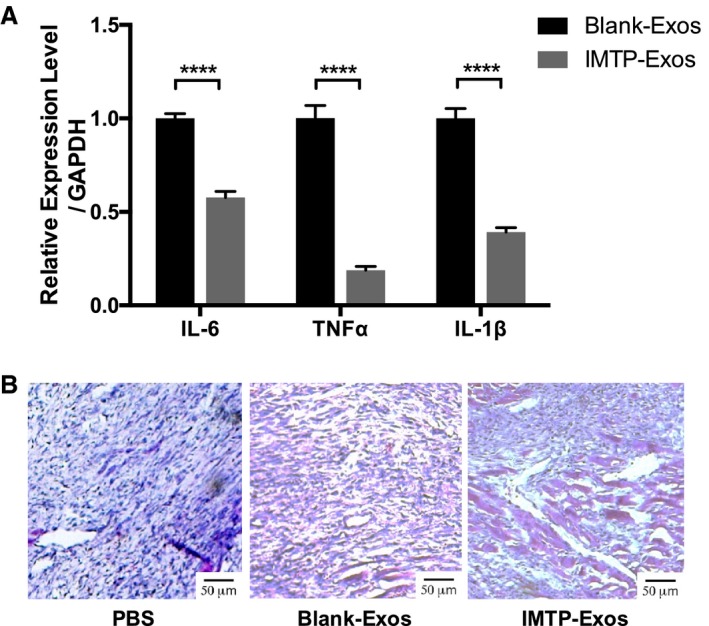
The effect of exosomes on inflammatory response. A, mRNA expression of interleukin (IL)‐6, tumor necrosis factor (TNF)‐α, and IL‐1β in both groups. Each bar represented as mean±SEM. n=3 mice. B, Hematoxylin and eosin staining of myocardial infarction region. n=3 mice. Bar=50 μm. Exos indicates exosomes; IMTP, ischemic myocardium‐targeting peptide CSTSMLKAC. ****P<0.0001.
Next, to detect the subsets of macrophage in the PBS group, the blank‐exosome group, and IMTP‐exosome group, heart slides were investigated for the recently identified M1/M2‐specific marker TNF‐α or CD206 (Figure 9A). TNF‐α+ M1 macrophages were dramatically increased in the PBS group and the blank‐exosome group, which was attenuated in the IMTP‐exosome group (Figure 9B). Moreover, CD206+ M2 macrophages were slightly increased in the IMTP‐exosome group compared with the other 2 groups (Figure 9C). CD68+ macrophages were reduced in ischemic area in the IMTP‐exosome group compared with the other 2 groups (Figure 9D).
Figure 9.
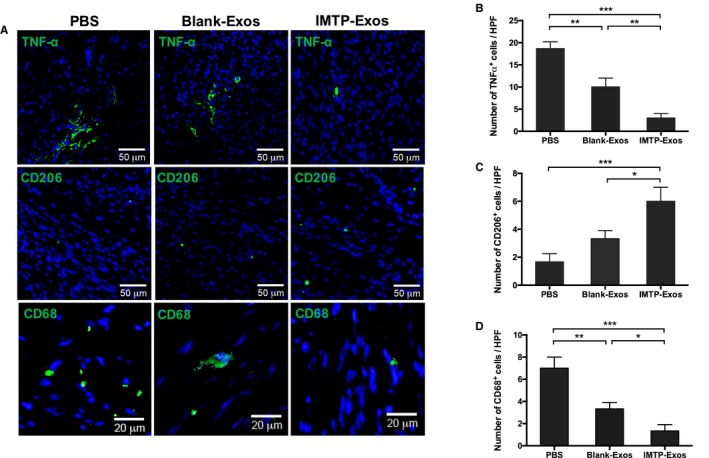
Detection of macrophage polarization and inflammatory marker in heart tissues by immunofluorescence. A, Green fluorescence indicated tumor necrosis factor (TNF)‐α+ M1 macrophages (top panels), CD206+ M2 macrophages (middle panels), and CD68+ cells (bottom panels). Nuclei were stained blue with 4′,6‐diamidino‐2‐phenylindole. n=3. Bars: 50 μm (top and middle panels) and 20 μm (bottom panels). B through D, Quantification of fluorescence signals in ischemic myocardium. n=3 Representative fields. Exos indicates exosomes; HPF, high‐power field; IMTP, ischemic myocardium‐targeting peptide CSTSMLKAC. *P<0.05, **P<0.01, ***P<0.001.
Increased Vasculature Was Observed After IMTP‐Exosome Treatment
For the observation of capillaries, BS1‐lectin staining was performed on paraffin‐embedded heart sections. The IMTP‐exosome group exhibited significantly increased capillary density in the ischemic border zone compared with the blank‐exosome group (65.0±5.5/high‐power field [HPF] versus 40.5±5.0/HPF; P<0.001) and the PBS group (65.0±5.5/HPF versus 24.5±4.2/HPF; P<0.001) (Figure 10A and 10B). Arterioles in myocardial tissues were also detected by immunohistochemistry using α‐smooth muscle actin antibody. Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole. The number of arterioles was highest in the IMTP‐exosome group compared with the blank‐exosome group (11.5±1.3/HPF versus 5.8±1.5/HPF; P<0.01) and the PBS group (11.5±1.3/HPF versus 3.5±1.3/HPF; P<0.001) (Figure 10C and 10D).
Figure 10.
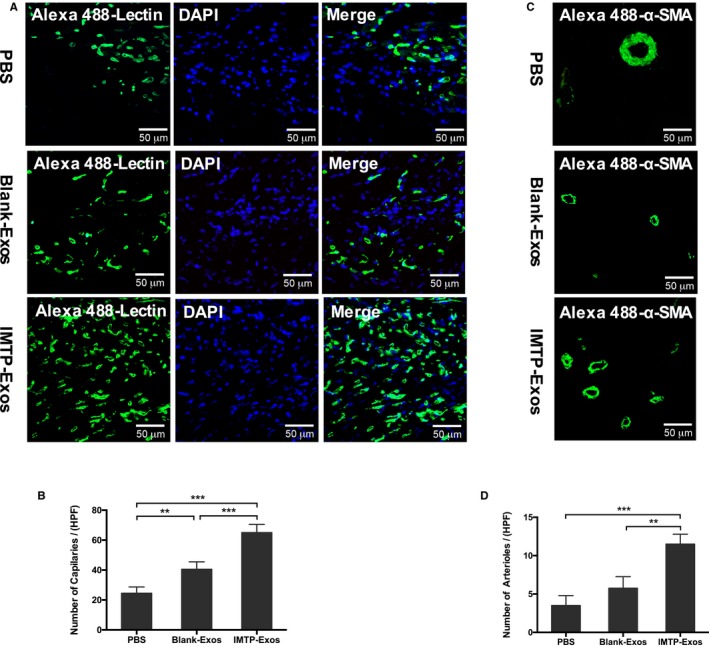
The effect of exosomes on revascularization. A, Capillaries that stained with Alexa 488–lectin (green) in the myocardial infarction (MI) border zone. Bar=50 μm. B, Statistical analysis of Alexa 488–lectin positive vessels per high‐power field (HPF). Each bar represented as mean±SEM. n=5 Representative fields. C, Detection of arterioles by α‐smooth muscle actin (SMA) (green) in the MI border zone. Bar=50 μm. D, Statistical analysis of α‐SMA–positive arterioles per HPF. Each bar represented as mean±SEM. n=5 Representative fields. DAPI indicates 4′,6‐diamidino‐2‐ phenylindole; Exos, exosomes; IMTP, ischemic myocardium‐targeting peptide CSTSMLKAC. **P<0.01, ***P<0.001.
Myocardial Apoptosis Was Reduced by IMTP‐Exosome Treatment
To illuminate whether the exosomes were able to restrain apoptotic cell death, we used terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling staining to identify chromosomal fragmentation and quantify apoptosis in heart paraffin‐embedded sections. As depicted in Figure 11A, a marked increase in the number of terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling–positive cells was observed in the PBS group, indicating the induction of apoptosis, and such an increase was mitigated after treatment with blank‐exosomes and IMTP‐exosomes. Quantitative results showed that, compared with the PBS group, blank‐exosomes and IMTP‐exosomes significantly inhibited myocardial apoptosis (IMTP‐exosomes, 4.5±1.3/HPF; blank‐exosomes, 11.3±2.6/HPF; PBS, 28.3±3.8/HPF; IMTP‐exosomes and blank‐exosomes versus PBS, P<0.001). IMTP‐exosomes were more effective in apoptosis resistance than blank‐exosomes (P<0.05) (Figure 11B).
Figure 11.
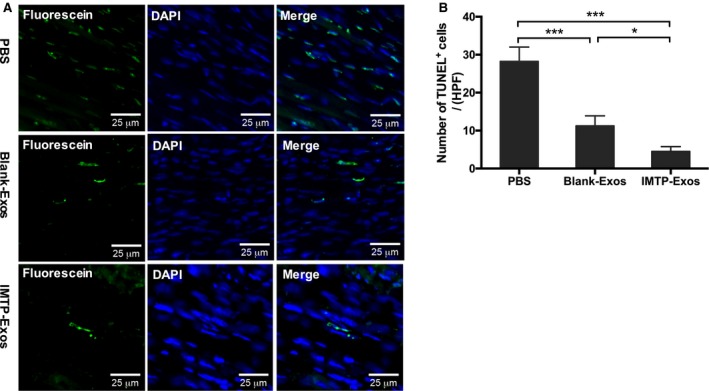
The effect of exosomes (Exos) on myocardial apoptosis. A, Representative images of terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling (TUNEL) staining (green) in the ischemic border zone in different groups. Bar=25 μm. B, Statistical analysis of TUNEL‐positive cells in each group. Data are described as mean±SEM. n=5 Representative fields. DAPI indicates 4′,6‐diamidino‐2‐phenylindole; HPF, high‐power field; IMTP, ischemic myocardium‐targeting peptide CSTSMLKAC. *P<0.05, ***P<0.001.
IMTP‐Exosome Treatment Significantly Reduced Infarct Size and Preserved Cardiac Function
We then detected the effect of exosomes on fibrosis and cardiac function. As depicted in Figure 12A and 12B, fibrosis length was remarkably reduced in the IMTP‐exosome–treated mice than in the blank‐exosome–treated mice (31.8±1.9% versus 47.9±1.1%; P<0.001). The PBS group had a significantly broader infarct length compared with the other 2 groups (55.9±2.0%; IMTP‐exosomes versus PBS, P<0.001; blank‐exosomes versus PBS, P<0.01).
Figure 12.
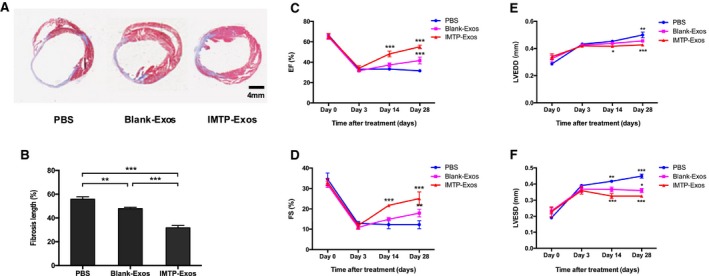
Assessment of fibrosis in the infarct area and cardiac function after induction of myocardial infarction. A, Myocardial sections stained with Masson's trichrome highlight the collagen fiber in blue. The number of mice in each group was 3. B, The scale bar showed the fibrosis length as mean±SEM. C through F, Temporal changes of ejection fraction (EF), fractional shortening (FS), left ventricular end‐diastolic diameter (LVEDD) and LV end‐systolic diameter (LVESD) were shown in the line chart. n=3 mice. In C and D, the star above the ischemic myocardium‐targeting peptide CSTSMLKAC–exosomes (IMTP‐Exos) group referred to difference against the PBS group and the blank‐Exos group. The star above the blank‐Exos group referred to difference against the PBS group. In E and F, the star below the IMTP‐Exos group referred to difference against the PBS group. The star above the blank‐Exos group referred to difference against the IMTP‐Exos group. The star above the PBS group referred to difference against the blank‐Exos group. *P<0.05, **P<0.01, ***P<0.001.
Cardiac function was examined using echocardiography. On postoperative day 0 and day 3, absolute values of ejection fraction, fractional shortening, LV end‐diastolic diameter, and LV end‐systolic diameter were basically the same among the PBS group, the blank‐exosomes group, and the IMTP‐exosomes group. Ejection fraction and fractional shortening in each group shared a similar downward tendency, whereas LV end‐diastolic diameter and LV end‐systolic diameter similarly experienced an increase. However, in the following days until the mice were euthanized, the group treated with IMTP‐exosomes resulted in a significantly upward tendency in ejection fraction and fractional shortening, whereas the blank‐exosomes group only had a slight increase in cardiac function and the PBS group even got worse (Figure 12C and 12D). At day 14 and day 28, LV end‐diastolic diameter and LV end‐systolic diameter recovered the best in the IMTP‐exosomes group, with the blank‐exosomes group at the medium level and the PBS group at the worst level (Figure 12E and 12F).
Discussion
The important findings of our study include the following: reconstruction of MSC exosomes with ischemic myocardium‐targeting peptide CSTSMLKAC for the first time; proving that IMTP‐exosomes can incorporate into hypoxic injured H9C2 cells more efficiently; and demonstration of the therapeutic efficacy by IMTP‐exosomes derived from MSCs in AMI.
Cholesterol‐rich and phospholipid exosomes can be uptaken by recipient cells so that cell‐cell communications are facilitated by transporting biomolecules, such as DNA, lipid, RNA, protein, and drugs. Several preclinical studies have elucidated the therapeutic potential of MSC‐derived exosomes for AMI treatment. Arslan et al27 and Lai et al27, 28 reported that exosomes extracted from human embryonic stem cell–derived MSCs obviously reduced myocardial infarct size, recovered cardiac function, and suppressed oxidative stress in a mouse AMI model. They further elaborated that MSCs‐exosomes had an antiapoptotic effect on AMI through activation of protein kinase B and glycogen synthase kinase 3 and inhibition of c‐Jun N‐terminal kinase. Bian et al29 found the same results when they treated the rats with AMI with exosome extracted from human BMSCs. Through a comparison of cardiac‐resident progenitor cell–secreted exosomes and BMSC‐secreted exosomes, Barile et al30 explained that the cardioprotection of exosomes may act via the pregnancy‐associated plasma protein‐A mediated insulin‐like growth factor‐1 release and subsequently modulate intracellular protein kinase B and extracellular signal regulated kinase 1/2 phosphorylation. Feng et al31 demonstrated that exosomes released from ischemic preconditioned MSCs contained an increased amount of miR‐22, which could exert a silence effect on methyl CpG binding protein 2 to ameliorate apoptosis in mice with AMI. Our group also proved that MSC‐derived exosomes could inhibit inflammation, augment vasculogenesis, and restore cardiac function in MI by activating and modifying the microRNA profiles of cardiac stem cells.32, 33 Despite these therapeutic effects of MSCs‐exosomes, targeting intended tissues or cells while avoiding nonintended delivery still remains challenging. Attempts have been made to genetically modify exosomes so that therapeutic effects can be enhanced. Ligands or homing peptides displayed on the external part of exosomes have been shown to promote the specificity and efficiency of delivery.
Compared with biological ligands, core ligand fragments or homing peptides are small molecules that can be easily and efficiently addressed to the external part of exosomes, and their interaction with targeting protein can be highly specific.34 Through phage display35 and in vivo biopanning techniques, lots of core ligand fragments or homing peptides targeting specific organs or tissues have been obtained and verified, so that targeting modification of exosomes can be realized. Displaying the tumor penetration peptide iRGD (CRGDKGPDC)36 on the surface of exosomes enabled them to specifically deliver doxorubicin to breast cancer cells expressing αv integrins, leading to inhibition of tumor growth.37 GE11 peptide (YHWYGYTPQNVI) fused to exosomes facilitates exosomes loaded with therapeutic microRNA targeting epidermal growth factor receptor–positive xenograft breast cancer cells.38 Rabies viral glycoprotein peptide (YTIWMPENPRPGTPCDIFTNSRGKRASNG) displayed on the external part of exosomes can specifically bind to acetylcholine receptor.39 In terms of the cardiovascular system, several homing peptides have been identified and applied to targeted therapy, including atherosclerosis,40, 41, 42 pulmonary arterial hypertension,43 and ischemia/reperfusion injured cardiomyocytes.44 In research by Won et al,44 they presented that a polymeric gene carrier conjugated with IMTP (CSTSMLKAC) was capable of targeting ischemic myocardium, resulting in high localization within the ischemic zone of the LV of an ischemia/reperfusion rat model on systemic administration. Vandergriff et al45 conjugated cardiac stem cell–derived exosomes with IMTP through a dioleoylphosphatidylethanolamine N‐hydroxysuccinimide linker, demonstrating an increasing retention of the IMTP‐exosomes within the ischemia/reperfusion injured heart tissues and a significant improvement in cardiac function.
In our study, we introduced the IMTP motif CSTSMLKAC onto exosomal surface to target ischemic myocardium. Our results demonstrated that effective targeting of exosomes to ischemic myocardium can be achieved by engineering exosomal enriched membrane protein (Lamp2b) fused with IMTP. In vitro experiment showed that IMTP‐exosomes could be internalized by hypoxia‐injured H9C2 cells more efficiently than blank‐exosomes. Compared with blank‐exosomes, a significant increase in targeting ischemic myocardium was observed in IMTP‐exosomes by in vivo tracking experiment. In addition, we confirmed that MSC‐derived IMTP‐exosomes could remarkably suppress inflammation and cardiomyocyte apoptosis, enhance angiogenesis, reduce infarct size, and improve cardiac function in a mouse MI model. To our knowledge, this is the first time MSC‐derived exosomes were reconstructed with IMTP and their therapeutic efficacy was elucidated in AMI.
We have to point out that we do not identify the target receptor for IMPT and the clear mechanism of interaction between them. Location, expression level, and profile of the receptors on cardiomyocyte surface may shift under hypoxia condition.46 Identification of the receptor bound by IMTP will reinforce the specificity and efficiency of transport. In the study, we creatively discovered that exosomes engineered with IMTP can preferentially target ischemic myocardium, and MSC‐derived IMTP‐exosomes play important roles in the treatment of AMI. The MSC‐derived exosomes targeting to ischemic myocardium could be supplied as a novel approach to carry therapeutic molecules, such as microRNAs, into ischemic heart.
Author Contributions
Z. Shen and J. Yang conceived and supervised the study; J. Yang, YH. Chen, and Wang designed experiments; X. Wang, Z. Zhao, Q. Meng, Y. Yu, Z. Yang, YQ. Chen, J. Li, T. Ma, and H. Liu performed experiments; Z. Zhao, Q. Meng, J. Sun, and Z. Li analyzed data; X. Wang wrote the manuscript; Z. Shen and J. Yang made manuscript revisions.
Sources of Funding
This work was supported by Jiangsu Clinical Research Center for CardiovascularSurgery (BL201451), National Clinical Key Specialty of Cardiovascular Surgery, National Natural Science Foundation of China (No. 81770260) and Natural Science Foundation of Jiangsu Province (BK20151212).
Disclosures
None.
(J Am Heart Assoc. 2018;7:e008737 DOI: 10.1161/JAHA.118.008737.)
Contributor Information
Junjie Yang, Email: junjieyang2009@gmail.com.
Zhenya Shen, Email: uuzyshen@aliyun.com.
References
- 1. Schuleri KH, Feigenbaum GS, Centola M, Weiss ES, Zimmet JM, Turney J, Kellner J, Zviman MM, Hatzistergos KE, Detrick B, Conte JV, McNiece I, Steenbergen C, Lardo AC, Hare JM. Autologous mesenchymal stem cells produce reverse remodelling in chronic ischaemic cardiomyopathy. Eur Heart J. 2009;30:2722–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amado LC, Saliaris AP, Schuleri KH, St John M, Xie JS, Cattaneo S, Durand DJ, Fitton T, Kuang JQ, Stewart G, Lehrke S, Baumgartner WW, Martin BJ, Heldman AW, Hare JM. Cardiac repair with intramyocardial injection of allogeneic mesenchymal stem cells after myocardial infarction. Proc Natl Acad Sci USA. 2005;102:11474–11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shake JG, Gruber PJ, Baumgartner WA, Senechal G, Meyers J, Redmond JM, Pittenger MF, Martin BJ. Mesenchymal stem cell implantation in a swine myocardial infarct model: engraftment and functional effects. Ann Thorac Surg. 2002;73:1919–1925. [DOI] [PubMed] [Google Scholar]
- 4. Toma C, Pittenger MF, Cahill KS, Byrne BJ, Kessler PD. Human mesenchymal stem cells differentiate to a cardiomyocyte phenotype in the adult murine heart. Circulation. 2002;105:93–98. [DOI] [PubMed] [Google Scholar]
- 5. Hare JM, Traverse JH, Henry TD, Dib N, Strumpf RK, Schulman SP, Gerstenblith G, DeMaria AN, Denktas AE, Gammon RS, Hermiller JB Jr, Reisman MA, Schaer GL, Sherman W. A randomized, double‐blind, placebo‐controlled, dose‐escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J Am Coll Cardiol. 2009;54:2277–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Perin EC, Silva GV, Henry TD, Cabreira‐Hansen MG, Moore WH, Coulter SA, Herlihy JP, Fernandes MR, Cheong BY, Flamm SD, Traverse JH, Zheng Y, Smith D, Shaw S, Westbrook L, Olson R, Patel D, Gahremanpour A, Canales J, Vaughn WK, Willerson JT. A randomized study of transendocardial injection of autologous bone marrow mononuclear cells and cell function analysis in ischemic heart failure (FOCUS‐HF). Am Heart J. 2011;161:1078–1087.e3. [DOI] [PubMed] [Google Scholar]
- 7. Hare JM, Fishman JE, Gerstenblith G, DiFede Velazquez DL, Zambrano JP, Suncion VY, Tracy M, Ghersin E, Johnston PV, Brinker JA, Breton E, Davis‐Sproul J, Schulman IH, Byrnes J, Mendizabal AM, Lowery MH, Rouy D, Altman P, Wong Po Foo C, Ruiz P, Amador A, Da Silva J, McNiece IK, Heldman AW, George R, Lardo A. Comparison of allogeneic vs autologous bone marrow‐derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: the POSEIDON randomized trial. JAMA. 2012;308:2369–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bartunek J, Behfar A, Dolatabadi D, Vanderheyden M, Ostojic M, Dens J, El Nakadi B, Banovic M, Beleslin B, Vrolix M, Legrand V, Vrints C, Vanoverschelde JL, Crespo‐Diaz R, Homsy C, Tendera M, Waldman S, Wijns W, Terzic A. Cardiopoietic stem cell therapy in heart failure: the C‐CURE (Cardiopoietic stem Cell therapy in heart failURE) multicenter randomized trial with lineage‐specified biologics. J Am Coll Cardiol. 2013;61:2329–2338. [DOI] [PubMed] [Google Scholar]
- 9. Lee JW, Lee SH, Youn YJ, Ahn MS, Kim JY, Yoo BS, Yoon J, Kwon W, Hong IS, Lee K, Kwan J, Park KS, Choi D, Jang YS, Hong MK. A randomized, open‐label, multicenter trial for the safety and efficacy of adult mesenchymal stem cells after acute myocardial infarction. J Korean Med Sci. 2014;29:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen SL, Fang WW, Ye F, Liu YH, Qian J, Shan SJ, Zhang JJ, Chunhua RZ, Liao LM, Lin S, Sun JP. Effect on left ventricular function of intracoronary transplantation of autologous bone marrow mesenchymal stem cell in patients with acute myocardial infarction. Am J Cardiol. 2004;94:92–95. [DOI] [PubMed] [Google Scholar]
- 11. Davidson SM, Riquelme JA, Takov K, Vicencio JM, Boi‐Doku C, Khoo V, Doreth C, Radenkovic D, Lavandero S, Yellon DM. Cardioprotection mediated by exosomes is impaired in the setting of type II diabetes but can be rescued by the use of non‐diabetic exosomes in vitro. J Cell Mol Med. 2018;22:141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thery C, Boussac M, Véron P, Ricciardi‐Castagnoli P, Raposo G, Garin J, Amigorena S. Proteomic analysis of dendritic cell‐derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol. 2001;166:7309–7318. [DOI] [PubMed] [Google Scholar]
- 13. Kanki S, Jaalouk DE, Lee S, Yu AY, Gannon J, Lee RT, Jaalouk DE, Lee S, Yu AY, Gannon J, Lee RT. Identification of targeting peptides for ischemic myocardium by in vivo phage display. J Mol Cell Cardiol. 2011;50:841–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–593. [DOI] [PubMed] [Google Scholar]
- 15. Gnecchi M, He H, Liang OD, Melo LG, Morello F, Mu H, Noiseux N, Zhang L, Pratt RE, Ingwall JS, Dzau VJ. Paracrine action accounts for marked protection of ischemic heart by Akt‐modified mesenchymal stem cells. Nat Med. 2005;11:367–368. [DOI] [PubMed] [Google Scholar]
- 16. Gnecchi M, He H, Noiseux N, Liang OD, Zhang L, Morello F, Mu H, Melo LG, Pratt RE, Ingwall JS, Dzau VJ. Evidence supporting paracrine hypothesis for Akt‐modified mesenchymal stem cell‐mediated cardiac protection and functional improvement. FASEB J. 2006;20:661–669. [DOI] [PubMed] [Google Scholar]
- 17. Mirotsou M, Zhang Z, Deb A, Zhang L, Gnecchi M, Noiseux N, Mu H, Pachori A, Dzau V. Secreted frizzled related protein 2 (Sfrp2) is the key Akt‐mesenchymal stem cell‐released paracrine factor mediating myocardial survival and repair. Proc Natl Acad Sci USA. 2007;104:1643–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ailawadi S, Wang X, Gu H, Fan GC. Pathologic function and therapeutic potential of exosomes in cardiovascular disease. Biochim Biophys Acta. 2015;1852:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Dommelen SM, Vader P, Lakhal S, Kooijmans SA, van Solinge WW, Wood MJ, Schiffelers RM. Microvesicles and exosomes: opportunities for cell‐derived membrane vesicles in drug delivery. J Control Release. 2012;161:635–644. [DOI] [PubMed] [Google Scholar]
- 20. El AS, Lakhal S, Mäger I, Wood MJ. Exosomes for targeted siRNA delivery across biological barriers. Adv Drug Deliv Rev. 2013;65:391–397. [DOI] [PubMed] [Google Scholar]
- 21. Ko YT, Hartner WC, Kale A, Torchilin VP. Gene delivery into ischemic myocardium by double‐targeted lipoplexes with anti‐myosin antibody and TAT peptide. Gene Ther. 2009;16:52–59. [DOI] [PubMed] [Google Scholar]
- 22. Laakkonen P, Vuorinen K. Homing peptides as targeted delivery vehicles. Integr Biol (Camb). 2010;2:326–337. [DOI] [PubMed] [Google Scholar]
- 23. Alvarez‐Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341–345. [DOI] [PubMed] [Google Scholar]
- 24. Blom JN, Lu X, Arnold P, Feng Q. Myocardial infarction in neonatal mice, a model of cardiac regeneration. J Vis Exp. 2016;24:54100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fischer AH, Jacobson KA, Rose J, Zeller R. Cryosectioning tissues. CSH Protoc. 2008;2008:pdb.prot4991. [DOI] [PubMed] [Google Scholar]
- 26. Fischer AH, Jacobson KA, Rose J, Zeller R. Cutting sections of paraffin‐embedded tissues. CSH Protoc. 2008;2008:pdb.prot4987. [DOI] [PubMed] [Google Scholar]
- 27. Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor EN, Timmers L, van Rijen HV, Doevendans PA, Pasterkamp G, Lim SK, de Kleijn DP. Mesenchymal stem cell‐derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013;10:301–312. [DOI] [PubMed] [Google Scholar]
- 28. Lai RC, Arslan F, Lee MM, Sze NS, Choo A, Chen TS, Salto‐Tellez M, Timmers L, Lee CN, El Oakley RM, Pasterkamp G, de Kleijn DP, Lim SK. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010;4:214–222. [DOI] [PubMed] [Google Scholar]
- 29. Bian S, Zhang L, Duan L, Wang X, Min Y, Yu H. Extracellular vesicles derived from human bone marrow mesenchymal stem cells promote angiogenesis in a rat myocardial infarction model. J Mol Med (Berl). 2014;92:387–397. [DOI] [PubMed] [Google Scholar]
- 30. Barile L, Cervio E, Lionetti V, Milano G, Ciullo A, Biemmi V, Bolis S, Altomare C, Matteucci M, Di Silvestre D, Brambilla F, Fertig TE, Torre T, Demertzis S, Mauri P, Moccetti T, Vassalli G. Cardioprotection by cardiac progenitor cell‐secreted exosomes: role of pregnancy‐associated plasma protein‐A. Cardiovasc Res. 2018;114:992–1005. [DOI] [PubMed] [Google Scholar]
- 31. Feng Y, Huang W, Wani M, Yu X, Ashraf M. Ischemic preconditioning potentiates the protective effect of stem cells through secretion of exosomes by targeting Mecp2 via miR‐22. PLoS One. 2014;9:e88685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Z, Yang J, Yan W, Li Y, Shen Z, Asahara T. Pretreatment of cardiac stem cells with exosomes derived from mesenchymal stem cells enhances myocardial repair. J Am Heart Assoc. 2016;5:e002856 DOI: 10.1161/JAHA.115.002856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Teng X, Chen L, Chen W, Yang J, Yang Z, Shen Z. Mesenchymal stem cell‐derived exosomes improve the microenvironment of infarcted myocardium contributing to angiogenesis and anti‐inflammation. Cell Physiol Biochem. 2015;37:2415–2424. [DOI] [PubMed] [Google Scholar]
- 34. Ladner RC, Sato AK, Gorzelany J, de Souza M. Phage display‐derived peptides as therapeutic alternatives to antibodies. Drug Discov Today. 2004;9:525–529. [DOI] [PubMed] [Google Scholar]
- 35. Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. [DOI] [PubMed] [Google Scholar]
- 36. Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, Hanahan D, Mattrey RF, Ruoslahti E. Tissue‐penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16:510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ, Wei J, Nie G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383–2390. [DOI] [PubMed] [Google Scholar]
- 38. Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, Fujita K, Mizutani T, Ohgi T, Ochiya T, Gotoh N, Kuroda M. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013;21:185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cooper JM, Wiklander PB, Nordin JZ, Al‐Shawi R, Wood MJ, Vithlani M, Schapira AH, Simons JP, El‐Andaloussi S, Alvarez‐Erviti L. Systemic exosomal siRNA delivery reduced alpha‐synuclein aggregates in brains of transgenic mice. Mov Disord. 2014;29:1476–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hong HY, Lee HY, Kwak W, Yoo J, Na MH, So IS, Kwon TH, Park HS, Huh S, Oh GT, Kwon IC, Kim IS, Lee BH. Phage display selection of peptides that home to atherosclerotic plaques: IL‐4 receptor as a candidate target in atherosclerosis. J Cell Mol Med. 2008;12:2003–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee GY, Kim JH, Oh GT, Lee BH, Kwon IC, Kim IS. Molecular targeting of atherosclerotic plaques by a stabilin‐2‐specific peptide ligand. J Control Release. 2011;155:211–217. [DOI] [PubMed] [Google Scholar]
- 42. Thapa N, Kim JH, Oh GT, Lee BH, Kwon IC, Kim IS. Identification of a peptide ligand recognizing dysfunctional endothelial cells for targeting atherosclerosis. J Control Release. 2008;131:27–33. [DOI] [PubMed] [Google Scholar]
- 43. Toba M, Alzoubi A, O'Neill K, Abe K, Urakami T, Komatsu M, Alvarez D, Järvinen TA, Mann D, Ruoslahti E, McMurtry IF, Oka M. A novel vascular homing peptide strategy to selectively enhance pulmonary drug efficacy in pulmonary arterial hypertension. Am J Pathol. 2014;184:369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Won YW, McGinn AN, Lee M, Bull DA, Kim SW. Targeted gene delivery to ischemic myocardium by homing peptide‐guided polymeric carrier. Mol Pharm. 2013;10:378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vandergriff A, Huang K, Shen D. Targeting regenerative exosomes to myocardial infarction using cardiac homing peptide. Theranostics. 2018;8:1869–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Muraguchi TA, Kawawa A, Kubota S. Prohibitin protects against hypoxia‐induced H9c2 cardiomyocyte cell death. Biomed Res. 2010;31:113–122. [DOI] [PubMed] [Google Scholar]