

Abstract
Background
Nonsyndromic thoracic aortic diseases (NS‐TADs) are often silent entities until they present as life‐threatening emergencies. Despite familial inheritance being common, screening is not the current standard of care in NS‐TADs. We sought to determine the incidence of aortic diseases, the predictive accuracy of available screening tests, and the effectiveness of screening programs in relatives of patients affected by NS‐TADs.
Methods and Results
A systematic literature search on PubMed/MEDLINE, Embase, and the Cochrane Library was conducted from inception to the end of December 2017. The search was supplemented with the Online Mendelian Inheritance in Man database. A total of 53 studies were included, and a total of 2696 NS‐TAD relatives were screened. Screening was genetic in 49% of studies, followed by imaging techniques in 11% and a combination of the 2 in 40%. Newly affected individuals were identified in 33%, 24%, and 15% of first‐, second‐, and third‐degree relatives, respectively. Familial NS‐TADs were primarily attributed to single‐gene mutations, expressed in an autosomal dominant pattern with incomplete penetrance. Specific gene mutations were observed in 25% of the screened families. Disease subtype and genetic mutations stratified patients with respect to age of presentation, aneurysmal location, and aortic diameter before dissection. Relatives of patients with sporadic NS‐TADs were also found to be affected. No studies evaluated the predictive accuracy of imaging or genetic screening tests, or the clinical or cost‐effectiveness of an NS‐TAD screening program.
Conclusions
First‐ and second‐degree relatives of patients affected by both familial and sporadic NS‐TADs may benefit from personalized screening programs.
Keywords: aortic disease, genetic testing, mortality, screening
Subject Categories: Cardiovascular Disease, Clinical Studies, Aortic Dissection, Aneurysm, Quality and Outcomes
Clinical Perspective
What Is New?
Imaging and/or genetic screening is not the current standard of care in relatives of patients affected by nonsyndromic thoracic aortic diseases (NS‐TADs).
Genetic and/or imaging screening of relatives of patients affected by NS‐TAD can detect more than 30% of patients newly affected by thoracic aortic diseases.
What Are the Clinical Implications?
Routine imaging and genetic testing of relatives of patients affected by nonsyndromic aortopathies should be encouraged.
The evidence suggests that screening of first‐ and second‐degree relatives of patients affected by familial NS‐TAD and first‐degree relatives of those affected by sporadic NS‐TADs will result in significant numbers of patients with otherwise undiagnosed disease.
Personalized screening programs determined by the subtype of NS‐TAD and its related genetic mutation have the potential to benefit these patients.
Introduction
Diseases of the thoracic aorta are increasing in prevalence, accounting for 1% to 2% of all deaths in Western countries.1, 2, 3, 4, 5 In the United States, diseases of the aorta account for more than 40 000 deaths per year.1, 4 Thoracic aortic diseases (TADs) are often silent entities with a mortality of almost 80% when presenting as life‐threatening emergencies.3, 6 Therefore, early diagnosis and treatment are likely to improve long‐term survival. TADs may be syndromic, associated with disorders involving other organs such as Marfan syndrome, or more commonly nonsyndromic, with manifestations restricted to the thoracic aorta.4, 5 Nonsyndromic TADs (NS‐TADs) may be familial, characterized by the presence of a family history and an autosomal dominant inheritance, or sporadic.4, 5, 6, 7 Unlike syndromic TADs, NS‐TADs are not evident from external physical features and abnormalities of other organ systems and are characterized by silent aneurysm formation and dissection.4, 5 Screening of first‐degree relatives (FDRs) of patients affected by NS‐TAD is therefore recommended for early detection and treatment of asymptomatic disease.4, 5 However, existing guidelines are based predominantly on the consensus of expert opinion, rather than high‐quality evidence, and the testing modality, frequency, and extent (FDRs versus second‐degree relatives [SDRs]) of screening are not defined.4, 5 As a consequence, there is widespread variation in the screening of family members of patients with NS‐TADs. To address this area of uncertainty, we performed a systematic review of the evidence for screening in the relatives of patients affected with NS‐TADs with reference to the prevalence of aortic disease, the predictive accuracy of genetic and imaging screening tests, and the effectiveness of screening programs in this high‐risk population.
Methods
The data, analytic methods, and study materials are available to other researchers for purposes of reproducing the results or replicating the procedure (Supplemental Material).
Protocol, Registration, and Search Strategy
The search strategy, objectives, study selection and eligibility, data collection, and assessment of study quality are published online and registered in the PROSPERO International Prospective Register of Systematic Reviews (PROSPERO registry—CRD42017064598).8 The protocol of the present systematic review is fully reported in Data S1. The review adhered to PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) guidelines (Table S1).9
We searched electronic databases (PubMed/MEDLINE, Embase, and Cochrane Library) without date or language restriction from inception to the end of December 2017. A systematic search in the Online Mendelian Inheritance in Man (OMIM) database10 on December 31, 2017, was also accomplished. To supplement the electronic search, the “first‐generation” reference lists of pertinent articles were reviewed. Search criteria, adopted keywords, and MeSH terms used in relevant combinations are reported in Data S1.
Participants
We included studies considering imaging and/or genetic screening tests in probands affected by diseases of the thoracic aorta (aneurysms and/or acute aortic syndrome), and their FDRs, SDRs, and third‐degree relatives (TDRs), with no restriction on ethnicity or age.
Target Condition
The target condition was disease of the thoracic aorta (aneurysm and/or acute aortic syndrome) defined by the international guidelines on diagnosis and management of patients with TAD.4, 5 Only NS‐TAD forms were considered in the present review; syndromic TADs or other forme fruste of syndromic TAD related to the transforming growth factor β pathway were excluded. Familial NS‐TAD forms were defined as those occurring in families with ≥2 members with a known TAD, but without a clinical diagnosis or history of a syndromic TAD or any other connective tissue disease.7 Sporadic TADs were defined as those occurring in patients apparently without a family history of TAD or evidence of syndromic TAD.4, 5, 7
Index Tests
For the purposes of the review, we included studies that phenotyped participants using the following imaging tests: transthoracic echocardiogram (TTE)/transoesophageal echocardiogram, computed tomography (CT), or magnetic resonance imaging (MRI) of the thoracic aorta, and genetic screening, individually or in combination with the acknowledgement that sensitivities and specificities of CT (100% and 100%, respectively) and MRI (95–100%) are higher when compared with those of transoesophageal echocardiogram and TTE (74–100% and 71–91%, respectively).11, 12, 13, 14, 15 In some studies, surgery for TAD, postmortem examination, or sudden death were used to assess the aortic phenotype. Molecular genetic testing approaches included a combination of gene‐targeted testing (multigene panel or single gene testing) and whole exome of genome sequencing.16, 17, 18
Study Selection, Data Collection, and Extraction
Two investigators (G.M. and R.D.) independently reviewed titles, abstracts, and full‐text articles against the specified inclusion criteria for studies regarding screening of relatives of patients with NS‐TADs. Discrepancies were resolved through consensus and consultation with a third investigator (G.J.M.). One reviewer extracted key data from the included studies using a standard dedicated pro forma; a second reviewer checked the collected data for completeness and accuracy. The Tables report full details on study design and quality, setting and population, details, and results of screening. Key study characteristics include details of the patient population (NS‐TAD form, ethnicity, family identification), participants undergoing screening (relatives eligible for screening; family pedigree; total number of screened relatives; numbers of FDRs, SDRs, and TDRs), TAD characteristics (new diagnosis of aortic disease, number/rate of newly diagnosed thoracic aortic aneurysms and/or dissection, rate of unexplained sudden death, age and aortic diameters at dissection, sex preponderance, and aortic disease penetrance), additional concomitant phenotype/clinical features (types and rates), and type of adopted screening modality (imaging and genetic test used, validation processes). The definitions of the extracted variables are fully reported in Data S1.
Quality Assessment, Data Synthesis, and Analysis
Two investigators (G.M., R.D.) independently appraised all articles that met inclusion criteria. Study quality was assessed using the Newcastle‐Ottawa Scale and the US Preventive Services Task Force.19, 20 The Cochrane Risk of Bias tool was also used to evaluate the methodological quality of all included studies.21
Because of the observational nature of the studies and their clinical heterogeneity, the analyses were largely descriptive, and a narrative and tabular synthesis of all included studies is provided. Inclusion and exclusion criteria for qualitative/quantitative analyses are summarized according to the PICOS (population, intervention, comparator, outcomes, and study design) approach (Table S2). Subgroup analysis considering type of NS‐TAD form, aortic disease (aneurysm and/or dissection), genetic mutation, and screening modality was also conducted. Categorical variables are reported as number and percentage, and continuous variables are reported as mean and SD or median and range, according to distribution. Analyses were performed with SPSS version 24.0 (IBM).
Results
Description of Studies and Quality Assessment
Of the 12 897 records identified, 53 studies were included in the systematic review, comprising a total of 2696 screened relatives. The studies were published between 1985 and 2017 (Figure S1).22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74 Regions of origin included North America (28 studies), Europe (17 studies), Asia (5 studies), and Australia (3 studies) (Table 1). No randomized trials were identified, and only 1 large cross‐sectional study was conducted including 581 at‐risk relatives.58 Study characteristics and collected outcomes are summarized in Tables S3 through S8 and study quality assessment in Table S9.
Table 1.
Details of Studies Included in the Systematic Review
| Study (Author/Y) | Country | NS‐TAD Form | Pedigree (Patients) | Relatives Affected | Penetrance, % | Inheritance (Modality) | Type of Screening | Related Gene | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total, No. | FDRs, No. | SDRs, No. | TDRs, No. | Probands, No. | No. | % | |||||||
| Barbier et al 201422 | France | FTAAD | 40 | 14 | 14 | 0 | 2 | 7 | 18 | 60 | AD | GEN+IMAG | MFAP5 |
| Bee et al 201223 | United States | FTAA | 54 | 37 | 3 | 0 | 9 | 12 | 22 | 100 | ··· | GEN | ACTA2, MYH11, TGFBR2 |
| Chamney et al 201524 | United Kingdom | FTAAD | 14 | 8 | 3 | 0 | 1 | 5 | 36 | 100 | AD | GEN+IMAG | ACTA2 |
| Disabella et al 201125 | Italy | FTAAD | 37 | 23 | 5 | 4 | 5 | 10 | 27 | 78 | AD | GEN+IMAG | ACTA2 |
| Disertori et al 199126 | Italy | FTAAD | 30 | 13 | 15 | 0 | 2 | 2 | 7 | na | ··· | IMAG | ··· |
| Dong et al 201427 | China | FTAAD | 64 | 5 | 9 | 30 | 1 | 8 | 13 | 64 | ··· | GEN+IMAG | TGFBR1 |
| Francke et al 199528 | United States | FTAAD | 26 | 15 | 9 | 0 | 1 | 9 | 35 | 67 | AD | GEN+IMAG | FBN1 |
| Gago‐Diaz et al 201429 | Spain | FTAAD | 31 | 3 | 10 | 13 | 1 | 6 | 19 | 60 | AD | GEN | TGFB2 |
| Gago‐Diaz et al 201630 | Spain | FTAAD | 30 | 12 | 14 | 3 | 1 | 10 | 33 | 88 | AD | GEN | PRKG1 |
| Guo et al 200131 | United Statesa | FTAAD | 219 | n/c | n/c | n/c | n/a | n/c | n/c | n/a | AD | GEN | Locus 5q13‐14b |
| Guo et al 200732 | United Statesa | FTAAD | 212 | n/c | n/c | n/c | n/a | n/c | n/c | 48 | AD | GEN | ACTA2 |
| Guo et al 200933 | United Statesa | FTAAD | 269 | n/c | n/c | n/c | n/a | n/c | n/c | 49 | AD | GEN | ACTA2 |
| Guo et al 201134 | United Statesa | FTAAD/pAA | 28 | 7 | 9 | 6 | 1 | 8 | 29 | 75 | AD | GEN | Locus 12q13‐14b |
| Guo et al 201335 | United Statesa | FTAAD | 89 | 40 | 18 | 12 | 6 | 31 | 35 | 100 | AD | GEN | PRKG1 |
| Guo et al 201536 | United Statesa | BAV/TAA | 48 | 10 | 14 | 15 | 1 | 7 | 15 | 44 | AD | GEN | MATA2 |
| Guo et al 201637 | United Statesa | FTAAD | 65 | 21 | 22 | 13 | 6 | 15 | 23 | 86 | AD | GEN | LOX |
| Hannuksela et al 201538 | Sweden | FTAAD | 270 | 60 | 89 | 55 | 7 | 37 | 14 | n/a | ··· | GEN+IMAG | ··· |
| Hannuksela et al 201639 | Sweden | FTAAD | 46 | n/c | n/c | n/c | 1 | n/c | n/c | 45 | ··· | GEN+IMAG | MYLK |
| Harakalova et al 201340 | Holland | TAAD/PDA | 75 | 6 | 15 | 34 | 2 | 13 | 17 | 45 | AD | GEN | MYH11 |
| Hasham et al 200341 | United Statesa | FTAAD | 69 | 4 | 5 | 39 | 1 | 16 | 23 | 75 | AD | GEN+IMAG | TGFBR2 |
| Kakko et al 200342 | Finland | FTAAD | 213 | n/c | n/c | n/c | n/a | n/c | n/c | n/a | ··· | GEN+IMAG | Locus 5q13‐14b |
| Kent et al 201343 | United States | BAV/TAA | 129 | 73 | 21 | 19 | 14 | 34 | 26 | n/a | AD | GEN+IMAG | NOTCH1 |
| Keramati et al 201044 | United States | FTAAD | 23 | 10 | 8 | 0 | 1 | 12 | 52 | 90 | AD | GEN+IMAG | Locus 15q21 (FBN1?) |
| Khau Van Kien et al 200445 | France | FTAAD/PDA | 68 | 13 | 21 | 24 | 1 | 7 | 10 | n/a | AD | GEN+IMAG | ··· |
| Khau Van Kien et al 200546 | France | FTAAD/PDA | 87 | 13 | 26 | 38 | 1 | 7 | 8 | 50 | AD | GEN+IMAG | MYH11 |
| Kuang et al 201647 | United Statesa | FTAAD | 40 | n/c | n/c | n/c | n/a | n/c | n/c | 75 | AD | GEN | FOXE3 |
| Loscalzo et al 200748 | United States | BAV/TAA | 194 | 72 | 37 | 65 | 13 | 44 | 23 | 88 | AD | GEN+IMAG | ··· |
| Marwick et al 198749 | Australia | FTADiss | 17 | 7 | 5 | 0 | 1 | 1 | 6 | n/a | ··· | IMAG | ··· |
| McManus et al 198750 | United States | FTADiss | 19 | 7 | 9 | 0 | 1 | 5 | 26 | n/a | ··· | IMAG | ··· |
| Milewicz et al 199851 | United Statesa | FTAAD | 123 | 44 | 44 | 7 | 6 | 24 | 20 | n/a | AD | GEN+IMAG | ···c |
| Morisaki et al 200952 | Japan | FTAAD | 47 | 10 | 6 | 27 | 3 | 11 | 23 | 100 | ··· | GEN | ACTA2 |
| Pannu et al 200553 | United Statesa | FTAAD | 235 | 18 | 35 | 121 | 4 | 54 | 23 | 79 | AD | GEN+IMAG | TGFBR2 |
| Pannu et al 200754 | United Statesa | FTAAD | 27 | 16 | 4 | 0 | 2 | 4 | 15 | 45 | ··· | GEN+IMAG | MYH11 |
| Regalado et al 201155 | United Statesa | FTAAD/ICA | 231 | 83 | 64 | 50 | 13d | 43 | 19 | n/a | AD | GEN | ACTA2, TGFBR1, TGFBR2 |
| Regalado et al 201156 | United Statesa | FTAAD/ICA/pAA | 106 | n/c | n/c | n/c | n/a | n/c | n/c | 65 | AD | GEN | SMAD3 |
| Regalado et al 201157 | United Statesa | FTAAD | 29 | 18 | 6 | 0 | 5 | 10 | 34 | n/a | ··· | GEN | FBN1 |
| Renard et al 201358 | Belgium | FTAAD | 97 | 34 | 30 | 7 | 8 | 21 | 22 | n/a | AD | GEN | ACTA2, MYH11 |
| Robertson et al 201659 | Australia | FTAAD | n/c | n/c | n/c | n/c | 270 | 341 | 56 | n/a | ··· | IMAG | ··· |
| Sherrah et al 201660 | Australia | FTAAD | n/c | n/c | n/c | n/c | n/a | n/c | n/c | n/a | ··· | IMAG | ··· |
| Takeda et al 201561 | Japan | FTAAD | 17 | 5 | 6 | 2 | 1 | 4 | 24 | 75 | ··· | GEN | MYH11 |
| Teixidó‐Turà et al 201462 | Spain | FTAAD | 36 | 8 | 5 | 15 | 1 | 2 | 6 | 10 | ··· | GEN | ACTA2 |
| Tortora et al 201763 | Italy | BAV/TAA | 97 | 77 | 0 | 0 | 20 | 5 | 7 | n/a | ··· | GEN+IMAG | ···e |
| Tran‐Fadulo et al 200664 | United Statesa | FTAAD | 153 | 14 | 45 | 63 | 3 | 18 | 12 | n/a | ··· | GEN | ··· |
| Tran‐Fadulo et al 200965 | United Statesa | FTAAD | 78 | 31 | 23 | 7 | 4f | 26 | 33 | 70 | AD | GEN | TGFBR1 |
| Vaughan et al 200166 | United Statesa | FTAA | 67 | 27 | 20 | 2 | 3 | 27 | 40 | n/a | AD | GEN+IMAG | Locus 11q23.3‐24b |
| Wang et al 201067 | United Statesa | FTADiss | 48 | n/c | n/c | n/c | n/a | n/c | n/c | 50 | AD | GEN | MYLK |
| Wang et al 201368 | China | FTAAD | 10 | 7 | 0 | 0 | 1 | 1 | 10 | n/a | ··· | GEN | ··· |
| Ware et al 201469 | United States | FTAAD | 7 | 4 | 0 | 0 | 2 | 0 | 0 | 100 | ··· | GEN | ACTA2 |
| Warnes et al 198570 | United States | FTAAD | 6 | 4 | 0 | 0 | 2 | 0 | 0 | n/a | ··· | IMAG | ··· |
| Weigang et al 200771 | Germany | FTAAD | 26 | n/c | n/c | n/c | n/a | n/c | n/c | n/a | AD | GEN+IMAG | ··· |
| Yoo et al 201072 | Korea | FTAAD | 20 | 7 | 7 | 0 | 1 | 4 | 20 | 67 | AD | GEN | ACTA2 |
| Zhu et al 200673 | France | FTAAD/PDA | 49 | n/c | n/c | n/c | n/a | n/c | n/c | 44 | AD | GEN+IMAG | MYH11 |
| Ziganshin et al 201574, g | United States | FTAAD | 27 | 7 | 11 | 2 | 1 | 3 | 11 | 70 | AD | GEN | MYLK |
| Ziganshin et al 201574, g | United States | FTAAD | 17 | 6 | 8 | 0 | 1 | 6 | 35 | 70 | ··· | GEN | TGFBR1 |
AD indicates autosomal dominant; BAV, bicuspid aortic valve; FDRs, first‐degree relatives; FTAA, familial thoracic aortic aneurysm; FTADiss, familial aortic dissection; FTAAD, familial thoracic aortic aneurysm and dissection; GEN, genetic; ICA, intracranial aneurysm; IMAG, imaging; n/a, not available; NS‐TAD, nonsyndromic thoracic aortic disease; n/c, not computable; pAA, peripheral artery aneurysm; PDA, patent ductus arteriosus; SDRs, second‐degree relatives; TAA, thoracic aortic aneurysm; TAAD, thoracic aortic aneurysm and/or dissection; TDRs, third‐degree relatives.
Study performed at University of Texas.
Mapped loci without identified gene.
No linkage to FBN1 or TAAD2.
Four probands not affected by aortic diseases (aortic aneurysm and/or dissections).
No linkage with ACTA2.
One proband not affected by aortic diseases (aortic aneurysm and/or dissection).
Data of 2 different screened families obtained from the same study.
Target Condition
Four main groups of familial NS‐TADs were identified: (1) those characterized by the presence of both aneurysms and dissections in the family pedigree (familial thoracic aortic aneurysm and dissection; 44 studies); (2) those characterized by aneurysmal disease only (familial thoracic aortic aneurysm; 2 studies); (3) those characterized by aortic dissection only (familial thoracic aortic dissection; 3 studies); and (4) thoracic aortic aneurysm forms associated with the presence of bicuspid aortic valve (4 studies). Among the familial thoracic aortic aneurysm and dissection forms, 3 additional subgroups were discovered based on the concomitant presence of patent ductus arteriosus (n=4), intracranial aneurysms (n=2), or peripheral arterial aneurysms (n=2) (Table 1).
Index Tests
Screening for TAD was performed using 2‐dimensional TTE in 27 (51%) studies, of which 15 (28%) employed 2‐dimensional TTE alone and the remaining 8 (15%) used 2‐dimensional TTE in association with CT and/or MRI. In 5 (9%) studies only, imaging screening included the simultaneous employment of 2‐dimensional TTE, CT, and MRI. In a further 26 (49%) studies, aortic phenotype (presence of an aortic aneurysm and/or dissection) was defined by reported clinical events including acute aortic syndrome, diagnosis made during routine diagnostic clinical care, or postmortem examination. The aortic diameter cutoff used for defining a critical dilation of the aorta varied among studies as the aortic site where the measurements were made (Table S7).
No study reported the sensitivity, specificity, or other measures of diagnostic accuracy for the index tests. One study reported 10‐year longitudinal follow‐up for relatives of patients with NS‐TAD.59 In this study, relatives with evidence of aortic dilatation were offered annual follow‐up imaging with prescription of β‐blockers or angiotensin receptor blockers at maximal tolerated doses. Relatives with no evidence of aortic dilatation (unaffected) were subjected to clinical review every 3 years. In the affected relatives (n=114) with serial aortic measurements over 4.5±4.4 years, a mean rate of increase in the aortic diameter of 0.56±0.76 mm per year was observed. No difference in the rate of aortic dilatation was observed between males and females or in patients receiving β‐blockers or angiotensin receptor blockers. No correlation with the age at diagnosis, the initial aortic diameter, and the systolic or diastolic blood pressure was documented. During 10‐year follow‐up, 9% of newly diagnosed relatives were affected by an aortic dissection, and 18% underwent elective aortic surgery. Six relatives (of 368) originally diagnosed as unaffected (initial aortic diameter with a Z score <2) experienced a subsequent aortic dissection.59
Results of Imaging Tests
A total of 1039 families underwent screening for NS‐TAD, with a median number of patients in each family pedigree of 48 (study range: 6–270) (Table S3). The proportion of potential eligible patients per family was 73% (study range: 50–100%), while the rate of relatives effectively screened was 54% (study range: 5–100%) (Table 2 and Table S4). FDRs, SDRs, and TDRs were variably screened throughout the studies. Twelve percent of FDRs, 24% of SDRs, and 18% of TDRs were not available for screening (Figure 1).
Table 2.
Details of Newly Diagnosed Diseases of the Thoracic Aorta in the Screened Relatives
| Study (Author/Y) | No. of Relatives Screened | Patients Affecteda (Aortic Aneurysm+Aortic Dissection) | Sudden Death (Unexplained) | Aortic Aneurysma | Aortic Dissectiona | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | % | Male | % | No. | % | No. | % | No. | % | Age at Dissection, y | Range (Age, y) | ||
| Barbier et al 201422 | 13 | 9 | 23 | 3 | 33 | n/a | ··· | 8 | 89 | 1 | 11 | 58 | n/a |
| Bee et al 201223 | 32 | 21 | 39 | 16 | 76 | n/a | ··· | 21 | 100 | 0 | 0 | ··· | ··· |
| Chamney et al 201524 | 6 | 6 | 43 | 4 | 67 | 0 | 0 | 3 | 50 | 3 | 50 | 49±10.4 | 37–55 |
| Disabella et al 201125 | 29 | 15 | 41 | 8 | 53 | 1 | 3 | 6 | 40 | 9 | 60 | 49.3±16.3 | 29–73 |
| Disertori et al 199126 | 14 | 4 | 13 | 4 | 100 | n/a | ··· | 2 | 50 | 2 | 50 | 46±2.8 | 44–48 |
| Dong et al 201427 | 39 | 9 | 14 | 7 | 78 | 1 | 2 | 6 | 67 | 3 | 33 | 39±6.9 | 35–47 |
| Francke et al 199528 | 23 | 10 | 38 | 6 | 60 | n/a | ··· | 8 | 80 | 2 | 20 | 55±14.1 | 45–65 |
| Gago‐Diaz et al 201429 | 12 | 7 | 23 | 5 | 71 | n/a | ··· | 5 | 71 | 2 | 29 | 37.5±4.9 | 34–41 |
| Gago‐Diaz et al 201630 | 14 | 11 | 37 | 6 | 55 | 1 | 3 | 5 | 45 | 6 | 55 | 34.2±12.9 | 15–48 |
| Guo et al 200131 | 121 | 73 | 33 | 47 | 64 | n/a | ··· | n/a | ··· | n/a | ··· | ··· | ··· |
| Guo et al 200732 | 130 | 53 | 25 | 33 | 62 | n/a | ··· | 8 | 15 | 45 | 85 | 37.3±13.9 | 13–67 |
| Guo et al 200933 | 163 | 66 | 25 | 39 | 59 | n/a | ··· | n/a | ··· | n/a | ··· | ··· | ··· |
| Guo et al 201134 | 18 | 9 | 32 | 9 | 100 | n/a | ··· | 8 | 89 | 1 | 11 | 32 | n/a |
| Guo et al 201335 | 39 | 37 | 42 | 16 | 43 | n/a | ··· | 15 | 41 | 22 | 59 | 31.1±10.3 | 17–51 |
| Guo et al 201536 | 34 | 8 | 17 | 5 | 63 | 1 | 2 | 8 | 100 | 0 | 0 | ··· | ··· |
| Guo et al 201637 | 21 | 21 | 32 | 17 | 81 | 2 | 3 | 17 | 81 | 4 | 19 | 44.8±15.1 | 25–60 |
| Hannuksela et al 201538 | 106 | 44 | 17 | 32 | 73 | 0 | 0 | 27 | 61 | 17 | 39 | 48b | 15–75 |
| Hannuksela et al 201639 | 19 | 6 | 13 | 4 | 67 | 0 | 0 | 0 | 0 | 6 | 100 | 53.2±21.1 | 23–75 |
| Harakalova et al 201340 | 40 | 15 | 20 | 10 | 67 | 3 | 4 | 4 | 37 | 11 | 73 | 46.6±19.5 | 18–70 |
| Hasham et al 200341 | 52 | 17 | 25 | 14 | 82 | n/a | ··· | 9 | 53 | 8 | 47 | 45.4±21.5 | 14–72 |
| Kakko et al 200342 | 115 | 39 | 18 | 25 | 64 | n/a | ··· | 26 | 67 | 13 | 33 | 53.2±15.5 | 26–80 |
| Kent et al 201343 | 93 | 48 | 37 | 37 | 77 | n/a | ··· | n/a | ··· | n/a | ··· | ··· | ··· |
| Keramati et al 201044 | 15 | 13 | 57 | 6 | 46 | n/a | ··· | 10 | 77 | 3 | 23 | n/a | n/a |
| Khau Van Kien et al 200445 | 49 | 8 | 12 | 6 | 75 | 3 | 4 | 4 | 50 | 4 | 50 | n/a | n/a |
| Khau Van Kien et al 200546 | 78 | 8 | 9 | 6 | 75 | 2 | 2 | 4 | 50 | 4 | 50 | n/a | n/a |
| Kuang et al 201647 | 16 | 11 | 28 | 11 | 100 | n/a | ··· | 0 | 0 | 11 | 100 | 44.3±22.6 | 9–88 |
| Loscalzo et al 200748 | 138 | 57 | 29 | 42 | 74 | n/a | ··· | n/a | ··· | n/a | ··· | ··· | ··· |
| Marwick et al 198749 | 4 | 2 | 12 | 1 | 50 | 0 | 0 | 0 | 0 | 2 | 100 | 26.5±3.5 | 24–29 |
| McManus et al 198750 | 8 | 6 | 32 | 5 | 83 | n/a | ··· | 0 | 0 | 6 | 100 | 33.5±14.9 | 22–62 |
| Milewicz et al 199851 | n/a | 30 | 24 | 18 | 60 | 9 | 7 | 12 | 40 | 18 | 60 | 42.9±11.3 | 22–62 |
| Morisaki et al 200952 | 9 | 14 | 30 | 10 | 71 | 5 | 11 | 3 | 21 | 11 | 79 | 36.8±10.1 | 25–52 |
| Pannu et al 200553 | 72 | 58 | 25 | 39 | 66 | n/a | ··· | 27 | 46 | 32 | 54 | 46.1±16.3 | 14–73 |
| Pannu et al 200754 | 23 | 6 | 22 | 4 | 67 | n/a | ··· | 1 | 17 | 5 | 83 | 45±8.8 | 37–56 |
| Regalado et al 201155 | 12 | 52 | 23 | 35 | 67 | 7 | 3 | 9c | 17 | 43b | 83 | 50.8±13.7 | 25–76 |
| Regalado et al 201156 | 36 | 23 | 22 | 14 | 61 | 1 | 1 | 9 | 39 | 14 | 61 | 42d | 25–54 |
| Regalado et al 201157 | 11 | 15 | 52 | 8 | 53 | n/a | ··· | 7 | 47 | 8 | 53 | 32.3±9.9 | 18–50 |
| Renard et al 201358 | 29 | 29 | 30 | 16 | 55 | 3 | 3 | 14 | 48 | 15 | 52 | 48.0±21.2 | 33–63 |
| Robertson et al 201659 | 581 | 486 | 38 | 266 | 72 | n/a | ··· | 370 | 76 | 116 | 24 | 50±13 | n/a |
| Sherrah et al 201660 | 119e | n/a | n/a | 68 | 76 | n/a | ··· | n/a | ··· | n/a | ··· | n/a | ··· |
| Takeda et al 201561 | 9 | 5 | 29 | 4 | 80 | 0 | 0 | 1 | 20 | 4 | 80 | 47.8±16.6 | 32–70 |
| Teixidó‐Turà et al 201462 | 10 | 3 | 8 | 2 | 67 | 1 | 3 | 1 | 33 | 2 | 67 | 46.5±12 | 38–55 |
| Tortora et al 201763 | 77 | 25 | 26 | 61 | 79 | n/a | ··· | 25 | 100 | 0 | 0 | ··· | ··· |
| Tran‐Fadulo et al 200664 | 9 | 21 | 14 | 7 | 33 | 0 | 0 | 4 | 19 | 17 | 81 | 32.0±12.3 | 16–55 |
| Tran‐Fadulo et al 200965 | 49 | 29 | 37 | 17 | 59 | 0 | 0 | 15 | 52 | 14 | 48 | n/a | 14–62 |
| Vaughan et al 200166 | 63 | 30 | 45 | 8 | 27 | n/a | ··· | n/a | ··· | n/a | ··· | ··· | ··· |
| Wang et al 201067 | 21 | 10 | 21 | 5 | 50 | 2 | 4 | 0 | 0 | 10 | 100 | 54.3±20.8 | 16–78 |
| Wang et al 201368 | 8 | 2 | 20 | 2 | 100 | 0 | 0 | 1 | 50 | 1 | 50 | n/a | n/a |
| Ware et al 201469 | 7 | 2 | 20 | 2 | 100 | 0 | 0 | 0 | 0 | 2 | 100 | 17 | ··· |
| Warnes et al 198570 | 2 | 2 | 33 | 2 | 100 | 0 | 0 | 0 | 0 | 2 | 100 | 35.0±18.4 | 22–48 |
| Weigang et al 200771 | 23 | 9 | 35 | 5 | 56 | 0 | 0 | 3 | 33 | 6 | 67 | 32c | 18–47 |
| Yoo et al 201072 | 6 | 5 | 25 | 1 | 20 | 0 | 0 | 0 | 0 | 5 | 100 | 32.5±12.9 | 20–46 |
| Zhu et al 200673 | 49 | 8 | 16 | 7 | 88 | n/a | ··· | 5 | 63 | 3 | 38 | n/a | n/a |
| Ziganshin et al 201574, f | 15 | 4 | 15 | 2 | 50 | n/a | ··· | 1 | 25 | 3 | 75 | n/a | n/a |
| Ziganshin et al 201574, f | 15 | 7 | 41 | 4 | 57 | n/a | ··· | 4 | 57 | 3 | 43 | n/a | n/a |
n/a indicates not available.
Percentage calculated in the family pedigree (as per protocol).
Median available only.
Data available from 4 families only (TAA288, TAA062, TAA549, TAA395).
Mean available only.
Comprehensive of patients affected by bicuspid aortic valve.
Data of 2 different screened families obtained from the same study.
Figure 1.
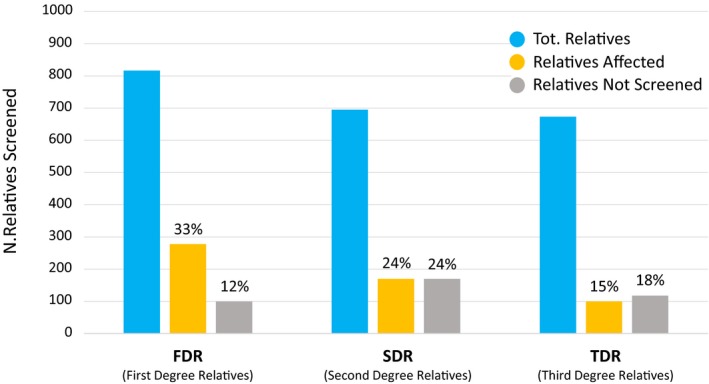
Relatives screened in the studies included in the systematic review. Details for newly affected and not screened individuals are provided for first‐, second‐, and third‐degree relatives (FDRs, SDRs, and TDRs, respectively).
A total of 893 FDRs, 695 SDRs, and 670 TDRs were identified in the family pedigrees of the included studies (Table S5). Of these, a total of 910 newly affected relatives were detected, with an average among studies of 22 newly diagnosed individuals. The percentage of newly diagnosed relatives was 23% (study range: 6–56%). Newly diagnosed individuals were male in 67% of the cases (study range: 20–100%). Sudden unexplained deaths were reported in 2% of the cases (study range: 0–9%). Detailed data about rates of newly affected and screened FDRs, SDRs, and TDRs are depicted in Figure 1.
The type of aortic diseases (aneurysm and dissection rates), male preponderance rate, and age at dissection are summarized in Table 2 and Table S4. Only 1 study screened the relatives of 53 probands identified as affected by a sporadic NS‐TAD form, identifying 83 of 321 newly affected relatives.59
Results of Genetic Tests
The techniques used in the genetic screening, the identified genes, and genetic mutations are listed in Table S8. Genetic screening was employed as the sole screening modality in 26 (49%) studies and in combination with imaging modalities in 21 (40%). A total of 14 known genes were identified as a causative mutation for NS‐TADs, while 3 mapped loci without an identified gene were also found (Table 3 and Table S11). Single‐gene testing was used in 24 (45%) studies, comprehensive genomic sequencing in 14 (26%), and a combination of the 2 approaches in 7 (13%), respectively (Figure 2, Tables S8 and S10).
Table 3.
Genetic Mutations and Correlations With Age and Size at Dissectiona
| Study (Author/Y) | Patients Affected (Aneurysm+Dissection) | Aortic Dissection | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | % | Patients, No. | Patients, % | Male No. | Male, % | Age at Dissection, y | Range, y | Size at Dissection, mm | Range, mm | Patients Available for Analysis | |
| ACTA 2 | |||||||||||
| Chamney et al 201524 | 6 | 43 | 3 | 50 | 3 | 100 | 49±10.4e | 37–55 | n/a | ··· | ··· |
| Disabella et al 201125 | 15 | 41 | 9 | 60 | 5 | 56 | 49.3±16.3e | 29–73 | 59.1±22.3e | 41–95 | 7 |
| Guo et al 200732 | 53 | 25 | 45 | 85 | 23 | 51 | 37.3±13.9e | 13–67 | 61.1±15.0e | 45–100 | 12 |
| Morisaki et al 200952 | 14 | 30 | 11 | 79 | 9 | 82 | 36.8±10.1e | 25–52 | n/a | ··· | ··· |
| Renard et al 201358 | 26 | 32 | 13 | 79 | 7 | 54 | 40.7±15.4e | 27–70 | n/a | ··· | ··· |
| Ware et al 201469 | 2 | 20 | 2 | 100 | 2 | 100 | 17 | ··· | 53±7.1e | 48–58 | 2 |
| Yoo et al 201072 | 5 | 25 | 5 | 100 | 1 | 20 | 32.5±12.9e | 20–46 | 35 | ··· | 1 |
| FBN1 | |||||||||||
| Francke et al 199528 | 10 | 38 | 3 | 30 | 2 | 67 | 55±14.1e | 45–65 | n/a | ··· | ··· |
| Regalado et al 201637 | 15 | 52 | 8 | 53 | 4 | 50 | 32.3±9.9e | 18–50 | 44 | ··· | 1 |
| FOXE3 | |||||||||||
| Kuang et al 201647 | 11 | 28 | 11 | 100 | 11 | 100 | 44.3±22.6e | 9–88 | n/a | ··· | ··· |
| LOX | |||||||||||
| Guo et al 201637 | 21 | 32 | 4 | 19 | 4 | 100 | 44.8±15.1e | 25–60 | n/a | ··· | ··· |
| MYH11 | |||||||||||
| Harakalova et al 201340 | 15 | 20 | 10 | 67 | 7 | 70 | 46.6±19.5e | 18–70 | 58.5±17.3e | 44–65 | 4 |
| Khau Van Kien et al 200546 | 8 | 9 | 4 | 50 | 3 | 75 | n/a | ··· | n/a | ··· | ··· |
| Pannu et al 200856 | 6 | 22 | 5 | 83 | 4 | 80 | 45±8.8e | 37–56 | 44 | ··· | 1 |
| Renard et al 201358 | 3 | 20 | 2 | 83 | 1 | 50 | 48.0±21.2e | 33–63 | n/a | ··· | ··· |
| Takeda et al 201561 | 5 | 29 | 4 | 80 | 4 | 100 | 47.8±16.6e | 32–70 | n/a | ··· | ··· |
| Zhu et al 200673 | 8 | 16 | 3 | 38 | 2 | 67 | n/a | ··· | 37.3±7.8b | n/a | 2 |
| MYLK | |||||||||||
| Hannuksela et al 201639 | 6 | 13 | 6 | 100 | 5 | 83 | 53.2±21.1e | 23–75 | 47.5±0.7e | 47–48 | 2 |
| Wang et al 201066 | 10 | 21 | 10 | 100 | 5 | 50 | 54.3±20.8e | 16–78 | 40 | ··· | 1 |
| Ziganshin et al 201574 | 4 | 15 | 3 | 75 | 1 | 33 | n/a | ··· | n/a | ··· | ··· |
| PRKG1 | |||||||||||
| Gago‐Diaz et al 201630 | 11 | 37 | 6 | 55 | 3 | 50 | 34.2±12.9e | 15–48 | 43±1.4e | 42–44 | 2 |
| Guo et al 201335 | 37 | 42 | 22 | 59 | 10 | 45 | 31.1±10.3e | 17–51 | 47±14.1e | 37–57 | 2 |
| SMAD3 | |||||||||||
| Regalado et al 201156 | 23 | 22 | 14 | 61 | n/a | n/a | 42c | 25–54 | 50 | 50 | 1 |
| TGFB2 | |||||||||||
| Gago‐Diaz et al 201429 | 6 | 19 | 2 | 33 | 2 | 100 | 37.5±4.9e | 34–41 | n/a | ··· | ··· |
| TGFBR1 | |||||||||||
| Dong et al 201427 | 9 | 14 | 3 | 33 | 3 | 100 | 39±6.9e | 35–47 | 51.3±17.9e | 40–72 | 3 |
| Tran‐Fadulo et al 200964 | 29 | 37 | 14 | 48 | 10 | 71 |
25.6±14.3 (male)d
38.6±9.7 (female) |
14–62 | 90.6±42.7e | 65–140 | 2 |
| Ziganshin et al 201574 | 7 | 41 | 3 | 43 | 2 | 67 | n/a | ··· | n/a | ··· | ··· |
| TGFBR2 | |||||||||||
| Hasham et al 200341 | 17 | 25 | 8 | 47 | 6 | 75 | 45.4±21.5e | 14–72 | n/a | ··· | ··· |
| Pannu et al 200553 | 59 | 25 | 32 | 54 | 22 | 69 | 46.1±16.3e | 14–73 | n/a | ··· | ··· |
| Tran‐Fadulo et al 200965 | n/a | ··· | n/a | ··· | n/a | ··· |
42.6±17.8 (male)d
51.3±17.1 (female) |
n/a | 44±2.8e | 42–46 | 2 |
n/a indicates not available.
No data available for patients affected by aortic dissection regarding the genes NOTCH1 (reference 22) and MFAP5 (reference 1), and patients with MAT2A mutation did not experience aortic dissections (reference 15).
Data available for dissection of the descending thoracic aorta only.
Average age onset of dissection as presented by the authors.
Derived from the entire cohort of patients with TGFBR1 and TGFBR2 mutations.
Expressed as mean±SD.
Figure 2.
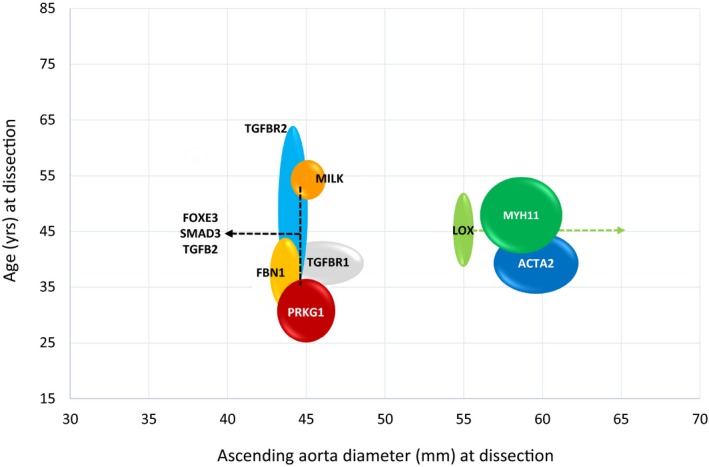
Schematic representation of genetic mutations with age and ascending aorta diameter at dissection. The widening of the circles/lines represents SD in terms of age and diameters. Data are obtained from studies included in the systematic review. No numerical data were available for patients affected by aortic dissection regarding the genes NOTCH1 and MFAP5, and patients with MAT2A mutation did not experience aortic dissections.1, 36, 43
The inheritance mode was essentially autosomal dominant (Table 1). Forty‐one (79%) studies reported on the penetrance of the NS‐TAD. Penetrance varied in relationship to the NS‐TAD form, with an average of 67% (study range: 20–100%) and was lower in females (Table 2). The age at dissection varied according to the underlying NS‐TAD form, with a mean age of presentation of 32 years for the familial thoracic aortic aneurysm and dissection forms associated with the mutations of the PRKG1 gene and of 54 years for those associated with the mutation of the MYLK gene (Figure 2 and Table 3). Ascending aortic diameters at the time of acute dissection were not reported for most of the individuals. Where this was reported, individuals affected by NS‐TADs showed stratification of the diameter of the thoracic aorta (aortic root, mid ascending, or descending aorta) at dissection and in the risk of progression to dissection by genetic mutation: from <4.5 cm for FBN1, FOXE2, MILK, PRKG1, SMAD3, TGFB2, TGFBR1, and TGFBR2, to >5.5 cm for ACTA2, LOX, and MYH11, respectively (Figure 2, Table S10). The identified NS‐TAD forms presented specific characteristics based on the causative genetic mutation (Tables 1 and 3, Table S10, and Figure S2).
Extra‐Aortic Manifestations of NS‐TAD
Concomitant cardiovascular diseases were diagnosed in 11% of the relatives undergoing screening, while concomitant physical abnormalities were observed in 18% of the cases. Full details of all described external physical features and abnormalities of other organ systems are reported in Table S6.
Resource Use and Cost‐Effectiveness
No information about resource use and the cost‐effectiveness of screening program in relatives of patients with NS‐TAD was reported in any of the identified studies. No studies address the psychological effect of screening in patients and their relative or its impact on quality of life of these families.
Discussion
Main Findings
The present study has identified an area of unmet clinical need with respect to screening of relatives of patients with NS‐TAD: familial NS‐TADs occur more frequently than previously recognized, affecting ≈30% of relatives with a male predominance (3:1). These are primarily inherited as single gene mutations, expressed in an autosomal dominant pattern with incomplete penetrance, which demonstrate variable expression with respect to age of presentation, sex, aneurysmal location, and aortic diameter before dissection. The risk of acute aortic syndrome is determined by the underlying genetic mutation and this risk extends not only to FDRs but also to SDRs and TDRs of patients affected by NS‐TADs. There is an overlap between nonsyndromic and syndromic TADs for some genetic mutations, as well as concomitant cardiovascular pathology in over 10% of screened patients. The review also identified knowledge gaps with respect to the predictive accuracy of commonly used screening tests across NS‐TAD populations, the optimal structure and extent of a screening program across families, and the effectiveness of a screening program with respect to clinical outcomes or cost.
Clinical Implications
Nonsyndromic aortopathies have poor prognosis if untreated and the lack of relevant physical features precludes identification based on a clinical characteristics alone.7, 75 As a consequence, NS‐TADs are asymptomatic, alerting clinicians to the underlining aortopathy only when sudden death or an acute aortic dissection occurs.7, 19, 72, 75 This review indicates that routine screening and surveillance programs in relatives of patients affected by NS‐TADs, similar to those of syndromic TAD, are likely to identify significant numbers of patients with asymptomatic NS‐TAD.4, 5, 76, 77 The overlap in genetic mutations between NS‐TAD and syndromic TAD identified in the review further support this assertion. It follows that diagnosis, surveillance, and treatment of NS‐TADs before clinical presentation, as is the standard of care for syndromic TAD, is likely to reduce premature deaths. The findings of this article also indicate that current guidelines which recommend treatment based predominantly on the aortic diameter are likely to result in the undertreatment of NS‐TADs.4, 5 Specifically, subtypes of NS‐TADs attributed to specific genetic mutations may progress to aortic dissection without aneurysm formation.78 Here, the treatment of affected relatives stratified by NS‐TAD subtype and genetic abnormality are likely to result in further clinical benefits (Figure 2).
In addition to defining an area of unmet need, the review has identified important knowledge gaps with respect to screening. Specifically, the diagnostic accuracy of existing screening tests, the optimal screening program, and the clinical, societal, or economic benefits of such a screening program in the relatives of patients with sporadic or familial NS‐TAD are unclear. Current guidelines for the diagnosis and treatment of aortic diseases do not specify the details of what screening tests should be used (Table S11).4, 5, 77 The 2014 European Society of Cardiology guidelines recommend investigating FDRs by genetic counseling for family investigation and molecular testing, with a 5‐year interval screening until diagnosis (clinical or molecular) is established or ruled out (class I, level of evidence C).5 The corresponding 2010 American guidelines suggest aortic imaging screening for FDRs along with counseling and testing whether a specific mutant gene (FBN1, TGFBR1, TGFBR2, COL3A1, ACTA2, MYH11) is identified in the affected probands (class I, level of evidence C).4 These recommendations are based on opinion of the experts and small group studies only.4, 5 Importantly, specific testing schedules, the requirement for screening of SDRs and TDRs, the need for sequencing of other less‐common mutant genes, the optimal screening interval and modality, or the need to investigate the entire arterial tree other than the thoracic aorta are not specified.4, 5 The results of the current study suggest that FDRs, SDRs, and possibly TDRs should be offered screening for TAD. Clarification of the family history regarding the location of the aortic disease, the specific nature of “sudden deaths,” or the presence of other concomitant cardiovascular disorders during clinical examination should represent the initial step of screening.75 Our results also suggest that genetic testing and cardiac imaging with at least TTE should be offered to all FDRs and SDRs of patients with suspected NS‐TADs. Mutation carriers should undergo further imaging (MRI or CT scan), focusing on thoracic aorta and/or other arterial trees based on the causative gene mutation.22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74 For example, ACTA2‐mutation carriers should be monitored for coronary artery disease and occlusive cerebrovascular disease, in addition to the currently recommended routine imaging tests.32 We suggest that complete aortic imaging at initial diagnosis and at 6 months for patients with a confirmed genetic aortopathy (eg, FBN1, FOXE3, MFAP5, MYLK, PRKG1, SMAD3, TGFB2, TGFBR1, and TGFBR2) should be obtained to establish whether aortic enlargement is occurring.4, 74 The final clinical management of patients with a specific gene mutation could be modified on the basis of these data, enabling personalized treatment that is independent of the native aortic diameters.4, 5, 41, 50 Only relatives in whom a causal mutation is excluded and the aortic size is within recommended diameters should reasonably undergo clinical and/or imaging screening every 2 to 5 years, until diagnosis is confirmed or ruled out.5, 76 The appropriate temporal interval for follow‐up imaging, as well as the starting age for aortic surveillance, are also poorly defined. Generally, patients with NS‐TAD are diagnosed on average 10 years older than patients affected by syndromic aortopathies, being also characterized by a lower annual aortic dilatation progression (0.5–0.7 mm/y).59, 60 It therefore seems reasonable to initiate the screening 15 to 10 years earlier than first aneurysm or when dissection or sudden death is recorded within the family.60, 79 A screening pathway based on these observations is proposed in Figure 3.
Figure 3.
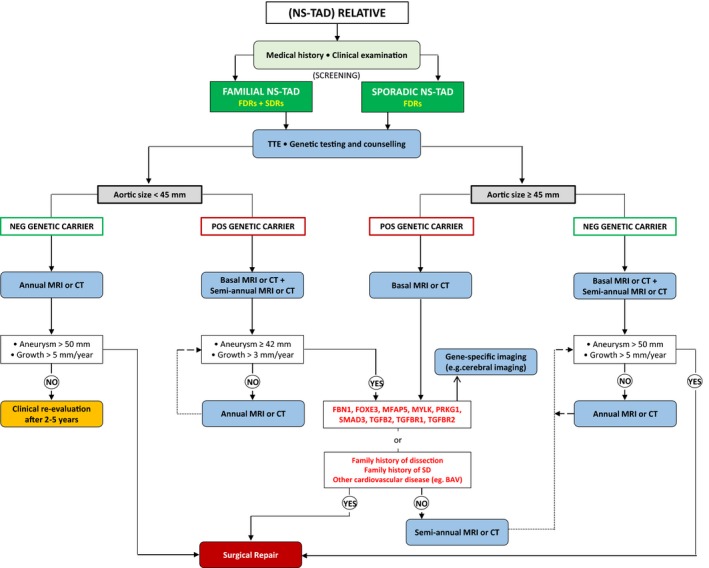
Proposed flow chart for a dedicated screening program for relatives of patients affected by nonsyndromic diseases of the thoracic aorta based on the authors’ extensive literature review. The figure represents the best judgement of the authors. BAV indicates bicuspid aortic valve; CT, computed tomography; FDRs, first‐degree relatives; MRI, magnetic resonance imaging; NS‐TAD, nonsyndromic thoracic aortic disease; SDRs, second‐degree relatives; TTE, transthoracic echocardiogram.
There are several additional factors that may influence our proposed screening algorithm. First, variable penetrance, which often characterizes NS‐TAD forms, is a potential confounder. This results in intrafamilial variability, which is evident not only with reference to the aortopathy itself (severity, age of onset), but also with regard to other phenotypic manifestations.65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81 The presence of associated features is certainly suggestive of having inherited the aortic condition along with the predisposition to the aortopathy, but the absence of these associated features does not eliminate the risk of having an underlying aortopathy. Second, women often demonstrate a lower lifetime risk of aortopathy, developing the condition at a later age than men.81 This phenomenon, known as sexual dimorphism, explains the apparent paradox of an affected teenager with an affected maternal grandfather but an unaffected mother with normal echocardiographic features. Third, the age at onset of the aortopathy may be important in the natural history of the disease. Ma et al82 recently demonstrated that age at onset of aortic dissection is lower in families with a positive history for aortic dissection, therefore suggesting a prompt and more aggressive screening pathway in these families. A positive family history with an aortopathy occurring at younger ages confers a significantly increased risk of developing a new dissection in apparently unaffected family members.81 The above findings are all important in guiding the proper screening and surveillance strategies.
Study Limitations
The most important limitation of the review is the uncertainty regarding the likely yield of new cases if a screening program were to be implemented. The studies identified in our searches were predominantly studies of familial aortopathy, and the prevalence of TAD in these populations will not reflect that for NS‐TAD overall. Conversely, sporadic NS‐TAD, which constitutes the majority (80%) of all cases also has a genetic component.7 Roberston et al59 investigating 321 NS‐TAD probands who had no family history of TAD identified 27% of newly affected relatives. It is likely that these patients are carriers of a de novo mutation, making these “sporadic” patients founders of a new nonsyndromic aortopathy. For example, recent studies have identified gene deletions and uniparental disomy, and genetic variations in LRP1 and ULK4 in sporadic NS‐TAD.83 This suggests that the relatives of patients affected by both familial and sporadic NS‐TADs may benefit from screening. It also argues for use of nontargeted genetic screening tests such as exome or whole genome sequencing that will detect de novo or as‐yet unrecognized common mutations. A second limitation is that there is currently no evidence to inform secondary prevention or intervention strategies in newly diagnosed NS‐TAD, particularly where the aorta is phenotypically normal. Although the evidence presented supports the routine implementation of combined imaging and genetic testing in relatives of patients with NS‐TAD, no study has proven that stratified treatment, independent of the native aortic diameter, will save lives. However, the stratified treatment of syndromic TAD is common practice, as this is known to prevent deaths from aortic disease. We suggest that the results of this review support the extension of similar programs to all patients with TAD. To address these limitations, we propose that further research should first establish the true prevalence of genetic abnormalities and phenotypic disease diagnosed by screening (genetic testing and imaging) all FDRs and SDRs of patients with both familial and sporadic NS‐TAD. Further studies will be required to address uncertainty with respect to effectiveness, psychological impact, and the costs of lifelong screening in these groups. Finally, the heterogeneity of the included studies, the large period of publication across 3 decades, and the familial‐based approach have limited our ability to analyze the impact of region or ethnicity in the risk of aortopathies and the related screening strategy.
Conclusions
The findings of this review support routine imaging and genetic testing of relatives of patients with nonsyndromic aortopathies. The evidence suggests that screening of FDRs and SDRs of patients affected by familial NS‐TAD and FDRs of those affected by sporadic NS‐TADs will result in significant numbers of patients with otherwise undiagnosed disease. Personalized screening programs determined by the subtype of NS‐TAD and its related genetic mutation have the potential to benefit these patients. However, the diagnostic yield of available screening tests is unclear, as are the details of a screening program, and there is no existing evidence that routine screening and stratified treatment will have clinical or economic benefits. Further studies are required to address these knowledge gaps.
Author Contributions
Mariscalco and Debiec had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Mariscalco, Debiec, Samani, and Murphy. Acquisition of data: Mariscalco and Debiec. Analysis and interpretation of data: Mariscalco, Debiec, Samani, and Murphy. Drafting of the article: Mariscalco, Elefteriades, Samani, and Murphy. Critical revision of the article for important intellectual content: Mariscalco, Debiec, Elefteriades, Samani, and Murphy. Article supervision: Mariscalco, Elefteriades, Samani, and Murphy. Statistical analysis: Mariscalco.
Disclosures
Mariscalco declares support from Vascutek, an aortic prosthesis manufacturer, to attend scientific meetings. Murphy declares support from BHF chair of cardiac surgery, Vascutek for attendance at scientific meetings and financial support for educational activities. The remaining authors have no disclosures to report.
Supporting information
Data S1. Systematic review protocol.
Table S1. PRISMA Checklist of Items to Include When Reporting a Systematic Review or Meta‐Analysis*
Table S2. PICOS Criteria for Inclusion and Exclusion of Studies Into Meta‐Analysis
Table S3. Full Details of the Screened Family Relatives With Number and ID of the Included Families
Table S4. Full Details of the Family Pedigree, Eligible, Screened, and Affected Patients and Relatives
Table S5. Full Details of the FDRs, SDRs, and TDRs of Evaluated Probands
Table S6. Full Details of the Screened Families and Relatives With Reference to Additional Observed Cardiovascular Diseases and Physical Features
Table S7. Details of the Adopted Imaging Modalities for the Screening of Relatives
Table S8. Details of the Adopted Screening Modalities in the Included Studies
Table S9. Quality Assessment of the Included Studies
Table S10. Genetic Architecture of Thoracic Aortic Diseases in Nonsyndromic Forms After Screening of the Family Relatives
Table S11. Current Guidelines for Diagnosis and Treatment of Aortic Diseases
Figure S1. PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) flow diagram of search strategy (through December 31, 2017).
Figure S2. Genes with established causative association with nonsyndromic thoracic aortic aneurysms and dissection identified in the present systematic review.
Acknowledgments
The authors thank Dr Tomomi Kimura‐Wozniak and Dr Tanaka Kayoko for the support in the analysis and translation of articles in Japanese language.
(J Am Heart Assoc. 2018;7:e009302 DOI: 10.1161/JAHA.118.009302.)
References
- 1. Sampson UK, Norman PE, Fowkes FG, Aboyans V, Yanna Song, Harrell FE Jr, Forouzanfar MH, Naghavi M, Denenberg JO, McDermott MM, Criqui MH, Mensah GA, Ezzati M, Murray C. Global and regional burden of aortic dissection and aneurysms: mortality trends in 21 world regions, 1990 to 2010. Glob Heart. 2014;9:171–180. [DOI] [PubMed] [Google Scholar]
- 2. von Allmen RS, Anjum A, Powell JT. Incidence of descending aortic pathology and evaluation of the impact of thoracic endovascular aortic repair: a population‐based study in England and Wales from 1999 to 2010. Eur J Vasc Endovasc Surg. 2013;45:154–159. [DOI] [PubMed] [Google Scholar]
- 3. Bottle A, Mariscalco G, Shaw MA, Benedetto U, Saratzis A, Mariani S, Bashir M, Aylin P, Jenkins D, Oo AY, Murphy GJ; UK Aortic Forum . Unwarranted variation in the quality of care for patients with diseases of the thoracic aorta. J Am Heart Assoc. 2017;6:e004913 DOI: 10.1161/JAHA.116.004913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE Jr, Eagle KA, Hermann LK, Isselbacher EM, Kazerooni EA, Kouchoukos NT, Lytle BW, Milewicz DM, Reich DL, Sen S, Shinn JA, Svensson LG, Williams DM; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine . 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121:e266–e369. [DOI] [PubMed] [Google Scholar]
- 5. Erbel R, Aboyans V, Boileau C, Bossone E, Bartolomeo RD, Eggebrecht H, Evangelista A, Falk V, Frank H, Gaemperli O, Grabenwöger M, Haverich A, Iung B, Manolis AJ, Meijboom F, Nienaber CA, Roffi M, Rousseau H, Sechtem U, Sirnes PA, Allmen RS, Vrints CJ; ESC Committee for Practice Guidelines . 2014 ESC guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2873–2926. [DOI] [PubMed] [Google Scholar]
- 6. Booher AM, Isselbacher EM, Nienaber CA, Trimarchi S, Evangelista A, Montgomery DG, Froehlich JB, Ehrlich MP, Oh JK, Januzzi JL, O'Gara P, Sundt TM, Harris KM, Bossone E, Pyeritz RE, Eagle KA; IRAD Investigators . The IRAD classification system for characterizing survival after aortic dissection. Am J Med. 2013;126:730.e19–24. [DOI] [PubMed] [Google Scholar]
- 7. Coady MA, Davies RR, Roberts M, Goldstein LJ, Rogalski MJ, Rizzo JA, Hammond GL, Kopf GS, Elefteriades JA. Familial patterns of thoracic aortic aneurysms. Arch Surg. 1999;134:361–367. [DOI] [PubMed] [Google Scholar]
- 8. Mariscalco G, Radoslaw D, Murphy G. A systematic review of screening for relatives of patients with non‐syndromic thoracic aortic diseases. PROSPERO 2017:CRD42017064598.
- 9. Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group . Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. BMJ. 2009;339:b2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. OMIM®. Online Mendelian Inheritance in Man® . An online catalog of human genes and genetic disorders. Available at: https://www.omin.org. Accessed June 30, 2017.
- 11. Evangelista A, Flachskampf FA, Erbel R, Antonini‐Canterin F, Vlachopoulos C, Rocchi G, Sicari R, Nihoyannopoulos P, Zamorano J; European Association of Echocardiography; Document Reviewers , Pepi M, Breithardt OA, Plonska‐Gosciniak E. Echocardiography in aortic diseases: EAE recommendations for clinical practice. Eur J Echocardiogr. 2010;11:645–658. [DOI] [PubMed] [Google Scholar]
- 12. Rodríguez‐Palomares JF, Teixidó‐Tura G, Galuppo V, Cuéllar H, Laynez A, Gutiérrez L, González‐Alujas MT, García‐Dorado D, Evangelista A. Multimodality assessment of ascending aortic diameters: comparison of different measurement methods. J Am Soc Echocardiogr. 2016;29:819–826.e4. [DOI] [PubMed] [Google Scholar]
- 13. Tamborini G, Galli CA, Maltagliati A, Andreini D, Pontone G, Quaglia C, Ballerini G, Pepi M. Comparison of feasibility and accuracy of transthoracic echocardiography versus computed tomography in patients with known ascending aortic aneurysm. Am J Cardiol. 2006;98:966–969. [DOI] [PubMed] [Google Scholar]
- 14. Mussa FF, Horton JD, Moridzadeh R, Nicholson J, Trimarchi S, Eagle KA. Acute aortic dissection and intramural hematoma: a systematic review. JAMA. 2016;316:754–763. [DOI] [PubMed] [Google Scholar]
- 15. Moore AG, Eagle KA, Bruckman D, Moon BS, Malouf JF, Fattori R, Evangelista A, Isselbacher EM, Suzuki T, Nienaber CA, Gilon D, Oh JK. Choice of computed tomography, transesophageal echocardiography, magnetic resonance imaging, and aortography in acute aortic dissection: International Registry of Acute Aortic Dissection (IRAD). Am J Cardiol. 2002;89:1235–1238. [DOI] [PubMed] [Google Scholar]
- 16. Milewicz DM, Regalado E. Heritable thoracic aortic disease overview In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, Bird TD, Ledbetter N, Mefford HC, Smith RJ, Stephens K, eds. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993. –2018. [PubMed] [Google Scholar]
- 17. Milewicz DM, Prakash SK, Ramirez F. Therapeutics targeting drivers of thoracic aortic aneurysms and acute aortic dissections: insights from predisposing genes and mouse models. Annu Rev Med. 2017;68:51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arslan‐Kirchner M, Arbustini E, Boileau C, Charron P, Child AH, Collod‐Beroud G, De Backer J, De Paepe A, Dierking A, Faivre L, Hoffjan S, Jondeau G, Keyser B, Loeys B, Mayer K, Robinson PN, Schmidtke J. Clinical utility gene card for: hereditary thoracic aortic aneurysm and dissection including next‐generation sequencing‐based approaches. Eur J Hum Genet. 2016;24:e1–e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wells GA, Shea B, O'Connell D, Peterson J, Welch V, Losos M, Tugwell P. The Newcastle‐Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta‐analyses. Ottawa Hospital Research Institute; Available at: http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp. Accessed June 30, 2017. [Google Scholar]
- 20. Harris RP, Helfand M, Woolf SH, Lohr KN, Mulrow CD, Teutsch SM, Atkins D; Methods Work Group, Third US Preventive Services Task Force . Current methods of the US Preventive Services Task Force: a review of the process. Am J Prev Med. 2001;20(3 suppl):21–35. [DOI] [PubMed] [Google Scholar]
- 21. Higgins JPT, Green S, eds. Cochrane Handbook for Systematic Reviews of Interventions. Chichester, UK: John Wiley and Sons; 2008. [Google Scholar]
- 22. Barbier M, Gross MS, Aubart M, Hanna N, Kessler K, Guo DC, Tosolini L, Ho‐Tin‐Noe B, Regalado E, Varret M, Abifadel M, Milleron O, Odent S, Dupuis‐Girod S, Faivre L, Edouard T, Dulac Y, Busa T, Gouya L, Milewicz DM, Jondeau G, Boileau C. MFAP5 loss‐of‐function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am J Hum Genet. 2014;95:736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bee KJ, Wilkes DC, Devereux RB, Basson CT, Hatcher CJ. TGFβRIIb mutations trigger aortic aneurysm pathogenesis by altering transforming growth factor β2 signal transduction. Circ Cardiovasc Genet. 2012;5:621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chamney S, McGimpsey S, McConnell V, Willoughby CE. Iris Flocculi as an ocular marker of ACTA2 mutation in familial thoracic aortic aneurysms and dissections. Ophthalmic Genet. 2015;36:86–88. [DOI] [PubMed] [Google Scholar]
- 25. Disabella E, Grasso M, Gambarin FI, Narula N, Dore R, Favalli V, Serio A, Antoniazzi E, Mosconi M, Pasotti M, Odero A, Arbustini E. Risk of dissection in thoracic aneurysms associated with mutations of smooth muscle alpha‐actin 2 (ACTA2). Heart. 2011;97:321–326. [DOI] [PubMed] [Google Scholar]
- 26. Disertori M, Bertagnolli C, Thiene G, Ferro A, Bonmassari R, Girardini D, Casarotto D. Familial dissecting aortic aneurysm. G Ital Cardiol. 1991;21:849–853. [PubMed] [Google Scholar]
- 27. Dong SB, Zheng J, Ma WG, Chen MJ, Cheng LJ, He L, Xing QH, Sun LZ. Identification and surgical repair of familial thoracic aortic aneurysm and dissection caused by TGFBR1 mutation. Ann Vasc Surg. 2014;28:1909–1912. [DOI] [PubMed] [Google Scholar]
- 28. Francke U, Berg MA, Tynan K, Brenn T, Liu W, Aoyama T, Gasner C, Miller DC, Furthmayr H. A Gly1127Ser mutation in an EGF‐like domain of the fibrillin‐1 gene is a risk factor for ascending aortic aneurysm and dissection. Am J Hum Genet. 1995;56:1287–1296. [PMC free article] [PubMed] [Google Scholar]
- 29. Gago‐Díaz M, Blanco‐Verea A, Teixidó‐Turà G, Valenzuela I, Del Campo M, Borregan M, Sobrino B, Amigo J, García‐Dorado D, Evangelista A, Carracedo A, Brion M. Whole exome sequencing for the identification of a new mutation in TGFB2 involved in a familial case of non‐syndromic aortic disease. Clin Chim Acta. 2014;437:88–92. [DOI] [PubMed] [Google Scholar]
- 30. Gago‐Díaz M, Blanco‐Verea A, Teixidó G, Huguet F, Gut M, Laurie S, Gut I, Carracedo Á, Evangelista A, Brion M. PRKG1 and genetic diagnosis of early‐onset thoracic aortic disease. Eur J Clin Invest. 2016;46:787–794. [DOI] [PubMed] [Google Scholar]
- 31. Guo D, Hasham S, Kuang SQ, Vaughan CJ, Boerwinkle E, Chen H, Abuelo D, Dietz HC, Basson CT, Shete SS, Milewicz DM. Familial thoracic aortic aneurysms and dissections: genetic heterogeneity with a major locus mapping to 5q13‐14. Circulation. 2001;103:2461–2468. [DOI] [PubMed] [Google Scholar]
- 32. Guo DC, Pannu H, Tran‐Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha‐actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. [DOI] [PubMed] [Google Scholar]
- 33. Guo DC, Papke CL, Tran‐Fadulu V, Regalado ES, Avidan N, Johnson RJ, Kim DH, Pannu H, Willing MC, Sparks E, Pyeritz RE, Singh MN, Dalman RL, Grotta JC, Marian AJ, Boerwinkle EA, Frazier LQ, LeMaire SA, Coselli JS, Estrera AL, Safi HJ, Veeraraghavan S, Muzny DM, Wheeler DA, Willerson JT, Yu RK, Shete SS, Scherer SE, Raman CS, Buja LM, Milewicz DM. Mutations in smooth muscle alpha‐actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84:617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo DC, Regalado ES, Minn C, Tran‐Fadulu V, Coney J, Cao J, Wang M, Yu RK, Estrera AL, Safi HJ, Shete SS, Milewicz DM. Familial thoracic aortic aneurysms and dissections: identification of a novel locus for stable aneurysms with a low risk for progression to aortic dissection. Circ Cardiovasc Genet. 2011;4:36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guo DC, Regalado E, Casteel DE, Santos‐Cortez RL, Gong L, Kim JJ, Dyack S, Horne SG, Chang G, Jondeau G, Boileau C, Coselli JS, Li Z, Leal SM, Shendure J, Rieder MJ, Bamshad MJ, Nickerson DA; GenTAC Registry Consortium; National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project , Kim C, Milewicz DM. Recurrent gain‐of‐function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet. 2013;93:398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo DC, Gong L, Regalado ES, Santos‐Cortez RL, Zhao R, Cai B, Veeraraghavan S, Prakash SK, Johnson RJ, Muilenburg A, Willing M, Jondeau G, Boileau C, Pannu H, Moran R, Debacker J; GenTAC Investigators, National Heart, Lung, and Blood Institute Go Exome Sequencing Project; Montalcino Aortic Consortium , Bamshad MJ, Shendure J, Nickerson DA, Leal SM, Raman CS, Swindell EC, Milewicz DM. MAT2A mutations predispose individuals to thoracic aortic aneurysms. Am J Hum Genet. 2015;96:170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guo DC, Regalado ES, Gong L, Duan X, Santos‐Cortez RL, Arnaud P, Ren Z, Cai B, Hostetler EM, Moran R, Liang D, Estrera A, Safi HJ; University of Washington Center for Mendelian Genomics , Leal SM, Bamshad MJ, Shendure J, Nickerson DA, Jondeau G, Boileau C, Milewicz DM. LOX mutations predispose to thoracic aortic aneurysms and dissections. Circ Res. 2016;118:928–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hannuksela M, Stattin EL, Johansson B, Carlberg B. Screening for familial thoracic aortic aneurysms with aortic imaging does not detect all potential carriers of the disease. Aorta (Stamford). 2015;3:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hannuksela M, Stattin EL, Klar J, Ameur A, Johansson B, Sörensen K, Carlberg B. A novel variant in MYLK causes thoracic aortic dissections: genotypic and phenotypic description. BMC Med Genet. 2016;17:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Harakalova M, van der Smagt J, de Kovel CG, Van't Slot R, Poot M, Nijman IJ, Medic J, Joziasse I, Deckers J, Roos‐Hesselink JW, Wessels MW, Baars HF, Weiss MM, Pals G, Golmard L, Jeunemaitre X, Lindhout D, Cuppen E, Baas AF. Incomplete segregation of MYH11 variants with thoracic aortic aneurysms and dissections and patent ductus arteriosus. Eur J Hum Genet. 2013;21:487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hasham SN, Willing MC, Guo DC, Muilenburg A, He R, Tran VT, Scherer SE, Shete SS, Milewicz DM. Mapping a locus for familial thoracic aortic aneurysms and dissections (TAAD2) to 3p24‐25. Circulation. 2003;107:3184–3190. [DOI] [PubMed] [Google Scholar]
- 42. Kakko S, Räisänen T, Tamminen M, Airaksinen J, Groundstroem K, Juvonen T, Ylitalo A, Uusimaa P, Savolainen MJ. Candidate locus analysis of familial ascending aortic aneurysms and dissections confirms the linkage to the chromosome 5q13‐14 in Finnish families. J Thorac Cardiovasc Surg. 2003;126:106–113. [DOI] [PubMed] [Google Scholar]
- 43. Kent KC, Crenshaw ML, Goh DL, Dietz HC. Genotype‐phenotype correlation in patients with bicuspid aortic valve and aneurysm. J Thorac Cardiovasc Surg. 2013;146:158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Keramati AR, Sadeghpour A, Farahani MM, Chandok G, Mani A. The non‐syndromic familial thoracic aortic aneurysms and dissections maps to 15q21 locus. BMC Med Genet. 2010;11:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Khau Van Kien P, Wolf JE, Mathieu F, Zhu L, Salve N, Lalande A, Bonnet C, Lesca G, Plauchu H, Dellinger A, Nivelon‐Chevallier A, Brunotte F, Jeunemaitre X. Familial thoracic aortic aneurysm/dissection with patent ductus arteriosus: genetic arguments for a particular pathophysiological entity. Eur J Hum Genet. 2004;12:173–180. [DOI] [PubMed] [Google Scholar]
- 46. Khau Van Kien P, Mathieu F, Zhu L, Lalande A, Betard C, Lathrop M, Brunotte F, Wolf JE, Jeunemaitre X. Mapping of familial thoracic aortic aneurysm/dissection with patent ductus arteriosus to 16p12.2‐p13.13. Circulation. 2005;112:200–206. [DOI] [PubMed] [Google Scholar]
- 47. Kuang SQ, Medina‐Martinez O, Guo DC, Gong L, Regalado ES, Reynolds CL, Boileau C, Jondeau G, Prakash SK, Kwartler CS, Zhu LY, Peters AM, Duan XY, Bamshad MJ, Shendure J, Nickerson DA, Santos‐Cortez RL, Dong X, Leal SM, Majesky MW, Swindell EC, Jamrich M, Milewicz DM. FOXE3 mutations predispose to thoracic aortic aneurysms and dissections. J Clin Invest. 2016;126:948–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Loscalzo ML, Goh DL, Loeys B, Kent KC, Spevak PJ, Dietz HC. Familial thoracic aortic dilation and bicommissural aortic valve: a prospective analysis of natural history and inheritance. Am J Med Genet A. 2007;143A:1960–1967. [DOI] [PubMed] [Google Scholar]
- 49. Marwick TH, Woodhouse SP, Birchley IN, Strong RW. Management of familial aortic dissection. Chest. 1987;92:954–956. [DOI] [PubMed] [Google Scholar]
- 50. McManus BM, Cassling RS, Soundy TJ, Wilson JE, Sears TD, Rogler WC, Buehler BA, Wolford JF, Duggan MJ, Byers PH, Fleming WH, Sanger WG. Familial aortic dissection in absence of ascending aortic aneurysms: a lethal syndrome associated with precocious systemic hypertension. Am J Cardiovasc Pathol. 1987;1:55–67. [PubMed] [Google Scholar]
- 51. Milewicz DM, Chen H, Park ES, Petty EM, Zaghi H, Shashidhar G, Willing M, Patel V. Reduced penetrance and variable expressivity of familial thoracic aortic aneurysms/dissections. Am J Cardiol. 1998;82:474–479. [DOI] [PubMed] [Google Scholar]
- 52. Morisaki H, Akutsu K, Ogino H, Kondo N, Yamanaka I, Tsutsumi Y, Yoshimuta T, Okajima T, Matsuda H, Minatoya K, Sasaki H, Tanaka H, Ishibashi‐Ueda H, Morisaki T. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD). Hum Mutat. 2009;30:1406–1411. [DOI] [PubMed] [Google Scholar]
- 53. Pannu H, Fadulu VT, Chang J, Lafont A, Hasham SN, Sparks E, Giampietro PF, Zaleski C, Estrera AL, Safi HJ, Shete S, Willing MC, Raman CS, Milewicz DM. Mutations in transforming growth factor‐beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–520. [DOI] [PubMed] [Google Scholar]
- 54. Pannu H, Tran‐Fadulu V, Papke CL, Scherer S, Liu Y, Presley C, Guo D, Estrera AL, Safi HJ, Brasier AR, Vick GW, Marian AJ, Raman CS, Buja LM, Milewicz DM. MYH11 mutations result in a distinct vascular pathology driven by insulin‐like growth factor 1 and angiotensin II. Hum Mol Genet. 2007;16:2453–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Regalado E, Medrek S, Tran‐Fadulu V, Guo DC, Pannu H, Golabbakhsh H, Smart S, Chen JH, Shete S, Kim DH, Stern R, Braverman AC, Milewicz DM. Autosomal dominant inheritance of a predisposition to thoracic aortic aneurysms and dissections and intracranial saccular aneurysms. Am J Med Genet A. 2011;155A:2125–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Regalado ES, Guo DC, Villamizar C, Avidan N, Gilchrist D, McGillivray B, Clarke L, Bernier F, Santos‐Cortez RL, Leal SM, Bertoli‐Avella AM, Shendure J, Rieder MJ, Nickerson DA; NHLBI GO Exome Sequencing Project , Milewicz DM. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011;109:680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Regalado ES, Guo DC, Santos‐Cortez RL, Hostetler E, Bensend TA, Pannu H, Estrera A, Safi H, Mitchell AL, Evans JP, Leal SM, Bamshad M, Shendure J, Nickerson DA; University of Washington Center for Mendelian Genomics , Milewicz DM. Pathogenic FBN1 variants in familial thoracic aortic aneurysms and dissections. Clin Genet. 2016;89:719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Renard M, Callewaert B, Baetens M, Campens L, MacDermot K, Fryns JP, Bonduelle M, Dietz HC, Gaspar IM, Cavaco D, Stattin EL, Schrander‐Stumpel C, Coucke P, Loeys B, De Paepe A, De Backer J. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFβ signaling in FTAAD. Int J Cardiol. 2013;165:314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Robertson EN, van der Linde D, Sherrah AG, Vallely MP, Wilson M, Bannon PG, Jeremy RW. Familial non‐syndromal thoracic aortic aneurysms and dissections—incidence and family screening outcomes. Int J Cardiol. 2016;220:43–51. [DOI] [PubMed] [Google Scholar]
- 60. Sherrah AG, Andvik S, van der Linde D, Davies L, Bannon PG, Padang R, Vallely MP, Wilson MK, Keech AC, Jeremy RW. Nonsyndromic thoracic aortic aneurysm and dissection: outcomes with Marfan syndrome versus bicuspid aortic valve aneurysm. J Am Coll Cardiol. 2016;67:618–626. [DOI] [PubMed] [Google Scholar]
- 61. Takeda N, Morita H, Fujita D, Inuzuka R, Taniguchi Y, Nawata K, Komuro I. A deleterious MYH11 mutation causing familial thoracic aortic dissection. Hum Genome Var. 2015;2:15028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Teixidó‐Turà G, Valenzuela I, Gutiérrez L, Borregan M, del Campo M, Evangelista A. Nonsyndromic familial aortic disease: an underdiagnosed entity. Rev Esp Cardiol (Engl Ed). 2014;67:861–863. [DOI] [PubMed] [Google Scholar]
- 63. Tran‐Fadulu V, Chen JH, Lemuth D, Neichoy BT, Yuan J, Gomes N, Sparks E, Kramer LA, Guo D, Pannu H, Braverman AC, Shete S, Milewicz DM. Familial thoracic aortic aneurysms and dissections: three families with early‐onset ascending and descending aortic dissections in women. Am J Med Genet A. 2006;140:1196–1202. [DOI] [PubMed] [Google Scholar]
- 64. Tortora G, Wischmeijer A, Berretta P, Alfonsi J, Marco L, Barbieri A, Marconi C, Isidori F, Rossi C, Leone O, Di Bartolomeo R, Seri M, Pacini D. Search for genetic factors in bicuspid aortic valve disease: ACTA 2 mutations do not play a major role. Interact Cardiovasc Thorac Surg. 2017;25:813–817. [DOI] [PubMed] [Google Scholar]
- 65. Tran‐Fadulu V, Pannu H, Kim DH, Vick GW III, Lonsford CM, Lafont AL, Boccalandro C, Smart S, Peterson KL, Hain JZ, Willing MC, Coselli JS, LeMaire SA, Ahn C, Byers PH, Milewicz DM. Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. J Med Genet. 2009;46:607–613. [DOI] [PubMed] [Google Scholar]
- 66. Vaughan CJ, Casey M, He J, Veugelers M, Henderson K, Guo D, Campagna R, Roman MJ, Milewicz DM, Devereux RB, Basson CT. Identification of a chromosome 11q23.2‐q24 locus for familial aortic aneurysm disease, a genetically heterogeneous disorder. Circulation. 2001;103:2469–2475. [DOI] [PubMed] [Google Scholar]
- 67. Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, Li L, Shete S, He WQ, Zhu MS, Offermanns S, Gilchrist D, Elefteriades J, Stull JT, Milewicz DM. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet. 2010;87:701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang WJ, Han P, Zheng J, Hu FY, Zhu Y, Xie JS, Guo J, Zhang Z, Dong J, Zheng GY, Cao H, Liu TS, Fu Q, Sun L, Yang BB, Tian XL. Exon 47 skipping of fibrillin‐1 leads preferentially to cardiovascular defects in patients with thoracic aortic aneurysms and dissections. J Mol Med (Berl). 2013;91:37–47. [DOI] [PubMed] [Google Scholar]
- 69. Ware SM, Shikany A, Landis BJ, James JF, Hinton RB. Twins with progressive thoracic aortic aneurysm, recurrent dissection and ACTA2 mutation. Pediatrics. 2014;134:e1218–e1223. [DOI] [PubMed] [Google Scholar]
- 70. Warnes CA, Kirkman PM, Roberts WC. Aortic dissection in more than one family member. Am J Cardiol. 1985;55:236–238. [DOI] [PubMed] [Google Scholar]
- 71. Weigang E, Chang XC, Munk‐Schulenburg S, Richter H, von Samson P, Goebel H, Frydrychowicz A, Geibel A, Ammann S, Schwering L, Brunner T, Severin T, Czerny M, Beyersdorf F. Actual management of patients with familial ascending aortic aneurysms and type‐A aortic dissections. Thorac Cardiovasc Surg. 2007;55:19–23. [DOI] [PubMed] [Google Scholar]
- 72. Yoo EH, Choi SH, Jang SY, Suh YL, Lee I, Song JK, Choe YH, Kim JW, Ki CS, Kim DK. Clinical, pathological, and genetic analysis of a Korean family with thoracic aortic aneurysms and dissections carrying a novel Asp26Tyr mutation. Ann Clin Lab Sci. 2010;40:278–284. [PubMed] [Google Scholar]
- 73. Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–349. [DOI] [PubMed] [Google Scholar]
- 74. Ziganshin BA, Bailey AE, Coons C, Dykas D, Charilaou P, Tanriverdi LH, Liu L, Tranquilli M, Bale AE, Elefteriades JA. Routine genetic testing for thoracic aortic aneurysm and dissection in a clinical setting. Ann Thorac Surg. 2015;100:1604–1611. [DOI] [PubMed] [Google Scholar]
- 75. Groenink M, Lohuis TA, Tijssen JG, Naeff MS, Hennekam RC, van der Wall EE, Mulder BJ. Survival and complication free survival in Marfan's syndrome: implications of current guidelines. Heart. 1999;82:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bradley TJ, Bowdin SC. Multidisciplinary aortopathy clinics should now be the standard of care in Canada. Can J Cardiol. 2016;32:8–12. [DOI] [PubMed] [Google Scholar]
- 77. Boodhwani M, Andelfinger G, Leipsic J, Lindsay T, McMurtry MS, Therrien J, Siu SC; Canadian Cardiovascular Society . Canadian Cardiovascular Society position statement on the management of thoracic aortic disease. Can J Cardiol. 2014;30:577–589. [DOI] [PubMed] [Google Scholar]
- 78. Pape LA, Tsai TT, Isselbacher EM, Oh JK, O'gara PT, Evangelista A, Fattori R, Meinhardt G, Trimarchi S, Bossone E, Suzuki T, Cooper JV, Froehlich JB, Nienaber CA, Eagle KA; International Registry of Acute Aortic Dissection (IRAD) Investigators ; Aortic diameter > or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation. 2007;116:1120–1127. [DOI] [PubMed] [Google Scholar]
- 79. Brown CR, Greenberg RK, Wong S, Eagleton M, Mastracci T, Hernandez AV, Rigelsky CM, Moran R. Family history of aortic disease predicts disease patterns and progression and is a significant influence on management strategies for patients and their relatives. J Vasc Surg. 2013;58:573–581. [DOI] [PubMed] [Google Scholar]
- 80. Richer J, Laberge AM. Screening children for familial aortopathies: tread with caution. Can J Cardiol. 2016;32:60–65. [DOI] [PubMed] [Google Scholar]
- 81. Ma WG, Chou AS, Mok SCM, Ziganshin BA, Charilaou P, Zafar MA, Sieller RS, Tranquilli M, Rizzo JA, Elefteriades JA. Positive family history of aortic dissection dramatically increases dissection risk in family members. Int J Cardiol. 2017;240:132–137. [DOI] [PubMed] [Google Scholar]
- 82. Prakash S, LeMaire SA, Bray M, Milewicz DM, Belmont JW. Large deletions and uniparental disomy detected by SNP arrays in adults with thoracic aortic aneurysms and dissections. Am J Med Genet A. 2010;152A:2399–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Guo DC, Grove ML, Prakash SK, Eriksson P, Hostetler EM, LeMaire SA, Body SC, Shalhub S, Estrera AL, Safi HJ, Regalado ES, Zhou W, Mathis MR; GenTAC Investigators; BAVCon Investigators , Eagle KA, Yang B, Willer CJ, Boerwinkle E, Milewicz DM. Genetic variants in LRP1 and ULK4 are associated with acute aortic dissections. Am J Hum Genet. 2016;99:762–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Systematic review protocol.
Table S1. PRISMA Checklist of Items to Include When Reporting a Systematic Review or Meta‐Analysis*
Table S2. PICOS Criteria for Inclusion and Exclusion of Studies Into Meta‐Analysis
Table S3. Full Details of the Screened Family Relatives With Number and ID of the Included Families
Table S4. Full Details of the Family Pedigree, Eligible, Screened, and Affected Patients and Relatives
Table S5. Full Details of the FDRs, SDRs, and TDRs of Evaluated Probands
Table S6. Full Details of the Screened Families and Relatives With Reference to Additional Observed Cardiovascular Diseases and Physical Features
Table S7. Details of the Adopted Imaging Modalities for the Screening of Relatives
Table S8. Details of the Adopted Screening Modalities in the Included Studies
Table S9. Quality Assessment of the Included Studies
Table S10. Genetic Architecture of Thoracic Aortic Diseases in Nonsyndromic Forms After Screening of the Family Relatives
Table S11. Current Guidelines for Diagnosis and Treatment of Aortic Diseases
Figure S1. PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) flow diagram of search strategy (through December 31, 2017).
Figure S2. Genes with established causative association with nonsyndromic thoracic aortic aneurysms and dissection identified in the present systematic review.