

Abstract
Background
Malaria rapid diagnostic tests (RDTs) represent an important antibody based immunoassay platform. Unfortunately, conventional monoclonal antibodies are subject to degradation shortening shelf lives of RDTs. The variable region of the receptor (VNAR) from shark has a potential as alternative to monoclonal antibodies in RDTs due to high thermal stability.
Methods
In this study, new binders derived from shark VNAR domains library were investigated. Following immunization of a wobbegong shark (Orectolobus ornatus) with three recombinant malaria biomarker proteins (PfHRP2, PfpLDH and Pvaldolase), a single domain antibody (sdAb) library was constructed from splenocytes. Target-specific VNAR phage were isolated by panning. One specific clone was selected for expression in Escherichia coli expression system, and study of binding reactivity undertaken.
Results
The primary VNAR domain library possessed a titre of 1.16 × 106 pfu/mL. DNA sequence analysis showed 82.5% of isolated fragments appearing to contain an in-frame sequence. After multiple rounds of biopanning, a highly dominant clone specific to PfHRP2 was identified and selected for protein production in an E. coli expression system. Biological characterization showed the recombinant protein expressed in periplasmic has better detection sensitivity than that of cytoplasmic proteins. Assays of binding activity indicated that its reactivity was inferior to the positive control mAb C1–13.
Conclusions
Target-specific bacteriophage VNARs were successfully isolated after a series of immunization, demonstrating that phage display technology is a useful tool for selection of antigen binders. Generation of new binding reagents such as VNAR antibodies that specifically recognize the malaria biomarkers represents an appealing approach to improve the performance of RDTs.
Electronic supplementary material
The online version of this article (10.1186/s12936-018-2531-y) contains supplementary material, which is available to authorized users.
Background
Malaria remains one of the most life-threatening infectious diseases in the world. Five species of Plasmodium cause malaria in humans. Of these species, infection with Plasmodium falciparum is the most prevalent and lethal, causing significant morbidity and mortality worldwide [1]. Most of the P. falciparum-detecting rapid diagnostic tests (RDTs) target histidine-rich protein 2 (PfHRP2) [2]. PfHRP2 is a water-soluble protein that is produced by all asexual stages of P. falciparum including gametocytes. This protein is abundantly expressed in the red cell, released during rupture of infected red cells and can remain in the blood for up to 28 days after the initiation of anti-malarial therapy, making it an excellent biomarker for diagnosing P. falciparum infections [3]. Plasmodium lactate dehydrogenase (pLDH), and fructose 1,6-biphosphate aldolase (Aldolase) are biomarkers commonly used for the detection of non-P. falciparum human malaria infections (species specific or PAN specific) and P. vivax infections, respectively [4, 5].
Unfortunately, the degradation of sensitive capture and detecting antibody reagents in malaria RDTs [6] can shorten the shelf lives of RDTs and may also result in false negative diagnosis and eventually delay the treatment time if undetected [7]. Antibodies with better stability profiles would improve the stability of RDTs. However, despite early attempts to engineer antibodies into more robust antibody fragments [8, 9], separating the VH and VL domains while retaining antibody specificity has proven to be difficult [10, 11]. In nature, sharks are the most ancient phylogenetic vertebrate group possessing the complete molecular components of an adaptive immune system [12, 13]. In contrast to immunoglobulin (Ig) isotypes in higher mammals, the immunoglobulin new receptor (IgNAR) of sharks are unusual antibodies that lack light chains and, therefore, exist as homodimers of a heavy chain [14]. Immune electron microscopy indicated that the IgNAR heavy chain contains one variable domain of (VNAR) and five constant (C) domains [15]. Similar to VHH in the camelid family, VNAR domains can function as soluble single domains which are capable of antigen binding [15]. These single domain fragments display excellent solubility and high thermostability due to substitutions of amino acids at VH–VL interaction, making the interface more hydrophilic compared to the hydrophobic interface present in conventional antibodies [14, 16].
Similar to the variable domains of conventional immunoglobulin scaffolds, shark VNAR have been determined to have four highly conserved framework regions (FR) and three highly variable complementary determining regions (CDRs). The deletion of a large portion of FR2–CDR2 has therefore made VNAR the smallest variable domain, with size of ~ 12 kDa [14]. In addition, shark VNAR domains possess an extraordinary CDR3 domain which is much longer than that of conventional antibodies. Therefore, the penetration capability of VNAR is perceived much easier to reach to the cleft region of the target antigen [17, 18]. Thus far, VNAR is recognized as the smallest natural single domain antibodies (sdAbs) found to date in the animal kingdom [14, 19].
The selection of a suitable expression system is vital to ensuring the solubility and correct folding required for expression of functional VNAR protein. Bacterial expression systems are often the first choice in the laboratories for the production of recombinant proteins for therapeutic and diagnostic applications [20–22]. It is amenable to produce recombinant proteins for a range of biological applications [23].
Due to the high numbers of cysteine residues, the soluble expression of VNAR constructs in bacterial hosts has been extensively studied by a range of laboratories [24, 25]. Thus far, VNAR proteins have been reportedly expressed in bacteria system using proprietary expression vectors such as pIMS100 [25], pIMS147 [26], Pecan22 [27, 28] pAK-300 [29], and pET-28a [30]. In addition, the PelB leader peptide has been commonly used as a fusion protein partner for bacterial periplasmic targeting [31, 32].
In the present work, new antigen binders against malaria biomarkers were investigated by constructing an immune shark VNAR domains library, selecting high affinity binders against malaria antigens by biopanning, followed by protein expression in various Escherichia coli expression systems. The binding ability of recombinant proteins to target antigens was then compared with a commercial mouse mAb.
Methods
Antigens and adjuvants
Recombinant malaria proteins rPfLDH (6 mg/mL), and rPvAldolase (1.6 mg/mL) were purchased from Standard Diagnostic Inc, Korea. Recombinant protein rPfHRP2 (5 mg/mL) was produced by Lee [33]. All three proteins were diluted in PBS to a final concentration of 250 µg/mL. Each diluted protein was then aliquoted (1 mL) in a fresh 1.5 mL tube and kept at − 20 °C until use. Complete Freund’s adjuvant (CFA) and incomplete Freund’s adjuvant (IFA) were used for shark immunization. CFA was only used during the first immunization and replaced by IFA during subsequent immune boosting of a wobbegong shark. Both CFA and IFA were purchased from Sigma-Aldrich (USA) and were kept at 4 °C until use.
Immunization of a wobbegong shark
All animal handling work was approved by Animal Ethics Committees of UQ and QIMR Berghofer MRI. A banded wobbegong shark (Orectolobus ornatus) weighing 2.1 kg, was housed at the Moreton Bay Research Station, University of Queensland. The three malaria recombinant soluble proteins (rPfHRP2, rPfLDH and rPvAldolase at 250 µg/mL each) were emulsified in equal amounts of CFA and subcutaneously injected in the pectoral fin as a mixed-antigens cocktail. Subsequent boosts were administered intravenously in the caudal vein. To enable production of the immune library, the wobbegong shark was given an initial injection with 2 mL mixed-antigen cocktail into its lateral fin. For subsequent boosts, 1 mL PBS containing 250 µg/mL of antigens cocktail was mixed with 200 µL IFA and was then injected into the caudal vein intravenously at 4-week intervals. To measure the humoral response of shark IgNAR against malaria antigens, 3 mL blood sample was drawn from the caudal vein. Serum was isolated to determine the reactivity of shark IgNAR plasma against malaria biomarkers by ELISA using a panel of IgNAR-specific monoclonal antibodies (kindly provided by Prof. Flajnik, University of Maryland, Baltimore). When sera titres rose to an appropriate level, the shark spleen and peripheral blood lymphocytes (PBL) were isolated for construction of the immune VNAR library.
Total RNA extraction and first strand cDNA synthesis
Total RNA extracted from shark spleen and peripheral blood lymphocytes (PBL) was pooled and approximately 20 ng/µL of purified mRNA extracted by Poly(A) Purist Kit (Ambion® Invitrogen, USA) was used as template for first strand cDNA synthesis with addition of Leader-F primer and C-domain R primer (Table 1). The pre-denaturation was set at 94 °C for 2 min. The PCR amplification conditions were set at 35 cycles of 30 s at 94 °C, 30 s at 55 °C, 1 min at 72 °C, and a final extension was at 72 °C for 10 min. The resultant amplicons were separated on an agarose gel and the target band was excised and purified for library construction.
Table 1.
Primers used for shark VNAR library construction and sequencing
| Primers description | Nucleotide sequence (5′→3′) | Applications |
|---|---|---|
| Leader-F |
ATGAATATTTTCTTGCYGTCAGTCC |
1st PCR |
| C-DomainR |
CCTCTCTGTTCTTCRGTTGCAGAGT |
1st PCR |
| FR1F1 |
RCAWGGGTRGACCAAACACC |
Nested PCR |
| FR4R |
TTTCACGGTYARTRCGGTGCC |
Nested PCR |
| T7SelectUP (For) |
GGAGCTGTCGTATTCCAGTC |
T7 Phage sequencing |
| T7SelectDOWN (Rev) |
AACCCCTCAAGACCCGTTTA |
T7 Phage sequencing |
| T7Seq_For |
TAATACGACTCACTATAGGG |
pET vector sequencing forward |
| T7Seq_Rev |
CTAGTTATTGCTCAGCGGTG |
pET vector sequencing reverse |
Association (ka), dissociation rates (kd), and equilibrium constants (KD) of mAbs D2, F9, and C1–13 against rPfHRP2. The analysis for mAbs D2 and F9 were undertaken using the Octet Red platform, whereas mAb C1–13 was determined by BIAcore
Construction of shark VNAR cDNA library
The purified PCR product was then used as a template for second PCR using nested primers set (FR1F1 and FR4R) shown in Table 1. The nested PCR products (VNAR cDNA) were ligated to EcoRI and HindIII linkers according to manufacturer’s protocol described in OrientExpress™ cDNA Cloning kit (Novagen, EMD Millipore Chemicals, USA). After treating with restriction enzymes EcoRI/HindIII, the digested product (100 µL) were purified by size fractionation through a mini column filled with gel filtration resin. The purified VNAR cDNA was subsequently ligated into EcoRI/HindIII digested T7Select®1-1b vector. For the ligation reaction, a 10 µL reaction mixture was set up according to manufacturer’s protocols. On the next day, in vitro Packaging of T7 Bacteriophage was performed by adding 5 µL of ligated VNAR/T7Select®1-1b product into 25 µL of Packaging Extract. After incubating at room temperature for 2 h, the packaging reaction was terminated by adding 270 µL sterile LB medium and 20 µL chloroform. The mixture was mixed well by inversion prior to undertaking the plaque assay.
T7 phage biopanning
Three immunotubes (Nunc) coated with 1 mL of recombinant rPfHRP2, rPfLDH and rPvAldolase with respective concentration of 100 µg/mL (Round 1), 50 µg/mL (Round 2), 25 µg/mL (Round 3) and 12.5 µg/mL (Round 4) were incubated overnight at 4 °C on a rotator. After washing and blocking, 1 mL of amplified T7 phage display library (1.5 × 1011 pfu/mL) was added to each immunotube and incubated for 1 h at room temperature in a rotator. The unbound phage was removed by washing 5 times with 0.1% PBST (PBS containing 0.1% Tween-20) in round one. The number of washes was increased to 10 times for subsequent rounds of panning. Bound phage were eluted with 1 mL of T7 elution buffer (1% SDS in TBS) of which 250 µL of eluted phage was used to infect host cell BLT5615 (OD 1.0 induced with 1 mM IPTG) for amplification. To enrich specific clones, the amplified phage was subjected to another three rounds of panning as described above. To monitor enrichment, the recovery rate of each round of panning was determined by plaque assay. The targeted T7 phage was scraped and dispersed in 300 µL of phage extraction buffer at 4 °C.
Determination of VNAR inserts with expected size and in-frame sequence
To determine the DNA sequence of VNAR insert in T7 phage, 2 µL of plaque lysate was used as template for PCR amplification with addition of T7SelectUP (For) and T7SelectDOWN (Rev) sequencing primers. When the amplification was completed, 10 µL of PCR reactions were mixed with 1 µL of 10× loading dye and separated by 1% agarose gel containing 0.5 µL/mL ethidium bromide. VNAR lengths were then determined visually from the agarose gel. PCR product were sequenced as described above. The deduced amino acids of VNAR were aligned using ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/) and the plaque containing intact VNAR region was determined using BLAST.
Expression of His6-tagged H8VNAR protein
Recombinant VNAR proteins were expressed in bacterial cytoplasmic and periplasmic compartment. Briefly, the selected VNAR gene fragment H8VNAR was cloned into the expression vectors pET-28a (cytoplasmic expression) and pDSB-28Y (periplasmic expression) prior to transformation in E. coli BL21(DE3) competent cells. After sequence confirmation of the constructs, the expression of His-tagged H8VNAR protein was initiated by inoculating 100 µL glycerol stock of transformed BL21 cells in 1 L of fresh 2YT medium containing 30 µg/mL kanamycin and incubating at 37 °C with 200 rpm shaking overnight. The cultures were then induced by IPTG at a final concentration of 0.1 mM, followed by an additional incubation at 20 °C, with shaking at 200 rpm for 16 h.
Extraction of protein fractions
Overnight cultures were centrifuged at 6000×g at 4 °C in an Ultra Centrifuge (Beckman, USA) for 40 min. To extract the soluble protein expressed in cytoplasmic compartment, pelleted bacteria were resuspended in 30 mL cold Tris–phosphate buffer and lysed by passing through a French press. To extract periplasmic proteins, pelleted bacteria were subjected to an osmotic shock. Briefly, pelleted bacteria were resuspended in 30 mL of ice-cold TES buffer (0.2 M Tris–HCl pH 8.0, 0.5 mM EDTA, 20% sucrose). The cell suspension was incubated on ice for 30 min. Cells were lysed by adding 30 mL of 5 mM MgSO4 to the cell suspension and further incubation for 30 min on ice. The slurry was then centrifuged at 20,000 rpm for 40 min at 4 °C. The resulting supernatant was collected and twice filtered through 0.45 µm filter. All supernatants collected were then dialyzed overnight against protein binding buffer (50 mM Tris–HCl pH 8.0, 3 mM MgCl2, 300 mM NaCl) at 4 °C. The purification of recombinant His6-tagged H8VNAR protein was done using a Nickel-NTA Agarose Resin (Sigma-Aldrich, USA), according to the manufacturer instructions.
rPfHRP2-specific VNAR protein sandwich ELISA
The purified H8VNAR proteins were analysed by SDS-PAGE (15%) and Coomassie staining. The concentration of protein was quantified by Bradford method [34] using the Bio-Rad Protein Assay (Bio-Rad Laboratories) according to manufacturer instructions. To determine the binding efficacy of recombinant antibodies, H8VNAR proteins (1 µg/mL or ~ 70 nM) purified from both cytoplasmic and periplasmic spaces were used as capture antibodies for the anti-rPfHRP2 assays in a sandwich ELISA. Tenfold serial diluted recombinant PfHRP2 proteins, with starting concentration at 10 µg/mL, were added to each H8VNAR. The commercial mouse mAb C1–13 (10 µg/mL or ~ 70 nM) (kindly provided by Dr. Martin Bubb, National Bioproducts Institute, South Africa) was used to detect captured antigens. The sandwich was detected by goat anti-mouse antibody conjugated to horseradish peroxidase (HRP) (Jackson, USA). Absorbance was read at 405 nm.
Dot blot analysis
A sandwich format of Dot Blot assay was used to determine the sensitivity of H8VNAR proteins against recombinant PfHRP2 protein. Briefly, H8VNAR proteins diluted in PBS (~ 70 nM) were transferred onto a pre-wetted nitrocellulose membrane (Hybond-C, Amersham Bioscience, UK) using Dot blotter (BioRad, USA). A twofold serial dilution of recombinant PfHRP2 protein was added to the H8VNAR proteins. The nitrocellulose membrane was then removed from the blotter and incubated with antibody C1–13 (~ 70 nM) at room temperature for 1 h. Detection of HRP-conjugated secondary antibody was performed using ECL chemiluminescence detection kit (Amersham Bioscience, USA), following the manufacturer’s instructions. The exposed film was developed by Kodak X-omat imager.
Results
Immunization of wobbegong shark and IgNAR ELISA screening
A wobbegong shark was immunized with three malaria antigens, rPfHRP2, rPfLDH, and rPvAldolase. After six immunizations over a 7 months period, the antibody titre specific to the three malaria biomarker had risen compared to the pre-bleed titer, and compared to titters in a naïve (non-immunized) wobbegong as a negative control. As shown in Fig. 1, IgNAR response against all three antigens increased markedly in bleed 4 and 5 compared to pre-bleed and bleed 1. However, antibody levels in bleed 5 were not different to those in bleed 4. The highest shark immune response in bleed 5 was observed against rPfLDH, followed by rPfHRP2, with the antibody titre over 1:51,200 and 1:25,600, respectively. The anti-rPvAldolase IgNAR titre was lower at 1:12,800. As the antibody response was no longer increasing after the fifth boost, the spleen and peripheral blood lymphocytes were harvested from the immunized shark and the total RNA was isolated.
Fig. 1.
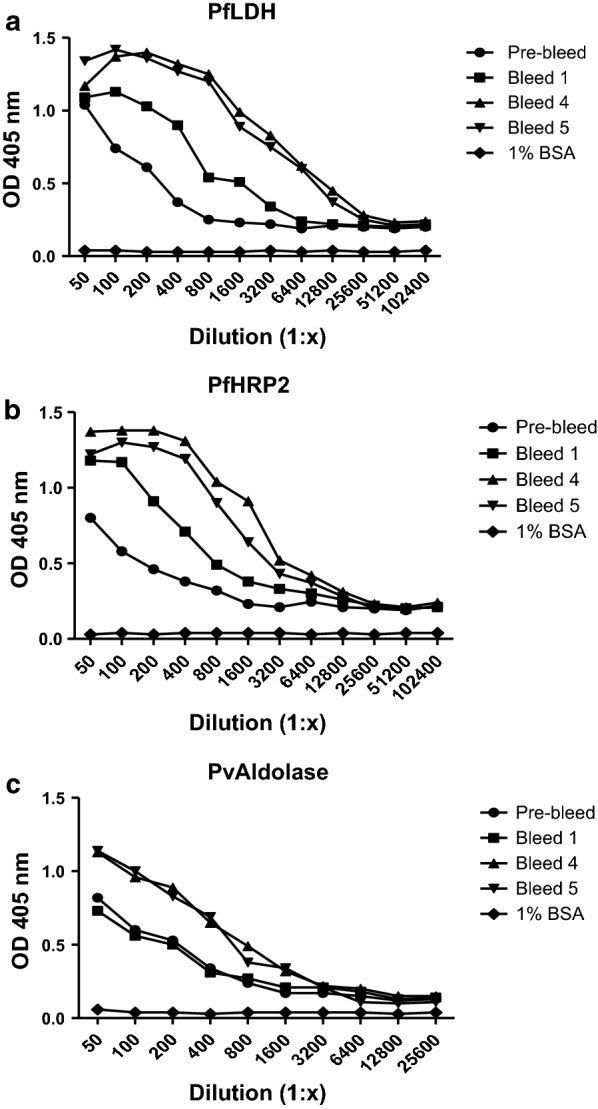
End-titre of shark plasma against three malaria recombinant proteins over course of immunization. Shark plasma reacted against PfLDH (a), PfHRP2 (b), and PvAldolase (c). The baseline antibody response (negative control) was represented by pre-bleed (naïve sera) of wobbegong shark. The immobilized 1% BSA was used as negative antigen and reacted with serial diluted pre-bleed sera
VNAR domains library construction
An immune IgNAR V region (VNAR) phage display library was constructed and screened for VNARs specific to malaria biomarkers. Total RNA was purified from splenocytes and peripheral blood lymphocytes of immunized shark. The successful amplification of VNAR region (nested PCR product) was clearly indicated with a strong band present at 340 bp. After digestion with appropriate restriction enzymes, the DNA was cloned into the T7Select®1-1b vector that facilitates expression of phage 10B-VNAR fusion proteins on the phage surface.
Library characterization
The size of primary library was determined to possess 1.16 × 106 pfu/mL. PCR screening of 132 randomly selected plaques from the primary library revealed that approximately 55% (72/132) of the clones contained inserts of the expected size at (~ 450 bp) (Additional file 1). Nucleotide sequence analysis showed that 82.5% (33/40) of isolated fragments contained an in-frame sequence, while 17.5% (7/40) contained frameshift mutations. Thus, it was estimated that over 80% of the VNAR library with a size of ~ 5.26 × 105 pfu/mL in 50 mL culture encoded functional inserts.
From the VNAR domains library, the amino acid sequences of 33 randomized clones were aligned and directly evaluated by comparison with three VNAR domain control genes derived from Orectolobus maculates (wobbegong shark) clones published in the protein database (PDB), namely AY261681 [32], AY261682 [32], and AF466395 [17]. The complete two signature CDR1 and CDR3 regions and four conserved framework regions (FR) were present in each of the sequenced domain of VNAR. In addition, the hypervariable (HV) 2 region with shorter length of the amino acid stretch also present. Furthermore, most of the clones demonstrated unique amino acid sequences, with no identical clones detected in the primary library.
Examination of the quality of VNAR domains library
Significant diversity in the VNAR domains was observed especially in their CDR3 regions. As expected for an adult shark, types 1 and 2 but not type 3 of IgNAR repertoires were represented in the library (Table 2). Considering the number of non-canonical cysteine (Cys) residue(s) encoded within CDR1 and CDR3 regions in each VNAR, approximately 88% of the sequenced clones in this library were classified as type 2, whereas 12% were classified as type 1. Generally, type 1 IgNAR V regions have two non-canonical Cys residues in the CDR3, and two additional in FR2 and FR4. Type 2, V regions have two non-canonical Cys residues, one each in CDR1 and CDR3. Unexpectedly, about 27% (9/33) of the clones in this library were found to possess three Cys residues in their CDR3 region.
Table 2.
Deduced amino acid sequences in hypervariable regions of VNAR clones randomly selected from primary library

The non-canonical cysteine residue is highlighted in red color. This table also indicates the length of CDR3, number of cysteine residue, and type of IgNAR family for each clone
In terms of the length of CDR3 regions, the encoded amino acid residues ranged from 10 to 24 in residue length, with an average of 17.2 amino acids. Most of the CDR3 regions appeared to generate long loops which predominantly consisted of 14, 16, 18 and 20 amino acid residues. The alignment also showed that a conserved canonical Cys residue at the position 22 was consistently observed in the C-terminal of CDR1 sequence, while mutations were found in the FR regions. Another canonical Cys residue was also detected at the C-terminal of CDR3 regions at position of 84 in all clones studied. The VNAR domains library was evaluated to be of good quality because of: (i) the highly variable CDR3 regions, (ii) the presence of type I and type II of IgNAR family, and (iii) the position of the conserved canonical C residues being similar to the VNAR sequences published in the protein database.
Biopanning of the immunized VNAR library against three malaria biomarkers
To screen for target clones, four rounds of panning were performed using an amplified library with the size of 1.5 × 1011 pfu/mL. Using this approach, target-specific clones were obtained for each malaria biomarker. The highest phage titre pattern was observed for rPfLDH, followed by rPfHRP2 and rPvAldolase as represented in Fig. 2. Following four rounds of panning, a total of 30 single phage plaques, targeting each antigen were separately picked for further analysis by PCR screening and sequencing. The results indicated that 20 anti-rPfHRP2 clones, 18 anti-rPfLDH clones, and 15 anti-rPvAldolase clones had the expected size and in-frame (ORF) sequences. Only the in-frame clones were chosen for further analysis.
Fig. 2.
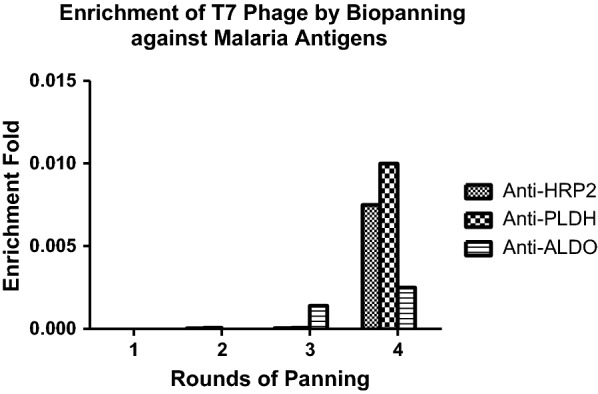
Selection and enrichment of malaria biomarker specific T7 phage after four rounds of biopanning. A plaque assay was performed to measure the size of eluted phage after each round of panning, and the size of amplified phage. The enrichment fold was calculated by dividing the eluted phage by input library size
Competitive biopanning
To avoid cross-reactivity of non-specific clones, a competitive biopanning was applied to further select the strongest unique binders. Briefly, three more rounds of biopanning were carried out by immobilizing the target proteins in immunotubes as described in section T7 Phage Biopanning. Of 53 selected clones isolated from the previous panning, 26 clones with distinct sequence were pooled and subsequently tested for competitive binding to either their putative target ligand or to the other two ligands, to which no binding should occur.
Competitive biopanning resulted in 26 clones retained after four rounds of panning. When inspecting the ratio of specific to non-specific binding of the resultant clones, four of the 26 clones were identified to be predominantly reacting to one specific antigen: H8 as an anti-PfHRP2 clone, P16 and P2–3 as anti-PfLDH clones, and A4–3 as an anti-PvAldolase clone (Fig. 3). The clones that showed binding to all antigens such as clones A5–3 and A7–3 were eliminated from further analysis.
Fig. 3.

Competitive biopanning between 26 selected T7 specific clones. Twenty-six cloned selected from initial four rounds of biopanning against three different malaria biomarkers were pooled. This pool was tested competitively to select the specific clones, based on high frequency of appearance and absence of cross-reactivity. The colour bars represent the clones isolated from biopanning against different malaria antigen proteins (blue: anti-PfHRP2; red: anti-PfLDH; green: anti-PvAldolase). Yellow arrows point to four clones identified to be specific (H8 as an anti-PfHRP2 clone, P16 and P2–3 as anti-PfLDH clones, and A4–3 as an anti-PvAldolase clone)
Sequence analysis of targeted clones
The amino acid sequence of the four final clones isolated following competitive biopanning were aligned (Fig. 4). The clones were found to lack Cys residues in FR2 and FR4 regions. However, inspection of CDR regions revealed that all clones had conventional type 2 IgNAR V regions. No major mutations were identified in FR1, FR2, FR3 and FR4 regions of clones P16, P2–3, and A4–3, except for the FR3 region of H8 clone.
Fig. 4.

Deduced amino acid residues of four clones isolated after competitive biopanning. The PfHRP2-specific clone was represented by H8; The PfLDH-specific clones were P16 and P2–3; the PvAldolase-specific clone was A4–P4. All FR regions are shown by orange bar. The CDR regions are shown by red box. The canonical cysteine residues in FR1 and FR3 regions are highlighted in green. The non-canonical cysteine residues in CDR1 and CDR3 regions are highlighted in yellow background. The mutation sites in FR3 region of clone H8 is highlighted in pink background
In terms of number of Cys residue, H8 contained four residues in CDR3 region, the largest numbers of Cys residues observed in this library. In comparison, three Cys residues were detected in clone P2–3, and only one in clones P16 and A4–3. All clones were observed to have longer amino acid residues stretches in CDR3 which were more than the average of 17 amino acids of the VNAR library (Fig. 5). The longest CDR3 were H8 and A4–3 with 20 residues, followed by P16 with 19 residues, and P2–3 with 17 residues. Being a highly dominant clone with an average appearance higher than 85% after each round of panning, H8 was selected for recombinant protein production.
Fig. 5.
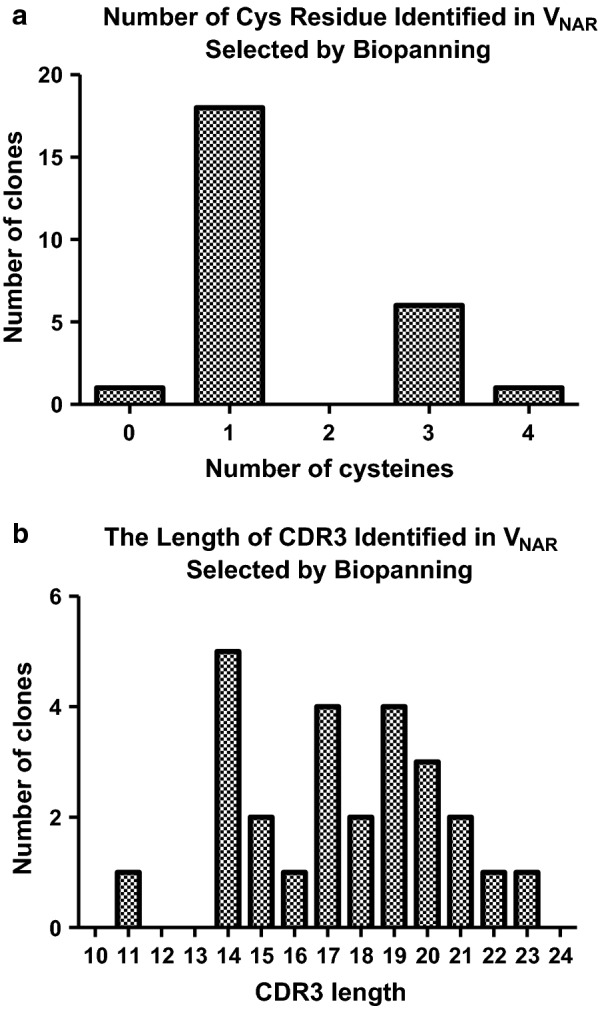
Analysis of deduced amino acids of VNAR clones selected after four rounds of biopanning. a The number of cysteine residues and b represents the length of the CDR3 region encoded within the VNAR clones
Protein expression and purification
The expression and purification of H8VNAR using two different expression vectors yielded a clearly visible band on SDS-PAGE stained with Coomassie Blue (Figs. 6, 7). All H8VNAR proteins were present as well defined single bands at around 14 kDa in size, corresponding to the predicted molecular mass of H8VNAR including the 6× His tag (13.57 kDa). In Western Blot analysis, a “split” band was detected in the IPTG-Induced total cell lysates of E. coli processed for periplasmic expression (Fig. 7). However, this phenomenon was not observed in the cytoplasmic expression system (Fig. 6). Nevertheless, only a single band was detected in both H8VNAR proteins after purification, suggesting the soluble of recombinant VNAR proteins can be obtained using bacterial expression system. No leakage of proteins from expression hosts could be observed. However, prolonged expression can lead to protein aggregation that eventually resulted in the formation of inclusion body in the E. coli expression host.
Fig. 6.

Expression and purification of recombinant cytosolic H8VNAR using pET-28a vector. Proteins were separated on 15% SDS-PAGE and stained with Coomassie Blue (left panel). H8VNAR was detected by Western Blot with anti-His tag antibody and a ~ 14 kDa band clearly apparent. Lane 1 represents total protein from non IPTG-induced bacteria cells; lane 2 represents total protein from IPTG-induced bacteria cells; and lane 3 represents purified cytosolic H8VNAR protein
Fig. 7.

Expression and purification of recombinant periplasmic DsbA-H8VNAR using pDSB-28Y vector. Proteins were separated on 15% SDS-PAGE and stained with Coomassie blue (left panel). DsbA-H8VNAR was detected by Western Blot with anti-His tag antibody and a ~ 14 kDa band clearly apparent. Lane 1 represents total protein from non IPTG-induced bacteria cells; lane 2 represents total protein from IPTG-induced bacteria cells; and lane 3 represents purified periplasmic DsbA-H8VNAR protein
Binding efficiency by sandwich ELISA
The binding ability of recombinant VNAR proteins was determined on ELISA plates in a sandwich format. The binding efficiency was compared with commercial mAb C1–13 that performed in an indirect ELISA format. From the screening assay, both VNAR proteins gave positive responses when read on plate readers at OD 450 nm. The recombinant H8VNAR protein purified from periplasmic space showed better performance than that of cytosolic recombinant H8VNAR in terms of absorbance (Fig. 8a). At a concentration of rPfHRP2 of 1 µg/mL, the OD reading of periplasmic VNAR protein (~ 0.58) was about twofold higher than that of protein produced by cytoplasmic expression (~ 0.32). However, these reactivities of the VNAR proteins are inferior to the reactivity of mAb C1–13, the positive control in this experiment (OD of ~ 1.4 at the same concentration of rPfHRP2) (Fig. 8b). All OD readings demonstrated decreasing signal proportionally with reducing concentrations of rHRP2. No specific binding to BSA was observed for both recombinant H8VNAR proteins or mAb C1–13.
Fig. 8.
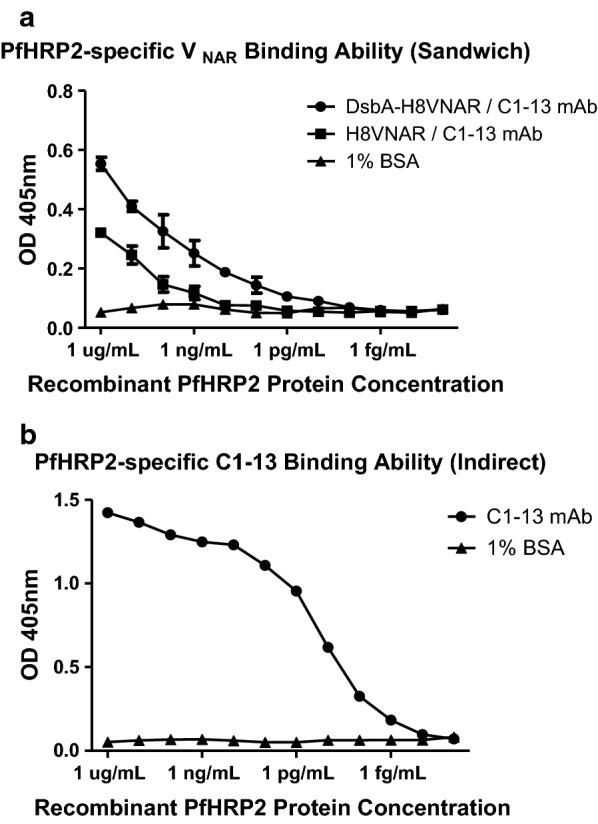
Comparison of binding efficiency of recombinant H8VNAR proteins with commercial mouse mAb C1–13 against tenfold serial diluted HRP2 proteins by ELISA. a The binding reactivity of purified cytosolic and periplasmic H8VNAR proteins was performed in a sandwich ELISA format. b The binding reactivity of positive control mAb C1–13 was performed in an indirect ELISA format. 1% BSA was used as negative antigen for rPfHRP2
Detection sensitivity by dot blot assay
To further determine the detection sensitivity of recombinant antibodies, both cytosolic and periplasmic H8VNAR proteins were deployed as capture antibodies and examined in Dot Blot assay (Fig. 9). All recombinant antibodies reacted with rPfHRP2 protein showing different intensities similar to that observed in ELISA. Mouse mAb C1–13 showed the highest response with detection below 5 ng/mL. In contrast, the H8VNAR proteins exhibited lower binding affinity, even different binding reactivity was observed in both cytosolic and perisplamic produced H8VNAR proteins. The H8VNAR proteins produced in periplasmic space showed binding to the rPfHRP2 at a lowest concentration of 78 ng/mL. In comparison, the detection limit for the H8VNAR protein expressed in cytoplasm was 312 ng/mL. As expected, none of the recombinant antibodies tested reacted with 1% BSA, the negative control. Based on this observation, it was concluded that the sensitivity of periplasmic produced H8VNAR protein was superior to that of cytosolic protein, however both were less reactive than mAb C1–13. More Western Blot assays, designed to investigate possible cross reactivity to other malaria biomarker proteins for all of the selected VNAR clones are planned. Further biological functions to investigate include reactivity for the native malaria protein, and thermostability will provide more insight in the diagnostic value of recombinant H8VNAR to rPfHRP2.
Fig. 9.

Comparison of binding reactivity of recombinant purified cytosolic and periplasmic H8VNAR against twofold serial diluted rPfHRP2 proteins in Dot Blot analysis. mAb C1–13 was a positive control. 1% BSA was a negative control
Discussion
The small antigen binding domains of shark IgNAR (VNAR) have some unique properties in terms of thermostability and refolding capacity, which make them ideal antibody-based diagnostic tools. Hence, they have gained the interest of researchers focused on developing new binders for targeting antigens from various diseases. This study describes shark VNAR antibodies that are specific against malaria antigens were successfully isolated and produced recombinantly in E. coli.
Following immunization of a wobbegong shark (Orectolobus ornatus) with three malaria biomarker proteins, the levels of IgNAR response against malaria recombinant PfLDH and PfHRP2 significantly increased in the wobbegong shark after the third immunization (Fig. 1), suggesting that the wobbegong shark could mount a humoral protective response to these antigens [35]. The fact that it took at least 3 boosts to observe an antibody response is likely due to sharks having a unique immune system where the antibody serum levels generally climb slowly compared to mammals. Therefore, it can take months until a significant antigen-specific titre is reached [36]. This phenomenon had also been indicated in work using hen egg lysozyme (HEL) to immunize nurse shark [25, 37–39]. This is presumably because pentameric IgM is responsible for providing early protection with high avidity interaction against invading pathogen in cartilaginous fish. In contrast, monomeric IgM likely IgG in mammals, and IgNAR are responsible for specific and high affinity binding response [35]. From these results, it is apparent that the immune system of the wobbegong shark studied herein was comparable to other shark families and appropriate IgNAR responses were identified.
After purification of mRNA from shark spleen, a VNAR phage display library was successfully constructed using a T7 phage display system. Using this system, VNAR displayed on the surface of T7 bacteriophage do not need to be extracted from the periplasmic layer of host cells which is a necessary step in M13 filamentous phage [40, 41]. Therefore, VNAR fusion phage particles could directly be obtained through cell lysis. The functional phage titer yielded in this VNAR domains primary library was estimated to have 5.3 × 105 independent clones which is slightly lower than that of the libraries derived from naïve [28] and immunized [25] shark using M13 phage display system, and synthetic sharks using T7 phage display system [39]. Therefore, it was assumed that the library size may partly be influenced by factors such as packaging efficiency and phage display system (M13 or T7). In contrast to VH and VL genes in conventional antibody libraries, the genes encoding VNAR repertoires were easier to clone [25, 41] with only two pairs of primers required to access the diversified shark immunoglobulin genes by PCR amplification of the VNAR fragments from the spleen and PBL of immunized shark.
Sequence analysis indicates that about 90% of the clones in this library belonged to type 2 NAR. These data are in agreement with other VNAR libraries derived from wobbegong sharks, indicating that the genes encoding IgNAR in the wobbegong family are prone to produce type 2 NAR domain [31]. However, genes encoding type 1 NAR representatives are commonly confined to nurse sharks [25]. In addition, about 27% of the clones in this library were found to possess three Cys residues in their CDR3 region. Although this number has rarely been described in IgNAR, Diaz et al. have previously reported it was found in some hypermutated clones in a nurse shark IgNAR [42].
In comparison with the human VH gene family, a higher degree of divergence in framework and hypervariable regions is exhibited in shark VNAR. First, the hypervariable regions of VNAR are on average longer than those in VH. The average CDR3 length in conventional human and mouse VH is 12 and nine amino acids, respectively [43], whereas the length of shark VNAR observed in this study was about 16–18 amino acids and in an agreement with those reported previously [25, 31, 42]. The hypervariability detected within CDR3 in VNAR against malaria antigens after repetitive rounds of panning, suggests that these residues are contacting the antigen and that the somatic hypermutations in this region will take place during the affinity maturation [14, 32]. Furthermore, the enrichment of disulfide bridge in the long CDR3 region may cause the formation of disulfide loops that resulted to type 1 or type 2 VNAR proteins configuration [44]. Therefore, to examine the quality of the VNAR domain library constructed, the diversity in the CDR3 region, the different isotypes, and the position of the conserved canonical Cys residues were analysed and compared with other VNAR domains that published in protein database.
Four rounds of biopanning resulted in the enrichment of antigen-specific phage antibodies directed against rPfHRP2, rPfLDH, and rPvAldolase proteins (Fig. 2). Interestingly, the pattern of enrichment was similar to the end titer of polyclonal IgNAR responses in vivo where the highest titers was rPfLDH-specific, rPfHRP2-specific, and followed by rPvAldolase-specific VNAR clones. From this point of view, it is postulated the immunization process is an important factor to generate the high affinity single domain clones [25, 45]. In contrast to “naïve” antibody libraries, lesser rounds of panning cycles were needed to produce a highly specific signal which is in agreement with that reported from other immune phage display libraries [46]. Thus, the rapid selection of specific VNAR-displayed phage suggests that antigen-specific clones are well represented within the VNAR population. The use of the T7 phage system has an advantage over M13 filamentous phage. In this system, phage amplification takes shorter time, and cell lysis can be observed by a visible reduction in OD of the culture after incubation.
The sequencing alignment revealed that all 26 clones specific to a malaria antigen protein were in-frame. Two distinct clones were identified targeting the recombinant rPfHRP2, whereas twelve clones each to targeting rPfLDH and rPvAldolase proteins. The amino acid residues found in the CDR3 regions differed for each antigen confirming that the CDR3 region is a major determining factor for the antigen binding specificity [47]. Moreover, the extraordinary length of CDR3 in shark VNAR has been exhibited potentially penetrating into the cavities of antigen [48]. All of the selected VNAR clones, except P9 and A9–3, appeared to contain at least one Cys residue in the CDR1 and CDR3 regions, suggesting that these clones belong to type 2 NAR family and are proposed to form an interloop of disulfide bond [49]. The length of the selected clones 10 to 24 amino acid residues with a mean of 17 amino acids (Additional file 2), indicating that longer hypervariable regions of the VNAR could aid enlarging the actual surface of the antigen binding site, and that this might compensate for the absence of the antigen-binding surface area provided by the VL domain in conventional Fv of IgG [14, 31, 42].
Despite most of the selected clones demonstrating a positive response to corresponding antigen in monoclonal ELISA screening, it was difficult to differentiate the genuine clones from “sticky” clones using the phage display system. This phenomenon is observed quite often with phage display [50, 51]. Indeed, selection of highly cross reactive clones could possibly lead to production of recombinant VNARs with inferior binding affinity. Another limitation observed with this T7 phage display system was the difficulty in isolating high affinity binders that contained disulfide bonds. The T7 phage display system is therefore more suitable for use in peptide discovery rather than antibody surface display [52, 53]. The polypeptides produced by T7 phage display system are synthesized in the reducing cytoplasmic environment of the host E. coli, which differs from phage secretion via the periplasmic space, as occurs in the filamentous M13 phage system. As a consequence, the proteins displayed on the surface of T7 phage may not be properly folded and therefore not ideally suited for functional screening for high affinity VNAR clones. Because misfolded VNAR presented on the surface of the T7 capsid could lead to false-positive plaques, additional rounds of competitive biopanning were undertaken to guarantee the specificity of targeted clones. In addition, the subsequent production of recombinant VNAR proteins was performed in an alternate expression system.
H8VNAR was one of the highest affinity clones, and also the most frequently isolated during panning. In this clone, seven Cys residues were observed and predicted to form three disulfide bridges in its molecular structure. This prediction was done by DiANNA 1.1 Web Server (http://clavius.bc.edu/~clotelab/DiANNA/), a bioinformatics tool which is available online. Formation of disulfide bonds is known to be problematic in bacterial expression systems for eukaryotic proteins, particularly antibody fragments [54]. To achieve optimal folding, the production of proteins containing disulfide bonds needs to be manipulated in an oxidative environment of E. coli cells.
To make this possible, a signal sequence DsbA peptide was added to express PfHRP2-specific H8VNAR protein in E. coli. The recombinant H8VNAR expressed in periplasmic and cytoplasmic was then examined for their yield and binding activity. A periplasmic protein with an expected band at ~ 14 kDa was visible on SDS-PAGE (Fig. 7), suggesting that adding the DsbA signal sequence has efficiently directed VNAR protein from cytoplasmic into the periplasmic space via the co-translational SRP pathway [55]. Thus, crystallography is planned to determine the conformation of recombinant H8VNAR and verify the presence of disulfide bond in this protein molecule.
When comparing the cytosolic H8VNAR and the periplasmic H8VNAR (or DsbA-H8VNAR), the binding capacity of DsbA-H8VNAR to recombinant PfHRP2 protein in Dot Blot assays was shown superior to that of cytosolic H8VNAR protein (Fig. 9). This insight indicated that periplasmic expression may potentially lead disulfide bond formation and proper folding of the protein thereby increasing the reactivity of recombinant antibody against the antigen epitopes [56, 57].
Conclusion
In this work, the ability to raise the specific IgNAR from shark against malaria biomarker proteins by immunization was demonstrated. Meanwhile, a phage display VNAR library was also successfully constructed by overlap extension PCR. Selection from this library resulted in several unique clones with specificity for each of three malaria antigens, suggesting the enrichment of affinity clones against malaria antigen proteins has been achieved by iterative biopanning and subsequent competitive biopanning. Also, the utilization of the DsbA signal peptide caused expression of soluble recombinant shark VNAR proteins by enhancing folding capability was evidenced in the screening assays. Despite further optimization conditions under investigation, these findings and insights imply the expression of shark VNAR is possible using the system designated in this study. The binding specificity and sensitivity of recombinant H8VNAR to the rPfHRP2 protein was demonstrated in a sandwich ELISA and a Dot Blot assay, respectively. Despite of that, further engineering including mutagenesis is planned in respect to improve the biological activity of H8VNAR towards malaria biomarker protein. Currently, a new semi-synthetic shark VNAR library using M13 phagemids is being constructed whereby the performance of new clones against malaria antigens will then be presented.
Additional files
Additional file 1. PCR amplification of randomly selected plaques from the VNAR domains primary library using T7SelectUP (For) and T7SelectDOWN (Rev) sequencing primers. Lane M represents 100 bp ladder; lane 1–20 represents insert of single plaque; lane −ve represents negative control (PCR product with no cDNA template).
Additional file 2. Deduced amino acid sequences in hypervariable regions of VNAR clones targeting to three malaria biomarkers. The non-canonical cysteine residue is highlighted in red colour. This table also indicates the length of CDR3, number of cysteine residue, and type of IgNAR family for each clone.
Authors’ contributions
All authors made contribution to this study. The objective of this project is to identify specific binders isolated from shark VNAR library against malaria biomarker PfHRP2. CHL, KB and CYL conceived the general project study. JM, QC and KF coordinated the project and led manuscript writing. All authors read and approved the final manuscript.
Acknowledgements
We gratefully thank Dr. Kathy Townsend, Emma Louise for all their help and cooperation as well as their advice on taking care of wobbegong shark in MBRS, UQ. Our high appreciation is also given to Dr. Joanna Stead and Dr. Susan Thesis from UQ on their kind advice and help on shark anatomy and dissection. We would like to thank Dr. Martin Bubb, National Products Institute, South Africa for providing mAb C1–13. The opinions expressed herein are those of the authors and do not necessarily reflect those of the Australian Defence Force Joint Health Command. Last but not least, we appreciate to part of the funding sponsored by Malaysian Government and University Sains Malaysia, including Malaysian Ministry of Higher Education the Higher Institutions Centre of Excellence Programme (Grant No.: 311/CIPPM/4401005), and USM RUI (Grant No.: 1001/CIPPM/811296) to complete this project.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- Aldolase
fructose 1,6-biphosphate aldolase
- CDRs
complementarity determining regions
- HRP
horseradish peroxidase
- IgG
immunoglobulin G
- IgNAR
immunoglobulin new antigen receptor
- mAb
monoclonal antibody
- PfHRP2
P. falciparum histidine-rich protein 2
- pLDH
Plasmodium lactate dehydrogenase
- RDTs
rapid diagnostic tests
- VNAR
variable domain of new antigen receptor
References
- 1.WHO . Malaria rapid diagnostic test performance. Geneva: World Health Organization; 2012. [Google Scholar]
- 2.Wellems TE, Howard RJ. Homologous genes encode two distinct histidine-rich proteins in a cloned isolate of Plasmodium falciparum. Proc Natl Acad Sci USA. 1986;83:6065–6069. doi: 10.1073/pnas.83.16.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tahar R, Sayang C, Ngane Foumane V, Soula G, Moyou-Somo R, Delmont J, et al. Field evaluation of rapid diagnostic tests for malaria in Yaounde, Cameroon. Acta Trop. 2013;125:214–219. doi: 10.1016/j.actatropica.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Bukirwa H. Assessing rapid diagnostic tests for malaria. Cochrane Database Syst Rev. 2011;9:ED000031. doi: 10.1002/14651858.ED000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kyabayinze DJ, Zongo I, Cunningham J, Gatton M, Angutoko P, Ategeka J, et al. HRP2 and pLDH-based rapid diagnostic tests, expert microscopy, and PCR for detection of malaria infection during pregnancy and at delivery in areas of varied transmission: a prospective cohort study in Burkina Faso and Uganda. PLoS One. 2016;11:e0156954. doi: 10.1371/journal.pone.0156954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murray CK, Gasser RA, Jr, Magill AJ, Miller RS. Update on rapid diagnostic testing for malaria. Clin Microbiol Rev. 2008;21:97–110. doi: 10.1128/CMR.00035-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reyburn H, Mbakilwa H, Mwangi R, Mwerinde O, Olomi R, Drakeley C, et al. Rapid diagnostic tests compared with malaria microscopy for guiding outpatient treatment of febrile illness in Tanzania: randomised trial. BMJ. 2007;334:403. doi: 10.1136/bmj.39073.496829.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niemi MH, Takkinen K, Amundsen LK, Soderlund H, Rouvinen J, Hoyhtya M. The testosterone binding mechanism of an antibody derived from a naive human scFv library. J Mol Recognit. 2011;24:209–219. doi: 10.1002/jmr.1039. [DOI] [PubMed] [Google Scholar]
- 9.Moon SA, Ki MK, Lee S, Hong ML, Kim M, Kim S, et al. Antibodies against non-immunizing antigens derived from a large immune scFv library. Mol Cells. 2011;31:509–513. doi: 10.1007/s10059-011-2268-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nuttall SD, Irving RA, Hudson PJ. Immunoglobulin VH domains and beyond: design and selection of single-domain binding and targeting reagents. Curr Pharm Biotechnol. 2000;1:253–263. doi: 10.2174/1389201003378906. [DOI] [PubMed] [Google Scholar]
- 11.Kortt AA, Guthrie RE, Hinds MG, Power BE, Ivancic N, Caldwell JB, et al. Solution properties of Escherichia coli-expressed VH domain of anti-neuraminidase antibody NC41. J Protein Chem. 1995;14:167–178. doi: 10.1007/BF01980329. [DOI] [PubMed] [Google Scholar]
- 12.Takezaki N, Figueroa F, Zaleska-Rutczynska Z, Klein J. Molecular phylogeny of early vertebrates: monophyly of the agnathans as revealed by sequences of 35 genes. Mol Biol Evol. 2003;20:287–292. doi: 10.1093/molbev/msg040. [DOI] [PubMed] [Google Scholar]
- 13.Dooley H, Flajnik MF. Antibody repertoire development in cartilaginous fish. Dev Comp Immunol. 2006;30:43–56. doi: 10.1016/j.dci.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 14.Greenberg AS, Avila, Hughes M, Hughes A, McKinney EC, Flajnik MF. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature. 1995;374:168–173. doi: 10.1038/374168a0. [DOI] [PubMed] [Google Scholar]
- 15.Roux KH, Greenberg AS, Greene L, Strelets L, Avila D, McKinney EC, et al. Structural analysis of the nurse shark (new) antigen receptor (NAR): molecular convergence of NAR and unusual mammalian immunoglobulins. Proc Natl Acad Sci USA. 1998;95:11804–11809. doi: 10.1073/pnas.95.20.11804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dumoulin M, Conrath K, Van Meirhaeghe A, Meersman F, Heremans K, Frenken LG, et al. Single-domain antibody fragments with high conformational stability. Protein Sci. 2002;11:500–515. doi: 10.1110/ps.34602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nuttall SD, Krishnan UV, Doughty L, Nathanielsz A, Ally N, Pike RN, et al. A naturally occurring NAR variable domain binds the Kgp protease from Porphyromonas gingivalis. FEBS Lett. 2002;516:80–86. doi: 10.1016/S0014-5793(02)02506-1. [DOI] [PubMed] [Google Scholar]
- 18.Wesolowski J, Alzogaray V, Reyelt J, Unger M, Juarez K, Urrutia M, et al. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med Microbiol Immunol. 2009;198:157–174. doi: 10.1007/s00430-009-0116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zielonka S, Empting M, Konning D, Grzeschik J, Krah S, Becker S, et al. The shark strikes twice: hypervariable loop 2 of shark IgNAR antibody variable domains and its potential to function as an autonomous paratope. Mar Biotechnol (NY). 2015;17:386–392. doi: 10.1007/s10126-015-9642-z. [DOI] [PubMed] [Google Scholar]
- 20.Chapman MD, Smith AM, Vailes LD, Arruda LK, Dhanaraj V, Pomes A. Recombinant allergens for diagnosis and therapy of allergic disease. J Allergy Clin Immunol. 2000;106:409–418. doi: 10.1067/mai.2000.109832. [DOI] [PubMed] [Google Scholar]
- 21.Babaei A, Zarkesh-Esfahani SH, Gharagozloo M. Production of a recombinant anti-human CD4 single-chain variable-fragment antibody using phage display technology and its expression in Escherichia coli. J Microbiol Biotechnol. 2011;21:529–535. doi: 10.4014/jmb.1010.10022. [DOI] [PubMed] [Google Scholar]
- 22.Kamionka M. Engineering of therapeutic proteins production in Escherichia coli. Curr Pharm Biotechnol. 2011;12:268–274. doi: 10.2174/138920111794295693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goulding CW, Perry LJ. Protein production in Escherichia coli for structural studies by X-ray crystallography. J Struct Biol. 2003;142:133–143. doi: 10.1016/S1047-8477(03)00044-3. [DOI] [PubMed] [Google Scholar]
- 24.Griffiths K, Dolezal O, Parisi K, Angerosa J, Dogovski C, Barraclough M, et al. Shark variable new antigen receptor (VNAR) single domain antibody fragments: stability and diagnostic applications. Antibodies. 2013;2:66–81. doi: 10.3390/antib2010066. [DOI] [Google Scholar]
- 25.Dooley H, Flajnik MF, Porter AJ. Selection and characterization of naturally occurring single-domain (IgNAR) antibody fragments from immunized sharks by phage display. Mol Immunol. 2003;40:25–33. doi: 10.1016/S0161-5890(03)00084-1. [DOI] [PubMed] [Google Scholar]
- 26.Shao CY, Secombes CJ, Porter AJ. Rapid isolation of IgNAR variable single-domain antibody fragments from a shark synthetic library. Mol Immunol. 2007;44:656–665. doi: 10.1016/j.molimm.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 27.Liu JL, Anderson GP, Goldman ER. Isolation of anti-toxin single domain antibodies from a semi-synthetic spiny dogfish shark display library. BMC Biotechnol. 2007;7:78. doi: 10.1186/1472-6750-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu JL, Anderson GP, Delehanty JB, Baumann R, Hayhurst A, Goldman ER. Selection of cholera toxin specific IgNAR single-domain antibodies from a naive shark library. Mol Immunol. 2007;44:1775–1783. doi: 10.1016/j.molimm.2006.07.299. [DOI] [PubMed] [Google Scholar]
- 29.Goodchild SA, Dooley H, Schoepp RJ, Flajnik M, Lonsdale SG. Isolation and characterisation of Ebolavirus-specific recombinant antibody fragments from murine and shark immune libraries. Mol Immunol. 2011;48:2027–2037. doi: 10.1016/j.molimm.2011.06.437. [DOI] [PubMed] [Google Scholar]
- 30.Ohtani M, Hikima J, Jung TS, Kondo H, Hirono I, Takeyama H, et al. Variable domain antibodies specific for viral hemorrhagic septicemia virus (VHSV) selected from a randomized IgNAR phage display library. Fish Shellfish Immunol. 2013;34:724–728. doi: 10.1016/j.fsi.2012.11.041. [DOI] [PubMed] [Google Scholar]
- 31.Nuttall SD, Krishnan UV, Hattarki M, De Gori R, Irving RA, Hudson PJ. Isolation of the new antigen receptor from wobbegong sharks, and use as a scaffold for the display of protein loop libraries. Mol Immunol. 2001;38:313–326. doi: 10.1016/S0161-5890(01)00057-8. [DOI] [PubMed] [Google Scholar]
- 32.Nuttall SD, Humberstone KS, Krishnan UV, Carmichael JA, Doughty L, Hattarki M, et al. Selection and affinity maturation of IgNAR variable domains targeting Plasmodium falciparum AMA1. Proteins. 2004;55:187–197. doi: 10.1002/prot.20005. [DOI] [PubMed] [Google Scholar]
- 33.Lee N. Towards the optimisation of malaria rapid diagnostic tests [PhD] Queensland: University of Queensland; 2010. [Google Scholar]
- 34.Kruger NJ. The Bradford method for protein quantitation. Methods Mol Biol. 1994;32:9–15. doi: 10.1385/0-89603-268-X:9. [DOI] [PubMed] [Google Scholar]
- 35.Dooley H, Flajnik MF. Shark immunity bites back: affinity maturation and memory response in the nurse shark, Ginglymostoma cirratum. Eur J Immunol. 2005;35:936–945. doi: 10.1002/eji.200425760. [DOI] [PubMed] [Google Scholar]
- 36.Shankey TV, Clem LW. Phylogeny of immunoglobulin structure and function–VIII. Intermolecular heterogeneity of shark 19S IgM antibodies to pneumococcal polysaccharide. Mol Immunol. 1980;17:365–375. doi: 10.1016/0161-5890(80)90057-7. [DOI] [PubMed] [Google Scholar]
- 37.Stanfield RL, Dooley H, Flajnik MF, Wilson IA. Crystal structure of a shark single-domain antibody V region in complex with lysozyme. Science. 2004;305:1770–1773. doi: 10.1126/science.1101148. [DOI] [PubMed] [Google Scholar]
- 38.Crouch K, Smith LE, Williams R, Cao W, Lee M, Jensen A, et al. Humoral immune response of the small-spotted catshark, Scyliorhinus canicula. Fish Shellfish Immunol. 2013;34:1158–1169. doi: 10.1016/j.fsi.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 39.Ohtani M, Hikima J, Jung TS, Kondo H, Hirono I, Aoki T. Construction of an artificially randomized IgNAR phage display library: screening of variable regions that bind to hen egg white lysozyme. Mar Biotechnol (NY). 2013;15:56–62. doi: 10.1007/s10126-012-9456-1. [DOI] [PubMed] [Google Scholar]
- 40.Russel M. Moving through the membrane with filamentous phages. Trends Microbiol. 1995;3:223–228. doi: 10.1016/S0966-842X(00)88929-5. [DOI] [PubMed] [Google Scholar]
- 41.Rosenberg A, Griffin K, Stiduer FW, McCormick LM, Berg J, Mierendorf R. T7 Phage Display System: a powerful new protein display system based on bacteriophage T7. Novations. 1998;6:1–6. [Google Scholar]
- 42.Diaz M, Stanfield RL, Greenberg AS, Flajnik MF. Structural analysis, selection, and ontogeny of the shark new antigen receptor (IgNAR): identification of a new locus preferentially expressed in early development. Immunogenetics. 2002;54:501–512. doi: 10.1007/s00251-002-0479-z. [DOI] [PubMed] [Google Scholar]
- 43.Wu TT, Johnson G, Kabat EA. Length distribution of CDR H3 in antibodies. Proteins Struct Funct Genet. 1993;16:1–7. doi: 10.1002/prot.340160102. [DOI] [PubMed] [Google Scholar]
- 44.Henderson KA, Streltsov VA, Coley AM, Dolezal O, Hudson PJ, Batchelor AH, et al. Structure of an IgNAR-AMA1 complex: targeting a conserved hydrophobic cleft broadens malarial strain recognition. Structure. 2007;15:1452–1466. doi: 10.1016/j.str.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 45.Swain MD, Anderson GP, Zabetakis D, Bernstein RD, Liu JL, Sherwood LJ, et al. Llama-derived single-domain antibodies for the detection of botulinum A neurotoxin. Anal Bioanal Chem. 2010;398:339–348. doi: 10.1007/s00216-010-3905-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Wyngaardt W, Malatji T, Mashau C, Fehrsen J, Jordaan F, Miltiadou D, et al. A large semi-synthetic single-chain Fv phage display library based on chicken immunoglobulin genes. BMC Biotechnol. 2004;4:6. doi: 10.1186/1472-6750-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohno S, Mori N, Matsunaga T. Antigen-binding specificities of antibodies are primarily determined by seven residues of VH. Proc Natl Acad Sci USA. 1985;82:2945–2949. doi: 10.1073/pnas.82.9.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simmons DP, Streltsov VA, Dolezal O, Hudson PJ, Coley AM, Foley M, et al. Shark IgNAR antibody mimotopes target a murine immunoglobulin through extended CDR3 loop structures. Proteins. 2008;71:119–130. doi: 10.1002/prot.21663. [DOI] [PubMed] [Google Scholar]
- 49.Streltsov VA, Varghese JN, Carmichael JA, Irving RA, Hudson PJ, Nuttall SD. Structural evidence for evolution of shark Ig new antigen receptor variable domain antibodies from a cell-surface receptor. Proc Natl Acad Sci USA. 2004;101:12444–12449. doi: 10.1073/pnas.0403509101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoogenboom HR, de Bruine AP, Hufton SE, Hoet RM, Arends JW, Roovers RC. Antibody phage display technology and its applications. Immunotechnol. 1998;4:1–20. doi: 10.1016/S1380-2933(98)00007-4. [DOI] [PubMed] [Google Scholar]
- 51.Muyldermans S, Lauwereys M. Unique single-domain antigen binding fragments derived from naturally occurring camel heavy-chain antibodies. J Mol Recognit. 1999;12:131–140. doi: 10.1002/(SICI)1099-1352(199903/04)12:2<131::AID-JMR454>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 52.Hust M, Dubel S. Phage display vectors for the in vitro generation of human antibody fragments. Methods Mol Biol. 2005;295:71–96. doi: 10.1385/1-59259-873-0:071. [DOI] [PubMed] [Google Scholar]
- 53.Beghetto E, Gargano N. Lambda-display: a powerful tool for antigen discovery. Molecules. 2011;16:3089–3105. doi: 10.3390/molecules16043089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Marco A. Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microb Cell Fact. 2009;8:26. doi: 10.1186/1475-2859-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schierle CF, Berkmen M, Huber D, Kumamoto C, Boyd D, Beckwith J. The DsbA signal sequence directs efficient, cotranslational export of passenger proteins to the Escherichia coli periplasm via the signal recognition particle pathway. J Bacteriol. 2003;185:5706–5713. doi: 10.1128/JB.185.19.5706-5713.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zapun A, Creighton TE. Effects of DsbA on the disulfide folding of bovine pancreatic trypsin inhibitor and alpha-lactalbumin. Biochemistry. 1994;33:5202–5211. doi: 10.1021/bi00183a025. [DOI] [PubMed] [Google Scholar]
- 57.Zapun A, Bardwell JC, Creighton TE. The reactive and destabilizing disulfide bond of DsbA, a protein required for protein disulfide bond formation in vivo. Biochemistry. 1993;32:5083–5092. doi: 10.1021/bi00070a016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. PCR amplification of randomly selected plaques from the VNAR domains primary library using T7SelectUP (For) and T7SelectDOWN (Rev) sequencing primers. Lane M represents 100 bp ladder; lane 1–20 represents insert of single plaque; lane −ve represents negative control (PCR product with no cDNA template).
Additional file 2. Deduced amino acid sequences in hypervariable regions of VNAR clones targeting to three malaria biomarkers. The non-canonical cysteine residue is highlighted in red colour. This table also indicates the length of CDR3, number of cysteine residue, and type of IgNAR family for each clone.