

Abstract
Over the last two decades, we have witnessed a revolution in the field of Parkinson’s disease (PD) genetics. Great advances have been made in identifying many loci that confer a risk for PD, which has subsequently led to an improved understanding of the molecular pathways involved in disease pathogenesis. Despite this success, it is predicted that only a relatively small proportion of the phenotypic variability has been explained by genetics. Therefore, it is clear that common heritable components of disease are still to be identified. Dissecting the genetic architecture of PD constitutes a critical effort in identifying therapeutic targets and although such substantial progress has helped us to better understand disease mechanism, the route to PD disease-modifying drugs is a lengthy one. In this review, we give an overview of the known genetic risk factors in PD, focusing not on individual variants but the larger networks that have been implicated following comprehensive pathway analysis. We outline the challenges faced in the translation of risk loci to pathobiological relevance and illustrate the need for integrating big-data by noting success in recent work which adopts a broad-scale screening approach. Lastly, with PD genetics now progressing from identifying risk to predicting disease, we review how these models will likely have a significant impact in the future.
Keywords: Parkinson’s disease, Genetics, Common risk factors, Genome-wide association study, Prediction
Introduction
Population aging is leading to an increased prevalence of neurodegenerative diseases. Parkinson’s disease (PD), the second most common neurodegenerative disease after Alzheimer’s disease and the most frequent movement disorder, represents a medical and economic challenge for our society; there are no available treatments that can stop or reverse the neurodegenerative process in PD. PD shows an age-dependent prevalence in which about 1% of the global population at 65 years of age and over and about 4–5% of individuals at 85 years of age and over are affected (de Lau and Breteler 2006).
Until two decades ago, PD was considered the archetypal example of a non-genetic disease. The first published cross-sectional series of twin studies supported this view (Ward et al. 1983; Eldridge and Ince 1984; Duvoisin et al. 1981). These results were supported by previous epidemiological studies which linked PD to environmental causes such as viral infection or neurotoxins. In fact, in 1918, a pandemic influenza virus was strongly associated with post-encephalitic parkinsonism, seen by many as evidence that viral infection may be a major cause of PD. Furthermore, the observation that drug users exposed to MPTP developed parkinsonian-like features strengthened the notion that PD was an environmental disease Langston and Ballard 1983).
An important step forward in favor of a genetic contribution to the etiology of PD was taken with the implementation of molecular genetics to dissect the underlying genetic cause of several families in which PD was inherited in an autosomal dominant or recessive manner. The first forms of monogenic PD were caused by highly penetrant mutations affecting multiple members per family and while the insights gained from such mutations were seen as very valuable, these families were extremely rare.
The year 1997 marked the starting point for PD genetics with the identification of mutations in SNCA (encoding α-synuclein) (Polymeropoulos et al. 1997). This key first finding to suggest a heritable component of PD was followed by the identification of additional rare recessive forms associated with early-onset disease such as those caused by mutations in PRKN, PINK1, and DJ-1 (Kitada et al. 1998; Polymeropoulos et al. 1997; Valente et al. 2001; Bonifati et al. 2003). In 2004, the identification of mutations in LRRK2 as a cause of autosomal dominant PD and the most common monogenic form of PD identified to date was a major advance in the field (Paisán-Ruíz et al. 2004; Zimprich et al. 2004).
To date, PD causing mutations have been identified in 15 genes responsible for monogenic forms. However, all these known monogenic forms combined explain only about 30% of monogenic and 3–5% of genetically complex cases. A detailed discussion of the monogenic forms causing PD is beyond the scope of this article and has been discussed in recent articles (Hernandez et al. 2016).
This review solely focuses on placing the currently known common genetic risk factors of PD in a future facing context. In addition, we focus on concepts that will help progress genetics from a more complex common variant prospective and we also touch on key areas such as multifactorial data integration, interactive risk assessments, and complex readouts of progression all leading us on a path towards developing and applying an intervention.
Parkinson’s disease is a complex genetic disorder
PD fits within the wide range of complex polygenic disorders influenced by both genetic and environmental factors. While just a small percentage of PD cases are monogenic forms sometimes exhibiting variable penetrance, the vast majority of cases are considered to be genetically complex presenting with multiple clinical presentations. It has been assumed that PD etiology lies on a continuum, ranging from the monogenic inheritance observed in monogenic disease to complex inheritance associated with an interplay of genetic risk and likely environmental influence..
The genetic portion of PD is often ascribed to two non-mutually exclusive ideas: the common disease common variant (CDCV) hypothesis and the common disease rare variant (CDRV) hypothesis (also known as the multiple rare variant hypothesis). On one side, the CVCD hypothesis would accept that the genetic basis of PD is a result of a large number of common variants that each exert relatively small effects on disease risk but that cumulatively confer substantial risk. On the other side, the CDRV hypothesis speculates that a contributing risk component for complex disease will be rare genetic variants of small or moderate/big effect where highly functional, deleterious alleles might exist. This phenomenon may be particularly pronounced in late-onset diseases such as genetically complex PD, where selective pressures are not as profound.
The emergence and improvement of technological approaches continues to test both paradigms by increasing the identification of very rare causative mutations underlying monogenic forms of disease through whole-genome and whole-exome sequencing (WES) approaches and of common variants with small effects contributing to genetically complex, late-onset disorders through genome-wide association studies (GWAS). The ultimate aim of these studies is to gain insight into the biology of disease, under the assumption that a better understanding will lead to translational advances enabling more effective prevention or potential treatments.
Common risk factors in PD
Both GWAS and candidate gene association studies continuously validate that the most statistically significant signals associated with PD are common variants located close to SNCA, LRRK2, and MAPT as well as low-frequency coding variants in GBA.
SNCA
Just after the discovery of SNCA mutations causing a rare monogenic form of PD, α-synuclein protein aggregates were identified as a major component of Lewy bodies, a primary pathological hallmark of PD (Spillantini et al. 1997). This finding tied together the pathogenesis of monogenic and genetically complex PD. Interestingly, in the context of risk for PD, SNCA is pleiomorphic; both rare mutations and common variation at this locus alter risk for disease. At one end of this risk spectrum, deleterious point mutations in and multiplications of this gene cause a severe early-onset form of PD with an autosomal dominant pattern of inheritance (Chartier-Harlin et al. 2004; A. B. Singleton 2003). At the other end of risk, it has been widely demonstrated that non-coding variability within this locus confers risk and predisposes to genetically complex PD. The first indication that the SNCA locus contained risk variants for idiopathic PD came from the association between the REP1 polymorphism in the promoter region of the gene and PD (Maraganore et al. 2006). Later on, GWAS signals at SNCA showed an association with PD from intron 4 to after the 3′ UTR region (Simón-Sánchez et al. 2009). Since then, SNCA has been overwhelmingly established in GWA studies identifying additional signals and providing further insights about the genetic risk at this locus (UK Parkinson’s Disease Consortium et al. 2011; Lill et al. 2012; Nalls et al. 2014; Chang et al. 2017). Current research suggests between 2 and 5 semi-independent association signals accounting for heritable risk at this locus (unpublished work, IPDGC). To definitively ascertain the number of independent risk factors within this region, high coverage sequencing and deep-resequencing in large and genetically diverse samples will be necessary.
LRRK2
Genetic variants in LRRK2 account for the majority of all known heritable PD. The most common pathogenic variant in LRRK2, p.G2019S, is responsible for about 1% of patients with genetically complex PD and 4% of patients with a family history of PD. LRRK2 p.G2019S exhibits incomplete and age-associated differences in penetrance. Collaborative research has shown that the risk of PD for individuals who inherit the LRRK2 p.G2019S variant varies from 28% at 59 years to 51% at 60 years reaching up to 75% at 80 years of age (Spatola and Wider 2014).
Notably, the frequency of this variant varies depending on ethnic background, with the highest frequencies among North African Arab and Jewish populations (Healy et al. 2008; Lesage et al. 2006). The frequency is also higher in the Middle East and in Southern Europe than in Northern Europe. Of note, a recent study reported a prevalence of 0.71% among Caucasians, 0.07% among Asians, and 30.2% among individuals of Arab origin with PD (Ross et al. 2011). It is thought that LRRK2 p.G2019S originated from a common founder in the North of Africa and spread globally with the Ashkenazi Diaspora (Lesage et al. 2005).
Similarly to SNCA, LRRK2 is a pleiomorphic locus. In addition to several disease-causing mutations characterized by segregation with PD in large families and by functional work, common variability has been continuously detected as a risk factor for PD. There are two lines of evidence supporting the idea that LRRK2 contains risk-modifying variants. Firstly, it has been widely reported that two polymorphic LRRK2 variants, p.G2385R and p.R1628P, are associated with a 2-fold risk of PD in Asian populations (but rarely found among Caucasians), with a frequency of approximately 6% in cases (Di Fonzo et al. 2006; Farrer et al. 2007; E. K. Tan and Schapira 2008; E.-K. Tan et al. 2007). Secondly, GWAS implicate non-coding variants close to LRRK2 with increased risk for PD ~ 1.2-fold suggesting that this risk may be mediated by an alteration in expression or splicing.
GBA
Homozygous mutations in GBA, encoding the enzyme glucocerebrosidase, lead to Gaucher’s disease, a lysosomal storage disorder with an autosomal recessive pattern of inheritance. The astute clinical observation in relatives of patients affected with Gaucher’s disease revealed that first- and second-degree family members manifested an increased incidence of PD, pointing to GBA as a risk factor for PD (Neudorfer et al. 1996; Halperin et al. 2006). Later on, a multicenter study conducted by Sidransky et al. showed that heterozygous GBA mutations are the largest genetic risk factor for developing PD, enhancing one’s risk approximately 5-fold and highlighting the importance of the lysosomal pathway in the pathogenesis of PD (Sidransky et al. 2009). Interestingly, because GBA variants can appear with frequencies <5%, it was initially omitted from GWAS analyses. It was only after a candidate gene approach were GWAS able to confirm its clear significance as a PD risk factor. A recent study has suggested that the GBA p.E326K variant could be the major driver of one of the GBA GWAS signals (Berge-Seidl et al. 2017). Notably, this variant is not sufficient to cause Gaucher’s disease (Duran et al. 2013) and it appears to have a lower odds ratio in PD compared to other Gaucher’s disease-linked mutations. Although this mutation on its own decreases glucocerebrosidase activity, residual activity is still better than other Gaucher’s disease mutations such as p.N370S and p.L444P (McNeill et al. 2014). GBA encodes a lysosomal glucocerebrosidase enzyme responsible for the synthesis of ceramide (Beutler 1992). A significant decrease in the enzyme activity and a reduced expression level of GBA have been found in PD patients carrying heterozygous mutations in GBA. Additionally, decreased rates in the glucocerebrosidase activity have been found in the substantia nigra of PD patients in comparison with other brain regions (Beutler 1992; Gegg et al. 2012).
Many studies suggest that around 5–10% of PD patients carry a GBA mutation, and it has been reported that the penetrance and lifetime risk of developing PD for GBA carriers varies in an age-dependent fashion from 20% at 70 years to 30% at 80 years (Anheim et al. 2012). GBA mutations are substantial common risk factors for PD; however, their frequency varies according to different ethnicities, being particularly frequent among Ashkenazi Jewish subjects. For example, the most common GBA variant, p.N370S, is present among those of European, American, or Middle Eastern origin but is not typically seen in Chinese or Japanese populations (Mitsui et al. 2009).
MAPT
Dominantly inherited mutations in MAPT were first associated with forms of frontotemporal dementia and parkinsonism linked to chromosome 17 (Hutton et al. 1998). The association between PD and the locus harboring MAPT is intriguing as MAPT mutations and tau pathology have been predominantly associated with dementias. Besides these monogenic forms, several studies have scrutinized MAPT for variability that may impart risk for PD. There are two major haplotypes at the MAPT locus: the directly oriented haplotype H1 and the H2 haplotype, which has an inverted chromosomal sequence (Baker et al. 1999). The H2 haplotype is present in approximately 20% of the European population (Evans et al. 2004) and shows very limited genetic variability contrary to the H1 haplotype. This locus represents the largest area of linkage disequilibrium known in the human genome (Pittman et al. 2005).
A growing body of evidence in the past decade has shown that MAPT H1 and its sub-haplotype H1c are associated with increased risk for PD, suggesting that haplotype-specific differences in expression and potentially alternative splicing of MAPT transcripts affect cellular functions at different levels, which eventually increases susceptibility (Skipper et al. 2004). Furthermore, MAPT is one of the top hits found in GWAS, although it seems to be limited to Europeans and not Asian populations. Determination of how common variation at the MAPT locus increases the risk for PD is challenging as this locus harbors many genes, and the extended linkage disequilibrium means that it is difficult to localize the genetic signal. Thus, while MAPT is the obvious candidate as the effector gene at this locus, we cannot be certain that this is the true biological mediator of risk; previous work in complex risk within the diabetes field has shown that such assumptions can be simply wrong (Smemo et al. 2014), (Claussnitzer et al. 2015).
Identifying risk loci from GWAS
The underlying idea of a GWAS is based on the CDCV paradigm with the objective of detecting common variants (MAF > 5%) in ethnically homogeneous populations. While critics suggest a genome-wide fishing expedition, the overwhelming majority of the genetics community would argue that the results gathered from such studies have marked a significant advancement from candidate gene studies and have driven the new era and concept of PD genetics. These advances are based on the premise that risk variants may occur within haplotype blocks shared with common variants through linkage disequilibrium. Since common variants can be tagged through genotyping marker arrays, risk variants in linkage disequilibrium should manifest an association by proxy with tagged common variants and ultimately with PD. By increasing sample size and genotype marker frequency, lower-risk variants with a lower population-attributable risk can be detected.
Along the way, several GWAS (Edwards et al. 2010; Hernandez et al. 2012; Pihlstrøm et al. 2013; International Parkinson Disease Genomics Consortium et al. 2011; Satake et al. 2009; Simón-Sánchez et al. 2009; Lill et al. 2012) and meta-analyses (Nalls et al. 2014; Chang et al. 2017) have been key at identifying common risk variability linked to genetically complex PD. To date, 43 loci have been associated at a genome-wide significant level with PD risk in individuals of European ancestry (Chang et al. 2017). However, despite this considerable success, only a relatively small proportion of the phenotypic variability has been explained. While the known loci only account for 6–7% of PD burden, it has been estimated that PD is roughly 27% heritable based on common variants in GWAS (Keller et al. 2012). Thus, it is evident that unknown common heritable components of disease remain to be identified so while we have come a long way, there is certainly a considerable distance left to travel before we reach a complete understanding of the genetic basis of PD. Additionally, it is highly likely that an important part of the missing heritability, defined as the phenotypic variance attributed to genotypic differences, exists in rare variants with low or high degrees of risk and structural variation, both of which are difficult to detect using traditional GWAS methods. In this review, we do not aim to provide a concurrent list of the known risk loci associated with PD, as this can be found in more detail elsewhere (Nalls et al. 2014; Chang et al. 2017) but aim to address (1) the challenges faced in progressing risk loci to disease-relevant molecular mechanism and (2) how using a pathway-based analysis to establish the pathobiology of PD may be a more successful approach.
Identifying candidate genes
A major challenge resulting from the success of GWAS is identifying the true candidate gene or genes at each locus that are causally and functionally associated with disease etiology. A recent study constructed a PD protein network from risk loci and identified over 500 candidate genes that could be implicated in the condition from the GWAS hits (Klemann et al. 2017). With identification of many more risk loci anticipated and the costly and time-intensive nature of dissecting risk loci, there is a focus on developing methods to more accurately determine the correct genes to pursue for further research, in order to unpin the molecular basis of PD. With the aim of identifying causative genes from GWAS signal, the most recent GWAS implemented a neurocentric strategy to nominate candidate genes. To annotate the risk loci, the approach combined seven sources of data and linked variants from PD-associated loci, which subsequently included expression quantitative trait loci (eQTL), GTEx expression data, and expression data from multiple mouse brain cell types. Annotating identified risk loci in this manner can be very informative, for example if the given hit is an already known eQTL, it determines that the disease-linked variation contributes to differential expression of a certain gene. The first stage of the most recent PD GWAS analysis assigned a candidate gene to a locus if the variant in question changed the protein sequence or if the variant was a cis-QTL for the gene. If this method failed to identify a candidate, the neighboring genes were then analyzed on the basis of neurologically related phenotypes and expression, and the highest scoring gene was assigned. Although these nominated genes require experimental functional validation, this method provides a more robust and defined list of genes to validate, enabling future research to focus efforts on this smaller candidate gene list, rather than the 500 plus genes that have been broadly associated in other studies (Chang et al. 2017).
A glimpse into much larger networks
Beyond discovering the causative genes, another challenge remains to understand the mechanisms by which these factors alter disease phenotype, including whether they act independently or interact within biological pathways. Considering that it is hypothesized that we only currently know a small proportion of the genes involved in the pathobiology of PD, looking at the identified risk loci systematically rather than individually is key. This approach has led to major biological pathways being implicated and validated in PD. Following the most recent meta-analysis, pathway analysis established that the nominated candidate genes were enriched in previously associated pathways including autophagy, endocytosis, mitochondrial biology, immune response, and lysosomal function (Fig. 1).
Fig. 1.
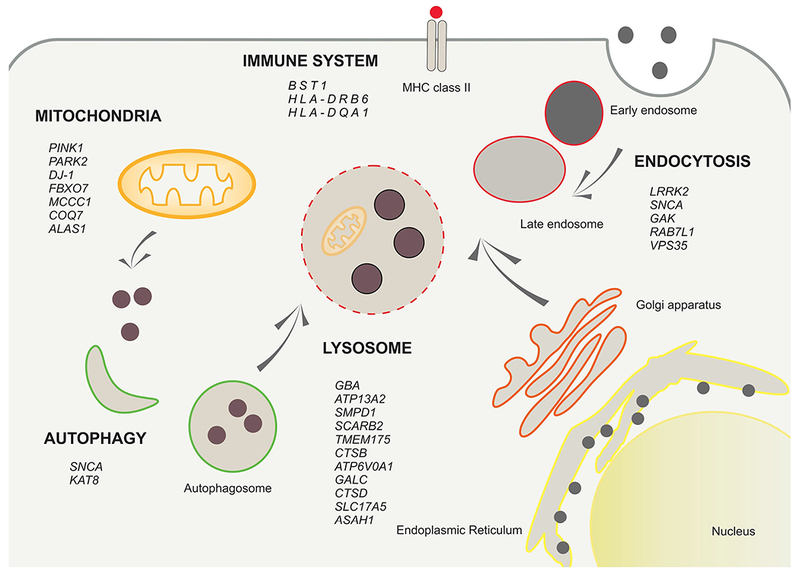
Overview scheme of the PD risk factor genes and the molecular pathway where they are involved. The genes encoded for proteins that mainly participate in mitochondrial turnover, autophagy, endocytosis, immune system and/or lysosomal function
Autophagy
Autophagy is the lysosome-mediated catabolic process in which dysfunctional organelles and proteins are degraded. This was one of the first pathways to be implicated in PD as it was an obvious choice following the breakthrough of identifying α-synuclein aggregation in the disease. This process is one of the main routes for the intracellular degradation of α-synuclein. A wealth of evidence from experimental, pathological, and genetic studies continues to support the role of autophagy in PD. A variant close to a gene encoding a major regulator of the process, KAT8 (lysine acetyltransferase 8), is significantly associated at a genome-wide level with PD risk, and notably, this variant is a strong cis-eQTL affecting KAT8 expression (Chang et al. 2017). In addition to the GWAS signal, studies have also associated lower levels of KAT8 mRNA with PD risk, and recently, when KAT8 was inhibited, a decrease in autophagic flux was demonstrated (Hale et al. 2016). Thus, recent genetic studies further suggest that dysfunctional autophagy in PD is a risk factor for the disease.
The immune response
The immune system is another pathway that has been strongly associated with PD susceptibility, either through inflammation or an autoimmune response. Multiple GWA studies report PD risk loci within key immune associated genes such as BST1 (bone marrow stromal cell antigen 1). BST1 has been proposed to play a role in neutrophil adhesion and migration, and it is also suggested that it could be a cause of selective vulnerability of dopaminergic neurons in PD (Zhang et al. 2006). Additionally, there are several hits at the HLA (human leukocyte antigen) locus (Chang et al. 2017; Nalls et al. 2014) which encodes the major histocompatibility complex class II (MHC-II). Although the association between PD and the HLA region is complex, it has been suggested that the hits at HLA-DRB6 and HLA-DQA1 could implicate regulation of antigen presentation as a potential mechanism by which the immune response links environmental factors to genetic susceptibility in conferring risk for PD (Kannarkat et al. 2015). Indeed, Sulzer et al. have recently reported that α-synuclein-derived fragments act as antigenic epitopes displayed by HLA receptors, where both helper and cytotoxic T-cell responses are present in a high percentage of patients when tested (Sulzer et al. 2017).
Mitochondrial biology
It has been widely suggested that proteins encoded by the recessive genes associated with early-onset PD, PINK1, PARK2, DJ-1, and FBXO7, are involved in the mitochondrial quality control system and its regulation (Mullin and Schapira 2015) including processes such as mitogenesis, mitophagy, and mitochondrial homeostasis and transport. In support of this, Burchell et al. identified that through direct interaction with PINK and Parkin, Fbxo7 mediates mitochondrial maintenance and is involved in Parkin-mediated mitophagy (Burchell et al. 2013). Hence, it is thought that mitochondrial dysfunction plays a pivotal role in the early events of genetically complex and genetic PD cases. The implication of mitochondrial dysfunction in PD susceptibility has been further supported by recent GWAS that have nominated mitochondrial-associated genes with PD risk. The MCCC1 (methylcrotonoyl-CoA carboxylase 1 (alpha)) gene is a robustly replicated example of this association as well as recent novel hits such as COQ7 (coenzyme Q7, hydroxylase) and the ALAS1 (delta-aminolevulinate synthase 1) genes.
Lysosomal dysfunction
Most significantly, the lysosomal pathway has also been strongly implicated in PD, as it is suggested that lysosomal impairment may contribute to α-synuclein aggregation (Dehay et al. 2013). In support of this theory, the last decade has witnessed an increasing amount of genetic evidence implicating the lysosomal system as a central mechanism in the pathobiology of PD (Kalia and Lang 2015; Moors et al. 2016; Vekrellis et al. 2011; Wong and Krainc 2016). As previously described, heterogeneous mutations in GBA confer high risk for PD, and pathogenic variants in ATP13A2, encoding a lysosomal ATPase, are responsible for a very rare cause of parkinsonism (Park et al. 2015). More recently, variation in SMPD1 encoding the lysosomal acid sphingomyelinase that is associated with Niemann-Pick type A and B disease, a rare autosomal-recessively inherited lysosomal storage disorder, was suggested as a novel susceptibility factor for PD in the Ashkenazi Jewish population, although this warrants robust replication (Zech et al. 2013).
This association has been further strengthened by the findings of the last two large-scale PD GWAS. The first PD metaanalysis confirmed a robust and potent risk signal at the GBA locus but also independently implicated common risk alleles at two other lysosomal genes: the scavenger receptor class B member 2 (SCARB2) and the transmembrane protein 175 (TMEM175) (Nalls et al. 2014). The most recent meta-analysis replicated the signals at these lysosomal-associated loci but also nominated three new lysosomal-associated genes: CTSB (cathepsin B), ATP6V0A1 (ATPase H+ transporting V0 subunit a1), and GALC (galactosylceramidase). Interestingly, a recent study has not only replicated the association between the outlined risk loci but also addressed an enrichment of rare variants in additional novel lysosomal genes (Robak et al. 2017, in press). Robak et al. tested the hypothesis that lysosomal gene variants are enriched in PD. WES from an initial 1167 PD cases and 1685 controls was interrogated for variant burden within 54 LSD genes. Independently of GBA, the analysis found a significant association between risk of PD and rare, likely damaging lysosomal variants, which was then replicated in an additional dataset. Remarkably, this approach identified new PD risk loci within lysosomal genes, implicating CTSD (cathepsin D), SLC17A5 (solute carrier family 17, member 5) and ASAH1 (N-acylsphingosine amidohydrolase 1) as new candidate PD susceptibility genes. This approach highlights the importance for the need of more high-power whole-genome analysis of PD in order to confirm these rare variants.
Endocytic pathway
There is growing evidence pointing to PD as an endosomal trafficking disease (Abeliovich and Gitler 2016). It is widely known that several PD-associated genes are related to the endocytic pathway. The link between PD and endocytosis was first described in an α-synuclein aggregation model in which α-synuclein was unable to be transported to the Golgi apparatus leading to Golgi apparatus fragmentation and thus a dysfunction in the endoplasmic reticulum(ER) to Golgi intracellular trafficking (Cooper 2006; García-Sanz et al. 2017). The phenotype is similar to the one observed in fibroblasts from patients carrying the mutation p.N370S in GBA, which also show a reduced level of the enzyme within the lysosomes of those individuals. It is thought that the lower level of the enzyme is due to a malfunction in the cellular traffic of the protein itself from the ER. As a consequence, GBA remains blocked in the ER and it cannot be sort to Golgi apparatus, which appears fragmented (García-Sanz et al. 2017).
The endocytosis mechanism mediated by clathrin has also linked to PD. GAK encodes a cyclin-G-associated kinase, which is a key protein in the clathrin uncoating mechanism at the synaptic terminal. Genetic variants in GAK have been recently confirmed as risk factors for PD disease (International Parkinson Disease Genomics Consortium et al. 2011) (Nagle et al. 2016). In addition, GAK have been recently proposed as a binding partner of LRRK2 (Beilina et al. 2014).
Evidently many pathways have been implicated in PD, illustrating the complexity of defining the molecular mechanism of the disease. With the progression from risk loci to molecular insights being slow and with the identification of many new risk loci on the horizon (which will likely implicate additional genes and pathways), there is an apparent lack of key foundational data that would aid in constructing networks from identified hits quickly. For swift development, substantial collaborative efforts need to be made mapping transcriptomic, epigenomic, and proteomic landscapes with genetic data. Overlaying high-throughput molecular screening in this way will not only help dissect the genetic risk factors in a biological sense for PD but this knowledge will also benefit the understanding of the etiology of other neurological disorders (Jain and Heutink 2010; Parikshak et al. 2015; A. Singleton and Hardy 2016).
Integrating genetics and functional work: challenges and achievements
Large-scale meta-analyses have identified multiple genetic loci associated to PD pointing out the complexity of the disease. As previously highlighted, scientists are facing the challenge to identify how those genetic risk variants may disrupt the molecular and cellular pathways within cells and thus unravel the molecular networks underlying the pathobiology of the disease (Parikshak et al. 2015). The fact that several of the pathogenic variants that have been described by GWAS as risk factors are also present in monogenic cases of PD leads to the idea that common pathways are involved in both forms and therefore several genes might interact to regulate downstream common targets (Chuang and Gitler 2013; Parikshak et al. 2015). However, functional studies are extremely complicated in the context of PD considering the complexity of the central nervous system and the lack of in vivo approaches, tools, and models to accurately reproduce the tremendous complexity of the human brain. Neurodegeneration associated with PD involves many features impossible to reproduce in a “less complex” system. Those involve tremendous cellular heterogeneity, preferential vulnerability of certain subset of neurons, and highly complex interconnected networks ((Parikshak et al. 2015; Surmeier et al. 2017). Nonetheless, in response to the requirements of the new era of big genetic data, cellular and molecular biologists are forced to turn towards unbiased high-throughput analysis.
As a result of the early discovery of monogenic forms of PD, the functions of the proteins encoded by these PD-associated genes have now been widely assessed in both in vivo and in vitro models using relatively traditional reductionist methods. Significant gains have been made in understanding the role of these genes and their proteins in PD pathogenesis; however, given the intense effort and length of time required for this process, it is clear that such a traditional approach will be insufficient for understanding the role of the large number of risk loci identified so far.
The sheer number and mechanism of action of risk loci requires that new approaches are needed to understand the risk. Notably, in the most usual scenario, while GWA studies nominate a risk-associated locus, the gene responsible for the pathogenic action is unknown. Thus, any functional work related to those variants needs to focus first in the identification and validation of the particular gene responsible for that variant. Secondly, the low- to moderate-risk variant alleles are extremely difficult to analyze in a biological setting. For the most part, these risk alleles are not associated with protein coding variability and thus their effects are mediated through expression changes in one form or another. In that sense, introducing an additional risk factor like aging, exposure to toxins, or oxidative stress may unmask the cellular mechanism underlying the pathology associated to those variants. However, this strategy adds another layer of complexity to the interpretation of the functional meaning and the pathobiology underlying the genetic risk variants.
Despite the challenges of using cellular and transgenic models to characterize PD risk loci, a method of combining unbiased and systematic genetic work with cellular biology has been successfully demonstrated by two overlapping efforts (Beilina et al. 2014), (MacLeod et al. 2013). For the first time in the field of PD, an effort to link different pathogenic variants to a common pathway has been made. To unravel the candidate gene for PARK16 loci, they compared the brain transcriptome from an unaffected carrier of the seven associated loci (SNCA, LRRK2, MAPT, HLA-DRA, PARK16, LAMP3, STK39) and concluded that LRRK2 and PARK16 variants provoke the same transcriptome signature. In addition, they confirmed the two variants interact genetically. Mechanistically, Rab7L1 rescue mutant LRRK2 neural shortening in rat cortical neurons. In addition, Rab7L1 knock-out induces dopaminergic cell loss in flies. By using cellular approaches, the group not only revealed a common pathway for PD but also resolves Rab7L1 as a candidate gene from PARK16 loci. The animal and cellular studies for those genes reveal Lrrk2 and Rab7L1 participates in the protein sorting and retromer function which vesicular trafficking malfunction, Golgi apparatus sorting, and retromer complex alterations via VPS35.
The work by Beilina et al., using a protein-protein interaction array, has not only confirmed this Lrrk2 and Rab7L1 interaction but identified another interactor: cyclin-G-associated kinase (encoded by GAK) as a binding partner to Lrrk2. GAK has been recently proposed as another risk loci for PD (MacLeod et al. 2013; Rhodes et al. 2011). Interestingly, those LRRK2 interactors function in the same pathway of vesicle trafficking (Beilina et al. 2014), pointing out the endolysosomal system as a key molecular clue to understand the pathobiology of the disease.
Although Chang et al. attempted to develop a much needed strategy to nominate candidate genes from GWAS loci, this had lead to discordant results. For example, the candidate genes nominated for the PARK16 locus through the neurocentric method adopted by Chang et al. suggested NUCKS1 and SLC4A9A1 as PD genes. Similarly, the authors nominated TMEM175 and DGKQ as candidate genes of the GAK/TMEM175/DGKQ locus. However, Beilina et al. identified GAK and RAB7L1 as LRRK2 interactors by performing an unbiased screen, raising the probability that such genes explain these GWAS signals (Beilina et al. 2014). This variance highlights the complexity of identifying the causal gene from a GWAS hit. Therefore, developing system-wide approaches that combine bioinformatic and functional data is key to addressing this issue (A. Singleton and Hardy 2016).
Understanding heterogeneity and complexity in Parkinson’s disease
PD is a heterogeneous disorder with varying manifestations of symptomatologies, from motor biased and tremor dominant to PD with dementia and the entire spectrum between. It is theoretically possible and a current topic of investigation that different genetic, demographic, and clinical risk factors may have a strong influence over the disease presentation at diagnosis as well as the trajectory and velocity of progression. Understanding the heterogeneity of PD requires a holistic view of the disease and will push clinical and computational scientists to work together closely to build accurate models of manifestation and progression to capture the unique flavors of this common disease. This attempt to redefine how PD progression and subtyping is carried out has been central to the missions of many research organizations such as the Michael J. Fox Foundation, who have offered great support for data-driven efforts in this area for a variety of research groups which are now beginning to yield high-quality results. From identifying PD mimics like subjects without evidence of dopaminergic dysfunction (SWEDDs) to delineating fast versus slow progressors, genetics will likely play a major role in future clinical trials (Nalls, McLean, et al., 2015). For example, GBA mutation carriers have been suggested to have earlier onset and more cognitive decline than typical PD patients (Liu et al. 2016).
Large studies with basic phenotyping have had moderate success such as a recent study that used cumulative genetic risk score calculations as predictor of age at onset, showing that generally greater genetic burden is concurrent with earlier age at onset (Lill et al. 2015; Nalls et al. 2015a, b). Although, in order to better understand how genetic variation affects PD etiology in greater detail, larger longitudinal cohorts of well clinically characterized patients are absolutely necessary. Additionally, as we look towards the next horizon in drug development, perspective and pre-diagnostic cohorts combining genetics and deep phenotypic data will be key.
The future of PD genetics
We have identified three key concepts that need to be addressed to speed progress in future PD genetic research ventures. These include identifying and refining heritable risk, predicting the course of disease, and focusing on general data-centric approaches, all of which will facilitate building a bridge to a future intervention.
Identifying and refining heritable risk
Current PD GWASs are up to 30,000 cases and 400,000 controls, although whole-genome sequencing studies in PD are less than 2% of that sample size but continue to grow rapidly. Future GWAS in PD will increase the number of risk alleles although each yielding smaller individual contributions to overall disease heritability. Since attempts to look at noncoding sequence for rare risk variants have been quite limited, whole-genome sequencing will likely be the main tool for discovery of additional rare risk loci. Knowledge of new genetic susceptibility variants may provide more precise estimates of risk and biological insight, shedding light on pathways disturbed during the pathogenesis of PD. Combining stepwise, conditional, and machine-learning approaches with large, deep-coverage sequencing-based datasets that are constantly growing in size and quality show promise as powerful tools to ultimately refine our estimates of local heritable risk. There is an increasing need and utility for fine-mapping GWAS-significant regions of PD originally identified in European-descent populations. Fine-mapping across populations of diverse continental ancestries has proven to be extremely valuable by leveraging allelic diversity and linkage disequilibrium patterns to identify putative functional variants. By identifying the functional variants within many GWAS loci, we can better approximate the local contribution to heritable risk while providing better insight into the biological processes underlying disease pathogenicity and finally improving the power of all predictive models. Whole-genome sequencing and similar technologies are critical because of denser coverage and greater genotype precision compared to imputation-based studies.
Thus, for progress in this domain, we need continued investment in genome-wide association studies, a concerted effort to execute genetics in non-Caucasian populations, and a committed investment in large-scale whole-genome sequencing approaches. It will also be important to address how specific gene-environment interactions influence PD susceptibility. Recent genome-wide gene-environment interaction studies for complex traits are generally thought to be underpowered for detecting most gene-environment interaction effects. Therefore, assessing the role of gene-environment interactions in disease risk will involve data collection efforts across multi-centric, carefully recruited, and well-characterized datasets (Biernacka et al. 2016).
Predicting the course of disease
Methods aimed at a deeper understanding of how PD will progress are emerging and will be key to future translational studies, the execution of clinical trials, and ultimately treatment. Predicting overall risk and age at onset at population scale has huge implications for drug development and clinical trial success as well as health economics and care planning. Fortunately, new research opportunities are emerging which will facilitate the development of novel therapeutics for such devastating condition by targeting the right patients at the right time, addressing relevant symptomatologies before irreparable damage has occurred. By using machine learning models in large population-based studies that combine clinical and genetic data, we can accurately predict who may likely get PD and when its onset will be. Prospective studies and the necessary resource triaging that is facilitated by predictive models such as these can only benefit from large data-centric efforts that will push the entire field of drug development. In PD, a few efforts that utilize predictive data to maximize study efficiency have already begun such as the prodromal aspects of the PPMI study in the USA [http://www.ppmi-info.org/] and predict PD in the UK [https://www.predictpd.com/]. Predicting onset and risk is useful, but predicting the precise molecular, clinical, and pathological flavor of PD from actual data as opposed to suggested clinical queries will prove invaluable. A number of labs have made great strides in this area including our own (Faghri et al., in press, https://github.com/ffaghri1/PD-progression-ML), attempting to not only partition PD into clusters of similar etiological manifestations but also predict the manner in which they progress, as well as the velocity in which they progress. Predicting these variable readouts of progression in such a complex and heterogenous disorder will likely have a huge impact on drug development in the future. For success in this domain, we require large increases in sample size for cohorts which also include multi-omics and detailed clinical data.
General data-centric approaches
Success in the new era of PD genetics requires extensive scientific collaboration and will not progress without teamwork in an open scientific environment. Open and easily accessible datasets that are both deep and wide are absolutely necessary, as are transparent and reproducible analytic methods. Genetics cannot operate in isolation; it will need to be combined with *-omics and curated clinical datasets to make great advances possible. Therefore, an international collaborative framework should be the priority to produce the best science possible. Stemming from these data-centric approaches, the field will leverage complex data and cutting edge analytics in a transparent public environment for research to build a bridge to an intervention. Complex problems like ours often require resource-intensive solutions. One perfect example of this is the Parkinson’s Disease Accelerating Medicines Partnership (PD-AMP). In the collaborative environment that is PD-AMP, cross-sections of academia, foundations, biotech, and technology industries will partner to investigate patterns of risk and de novo etiological pathways in the most well-characterized PD samples available, from a genetic to clinical level and back again. Projects like these that seek to use deep learning and artificial intelligence to investigate perturbed iPSC *-omics datasets anchored to wealth of clinical data via genomics will be the sources of discovery for the future.
For success in this domain, we require huge public facing projects that provide foundational data for the understanding of the molecular pathogenesis of PD; an EnCode-style/scale project for PD would provide such a resource.
In closing, there has been tremendous and most recently, rapid, progress in our understanding of the genetics of PD. This has, albeit slowly, revealed critical molecular mechanisms in the disease process, some of which are amenable to therapeutic intervention and are being targeted by the pharmaceutical and biotech industry now. We believe however that there is much to learn, and that critical to future success is the continued investigation of genetics in PD, expanding upon current gains in defining risk and diversifying to an understanding of disease progression. Critical to the successful translation of these findings will be the development of companion resources that afford rapid understanding of the biological consequences of these genetic factors. This is a difficult and expensive approach; however, for the first time, we have the technological means with which to pursue this goal.
References
- Abeliovich A, Gitler AD (2016) Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539(7628):207–216 [DOI] [PubMed] [Google Scholar]
- Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F, … French Parkinson Disease Genetic Group. (2012). Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology, 78(6), 417–420 [DOI] [PubMed] [Google Scholar]
- Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, … Hutton M (1999). Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet, 8(4), 711–715 [DOI] [PubMed] [Google Scholar]
- Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, … Cookson MR (2014). Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A, 111(7), 2626–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge-Seidl V, Pihlstrøm L, Maple-Grødem J, Forsgren L, Linder J, Larsen JP, … Toft M (2017). The GBA variant E326K is associated with Parkinson’s disease and explains a genome-wide association signal. Neurosci Lett, 658, 48–52 [DOI] [PubMed] [Google Scholar]
- Beutler E (1992) Gaucher disease: new molecular approaches to diagnosis and treatment. Science 256(5058):794–799 [DOI] [PubMed] [Google Scholar]
- Biernacka JM, Chung SJ, Armasu SM, Anderson KS, Lill CM, Bertram L, … Maraganore DM (2016). Genome-wide gene-environment interaction analysis of pesticide exposure and risk of Parkinson’s disease. Parkinsonism Relat Disord, 32, 25–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, Squitieri F, Krieger E, Vanacore N, van Swieten JC, … Heutink P (2003). DJ-1( PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurological Sciences: Official Journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology, 24(3), 159–160 [DOI] [PubMed] [Google Scholar]
- Burchell VS, Nelson DE, Sanchez-Martinez A, Delgado-Camprubi M, Ivatt RM, Pogson JH, … Plun-Favreau H (2013). The Parkinson’s disease–linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci, 16(9), 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, … Graham. (2017). A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet, 49(10), 1511–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, … Destée A(2004). Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet, 364(9440), 1167–1169 [DOI] [PubMed] [Google Scholar]
- Chuang RS, Gitler AD (2013) Parallel PARKing: Parkinson’s genes function in common pathway. Neuron 77(3):377–379 [DOI] [PubMed] [Google Scholar]
- Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, … Kellis M (2015). FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med, 373(10), 895–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA (2006) Synuclein blocks ER-golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313(5785):324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau LML, Breteler MMB (2006) Epidemiology of Parkinson’s disease. Lancet Neurol 5(6):525–535 [DOI] [PubMed] [Google Scholar]
- Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, … Bezard E (2013). Lysosomal impairment in Parkinson’s disease. Movement Disorders: Official Journal of the Movement Disorder Society, 28(6), 725–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fonzo A, Wu-Chou YH, Lu CS, van Doeselaar M, Simons EJ, Rohé CF, … Bonifati V (2006). A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson’s disease risk in Taiwan. Neurogenetics, 7(3), 133–138 [DOI] [PubMed] [Google Scholar]
- Duran R, Mencacci NE, Angeli AV, Shoai M, Deas E, Houlden H, … Foltynie T (2013). The glucocerobrosidase E326K variant predisposes to Parkinson’s disease, but does not cause Gaucher’s disease. Mov Disord, 28(2), 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvoisin RC, Eldridge R, Williams A, Nutt J, Calne D (1981) Twin study of Parkinson disease. Neurology 31(1):77–80 [DOI] [PubMed] [Google Scholar]
- Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, … Martin ER (2010). Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet, 74(2), 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge R, Ince SE (1984) The low concordance rate for Parkinson’s disease in twins: a possible explanation. Neurology 34(10): 1354–1356 [DOI] [PubMed] [Google Scholar]
- Evans W, Fung HC, Steele J, Eerola J, Tienari P, Pittman A, … Hardy J (2004). The tau H2 haplotype is almost exclusively Caucasian in origin. Neurosci Lett, 369(3), 183–185 [DOI] [PubMed] [Google Scholar]
- Farrer MJ, Stone JT, Lin CH, Dachsel JC, Hulihan MM, Haugarvoll K, … Wu RM (2007). Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinsonism Relat Disord, 13(2), 89–92 [DOI] [PubMed] [Google Scholar]
- García-Sanz P, Orgaz L, Bueno-Gil G, Espadas I, Rodriguez-Traver E, Kulisevsky J et al. (2017) N370S-GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson’s disease. Mov Disord 32(10):1409–1422 [DOI] [PubMed] [Google Scholar]
- Gegg ME, Burke D, Heales SJR, Cooper JM, Hardy J, Wood NW, Schapira AHV (2012) Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann Neurol 72(3):455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CM, Cheng Q, Ortuno D, Huang M, Nojima D, Kassner PD, … Carlisle HJ (2016). Identification of modulators of autophagic flux in an image-based high content siRNA screen. Autophagy, 12(4), 713–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halperin A, Elstein D, Zimran A (2006) Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol Dis 36(3):426–428 [DOI] [PubMed] [Google Scholar]
- Healy DG, Wood NW, Schapira AHV (2008) Test for LRRK2 mutations in patients with Parkinson’s disease. Pract Neurol 8(6):381–385 [DOI] [PubMed] [Google Scholar]
- Hernandez DG, Nalls MA, Ylikotila P, Keller M, Hardy JA, Majamaa K, Singleton AB (2012) Genome wide assessment of young onset Parkinson’s disease from Finland. PLoS One 7(7):e41859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez DG, Reed X, Singleton AB (2016) Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem 139(Suppl 1):59–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, … Heutink P (1998). Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature, 393(6686), 702–705 [DOI] [PubMed] [Google Scholar]
- International Parkinson Disease Genomics Consortium, Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, … Wood NW (2011). Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet, 377(9766), 641–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Heutink P (2010) From single genes to gene networks: high-throughput-high-content screening for neurological disease. Neuron 68(2):207–217 [DOI] [PubMed] [Google Scholar]
- Kalia LV, Lang AE (2015) Parkinson’s disease. Lancet 386(9996):896–912 [DOI] [PubMed] [Google Scholar]
- Kannarkat GT, Cook DA, Lee JK, Chang J, Chung J, Sandy E, … Tansey MG (2015). Common genetic variant association with altered HLA expression, synergy with pyrethroid exposure, and risk for Parkinson’s disease: an observational and case-control study. Npj Parkinson’s Disease, 1(1). 10.1038/npjparkd.2015.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller MF, Saad M, Bras J, Bettella F, Nicolaou N, Simón-Sánchez J, … Wellcome Trust Case Control Consortium 2 (WTCCC2). (2012). Using genome-wide complex trait analysis to quantify “missing heritability” in Parkinson’s disease. Hum Mol Genet, 21(22), 4996–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, … Shimizu N (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature, 392(6676), 605–608 [DOI] [PubMed] [Google Scholar]
- Klemann CJHM, Martens GJM, Sharma M Martens MB, Isacson O, Gasser T, … Poelmans G (2017). Integrated molecular landscape of Parkinson’s disease. NPJ Parkinson’s Disease. 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston JW, Ballard PA Jr (1983) Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N Engl J Med 309(5):310. [DOI] [PubMed] [Google Scholar]
- Lesage S, Leutenegger AL, Ibanez P, Janin S, Lohmann E, Dürr A., … French Parkinson’s Disease Genetics Study Group. (2005). LRRK2 haplotype analyses in European and North African families with Parkinson disease: a common founder for the G2019S mutation dating from the 13th century. Am J Hum Genet, 77(2), 330–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, … French Parkinson’s Disease Genetics Study Group. (2006). LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med, 354(4), 422–423 [DOI] [PubMed] [Google Scholar]
- Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BMM, Bertram L (2012). Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: the PDGene database. PLoS Genet, 8(3), e1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill CM, Hansen J, Olsen JH, Binder H, Ritz B, Bertram L (2015) Impact of Parkinson’s disease risk loci on age at onset. Mov Disord 30(6): 847–850 [DOI] [PubMed] [Google Scholar]
- Liu G, Boot B, Locascio JJ, Jansen IE, Winder-Rhodes S, Eberly S, … for the International Genetics of Parkinson Disease Progression (IGPP) Consortium. (2016). Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol, 80(5), 674–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD et al. (2013) RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77(3):425–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Krüger R, … Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium. (2006). Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA, 296(6), 661–670 [DOI] [PubMed] [Google Scholar]
- McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, … Schapira AHV (2014). Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain J Neurol, 137(Pt 5), 1481–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui J, Mizuta I, Toyoda A, Ashida R, Takahashi Y, Goto J, … Tsuji S (2009). Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol, 66(5), 571–576 [DOI] [PubMed] [Google Scholar]
- Moors T, Paciotti S, Chiasserini D, Calabresi P, Parnetti L, Beccari T, van de Berg WDJ (2016) Lysosomal dysfunction and α-synuclein aggregation in Parkinson’s disease: diagnostic links. Mov Disord 31(6):791–801 [DOI] [PubMed] [Google Scholar]
- Mullin S, Schapira AHV (2015) Pathogenic mechanisms of neurodegeneration in Parkinson disease. Neurol Clin 33(1):1–17 [DOI] [PubMed] [Google Scholar]
- Nagle MW, Latourelle JC, Labadorf A, Dumitriu A, Hadzi TC, Beach TG, Myers RH (2016) The 4p16.3 Parkinson Disease Risk Locus Is Associated with GAK Expression and Genes Involved with the Synaptic Vesicle Membrane. PloS One 11(8):e0160925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, … Singleton AB (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet, 46(9), 989–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Escott-Price V, Williams NM, Lubbe S, Keller MF, Morris HR,… on behalf of the International Parkinson’s Disease Genomics Consortium (IPDGC). (2015a). Genetic risk and age in Parkinson’s disease: continuum not stratum. Movement Disorders: Official Journal of the Movement Disorder Society, 30(6), 850–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, McLean CY, Rick J, Eberly S, Hutten SJ, Gwinn K, … Parkinson’s Disease Biomarkers Program and Parkinson’s Progression Marker Initiative investigators. (2015b). Diagnosis of Parkinson’s disease on the basis of clinical and genetic classification: a population-based modelling study. Lancet Neurol, 14(10), 1002–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, … Zimran A (1996). Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM, 89(9), 691–694 [DOI] [PubMed] [Google Scholar]
- Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M et al. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44(4):595–600 [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Gandal MJ, Geschwind DH (2015) Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Genetics 16(8):441–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J-S, Blair NF, Sue CM (2015) The role of ATP13A2 in Parkinson’s disease: clinical phenotypes and molecular mechanisms. Mov Disord 30(6):770–779 [DOI] [PubMed] [Google Scholar]
- Pihlstrøm L, Axelsson G, Bjørnarå KA, Dizdar N, Fardell C, Forsgren L, … Toft M (2013). Supportive evidence for 11 loci from genomewide association studies in Parkinson’s disease. Neurobiology of Aging, 34(6), 1708.e7–13 [DOI] [PubMed] [Google Scholar]
- Pittman AM, Myers AJ, Abou-Sleiman P, Fung HC, Kaleem M, Marlowe L, … de Silva R (2005). Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet, 42(11), 837–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, … Nussbaum RL (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science, 276(5321), 2045–2047 [DOI] [PubMed] [Google Scholar]
- Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, … Shulman (2017). Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain: J Neurol, 140(12), 3191–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes SL, Sinsheimer JS, Bordelon Y, Bronstein JM, Ritz B (2011) Replication of GWAS associations for GAK and MAPT in Parkinson’s disease. Ann Hum Genet 75(2):195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross OA, Soto-Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G, … Genetic Epidemiology Of Parkinson’s Disease (GEO-PD) Consortium. (2011). Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: a case-control study. Lancet Neurol, 10(10), 898–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, … Toda T (2009). Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet, 41(12), 1303–1307 [DOI] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, … Ziegler SG (2009). Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med, 361(17), 1651–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, … Gasser T (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet, 41(12), 1308–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB (2003) Synuclein locus triplication causes Parkinson’s disease. Science 302(5646):841–841 [DOI] [PubMed] [Google Scholar]
- Singleton A, Hardy J (2016) The evolution of genetics: Alzheimer’s and Parkinson’s diseases. Neuron 90(6):1154–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skipper L, Wilkes K, Toft M, Baker M, Lincoln S, Hulihan M, … Farrer M (2004). Linkage disequilibrium and association of MAPT H1 in Parkinson disease. Am J Hum Genet, 75(4), 669–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gómez-Marín C, … Nóbrega MA (2014). Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature, 507(7492), 371–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatola M, Wider C (2014) Genetics of Parkinson’s disease: the yield. Parkinsonism Relat Disord 20(Suppl 1):S35–S38 [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Aloe L, Alleva E, De Simone R, Goedert M, and Levi-Montalcini R (1997). Nerve growth factor mRNA and protein increase in hypothalamus in a mouse model of aggression. In World Scientific Series in 20th Century Biology (pp. 374–378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J et al. (2017) T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 546(7660):656–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Obeso JA, Halliday GM (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18(2): 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan EK, Schapira AH (2008) Uniting Chinese across Asia: the LRRK2 Gly2385Arg risk variant. Eur J Neurol 15(3):203–204 [DOI] [PubMed] [Google Scholar]
- Tan E-K, Zhao Y, Tan L, Lim H-Q, Lee J, Yuen Y et al. (2007) Analysis of LRRK2 Gly2385Arg genetic variant in non-Chinese Asians. Mov Disord 22(12):1816–1818 [DOI] [PubMed] [Google Scholar]
- UK Parkinson’s Disease Consortium, Wellcome Trust Case Control Consortium 2, Spencer CCA, Plagnol V, Strange A, Gardner M et al. (2011) Dissection of the genetics of Parkinson’s disease identifies an additional association 5’ of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet 20(2):345–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, … Wood NW (2001). Localization of a novel locus forautosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet, 68(4), 895–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vekrellis K, Xilouri M, Emmanouilidou E, Rideout HJ, Stefanis L (2011) Pathological roles of α-synuclein in neurological disorders. Lancet Neurol 10(11):1015–1025 [DOI] [PubMed] [Google Scholar]
- Ward CD, Duvoisin RC, Ince SE, Nutt JD, Eldridge R, Calne DB (1983) Parkinson’s disease in 65 pairs of twins and in a set of quadruplets. Neurology 33(7):815–824 [DOI] [PubMed] [Google Scholar]
- Wong YC, Krainc D (2016) Lysosomal trafficking defects link Parkinson’s disease with Gaucher’s disease. Mov Disord 31(11): 1610–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zech M, Nübling G, Castrop F, Jochim A, Schulte EC, Mollenhauer B, Winkelmann J (2013). Niemann-pick C disease gene mutations and age-related neurodegenerative disorders. PLoS One, 8(12), e82879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Stumpo DJ, Graves JP, DeGraff LM, Grissom SF, Collins JB, … Blackshear PJ (2006). Identification of potential target genes for RFX4_v3, a transcription factor critical for brain development. J Neurochem, 98(3), 860–875 [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, … Gasser T (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology Neuron, 44(4), 601–607 [DOI] [PubMed] [Google Scholar]