

Abstract
De novo design of protein nano‐cages has potential applications in medicine, synthetic biology, and materials science. We recently developed a modular, symmetry‐based strategy for protein assembly in which short, coiled‐coil sequences mediate the assembly of a protein building block into a cage. The geometry of the cage is specified by the combination of rotational symmetries associated with the coiled‐coil and protein building block. We have used this approach to design well‐defined octahedral and tetrahedral cages. Here, we show that the cages can be further elaborated and functionalized by the addition of another protein domain to the free end of the coiled‐coil: in this case by fusing maltose‐binding protein to an octahedral protein cage to produce a structure with a designed molecular weight of ~1.8 MDa. Importantly, the addition of the maltose binding protein domain dramatically improved the efficiency of assembly, resulting in ~ 60‐fold greater yield of purified protein compared to the original cage design. This study shows the potential of using small, coiled‐coil motifs as off‐the‐shelf components to design MDa‐sized protein cages to which additional structural or functional elements can be added in a modular manner.
Keywords: protein nano‐cages, protein design, computational modeling, coiled coils
Introduction
Many proteins naturally assemble into well‐defined, higher‐order oligomeric structures that exhibit a high degree of symmetry. The assembly process is often essential to the protein's biological function. Examples include actin and tubulin fibers,1, 2 viral capsids,3 pyruvate dehydrogenase complex (PDH),4 ferritin,5 and the bacterial carboxysome.6 The ability of proteins to assemble into such diverse higher‐order architectures has made them attractive systems for designing biological nano‐materials with applications in medicine, synthetic biology, and materials science.7, 8, 9, 10, 11, 12, 13, 14
Yeates et al.15 were among the first to realize the potential of symmetry‐based methods to design protein cages. They recognized that protein assemblies with many different geometries could, in principle, be specified by precisely controlling the angle between two rotational symmetry axes (typically specified by the quaternary structure of the protein). The development of computational approaches utilizing the Rosetta suite of programs to design new, rigid protein–protein interfaces into existing oligomeric proteins subsequently allowed the requisite pairs of protein rotational symmetry axes to be oriented with the precision necessary to implement this strategy. This has resulted in some impressive successes, including the assembly of well‐defined protein cages with tetrahedral, octahedral, dodecahedral, and icosahedral symmetries, the largest of which rival some viral capsids in size.15, 16, 17, 18, 19, 20, 21
Our approach to designing protein cages differs from that described above in that we relax the requirement for the symmetry axes to be explicitly oriented at precise angles. Our design strategy utilizes small coiled‐coil domains as modular, off‐the‐shelf assembly domains that are fused to a homo‐oligomeric building block protein through a short, flexible linker sequence (Fig. 1). The geometry of the protein cage is, thus, primarily specified by the rotational symmetries of the coiled‐coil and building block protein. This flexible approach to protein assembly produces cages that are more conformationally labile than those assembled using designed protein interfaces, but has the advantage that it does not require extensive computational modeling. A further feature of our design strategy is the potential to elaborate protein cages in a modular fashion by the addition of further protein domains to the free end of the coiled‐coil domain [Fig. 1(D)]. We note that coiled coils have also been used to assemble a tetrahedral cage from a single protein chain encoding 12 concatenated coiled‐coil‐forming segments separated by flexible peptide hinges.22 In this rather different approach, the coiled coils form the edges of the polyhedron, rather than the vertices as here.
Figure 1.

Outline of the strategy used to construct octahedral cages. (A) and (B) display linear maps of the protein sequences of Oct‐4 and Oct‐MBP, respectively. (C) and (D) show cartoons illustrating the coiled‐coil‐mediated oligomerization of Oct‐4 and Oct‐MBP, respectively, to form octahedral protein cages.
Using this coiled‐coil‐mediated design approach, we successfully assembled octahedral (Oct‐4) and tetrahedral (Tet8‐5H) protein cages by linking a C3‐symmetric esterase (Tri‐EST; PDB ID: http://firstglance.jmol.org/fg.htm?mol=1ZOI) to C4‐symmetric and C3‐symmetric coiled‐coils, respectively.23, 24 Whereas the tetrahedral cages could be purified in good yields (~20 mg/L of culture), the octahedral cages could only be purified in low yields (~1 mg/L of culture). The octahedral cages bound poorly to the Ni‐NTA affinity column used to purify them, and a significant amount of the protein formed inclusion bodies. Furthermore, although cryo‐EM reconstruction of the tetrahedral cage resolved the coiled‐coil domains, showing them as protruding from the surface of the cage, we were unable to resolve these domains in a similar reconstruction of the octahedral cage. This suggested that in the octahedral design there was heterogeneity in the orientation of the coiled‐coil domains. We reasoned that we could take advantage of our design strategy to elaborate the octahedral cage by attaching an additional large protein domain to the C‐terminal ends of the coiled‐coil domains. This would force the large protein to point outwards, potentially reducing the formation of inclusion bodies and improving the homogeneity of the resulting elaborated octahedral cage.
To test this reasoning, maltose binding protein (MBP), a ~40‐kDa, monomeric protein commonly used to help solubilize and affinity purify proteins, was appended to the C‐terminal end of the coiled‐coil of our previous Oct‐4 octahedral cage construct.23 The resulting complex protein assembly, Oct‐MBP (Fig. 1), retained an octahedral structure, was more homogenous than the original Oct‐4 protein cage, and could be purified in ~60‐fold greater yield.
Results
Construction of the elaborated octahedral protein cage, Oct‐MBP
MBP was genetically fused to the C‐terminus of Oct‐4 through a 13‐residue spacer sequence (GGENLYFQGGGGG) that incorporates a TEV protease cleavage site, which potentially allows the MBP domain to be removed from the cage, and a series of glycine residues designed to impart flexibility. This design was designated Oct‐MBP (the full sequence is provide in Fig. S1). The choice of this relatively long spacer sequence was informed by our previous studies, which have shown that coiled‐coil oligomerization states can be affected, leading to aggregation, by the proteins attached to them if the spacer is too short.25
Oct‐MBP was over‐expressed in Escherichia coli and purified to homogeneity, as judged by SDS‐PAGE, through MBP‐affinity chromatography utilizing a maltose column [Fig. 2(B)]. The yield of purified protein (~60 mg/L culture) represented a dramatic improvement over the yields obtained with the original Oct‐4 design. We were unable to purify the Oct‐MBP protein using its N‐terminal His‐tag, suggesting that the N‐terminus is buried within the protein cage. The esterase activity of Oct‐MBP, measured as turn‐over number with 2,4‐dinitrophenol acetate as substrate, was 12.4 ±0.7 min−1, which is very similar to the activity of the unassembled trimeric esterase (14.2 ±2.3 min−1). This indicates that the assembly process did not result in any gross structural perturbation of the protein building blocks.
Figure 2.

Initial characterization of Oct‐MBP. (A) Size exclusion chromatographs (Superose 6 300/10 column) of, top to bottom: Oct‐MBP, Oct‐4, TEV protease‐cleaved OCT‐MBP and Tri‐EST. (B) SDS‐PAGE analysis of Oct‐MBP. Lane 1: marker proteins; Lane 2: purified Oct‐MBP; Lane 3: cross‐linked and TEV‐cleaved Oct‐MBP. For details see the text. (C) Native PAGE analysis of Oct‐MBP and Oct‐4. Lane 1 Oct‐4; Lane 2: Oct‐MBP; Lane 3: Oct‐MBP after cross‐linking and TEV protease cleavage. For details, see the text.
Characterization of the elaborated octahedral cage
To examine whether the construct assembled into the intended octahedral cage geometry, we characterized the complex using size exclusion chromatography (SEC), native PAGE, sedimentation velocity analytical ultracentrifugation (SV‐AUC), negative‐stain transmission electron microscopy (TEM), and dynamic light scattering (DLS). These techniques provide complementary information on the size and shape of protein complexes.
Size exclusion chromatography
We initially used SEC to examine whether Oct‐MBP assembled into a high molecular weight complex. When subjected to FPLC chromatography on a Superose 6 10/300 SEC column, (molecular cutoff ~5 × 106 Da; void volume ~8 mL), purified Oct‐MBP eluted a single, symmetrical peak, with an approximate elution volume of 10 mL [Fig. 2(A)]. For comparison, Oct‐4 (~854 kDa), the unelaborated octahedral protein cage, eluted at ~12 mL, Tet8‐5H (~439 kDa), a tetrahedral protein cage, eluted at ~14 mL and the unassembled trimeric protein building block (Tri‐EST, ~97‐kDa) eluted at 18 mL.23, 24 These indicated that, as expected, Oct‐MBP assembles into a complex that is significantly larger than Oct‐4, from which it is derived.
Native PAGE
The approximate size and homogeneity of Oct‐MBP was further analyzed by native PAGE on a 3–12% gradient gel. Oct‐MBP migrated as a smeared band that ran significantly more slowly than Oct‐4 [Fig. 2(C)]. The observed smearing may result from interactions of the large protein complex with the gel matrix, or possibly the presence incorrectly assembled material. The electrophoretic behavior of Oct‐MBP is also consistent with Oct‐MBP forming fairly homogenous cages that are significantly larger than Oct‐4.
Dynamic light scattering
The size of the particles formed by Oct‐MPB was further investigated by dynamic light scattering. These revealed the particles formed by Oct‐MBP to be homogeneous and, intensity distribution analysis gave a hydrodynamic diameter of 28.1 ± 0.8 nm (Fig. 3). This value is in good agreement with diameter of ~24 nm for Oct‐MBP, which was simply estimated by modeling the structure of MBP on to the vertices of the Oct‐4 protein cage.
Figure 3.
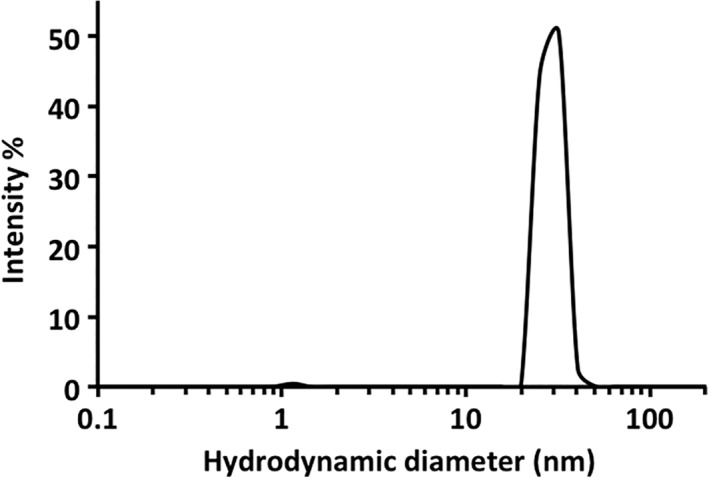
Characterization of Oct‐MBP using dynamic light‐scattering analysis of Oct‐MBP. For details see the text.
Sedimentation‐velocity analytical ultracentrifugation
Analytical ultracentrifugation is a powerful analytical tool with which to characterize protein species in solution, as, in favorable cases, it provides information on both shape and molecular weight. Sedimentation traces for Oct‐MBP were recorded at 22,500 rpm and 6°C on samples with initial protein concentrations ranging between 0.014 and 0.07 mg/mL. The sedimentation velocity experiments were first analyzed by the enhanced van‐Holde Weischet method26, 27 to assess the homogeneity of the sample [Fig. 4(A)]. At higher Oct‐MBP concentrations, the observed sedimentation coefficient, s, decreased, which indicates non‐ideal behavior. This non‐ideal behavior likely arises from non‐specific interactions between protein cages at higher concentrations, a phenomenon that is generally more pronounced for larger macromolecules. At lower concentrations, the samples were characterized by a major component with a sedimentation coefficient (s) of ~23 S and another less abundant component comprising faster sedimenting species.
Figure 4.
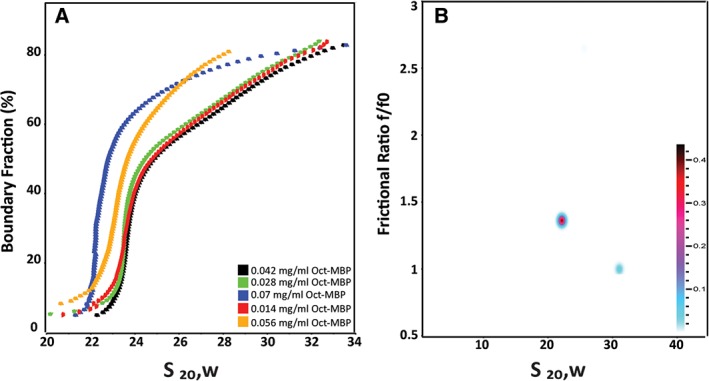
Characterization of Oct‐MBP using sedimentation velocity‐analytical ultracentrifugation: (A) enhanced van Holde–Weischet analysis of Oct‐MBP sedimenting at a range of initial concentrations, as indicated on plot. (B) Representative plot obtained by two‐dimensional sedimentation analysis of Oct‐MBP initial protein concentration 0.028 mg/mL.
We further analyzed the data from the lower concentration samples by two‐dimensional sedimentation spectrum analysis (2DSA) followed by a genetic algorithm analysis, which was validated by a Monte Carlo analysis (GA‐MC). This is a model‐independent analysis approach to fit sedimentation boundaries that allows the shape and molecular mass distribution of macromolecular mixtures to be determined.28 We have previously used this methodology to characterize smaller protein cages and determine their molecular weights.23, 24, 29 In this case, 2DSA analysis of Oct‐MBP traces recorded at initial concentrations ranging between 0.014 and 0.042 mg/mL consistently resolved two sedimenting species [Fig. 4(B)]. The principle component (comprising ~75% of the sample) had s = 23 ± 1 S, consistent with the van‐Holde Weischet analysis, and the minor component had s = 30 ± 1 S. The frictional ratio (f/f 0) of the major 23 S species was 1.7 ±0.04 for the major component, although this varied somewhat dependent upon the sample preparation and concentration. The minor 30 S species has a surprisingly small frictional ratio, f/f 0 = 1.005 ±0.03, that might indicate that this is a more compact cage conformation or aggregated form of the protein. Overall, the AUC analyses indicate that Oct‐MBP is predominantly a single species that appears significantly larger than Oct‐4 for which s = 17.5 S. The range of frictional ratios associated with major Oct‐MBP species falls within the range reported for other porous protein cages such as ferritin, f/f 0 = 1.3,30 the E2 complex of pyruvate dehydrogenase, f/f 0 = 2.531 and Oct‐4, f/f 0 = 1.9,23 suggesting that it too maintains an open, porous structure.
Electron microscopy
The assemblies formed by Oct‐MBP were imaged by negative‐stain TEM [Fig. 5(A) and (B)]. The protein cages appeared as discrete particles with a fairly uniform size distribution. Analysis of ~300 particle images using the program Image J gave average diameter of 23.4 ± 2.9 nm (Fig. S2) and which closely matches with the modeled diameter of ~24 nm. Comparison of the images with those obtained from Oct‐4 showed that they appeared less well defined and in particular, unlike the parent Oct‐4 design, no symmetry was apparent in the particle images. This is most likely a result of the appended monomeric MBP domains masking the core octahedral cage. In most images, poorly defined peripheral structures were discernable surrounding the core particle that may represent individual MBP domains.
Figure 5.
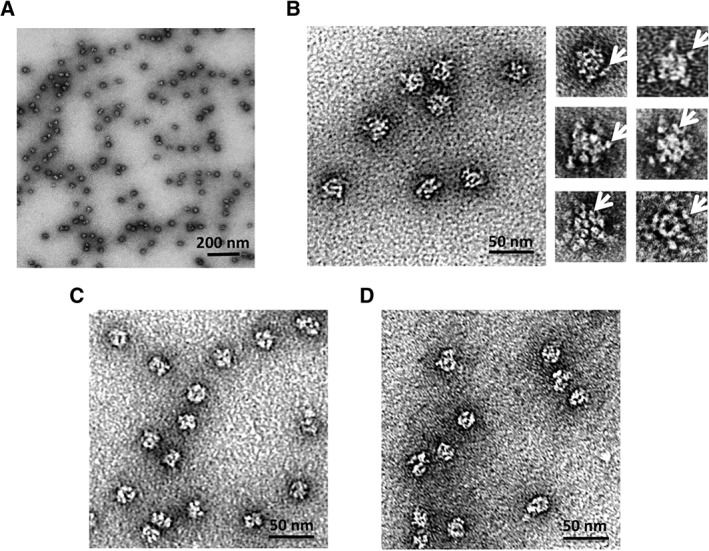
Negative‐stain transmission electron microscopy images of Oct‐MBP. (A) Wide field view of particles formed by Oct‐MBP (B) left: Oct‐MBP particles at higher magnification exhibit a less compact structure that Oct‐4 (compare with Panel C) right: individual particles at high magnification; arrows indicate peripheral structures that likely represent MBP domains. (C) Images of Oct‐4 particles lack peripheral structures (compare with Panel B) (D) images of Oct‐MBP after removal of MBP domains by treatment with TEV protease. These particles more closely resemble the compact structures formed by Oct‐4 shown in Panel C.
Cleavage of the MBP domain
To further establish the multidomain nature of Oct‐MBP, we used tobacco etch virus protease (TEV) to cleave the maltose‐binding domain from the octahedral core. Initial attempts at proteolysis resulted in the dissociation of Oct‐MBP to its component trimers (data not shown). The reason for this is unclear. Therefore, we first covalently cross‐linked the Oct‐MBP subunits using the lysine‐reactive cross linker, bis(sulfosuccinimidyl)suberate (BS3). The protein was then incubated with TEV protease overnight and the material re‐purified by SEC. The cross‐linking preserved the structure of the cages and the TEV‐cleaved material chromatographed with an elution volume similar to Oct‐4 [Fig. 2(A)]. Analysis by native PAGE showed that most of the TEV‐cleaved material migrated similarly to Oct‐4, although some higher molecular weight bands are evident [Fig. 2(C)]. These may represent cages from which the MBP domain has been incompletely cleaved or cages in which the MBP domain was covalently cross‐linked to the Oct core. Examination of the cleaved cages by TEM showed that the particles have a similar appearance and size to Oct‐4, consistent with the removal of the outer MBP domain [Fig. 4(C) and (D)]. The cross‐linked and TEV‐cleaved cages retained their catalytic activity (14.8 ±1.8 min−1), indicating that the structure of the esterase building block was not significantly altered by these manipulations.
Discussion
Growing interest in the use of protein nanoparticles for applications in medicine and materials science has stimulated efforts to design new protein cages from a range of protein building blocks.7, 8, 9, 10, 11, 12, 13, 14 Our efforts have focused on the use of de novo‐designed coiled coils as “off‐the‐shelf” components to assemble proteins into cages.23, 24, 29, 32 In this study, we demonstrated the potential of our design strategy for the modular expansion of a protein cage by fusing a relatively large monomeric protein, MBP, to the free end of the coiled‐coil domain used to mediate cage assembly. The resulting construct self‐assembled into a fairly homogeneous protein cage in which MBP was displayed on the exterior. The addition of the MBP domain both improved protein folding and allowed the cage to be easily purified by affinity chromatography. Furthermore, it was possible to remove the MBP domain by TEV protease cleavage and recover the core octahedral protein cage.
A valuable consequence of appending MBP to the octahedral cage was to increase the purified yield of protein by ~60‐fold. This is important for real‐world applications where proteins need to be produced in high yields. The purified yield of our initial Oct‐4 design was only about 1 mg/L culture, both because a significant fraction of the protein was produced as inclusion bodies, presumably because it was misfolded, and because the His‐tag‐based affinity chromatography used during its purification was inefficient. We suspect that this inefficiency arose because the N‐terminus of the trimeric esterase, which contained the His‐tag, was not exposed to the exterior of the cage, so it could not bind to the Ni‐NTA affinity column material. Thus, elaborating the original octahedral cage with MBP domains has provided three benefits over the original design. First, it allowed an alternative method of affinity purification that resulted in increased efficiency. Second, it forced the coiled‐coil domains to point outwards from the octahedral cage (because there is insufficient space within the cage to accommodate the MBP domain) thereby improving homogeneity. Third, it improved folding and assembly, thereby largely eliminating the formation of inclusion bodies and improving the yield of protein.
The successful elaboration of Oct‐MBP supports the idea that the coiled‐coil‐mediated assembly of protein superstructures based primarily on symmetry considerations is a generalizable and robust strategy. By appending a large protein domain at each end of the small, coiled‐coil domain (~3 kDa) we have constructed discrete cage‐like assemblies that extend to the mega‐Dalton size range. The domain fusion strategy that we demonstrate here potentially allows the cages to be elaborated toward various applications such as the polyvalent display of antigens for vaccine development or the construction of artificial multi‐enzyme complexes. More generally, this design strategy provides a conceptually simple route to design complex, higher order protein assemblies that is largely independent of the structure of the proteins and does not require extensive computational modeling and protein re‐design.
Materials and Methods
Construction of genes encoding fusion proteins
The MBP gene was PCR amplified from pMAL‐c5X using commercially synthesized primers. The 5′ end of the forward primer was designed to overlap with the sequence encoding the C‐terminus of the coiled‐coil and 13‐residue linker in the middle. The reverse primer incorporated two stop codons and overlapped the T7 terminator region of pET‐28B at its 5′ end. The codon‐optimized gene encoding the tetrameric coiled‐coil and the Gly spacer units was commercially synthesized. Both the tetrameric coiled‐coil and the MBP construct were introduced into the expression vector pET‐28B by Gibson assembly using the NEB Gibson assembly protocol. The complete sequence of the Oct‐MBP design is shown in Table S1.
Protein expression and purification
Proteins were expressed in E. coli BL21 by standard methods, as previously described,22 and cell pellets were stored at −80°C. All purification steps were performed at 4°C and all the buffers used were at pH 7.5. Cell pellets (~8 g) were thawed on ice for 20–30 min, then resuspended in buffer containing 50 mM HEPES, 100 mM ammonium acetate, 5% glycerol, 1 FAST protease inhibitor tablet (Sigma), and 50 mg of hen egg lysozyme (Sigma). This suspension was kept on ice and gently agitated for 20 min. Cells were lysed by sonication. The lysate was clarified by centrifugation at 40,000g for 30 min and filtered through a 0.22 μm filter. The supernatant was loaded on to a 5 mL MBP‐trap column (GE Healthcare) at a flow rate of 0.5 mL/min. The column was subsequently washed with 60–80 mL of the same buffer at a flow rate of 1 mL/min and then eluted with buffer containing 10 mM maltose, 50 mM HEPES, 100 mM ammonium acetate, and 5% glycerol. Fractions containing the desired protein (approximately 15 mL) were combined and treated with benzonase (1 μL). The resulting solution was dialyzed against buffer containing 20 mM HEPES, 100 mM ammonium acetate, 2 mM EDTA, and 10% glycerol for 2 days.
Size exclusion chromatography
SEC was used as an additional purification step and also an analytical step. SEC was performed on Superose 6 300/10 column equilibrated with buffer containing 20 mM HEPES, 100 mM ammonium acetate, and 2 mM EDTA at 4°C. Injections comprised 400 μL of sample at 1 mg/mL. Fractions with desired oligomeric state were pooled and kept in the column elution buffer for further characterization using the techniques described below. If needed, protein was concentrated using 100‐kDa Amicon ultra‐centrifugal filter units.
Analytical ultracentrifugation
Sedimentation velocity analysis was performed using a Beckman Proteome Lab XL‐I analytical ultracentrifuge (Beckman Coulter, Indianapolis, IN) equipped with an AN60TI rotor, as previously described. For AUC, samples were prepared by buffer exchanging into PBS, 100 mM NaCl, pH 7.4 by SEC on a Superose 6 10/300 FPLC column equilibrated with the above buffer. Proteins were sedimented at 22,500 rpm at 6°C. Absorbance data were collected at a wavelength of 220 nm.
Negative‐stain TEM imaging
Protein samples were adjusted to a concentration 0.04 mg/mL and fixed on Formvar/Carbon 400 Mesh, Cu grids using conventional negative staining procedures. Imaging was performed using a JEOL 1500 electron microscope equipped with tungsten filament, XR401 high sensitivity sCMOS camera and operated at 90 kV.
Activity assays
Catalytic activity for Tri‐EST, Oct‐MBP, and cross‐linked and TEV‐cleaved OCT‐MBP was assessed as previously described23 (1,2) using 1 mM 2,4‐dinitrophenyl acetate (2,4‐DNPA) as the substrate at 45°C. Changes in absorbance were measured at 405 nm.
Dynamic light scattering
DLS was performed using a DynaPro NanoStar ZS instrument using a standard 90° scattering geometry. The samples were centrifuged to remove any suspended particles before being analyzed in 10 μL cuvettes at 25°C. The refractive index and absorption coefficient for the particles were set at 1.45 and 0.001, respectively. Runs were performed in triplicate and each run is an average of 15 scans. At the concentration, ranges used non‐ideality effects were assumed to be negligible.
Protein crosslinking and TEV cleavage
The Oct‐MBP protein cages were cross‐linked using bis(sulfosuccinimidyl)suberate (BS3). BS3 concentrations were varied between 1 mM and 2 mM whereas the protein concentrations were varied between 5 and 10 μM. The reaction was performed in dialysis buffer at pH 7.5 and the sample was kept at 4°C for 3–4 days to ensure that the cross‐linker is fully destabilized. Approximately 0.5 μM TEV protease was added and incubated over night at 4°C. The cleaved material was purified by SEC and the efficiency of cleavage was assessed by SDS‐PAGE.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Supporting information
Figure S1 Protein sequence of Oct‐MBP: trimeric esterase domain (PDB ID: http://firstglance.jmol.org/fg.htm?mol=1ZOI) is shown in black; spacer sequence 1 in red; tetrameric coiled‐coil domain (PDB ID: http://firstglance.jmol.org/fg.htm?mol=3R4A) in blue; spacer sequence 2 in green; maltose binding protein domain (PDB ID: http://firstglance.jmol.org/fg.htm?mol=1ANF) I orange.
Figure S2 Size distribution analysis of protein cages formed by Oct‐MBP obtained from measurement of ~ 300 particles imaged by negative stain TEM using the software package Image J.
Acknowledgments
This research was supported in part by a grant from the Army Research Office ARO W911NF‐16‐1‐0147 to E. N. G. M. Electron microscopy was performed at the Microscope Imaging Laboratory (MIL) core facility at University of Michigan. Dynamic Light Scattering was performed at Biophysics Research Facility at University of Michigan.
References
- 1. Dominguez R, Holmes KC (2011) Actin structure and function. Ann Rev Biophys 40:169–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abal M, Andreu JM, Barasoain I (2003) Taxanes: microtubule and centrosome targets, and cell cycle dependent mechanisms of action. Curr Cancer Drug Targets 3:193–203. [DOI] [PubMed] [Google Scholar]
- 3. Caspar DLD, Klug A (1962) Physical principles in the construction of regular viruses. Cold Spring Harb Symp Quant Biol 27:1–24. [DOI] [PubMed] [Google Scholar]
- 4. Wieland OH (1983) The mammalian pyruvate dehydrogenase complex: structure and regulation. Rev Physiol Biochem Pharmacol 96:123–170. [DOI] [PubMed] [Google Scholar]
- 5. Theil EC (1987) Ferritin: structure, gene regulation, and cellular function in animals, plants, and microorganisms. Ann Rev Biochem 56:289–315. [DOI] [PubMed] [Google Scholar]
- 6. Yeates TO, Kerfeld CA, Heinhorst S, Cannon GC, Shively JM (2008) Protein‐based organelles in bacteria: carboxysomes and related microcompartments. Nat Rev Microbiol 6:681–691. [DOI] [PubMed] [Google Scholar]
- 7. Cohen B, Dafni H, Meir G, Harmelin A, Neeman M (2005) Ferritin as an endogenous MRI reporter for noninvasive imaging of gene expression in C6 glioma tumors. Neoplasia 7:109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klem MT, Willits D, Solis DJ, Belcher AM, Young M, Douglas T (2005) Bio‐inspired synthesis of protein‐encapsulated CoPt nanoparticles. Adv Funct Mater 15:1489–1494. [Google Scholar]
- 9. Kaba SA, Brando C, Guo Q, Mittelholzer C, Raman S, Tropel D, Aebi U, Burkhard P, Lanar DE (2009) A nonadjuvanted polypeptide nanoparticle vaccine confers long‐lasting protection against rodent malaria. J Immunol 183:7268–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yildiz I, Shukla S, Steinmetz NF (2011) Applications of viral nanoparticles in medicine. Curr Opin Biotechnol 22:901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamada K, Yoshii S, Kumagai S, Miura A, Uraoka Y, Fuyuki T, Yamashita I (2006) Floating gate metal–oxide–semiconductor capacitor employing array of high‐density nanodots produced by protein dupramolecule. Jpn J Appl Phys 45:8946–8951. [Google Scholar]
- 12. Lawrence AD, Frank S, Newnham S, Lee MJ, Brown IR, Xue W‐F, Rowe ML, Mulvihill DP, Prentice MB, Howard MJ, Warren MJ (2014) Solution structure of a bacterial microcompartment targeting peptide and its application in the construction of an ethanol bioreactor. ACS Synth Bio 3:454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Raeeszadeh‐Sarmazdeh M, Hartzell E, Price JV, Chen W (2016) Protein nanoparticles as multifunctional biocatalysts and health assessment sensors. Curr Opin Chem Eng 13:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhattacharya P, Du D, Lin Y (2014) Bioinspired nanoscale materials for biomedical and energy applications. J R Soc Interface 11:20131067–20131067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Padilla JE, Colovos C, Yeates TO (2001) Nanohedra: using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc Natl Acad Sci USA 98:2217–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsia Y, Bale JB, Gonen S, Shi D, Sheffler W, Fong KK, Nattermann U, Xu C, Huang P‐S, Ravichandran R, Yi S, Davis TN, Gonen T, King NP, Baker D (2016) Design of a hyperstable 60‐subunit protein icosahedron. Nature 535:136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. King NP, Sheffler W, Sawaya MR, Vollmar BS, Sumida JP, Andre I, Gonen T, Yeates TO, Baker D (2012) Computational design of self‐assembling protein nanomaterials with atomic level accuracy. Science 336:1171–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. King NP, Bale JB, Sheffler W, McNamara DE, Gonen S, Gonen T, Yeates TO, Baker D (2014) Accurate design of co‐assembling multi‐component protein nanomaterials. Nature 510:103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lai Y‐T, Cascio D, Yeates TO (2012) Structure of a 16‐nm cage designed by using protein oligomers. Science 336:1129–1129. [DOI] [PubMed] [Google Scholar]
- 20. Lai Y‐T, Reading E, Hura GL, Tsai K‐L, Laganowsky A, Asturias FJ, Tainer JA, Robinson CV, Yeates TO (2014) Structure of a designed protein cage that self‐assembles into a highly porous cube. Nat Chem 6:1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bale JB, Gonen S, Liu Y, Sheffler W, Ellis D, Thomas C, Cascio D, Yeates TO, Gonen T, King NP, Baker D (2016) Accurate design of megadalton‐scale two‐component icosahedral protein complexes. Science 353:389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gradisar H, Bozic S, Doles T, Vengust D, Hafner‐Bratkovic I, Mertelj A, Webb B, Sali A, Klavzar S, Jerala R (2013) Design of a single‐chain polypeptide tetrahedron assembled from coiled‐coil segments. Nat. Chem. Bio. 9:362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sciore A, Su M, Koldewey P, Eschweiler JD, Diffley KA, Linhares BM, Ruotolo BT, Bardwell JCA, Skiniotis G, Marsh ENG (2016) Flexible, symmetry‐directed approach to assembling protein cages. Proc Natl Acad Sci USA 113:8681–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Badieyan S, Sciore A, Eschweiler JD, Koldewey P, Cristie‐David AS, Ruotolo BT, Bardwell JCA, Su M, Marsh ENG (2017) Symmetry‐directed self‐assembly of a tetrahedral protein cage mediated by de novo‐designed coiled coils. ChemBioChem 19:188–1892. [DOI] [PubMed] [Google Scholar]
- 25. Cristie‐David AS, Sciore A, Badieyan S, Escheweiler JD, Koldewey P, Bardwell JCA, Ruotolo BT, Marsh ENG (2017) Evaluation of de novo‐designed coiled coils as off‐the‐shelf components for protein assembly. Mol Syst Des Eng 2:140–148. [Google Scholar]
- 26. Demeler B, Brookes E, Nagel‐Steger L (2009) Analysis of heterogeneity in molecular weight and shape by analytical ultracentrifugation using parallel distributed computing. Methods Enzymol 454:87–113. [DOI] [PubMed] [Google Scholar]
- 27. Demeler B, van Holde KE (2004) Sedimentation velocity analysis of highly heterogeneous systems. Anal Biochem 335:279–288. [DOI] [PubMed] [Google Scholar]
- 28. Brookes E, Cao W, Demeler B (2010) A two‐dimensional spectrum analysis for sedimentation velocity experiments of mixtures with heterogeneity in molecular weight and shape. Eur Biophys J 39:405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patterson DP, Su M, Franzmann TM, Sciore A, Skiniotis G, Marsh ENG (2014) Characterization of a highly flexible self‐assembling protein system designed to form nanocages. Protein Sci 23:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jutz G, van Rijn P, Santos Miranda B, Böker A (2015) Ferritin: a versatile building block for bionanotechnology. Chem Rev 115:1653–1701. [DOI] [PubMed] [Google Scholar]
- 31. Bosma HJ, Kok A, Markwijk BW, Veeger C (1984) The size of the pyruvate dehydrogenase complex of Azotobacter vinelandii. Association phenomena. Eur J Biochem 140:273–280. [DOI] [PubMed] [Google Scholar]
- 32. Patterson DP, Desai AM, Holl MMB, Marsh ENG (2011) Evaluation of a symmetry‐based strategy for assembling protein complexes. RSC Adv 1:1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Protein sequence of Oct‐MBP: trimeric esterase domain (PDB ID: http://firstglance.jmol.org/fg.htm?mol=1ZOI) is shown in black; spacer sequence 1 in red; tetrameric coiled‐coil domain (PDB ID: http://firstglance.jmol.org/fg.htm?mol=3R4A) in blue; spacer sequence 2 in green; maltose binding protein domain (PDB ID: http://firstglance.jmol.org/fg.htm?mol=1ANF) I orange.
Figure S2 Size distribution analysis of protein cages formed by Oct‐MBP obtained from measurement of ~ 300 particles imaged by negative stain TEM using the software package Image J.