

Abstract
DNA and RNA nanotechnology has been used for the development of dynamic molecular devices. In particular, programmable enzyme-free nucleic acid circuits, such as catalytic hairpin assembly, have been demonstrated as useful tools for bioanalysis and to scale up system complexity to an extent beyond current cellular genetic circuits. However, the intracellular functions of most synthetic nucleic acid circuits have been hindered by challenges in the biological delivery and degradation. On the other hand, genetically encoded and transcribed RNA circuits emerge as alternative powerful tools for long-term embedded cellular analysis and regulation. Herein, we reported a genetically encoded RNA-based catalytic hairpin assembly circuit for sensitive RNA imaging inside living cells. The split version of Broccoli, a fluorogenic RNA aptamer, was used as the reporter. One target RNA can catalytically trigger the fluorescence from tens-to-hundreds of Broccoli. As a result, target RNAs can be sensitively detected. We have further engineered our circuit to allow easy programming to image various target RNA sequences. This design principle opens the arena for developing a large variety of genetically encoded RNA circuits for cellular applications.
Graphical Abstract
INTRODUCTION
The ability of DNA and RNA to fold into precise nanostructures has been exploited for various biological and biomedical applications.1−3 The precise self-assembly of these nucleic acid nanostructures has created exciting platforms for the development of programmable molecular recognition tools.3–5 During the past decade, a complex array of nucleic acid-based circuits has been engineered and applied for bioanalytical detections.6–9 One of the key mechanisms enabling the design of nucleic acid circuits is so-called toehold-mediated strand displacement, in which an invading oligonucleotide strand binds to its complementary strand in a duplex via a single-stranded toehold region and branch migration.10–12 Based on this mechanism, several nonenzymatic isothermal circuits have been developed, including entropy-driven catalysis, hybridization chain reaction, and catalytic hairpin assembly.6,13−15
Catalytic hairpin assembly (CHA), which was originally developed by the Pierce lab, has proven to be an extraordinarily versatile tool.15 In CHA, two complementary nucleic acid hairpins are designed, such that their spontaneous hybridization is kinetically hindered by embedding the complementary regions within the hairpin stems. Only in the presence of a target input strand, one of the hairpins can be opened based on the toehold-mediated strand displacement, which will further enable the assembly of both hairpins. Eventually the target strand can be spontaneously displaced and recycled, similar to a catalyst, to induce more hairpin assembly events.6,15 These enzyme-free isothermal CHA circuits can provide rapid and effcient signal amplification with low background and good turnover rates.8,13 DNA circuits based on these highly sensitive and target-specific CHA reactions have been applied for various in vitro cell-free analysis, including the detection and quantification of nucleic acids, small molecules, and proteins..13,14,16–18
With advanced sensitivity and programmability, nucleic acid- based circuits have the potential to revolutionize biological imaging and genetic regulation in living systems. Unfortunately, the intracellular functions of most DNA-based circuits have been hindered by difficulties in biological delivery and degradation.2,19 In contrast, RNA molecules can be genetically encoded and transcribed within living systems. As a result, embedded RNA-based circuits and devices may hold great potential for the intracellular applications. Here, we report our effort in designing and applying genetically encoded RNA-based CHA circuits for sensitive RNA imaging within live cells. CHA circuits are well-suited for these genetically encoded sensor developments. In addition to their high effciency, enzyme-free, and isothermal properties, the requirement of only self-folded hairpin structures is critical in reducing the leakage in the absence of target molecules. Indeed, the cotranscriptional folding of these hairpins is much more effcient and accurate than the interstrand hybridizations that are normally required in other nucleic acid circuits. Interstrand kinetically trapped nucleic acid duplexes can be separately prepared in vitro; however, in the live cell circumstances, interstrand hybridizations will result in high background signals.
The cellular application of RNA circuits is still limited, which is partially due to the lack of a genetically encoded reporting system. The fluorophores and quenchers that are normally employed for the in vitro characterization of nucleic acid circuits cannot be genetically encoded, while a fluorescent protein-based reporting system requires a time-consuming transcription, translation, and maturation process. It has been demonstrated that fluorescent RNA molecules, named Spinach and Broccoli, can be employed as genetically encoded reporters for RNAimaging.20–22 Spinach and Broccoli are RNA aptamers that can bind and activate the fluorescence of small-molecule dyes, such as 3,5-difluoro-4-hydroxybenzylidene-1-trifluoroethyl-imidazolinone (DFHBI-1T).23–25 Recently, the Ellington group has successfully demonstrated that Spinach can function as a reporter in an RNA-based CHA reaction in vitro.26 Unfortunately the limited signal-to-background ratio and the slow kinetics of the system have prevented the further usage of Spinach CHA for intracellular applications
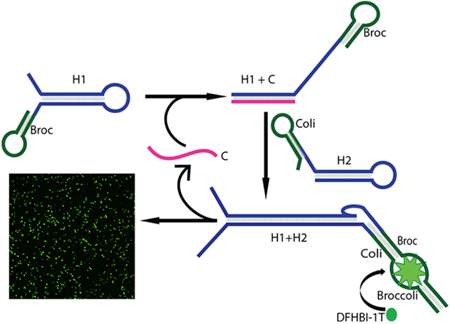
In this report, we demonstrated a novel design principle of Catalytic Hairpin Assembly RNA circuit that is Genetically Encoded, termed CHARGE. This new system provides a robust fluorescence signal and can be used for RNA imaging in live cells with high sensitivity. In our design, Broccoli was separated into two nonfluorescent parts, termed Broc and Coli, which was conjugated to the end of two CHA hairpins, H1 and H2, respectively.27 The active fluorogenic form of Broccoli can only be formed upon target-driven H1 and H2 hybridization (Figure 1). One target RNA can catalytically activate tens-to-hundreds of Broccoli fluorescences. We have further demonstrated that CHARGE can be programmed to sensitively detect various RNA targets within the live cell environment.
Figure 1.
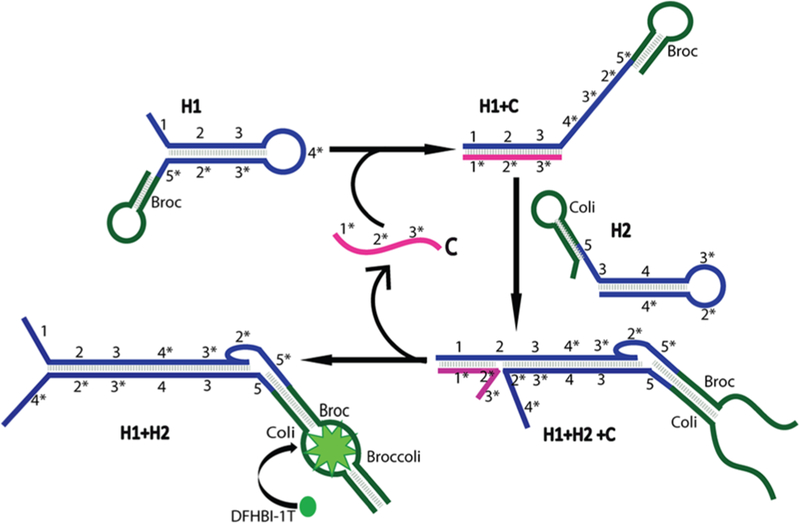
Schematic of the CHARGE circuit. The H1 and H2 strands in the circuit were modified with Broc and Coli, respectively. Target (C) will initiate and catalyze the assembly of H1 and H2 into an H1+H2 duplex, and C is further recycled. Recombined Broccoli in the H1+H2 duplex will activate the fluorescence of DFHBI-1T dye
RESULTS
Design and Optimization of the CHARGE System.
To design a CHA system using split Broccoli as the reporter, we started with RNA sequences modified from a CHA system that has been used for RNA detection in vitro.26 As shown in Figure 1, two complementary RNA hairpins, H1 and H2, are kinetically trapped until a specific RNA target strand, C, initiates their hybridization. In our work, the Broccoli aptamer is divided into two parts, “Broc” and “Coli”. When separated in solution, Broc and Coli provide a low fluorescence signal. Only when they are reunited into the original Broccoli structure, the fluorescence signal of DFHBI-1T can be recovered. We fused Broc to the 3′ end of the H1 hairpin and Coli to the 5′end of the H2 hairpin. As a result, the complete Broccoli aptamer structure is reformed upon target (C)-driven hybridization of H1 and H2 (Figure 1). Indeed, our initial trial(D1)using a previously reported CHA26 and split Broccoli sequences can be used to detect low abundance target RNA (Figure 2 and Table S1).
Figure 2.
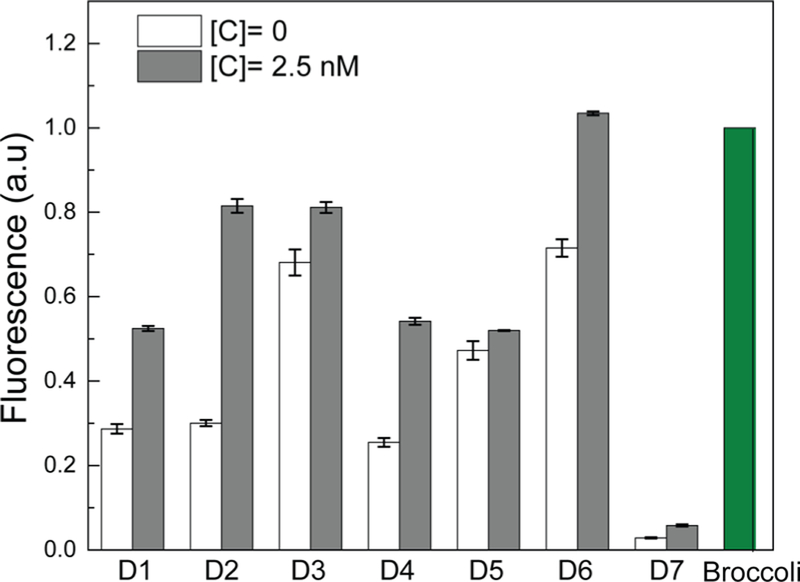
Optimizing the signal-to-background ratio of CHARGE based on different sequence designs. Shown are normalized fluorescence values of three independent replicates as measured in a solution containing 0 or 2.5 nM target (C), 250 nM H1 and H2 (or Broccoli itself), and 5 μM DFHBI-1T. All the RNA sequences were PAGE gel-purified, and the fluorescence values were normalized to the Broccoli intensity.
Next, we asked how to improve the signal-to-background ratio (1.5-fold) of our initial design, D1 (Figure 2). In the absence of target, two mechanisms can result in the nonspecific hybridization or leakiness, i.e., initial leakage and asymptotic leakage.8 The initial leakage is due to the fraction of hairpin that is malformed and can be minimized by gel purification and pre- annealing of each initial RNA hairpin strand. On the other hand, the asymptotic leakage occurs when hairpins undergo conformational fluctuations that will eventually drive the formation of uncatalyzed H1+H2 (without target) hybridization. Both types of leakage will induce nonspecific, partial hybridization of H1+H2 duplexes and reform active Broccoli aptamer to produce fluorescence. Having already gel-purified and pre-annealed H1 and H2 hairpins to minimize the initial leakage, we focused on reducing the asymptotic leakage. Asymptotic leakage could arise from the following two mechanisms: (a) low percentage of GC base pairs or low total base pair numbers in the H1 or H2 hairpin stem region, which makes H1 and H2 unstable, leading to spontaneous hybridization without target, and (b) spontaneous reassembly of the terminal Broc and Coli regions into the complete Broccoli structure.
To study the effect of stem region GC% and number of base pairs on the asymptotic leakage (note that all sequence design was analyzed and optimized using a NUPACK software), we mutated the stem region of H1 hairpin to increase the GC% from 42% (D1) to 53% (D2) while maintaining the same number of base pairs as in our initial trial design (Tables S1 and S2). Indeed, an increased fluorescence signal-to-background ratio was observed for D2 (2.2-fold), as compared to that of D1 (1.5-fold) (Figure 2). We have further increased the GC% in the H1 loop and the number of base pairs in the H2 stem (D3). Unfortunately, the fluorescence signal increased in both the presence and absence of the target RNA, which resulted in a reduced signal-to-background ratio (Figure 2). After careful examination of the D3 sequences, we realized that the GC% in the H2 loop might be too low in D3 (Table S2). A new design D4 was made to further increase the GC% in the H2 loop to make hairpin design more symmetrical (Tables S1 and S2). Indeed, the signal leakage was dramatically reduced in D4, even though D3 and D4 shared almost identical sequences except for two base pair mutations (Figure 2 and Table S1).
To study the effect of the terminal Broc and Coli regions on the asymptotic leakage, we have designed other mutations based on D2 sequence. We have first removed either the 5′ end free region of H2 (D5) or the terminal loop region of Broc (D6) (Table S1 and Figure S1). Unexpectedly, both designs resulted in even larger leakage. In another attempt to decrease Broc- and Coli-induced leakage, three nucleotides of Broc were incorporated into the stem region of H1 (D7). As expected, this modification in D7 dramatically reduced the leakage (Figure 2). However, the overall sensitivity of the system was reduced as well. It suggests that the asymptotic leakage might be challenging to be eliminated by only modifying the terminal Broc and Coli regions. It requires careful thermodynamic consideration in both hairpin and sequence designing.
Taken together, these data indicated that an RNA-based CHA system could be developed with split Broccoli as the reporter. It requires careful thermodynamic considerations in the design of both hairpins and terminal Broccoli sequences to reduce the leakage. Because D2 yielded the optimal signal-to-background ratio (Figure 2), subsequent experiments were conducted using D2 as the model.
In Vitro RNA Detection with CHARGE.
To determine the sensitivity of the approach, different concentrations of target RNA were added to solutions containing 250 nM H1 and 250 nM H2. Similar to previous reports on DNA-based CHA systems,13 CHARGE was able to detect as low as 2.5 nM RNA target, i.e., 100-fold lower concentration than the hairpin (Figure 3a). Interestingly, CHARGE behaves as a “digital” sensor when the fluorescence intensity is measured after 2 h incubation (Figure S2). A similar intensity of ON fluorescence signal was observed when 2.5−250 nM target RNA was added, and an OFF fluorescence was observed only at target concentrations below 0.5 nM (Figure 3b). Indeed, RNA target can function as a catalyst in CHARGE and result in the signal amplification for low abundance RNA detection.
Figure 3.
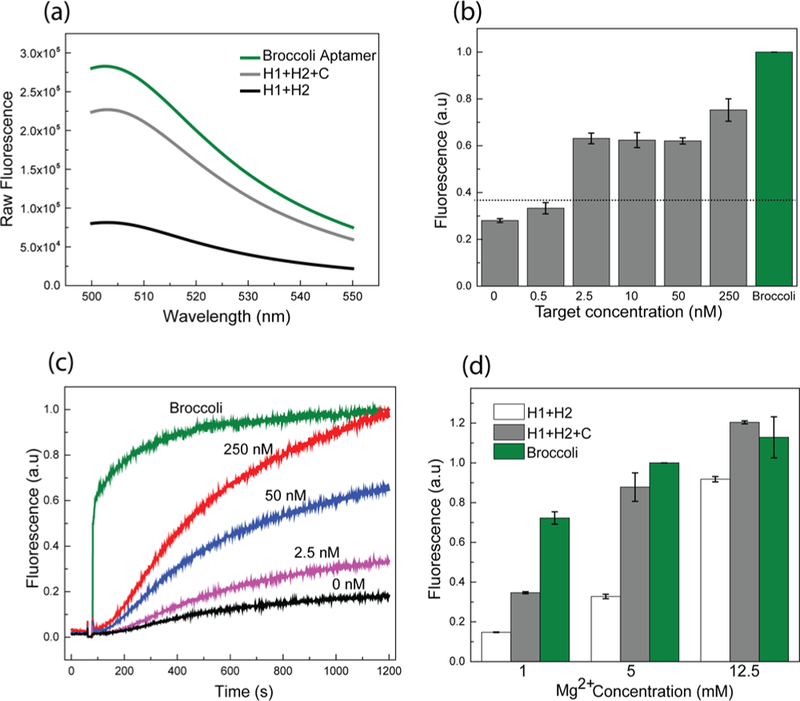
In vitro characterization of the D2 CHARGE system. (a) Fluorescence emission spectra of Broccoli aptamer and CHARGE circuit in the presence or absence of target, C. Spectra were measured in a solution containing 250 nM Broccoli or H1 and H2, 2.5 nM target C, and 5 μM DFHBI-1T. (b) Fluorescence response of D2 CHARGE on different concentrations of targets. Sensitive detection of 2.5 nM (100- fold less than H1 and H2) target was observed. (c) Kinetics of CHARGE circuit when adding various amounts of target. (d) Effect of Mg2+ concentration on CHARGE circuit. All the RNA sequences in these experiments were PAGE gel-purified, and all the fluorescence values in (b−d) were normalized to Broccoli intensity.
We next determined the response kinetics of CHARGE (Figure 3c). By varying the target concentrations, the initial rate of fluorescence signal increase varied significantly. When the target concentration was similar to H1 and H2 (250 nM each), the maximum signal was reached within 20 min. When 2.5 nM RNA target was added, i.e., 100-fold lower concentration than the hairpin, half-maximum signal was observed in around 35 min, with 90% maximum signal in ∼60 min. We have further measured the turnover rate of the catalyzed hybridization reaction by changing the H1 concentrations (Figure S2). The observed turnover rate was 0.6−1/s in the presence of 100−250 nM H1. These kinetic data indicated that CHARGE can provide a fast and efficient response toward the target RNA.
It is known that magnesium concentration plays important roles in the efficiency of CHA.13,14 We want to further test if CHARGE can function in the presence of physiological concentration of magnesium ions, i.e., less than 5 mM.28,29 As shown in Figure 3d, the maximum fluorescence intensity of CHARGE was enhanced at higher magnesium concentrations; however, the background signal from leakage also increased. Optimal signal-to-background ratio was observed at magnesium concentration between 1 and 5 mM. Indeed, CHARGE can perform well with physiological concentrations of magnesium ions.
We further studied the effect of different environmental conditions on CHARGE. The performance of our RNA circuit was first tested over a temperature range of 15−37 °C. An optimal in vitro CHARGE assembly efficiency was observed at around 22 °C, with larger signal leakage shown at higher temperatures (Figure S3a). After further optimization of the buffer conditions (Figure S3b), our data indicated that most efficient D2 CHARGE appeared at 22 °C in a buffer consisting of 10 mM Tris, 5 mM MgCl2, 100 mM KCl, and 10 mM NaCl at pH = 7.5, with a signal-to-background ratio of 3.0. Taken together, these data suggest that CHARGE exhibits character- istics that may be useful for intracellular applications.
CHARGE-Based RNA Imaging Inside Live Cells.
After demonstrating the in vitro performance of CHARGE, the RNA- based sensor system was tested for target detection inside live cells. We used E. coli BL21 (DE3)* cells as the model system. The H1 and H2 sequences were first cloned into a dual expression vector, pETDuet, which will express similar levels of the two hairpins simultaneously. Target was cloned into another vector, pCDFDuet. Both pCDFDuet and pETDuet were transformed in the same E. coli cell and were induced by the addition of isopropyl β-D-1-thiogalactopyranoside, IPTG. After 2 h IPTG induction, DFHBI-1T was added, and cellular fluorescence was measured using a confocal fluorescence microscope.
As shown in Figure 4, an obvious fluorescence signal was observed after the cellular transcription of H1, H2, and target (C) RNAs. The cellular fluorescence intensity is comparable with that of Broccoli-expressing cells (Figure 4). As a control, we measured the target-free CHARGE fluorescence by expressing only H1- and H2-containing pETDuet vectors. Indeed, much lower cellular fluorescence was observed (Figure 4). Similarly, with only H1 or H2 hairpin being co expressed with the target C, the cellular fluorescence signals were too low to be determined (Figure S4).
Figure 4.
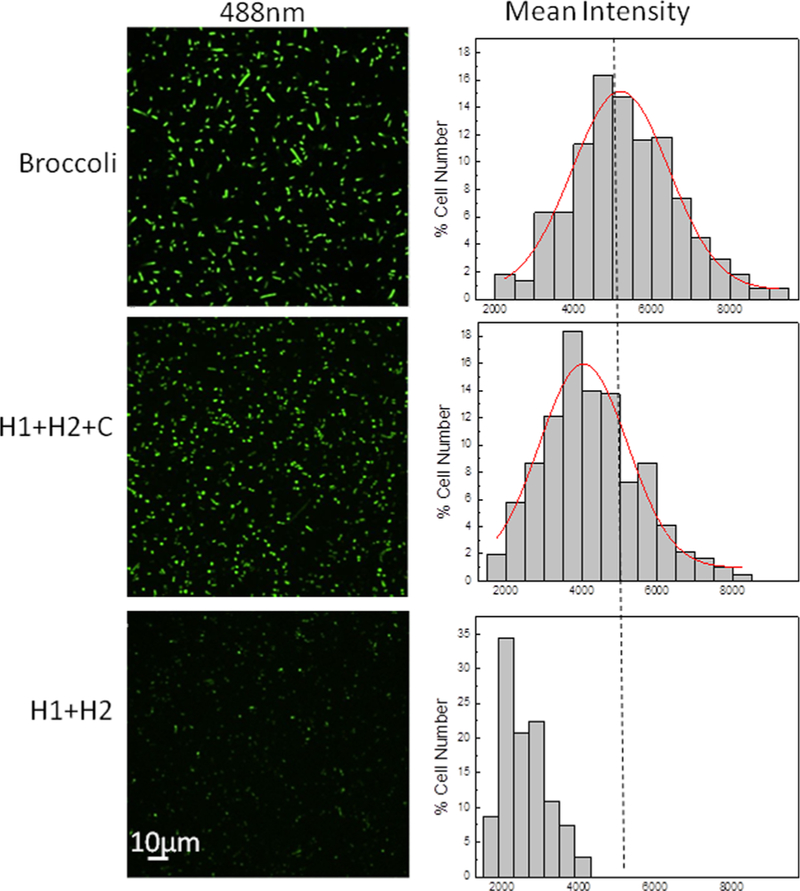
Confocal fluorescence imaging with live BL21 (DE3)* cells. After 2 h IPTG induction, 200 μM DFHBI-1T was added for 30 min before imaging of cells coexpressing pETDuet-H1/H2 and pCDFDuet- target (H1+H2+C), or expressing only pETDuet-H1/H2 (H1+H2), or expressing only pETDuet-Broccoli (Broccoli). Distribution of cellular fluorescence levels was shown on the right. Here, individual cells were binned according to their brightness. The percentage of cells in each bin was plotted. A total of 300 cells were measured in each case from three experimental replicates.
In addition, cellular activation of CHARGE can be observed at both 25 and 37 °C (Figure S5). Slightly enhanced cellular signal as well as improved signal-to-background ratio were observed at 25 °C compared to that of 37 °C, which is consistent with our in vitro data (Figure S3a). Moreover, CHARGE fluorescence was still clearly active after our whole imaging period of 6 h since the addition of IPTG, while it appears that at longer incubation time the background signal leakage was also increased for target-free cells expressing only H1 and H2 RNAs (Figure S6). Taken together, these data suggested that the genetically encoded CHARGE system functions well inside living E. coli cells. Activated Broccoli signals can be used for the target detection.
Detecting Biologically Relevant RNA Targets with a Modular CHARGE System.
As shown above, careful thermodynamic considerations of RNA sequences are usually needed to obtain an optimized CHARGE for a particular RNA target. Because designing different H1 and H2 sequences for each RNA target would be challenging, we developed here a modular CHARGE system to image different RNA targets without redesigning the entire RNA circuit each time. For this purpose, we engineered a genetically encoded RNA-based molecular beacon probe (Figure 5a). The stem region of this molecular beacon contains the 1* domain of the target, and the freedom of which domain is required to initiate the catalytic activity of the CHA circuit. The loop region of the molecular beacon was designed to bind with the RNA target of interest. Upon target binding, the stem is disrupted and releases the 1* domain to activate CHARGE fluorescence.13 Based on D2 CHARGE, we first designed two molecular beacons to detect microRNA 21 (miR21) and a bacterial sugar transport-related small RNA (SgrS), respectively. Indeed, by simply changing the loop region of molecular beacon, both miR21- and SgrS- selective RNA circuits can be developed using the original D2 H1 and H2 hairpins. For both RNA targets, a similar level of fluorescence signal enhancement was observed as that of the original CHARGE (Figure 5b). As a result, molecular beacon- incorporated CHARGE functions as a modular system for detecting various RNA targets.
Figure 5.
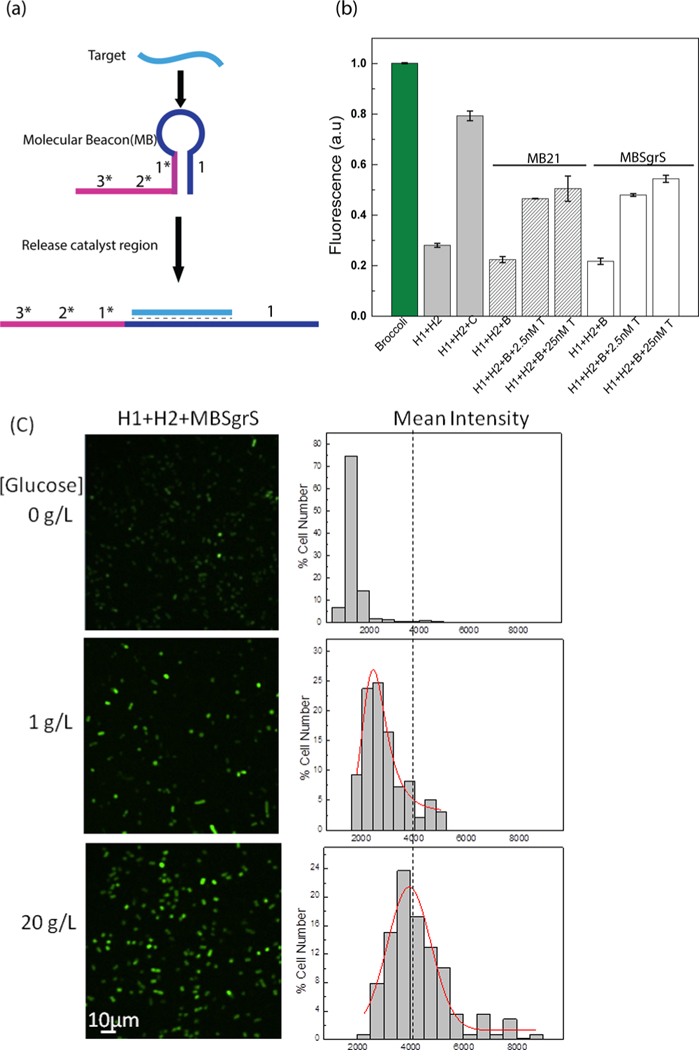
Modular CHARGE system to detect various RNA targets. (a) Schematic of molecular beacon-based target detection. Here, the binding of target RNA induces the opening of the molecular beacon, which further activates the 1* domain of the original catalyst. (b) In vitro detection of miR21 (striped bars) and SgrS (white bars) with molecular beacon-incorporated D2 CHARGE. B: beacon, C: original catalyst target in D2, T: miR21 or SgrS target. (c) Confocal fluorescence imaging of cells expressing H1+H2+MBSgrs (SgrS- targeting molecular beacon) after adding different amounts of glucose and their cellular fluorescence intensity distributions. After 1.5 h IPTG induction, cells were exposed to glucose stress for 30 min. 200 μM DFHBI-1T was added for 30 min before imaging, and images were taken 45 min after adding DFHBI-1T. Here, individual cells were binned according to their brightness. The percentage of cells in each bin was plotted. A total of 200 cells were measured in each case from three experimental replicates.
Next, we used this modular system to image SgrS expression in live bacterial cells. SgrS is an Hfq protein-dependent small RNA, whose expression level can be regulated by glucose phosphate.30–33 We first cloned the SgrS-targeting molecular beacon into a pCDFDuet vector and coexpressed it with H1 and H2 in the above-mentioned BL21 (DE3)* cells. The addition of glucose increases the expression level of SgrS in these BL21 strains.34 Indeed, obvious cellular fluorescence signal enhance- ment was observed upon increasing the glucose concentrations (Figure 5c). As a control, the fluorescence level of both Broccoli and original CHARGE did not respond to the variations in the glucose concentrations (Figure S7). Altogether, these data suggested that our molecular beacon−CHARGE system can be easily programmed to detect different RNA targets both in vitro and in live cells.
Measuring Intracellular CHARGE Efficiency with Hammerhead Ribozyme.
Our next goal was to determine if the CHARGE system could be used to image low abundance cellular RNAs in live cells. According to our in vitro data, CHARGE can sensitively detect 100-fold lower target RNA concentration compared to the H1 and H2 concentrations (Figure 3b). To selectively tune the cellular concentration of the target while maintaining the similar high expression level of H1 and H2, we constructed a theophylline-regulated target RNA expressing system based on a hammerhead ribozyme (HHR) (Figure 6a). Theophylline aptamer-fused HHR has been used as a switch for genetic regulation.35–37 Herein, the binding of theophylline with aptamer will induce the self-cleavage of HHR. The cleavage efficiency can be quantitatively regulated by adding different amounts of theophylline. By introducing the D2 target sequence into the HHR region that will be cleaved, the intracellular concentration of free target RNA can be controlled by theophylline (Figures 6a and 6c).
Figure 6.
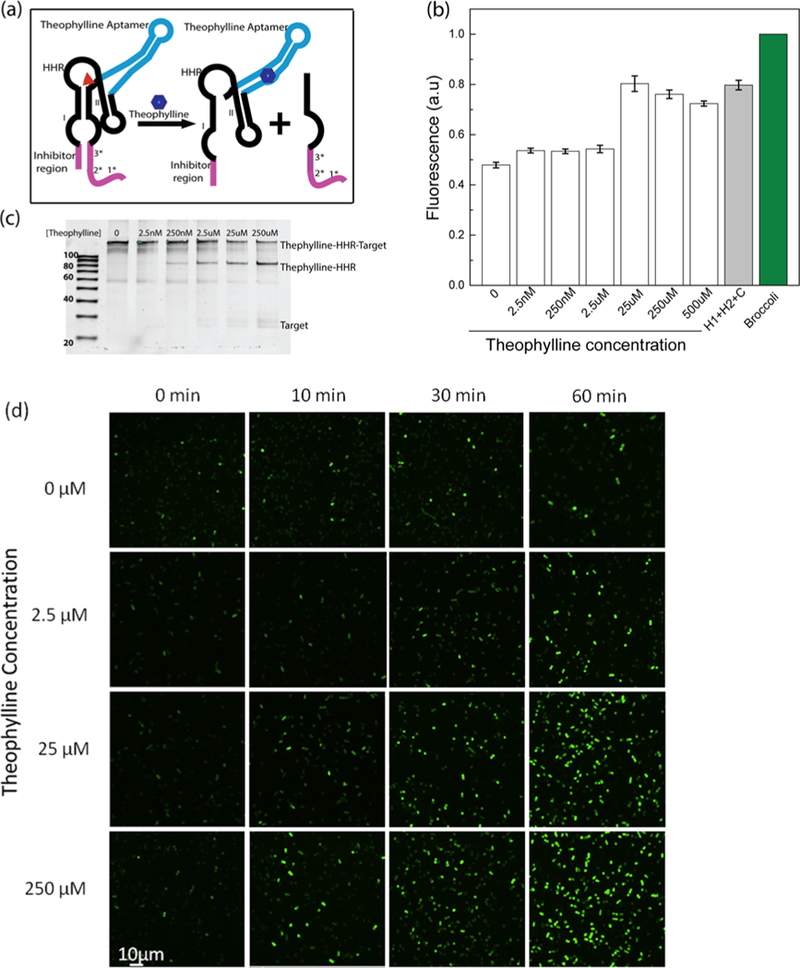
Theophylline-regulated cellular RNA levels based on a hammerhead ribozyme (HHR) system. (a) Schematic of theophylline- induced HHR self-cleavage and target RNA release. The red triangle denotes the cleavage site of HHR. (b) In vitro fluorescence assay data after adding different concentrations of theophylline. (c) 10% denaturing PAGE gel characterization of the 10-base-long inhibitor- incorporated HHR system at 2 h after adding different concentrations of theophylline. (d) Confocal fluorescence imaging of the theophylline−HHR construct treated with different concentrations of theophylline. These BL21 (DE3)* cells were induced by IPTG for 1 h before imaging.
In order to prevent the target sequence from activating CHA before the addition of theophylline, an inhibitor sequence was introduced to hybridize with the target RNA. This inhibitor sequence was optimized to form 8, 9, or 10 base pairs with the target. An optimal signal-to-background ratio was observed when a 10-base-long inhibitor was introduced (Figures 6b and S8). Using this inhibitor, tunable 6−72% of target RNA can be released 1 h after adding 0−250 μM theophylline (Figure 6c). Here, a relatively high signal leakage was observed in the absence of theophylline, which may result from the spontaneous self- cleavage of HHR. Such self-cleavage and leakage were not reduced by simply adding a lower concentration of magnesium ions, e.g., 1 mM vs 5 mM (data not shown).
We have further cloned this HHR probe into a pCDFDuet vector to test its intracellular behavior. After 1 h IPTG induction, cells were treated with different concentrations of theophylline, and cellular fluorescence intensity was then monitored. As shown in Figure 6d, obvious cellular fluorescence was observed within 10 min when a large amount of theophylline was added. Furthermore, within the 30−60 min detection window, the fluorescence signal could be observed even in cells containing low concentrations of theophylline (Figure 6d). Our gel results indicated that the addition of theophylline indeed induced the intracellular cleavage to release the target RNA accordingly. In the presence of 0−250 μM theophylline, under our experimental conditions, 0.11−17 μM concentration of cellular target RNA was generated (Figure S9). Based on the Gaussian distributions of cellular fluorescence level at each target RNA concentration, CHARGE can detect as low as 110 copies of target RNA per cell at 30 min after activating the circuit (Figure S10). These results suggested that our CHARGE design indeed can be used to efficiently detect low abundance target RNA.
DISCUSSION
Nucleic acid-based circuits are finding increasing applications in bioanalysis and medicine. Although most of these circuits are developed with DNA as the main operator, RNA molecules can also provide an alternative versatile platform, especially in realizing genetically encoded circuits for long-term cellular analysis and regulation. Here, we demonstrated a novel RNA- based signal amplification circuit, termed CHARGE. Our results indicated that CHARGE can be used for sensitive RNA detection and imaging in live cells.
There are several advantages of the CHARGE system in comparison with the previously reported Spinach-based CHA circuit (Figure S11).26 (1) Broccoli is an improved version of Spinach with higher intracellular folding efficiency and bright- ness. In addition, Broccoli has lower dependence on the concentrations of magnesium ions than Spinach.23 As a result, CHARGE can potentially provide better performance under physiological conditions. (2) Broccoli can efficiently self- assemble into the fluorogenic active form even without adding the tRNA scaffold. As a result, CHARGE does not have the structurally complicated tRNA scaffold needed for Spinach- CHA. The asymptotic leakage from these tRNA scaffolds can thus be avoided. (3) By splitting Broccoli into two non- fluorescent parts, Broc and Coli, the fluorescence signal can be observed directly after the assembly of two CHA hairpins. As a result, the step of Spinach displacement reaction in Spinach- CHA is not needed (Figure S11). This is significant because the kinetics and efficiency of these RNA circuits will be reduced with increasing layers of toehold-mediated strand displacement.
As shown above, we achieved moderate progress in our in vitro attempt to reduce signal leakage and optimize CHA efficiency. The design of the RNA circuit requires careful thermodynamic and kinetic considerations, especially in pathways to avoid nonspecific hybridization between RNA hairpins. Even though DNAs and RNAs share a similar pattern of base pairing, DNAs mainly stay in a B-form helical structure, while RNAs prefer an A-form helix. B-formed DNA duplexes have more flexibility and pay a greater entropic penalty in the formation of helical structures.38–40 As a result, more base pairing (enthalpy contribution) is required for DNAs to hybridize with each other compared to RNAs. On the other hand, RNA hybridization is less specific with the formation of some unique loop−loop pseudoknots and wobble base pairs. Indeed, to engineer more accurate and efficient RNA circuits, further development in the biophysical theories and software is still needed.
One key feature of the CHARGE sensor is its high sensitivity and digital-like behavior. Using target RNA as the catalyst, CHARGE can detect low abundance RNA molecules and high abundance RNAs with similar sensitivities. The switch between low and high fluorescence output occurs over a short range of target concentration (Figure 3b). On one hand, the digital output is advantageous if the goal is to determine the presence or absence of a particular RNA target or if there is a specific threshold value for the cellular functions of RNA target. However, in order to quantify the cellular levels of target RNA, these digital sensors could be less favored with small detection range. Here, we argue that CHARGE can function well under both circumstances when the signal is detected at the proper time window. Prolonged incubation time is favorable for the digital imaging of CHARGE, which allows the target RNAs to be recycled and catalyze all the possible hybridization events. Alternatively, to differentiate the cellular levels of target RNAs, fluorescence signals should be detected directly after the cellular introduction of the circuit, when the CHARGE efficiency still depends on the initial rate of target-binding-induced activation. For example, as shown above in the cellular detection of glucose-induced SgrS expression, similar fluorescence signals were observed 2−3 h after the addition of different glucose concentrations (Figure S7). In contrast, variations in the target RNA concentrations can be measured within 15−30 min after the activation of the CHARGE circuit (Figures 5c and 6c).
In summary, we have introduced a novel genetically encoded RNA circuit that can detect cellular RNA targets with high sensitivity. These CHARGE circuits can be easily programmed to image various RNA targets in living cells. Even though the current circuit is demonstrated in bacterial cells, CHARGE indeed has the potential for eukaryotic cell studies as well. Similar to the versatile applications of catalytic hairpin assembly circuit for in vitro analysis, various CHARGE could be potentially developed to further detect other cellular RNAs, proteins, and small molecules, as well as to function as switches for genetic regulation. This study opens a new opportunity to use genetically encoded RNA circuits for cellular applications.
Supplementary Material
ACKNOWLEDGMENTS
The authors gratefully acknowledge the start-up grant from UMass Amherst and NIH R01AI136789. We are grateful to Dr. Craig Martin for valuable comments, Dr. James Chambers for the assistance in fluorescence imaging, and Dr. Kathryn R. Williams for help with manuscript preparation. The authors also thank Yousef Bagheri and other members in the You and Martin lab for useful discussion and valuable comments.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Zhang J; Narayan RJ DNA Nanotechnology. In NanoScience in Biomedicine; 2009; pp 405–427. [Google Scholar]
- (2).Chen YJ; Groves B; Muscat RA; Seelig G Nat. Nanotechnol 2015, 10, 748–760. [DOI] [PubMed] [Google Scholar]
- (3).Pinheiro AV; Han D; Shih WM; Yan H Nat. Nanotechnol 2011, 6, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chao J; Zhu D; Zhang Y; Wang L; Fan C Biosens. Bioelectron 2016, 76, 68–79. [DOI] [PubMed] [Google Scholar]
- (5).Rinker S; Ke Y; Liu Y; Chhabra R; Yan H Nat. Nanotechnol 2008, 3 (7), 418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Jung C; Ellington AD Acc. Chem. Res 2014, 47 (6), 1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Li B; Chen X; Ellington AD Anal. Chem 2012, 84 (19), 8371–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chen X; Briggs N; McLain JR; Ellington AD Proc. Natl. Acad. Sci. U. S. A 2013, 110 (14), 5386–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Strack RL; Song W; Jaffrey SR Nat. Protoc 2014, 9 (1), 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang DY; Seelig G Nat. Chem 2011, 3 (2), 103–113. [DOI] [PubMed] [Google Scholar]
- (11).Srinivas N; Ouldridge TE; Šulc P; Schaeffer JM; Yurke B; Louis AA; Doye JPK; Winfree E Nucleic Acids Res 2013, 41 (22), 10641–10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Qian L; Winfree E; Bruck J Nature 2011, 475 (7356), 368–372. [DOI] [PubMed] [Google Scholar]
- (13).Li B; Ellington AD; Chen X Nucleic Acids Res 2011, 39 (16), e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jiang Y; Li B; Milligan JN; Bhadra S; Ellington AD J. Am. Chem. Soc 2013, 135 (20), 7430–7433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yin P; Choi HMT; Calvert CR; Pierce NA Nature 2008, 451 (7176), 318–322. [DOI] [PubMed] [Google Scholar]
- (16).Chen W; Yan Y; Zhang Y; Zhang X; Yin Y; Ding S Sci. Rep 2015, 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li D; Cheng W; Li Y; Xu Y; Li X; Yin Y; Ju H; Ding S Anal. Chem 2016, 88 (15), 7500–7506. [DOI] [PubMed] [Google Scholar]
- (18).Li C; Li Y; Xu X; Wang X; Chen Y; Yang X; Liu F; Li N Biosens. Bioelectron 2014, 60, 57–63. [DOI] [PubMed] [Google Scholar]
- (19).Wu C; Cansiz S; Zhang L; Teng IT; Qiu L; Li J; Liu Y; Zhou C; Hu R; Zhang T; et al. J. Am. Chem. Soc 2015, 137 (15), 4900–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhang J; Fei J; Leslie BJ; Han KY; Kuhlman TE; Ha T Sci. Rep 2015, 5, 17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Filonov GS; Kam CW; Song W; Jaffrey SR Chem. Biol 2015, 22 (5), 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bhadra S; Ellington AD RNA 2014, 20 (8), 1183–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Filonov GS; Moon JD; Svensen N; Jaffrey SR J. Am. Chem. Soc 2014, 136 (46), 16299–16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Huang H; Suslov NB; Li NS; Shelke SA; Evans ME; Koldobskaya Y; Rice PA; Piccirilli JA Nat. Chem. Biol 2014, 10 (8), 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Paige JS; Wu KY; Jaffrey SR Science 2011, 333 (6042), 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bhadra S; Ellington AD Nucleic Acids Res 2014, 42 (7), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Alam KK; Tawiah KD; Lichte MF; Porciani D; Burke DH ACS Synth. Biol 2017, 6 (9), 1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Moncany MLJ; Kellenberger E Experientia 1981, 37 (8), 846–847. [DOI] [PubMed] [Google Scholar]
- (29). Grubbs RD BioMetals 2002, 15 (3), 251–259. [DOI] [PubMed] [Google Scholar]
- (30).Bobrovskyy M; Vanderpool CK Front. Cell. Infect. Microbiol 2014, 4, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Horler RSP; Vanderpool CK Nucleic Acids Res 2009, 37 (16), 5465–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Richards GR; Patel MV; Lloyd CR; Vanderpool CK J. Bacteriol 2013, 195 (21), 4816–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Negrete A; Shiloach J Microb. Cell Fact 2017, 16 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Negrete A; Ng WI; Shiloach J Microb. Cell Fact 2010, 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).You M; Litke JL; Jaffrey SR Proc. Natl. Acad. Sci. U. S. A 2015, 112 (21), E2756–E2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Litke JL; You M; Jaffrey SR Methods Enzymol 2016, 572, 315–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wieland M; Hartig JS Angew. Chem., Int. Ed 2008, 47 (14), 2604–2607. [DOI] [PubMed] [Google Scholar]
- (38).Rauzan B; McMichael E; Cave R; Sevcik LR; Ostrosky K; Whitman E; Stegemann R; Sinclair AL; Serra MJ; Deckert AA Biochemistry 2013, 52 (5), 765–772. [DOI] [PubMed] [Google Scholar]
- (39).Jasinski D; Haque F; Binzel DW; Guo P ACS Nano 2017, 11 (2), 1142–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sugimoto N; Nakano SI; Yoneyama M; Honda KI Nucleic Acids Res 1996, 24 (22), 4501–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.