

Abstract
N-Glycans are modified as part of a quality control mechanism during glycoprotein folding in the endoplasmic reticulum (ER). Glucosidase II (GII) plays a critical role by generating monoglucosylated glycans that are recognized by lectin chaperones, calnexin and calreticulin. To understand how the hydrolytic activity of GIIα is enhanced by the mannose 6-phosphate receptor (MPR) homology domain (MRH domain) of its β subunit, we now report a 1.6 Å resolution crystal structure of the MRH domain of GIIβ bound to mannose. A comparison of ligand-bound and unbound structures reveals no major difference in their overall fold, but rather a repositioning of side chains throughout the binding pocket, including Y372. Mutation of Y372 inhibits GII activity, demonstrating an important role for Y372 in regulating GII activity. Comparison of the MRH domains of GIIβ, MPRs, and the ER lectin OS-9 identified conserved residues that are critical for the structural integrity and architecture of the carbohydrate binding pocket. As shown by nuclear magnetic resonance spectroscopy, mutations of the primary binding pocket residues and adjacent W409, all of which inhibit the activity of GII both in vitro and in vivo, do not cause a significant change in the overall fold of the GIIβ MRH domain but impact locally the stability of the binding pocket. W409 does not directly contact mannose; rather, its indole ring is stabilized by binding into a hydrophobic pocket of an adjacent crystallographic neighbor. This suggests that W409 interacts with a hydrophobic region of the GIIβ or GIIα subunit to modulate its effect on GII activity.
The endoplasmic reticulum (ER) has a quality control mechanism that ensures the correct folding of glycoproteins within the secretory pathway. Recognition of specific N-glycan structures serves critical roles during this process to direct glycoproteins into either a chaperone-mediated folding pathway or a proteasomal degradation pathway termed ER-associated degradation (ERAD) (for reviews, see refs 1–4). A major hub in this folding pathway revolves around monoglucosylated N-glycans. During N-glycosylation (Figure 1A), a glycan [Glc3Man9GlcNAc2 (Figure 1B)] is transferred by the oligosaccharyltransferase complex from a dolichol phosphate derivative to the asparagine-X-serine/threonine sequence of nascent polypeptides, a process that is conserved among animal, plant, and fungal species.5–7 The formation of monoglucosylated glycan-bearing glycoproteins occurs either by (1) rapid deglucosylation of the transferred N-glycan by glucosidase I followed by glucosidase II (GII) or (2) reglucosylation of deglucosylated species via UDP-Glc:glycoprotein glucosyltransferase. UDP-Glc:glycoprotein glucosyltransferase is a glycoprotein folding status sensor that transfers a glucose residue to glycoproteins that have not yet acquired their native structure.8 The resulting monoglucosylated glycoproteins are recognized by the type I transmembrane lectin calnexin and/or its soluble counterpart calreticulin, each of which is associated with protein disulfide isomerase ERp57. This lectin–chaperone complex facilitates glycoprotein folding and prevents exit from the ER of folding intermediates or misfolded species9 (reviewed in refs 10 and 11). Cycles of deglucosylation and reglucosylation occur by the opposing activities of GII and UDP-Glc:glycoprotein glucosyltransferase, allowing opportunities for a nascent glycoprotein to attain its native conformation. Glycoproteins with a long residence time in the ER may encounter α-mannosidase(s) (ERManI/EDEM) that sequentially removes mannose residues. Demannosylated glycoproteins have a lower affinity for GII12 and, conversely, are better ligands for lectins (e.g., OS-9) in the ERAD pathway.13–15 OS-9 recognizes Manα1,6Man on the trimmed C arm14,16,17 and facilitates entry of misfolded glycoproteins into the ERAD pathway.
Figure 1.
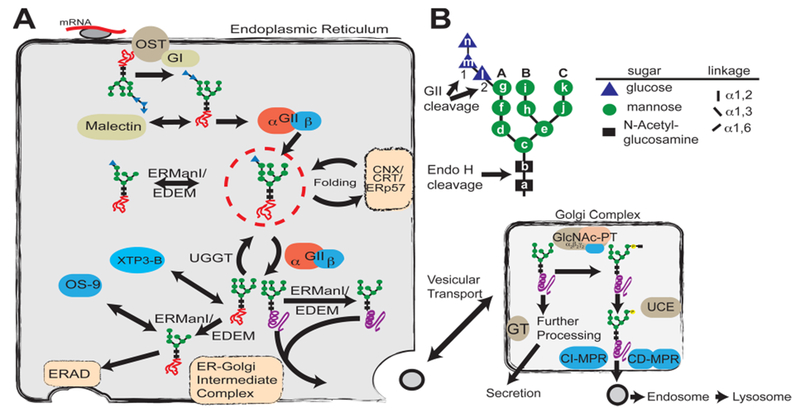
Glycan processing in the secretory pathway. (A) Role of glycosylation in the determination of protein fate in the ER and Golgi complex. A preassembled Glc3Man9GlcNAc2 (G3M9) is transferred by oligosaccharyltransferase (OST) from a dolichol-PP-glycan donor to a consensus sequence asparagine-X-serine/threonine (X can be any amino acid except proline) in a nascent polypeptide chain (red line). Glucosidase I (GI) removes the first glucose residue (residue n), producing a G2M9 glycan that is a substrate for malectin as well as glucosidase II (GII). GII catalyzes the hydrolysis of the second glucose (residue m), leaving G1M9 on the newly synthesized glycoprotein. The protein is then recognized by calnexin/calreticulin (CNX/CRT), which in turn recruits ERp57, a disulfide protein isomerase. GII cleaves the innermost glucose (residue l), making it a target for UDP-Glc:glycoprotein glucosyltransferase (UGGT), which may add residue l depending on the folding status of the glycoprotein. Throughout the folding process, proteins are also targets for ER mannosidases. Folded proteins (purple helix) shuttle to the ER–Golgi intermediate complex for subsequent vesicular transport, whereas non-native structures (orange scrambled lines) are reglucosylated by UGGT for recognition by the CNX/CRT cycle or further processed by mannosidases for removal by the endoplasmic reticulum-associated degradation (ERAD) pathway. (B) Structure of the glycan transferred to proteins during N-glycosylation. Individual hexose moieties are labeled a–n in the order of synthesis of the dolichol-PP-glycan. First and second cleavages by GII, the endo-β-N-acetylglucosaminidase H (Endo H) cleavage site, and arms A–C of the glycan are indicated.
GII is a key player in the regulation of the cycles occurring during the quality control of glycoprotein folding. GII is a heterodimer formed by a catalytic GIIα subunit and a regulatory GIIβ subunit. In vitro and in vivo studies have revealed that the hydrolytic activity of GII is enhanced by the mannose binding activity of the mannose 6-phosphate receptor (MPR) homology domain (MRH domain) contained within its β subunit (Figure 2A). The lack of the GIIβ subunit abolishes trimming of both the second and third glucose by GII (Figure 1B) in vitro and significantly delays trimming of glycoproteins bearing such species in vivo.18,19 The GIIβ MRH domain recognizes mannose residues in arm B and/or arm C of the transferred N-glycan,18,20,21 and as shown in vivo, the absence of these terminal mannose residues prevents GII from trimming Glc in arm A in an MRH-dependent manner.12 Together, these studies show that optimal GII deglucosylation activity requires a functional GIIβ MRH domain and nascent glycoproteins bearing Man9-containing glycans. The importance of proper deglucosylation by GII, and specifically the role of the MRH containing the β subunit, has been illustrated in a study of transient receptor potential channel polycystin 2 in which insufficient glycosylation results in a reduced level of interaction with GIIβ, leading to degradation of transient receptor potential channel polycystin 2 and manifestation of autosomal dominant polycystic kidney disease.22 Furthermore, mutation of the PRKCSH gene, encoding GIIβ, is one of two genes in which defects cause autosomal dominant polycystic liver disease.23,24
Figure 2.
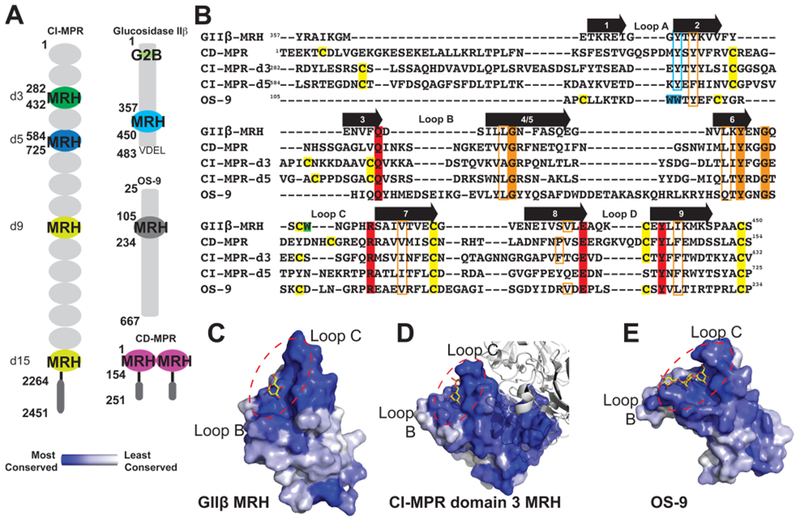
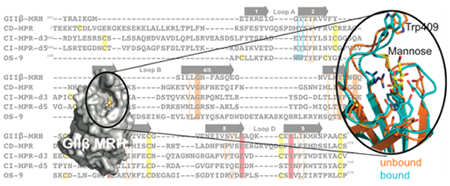
Comparison of glycan binding MRH domains from GIIβ, CD-MPR, CI-MPR, and OS-9. (A) Schematic representation comparing the domain organization of the ER resident protein GIIβ subunit from Schizosaccharomyces pombe and human OS-9 with the bovine CD-MPR and CI-MPR that recycle between the Golgi complex, endosomes, and cell surface. The transmembrane and cytosolic region of the CD-MPR and CI-MPR are indicated by a horizontal black line and dark gray oval, respectively. The mature proteins, lacking the signal sequence, are depicted. The numbering of residues begins at the mature protein for GIIβ, CD-MPR, and CI-MPR and at the initiator methionine for OS-9. The carbohydrate binding MRH domains of CI-MPR are highlighted (domain 3, d3; domain 5, d5; domain 9, d9; domain 15, d15). The structures are known for the MRH domains of domains 3 and 5 of CI-MPR, CD-MPR, OS-9, and GIIβ, and their amino acid sequences are shown in panel B. (B) Structure-based sequence alignment of MRH domains from MRH-bearing proteins. Residues essential for Man-6-P binding (Q, R, E, and Y) in MPRs are highlighted in red, and tryptophan residues of OS-9 involved in Manα1,6Man linkage recognition are highlighted in cyan. The corresponding tyrosine residue in GIIβ-MRH, domain 3 and 5 of the CI-MPR, and the CD-MPR are boxed in cyan. W409 of GIIβ is highlighted in green. The newly identified conserved glycine and tyrosine residues are highlighted in orange, while conserved hydrophobic residues are boxed in orange. Cysteine residues are highlighted in yellow. The molecular surface of the MRH domain of GIIβ (C), domain 3 of CI-MPR (D), and OS-9 (E) are shown colored by species conservation as determined by ConSurf (http://consurf.tau.ac.il/).50 The ligand binding region in panels C–E is circled with a dashed red line.
The MRH domain of GIIβ (Figure 2A,B) shares homology with other MRH family members (for a complete listing, see ref 25). Structural studies have been undertaken on several members, including (1) mannose 6-phosphate receptors (CI-MPR and CD-MPR), type I membrane proteins involved in the transport of lysosomal enzymes from the Golgi to lysosomes through recognition of the Man-6-P signal that are the founding members of the MRH family of proteins,26,27 (2) OS-9, and (3) GIIβ (see Figure 1A). The crystal/nuclear magnetic resonance (NMR) solution structures of the CD-MPR,28,29 MRH domains 1–330, 31 and MRH domain 532 of the CI-MPR, the MRH domain of OS-9,17 and, in this report, the MRH domain of GIIβ have been determined in the presence of ligand. The binding site region of each of these MRH domains is highly conserved among species (Figure 2C–E). Importantly, the four conserved residues (glutamine, arginine, glutamate, and tyrosine) essential for Man-6-P recognition by the MRH domains of the MPRs are also present in GIIβ (Figure 2B), and mutagenesis of these residues in the Schizosaccharomyces pombe GIIβ MRH domain abolished the ability of the GIIβ subunit to enhance glycan trimming by GII in live cells.18
We previously reported the structure of the MRH domain of S. pombe GIIβ in a ligand-unbound state.33 This structure revealed a tryptophan residue, W409 (Figure 2C), located close to the primary mannose binding pocket (which includes the four essential glutamine, arginine, glutamate, and tyrosine residues) that is conserved in GIIβ among all species. Mutation of W409 decreased the efficiency of GII’s removal of glucose residues from G2M9 and G1M9 both in vivo and in vitro, although not to the extent observed for GII harboring a mutation of either glutamate or tyrosine in the primary mannose binding pocket.18,33 However, contrary to the mutations within the primary mannose binding pocket, we observed that the W409A mutant was still able to bind Manα1,2Man as monitored by 15N–1H HSQC and surface plasmon resonance analyses using the lysosomal enzyme acid α-glucosidase. To further investigate the mechanism by which GIIβ binds glycans to enhance the catalytic activity of GII, structural studies of wild-type and mutant forms of the MRH domain of GIIβ subunit were performed and evaluated by in vitro and in vivo GII activity assays. In the report presented here, we present the crystal structure of the MRH domain of GIIβ bound to mannose at 1.6 Å resolution and probe the structure of mutants bearing single-amino acid substitutions by NMR spectroscopy that includes a solution structure of the W409A mutant. Comparisons to our previously reported apo GIIβ MRH structure, as well as to the structures of other MRH domains reported to date, provide insight into how this family of proteins is able to recognize and bind a diverse set of carbohydrate ligands.
MATERIALS AND METHODS
Materials.
Yeast extract and bactopeptone were from Difco. Endo-β-N-acetylglucosaminidase H, porcine trypsin, p-nitrophenyl α-d-glucopyranoside (pNPG), dithiothreitol, amino acids and supplements for culture media, and protease inhibitors were from Sigma. [14C]Glucose (301 Ci/mol) was from PerkinElmer Life Sciences. [15N]Ammonium chloride, [13C]glucose, mannose, and supplements for culture media were from Sigma. Manα1,2Man was purchased from Dextra Laboratories.
Strains and Media.
Escherichia coli JA226 was used for cloning purposes. Bacteria were grown at 37 °C in LB medium (0.5% NaCl, 1% tryptone, and 0.5% yeast extract) supplemented with 200 μg/mL ampicillin or 50 μg/mL kanamycin. S. pombe cells were grown at 28 °C in YES medium (0.5% yeast extract, 3% glucose, and 75 μg/L adenine) or EMM minimal medium34 supplemented with adenine (75 μg/L), uracil (75 μg/L), and/or leucine (250 μg/L) as needed. The S. pombe ΔGIIβ and ΔGIIα strains used were ADmIIβ (h−, leu1-32, ade6-M210, ura4-D18, Δgls2β::ura4+) described in ref 18 and Sp61IIα (h−, gls2α::ura4+, leu1-32, ade6-M210, ura4-D18, ade1) described in ref 35, respectively.
Generation, Expression, and Purification of GIIβ MRH Constructs.
The cDNA encoding residues 357–450 of the mature glucosidase IIβ domain was cloned into a modified pQE30 vector harboring the sequence for the small ubiquitin-like modifier (SUMO) protein with an N-terminal hexahistidine tag and transformed into BL21(pREP4) cells. Mutant cDNAs were generated using DpnI-mediated site-directed mutagenesis and confirmed by DNA sequencing. Proteins were grown, purified from inclusion bodies, and refolded as previously described.33
Crystallization, Structure Determination, and Refinement of the MRH Domain of GIIβ in Complex with Mannose.
Crystallization conditions were originally screened by the sitting drop vapor diffusion method using an in-house sparse matrix screen kit consisting of 96 conditions. Crystals were grown at 19 °C utilizing the hanging drop vapor diffusion method by mixing protein [10 mg/mL GIIβ MRH domain protein in 20 mM Tris (pH 7.5), 150 mM NaCl, and 100 mM mannose] with well solution [100 mM triethanolamine hydrochloride (pH 8.25), 200 mM potassium glutamate, and 28% PEG 4000] in a 1:1 ratio. Crystals were passed through a cryo-solution containing well solution and 20% glycerol prior to being flash-frozen in liquid nitrogen. Diffraction data were collected (720 frames using 0.5° oscillations in 10° wedges 180° apart) using a Rigaku Micromax 007 generator with an R-AXIS IV++ detector. Data processing was conducted using HKL2000.36
The structure of the MRH domain was determined by single-wavelength anomalous dispersion (SAD) using the SHELX suite of programs37 for identification of the native sulfur substructure (five sites) and polyalanine model building. Final model building and refinement were conducted with the Phenix suite of programs.38 Iterative rounds of model refinement were conducted with COOT.39 The crystals belong to space group I4 and contain one polypeptide chain per asymmetric unit. Final model [Protein Data Bank (PDB) entry 4XQM] statistics are listed in Table 1.
Table 1.
X-ray Diffraction, Phasing, and Model Statistics
| Data Collection | |
| wavelength (Å) | 1.54 |
| resolution range (Å) | 29.16–1.625 (1.683–1.625) |
| space group | I4 |
| unit cell | 57.57 Å, 57.57 Å, 58.32 Å, 90°, 90°, 90° |
| total no. of reflections | 153737 (5953) |
| no. of unique reflections | 12000 (1146) |
| multiplicity | 12.8 (5.2) |
| completeness (%) | 99.59 (95.82) |
| mean I/σ(I) | 35.09 (4.07) |
| Wilson B factor (Å2) | 15.32 |
| Rmerge | 0.063 (0.406) |
| Phasing | |
| no. of sulfurs | |
| disulfide | 2 |
| methionine | 1 |
| SHELXD CC all/weak | 56.6/36.17 |
| Refinement | |
| Rwork, Rfree | 0.167 (0.298), 0.197 (0.324) |
| total no. of atoms | 891 |
| no. of macromolecules | 737 |
| no. of ligands | 12 |
| no. of waters | 142 |
| no. of protein residues | 94 |
| rmsd for bonds (Å) | 0.009 |
| rmsd for angles (deg) | 1.4 |
| Ramachandran favored (%) | 97 |
| Ramachandran outliers (%) | 0 |
| Clashscore | 3.37 |
| average B factor (Å2) | 17.7 |
| macromolecules (Å2) | 15.8 |
| ligands (Å2) | 18.6 |
| solvent (Å2) | 27.2 |
| PDB entry | 4XQM |
GIIβ Mutagenesis and Expression in S. pombe.
The S. pombe GIIβ subunit cloned in the gateway pDONR201 plasmid, obtained from the RIKEN DNA Bank,40 was used as the template for single-amino acid polymerase chain reaction mutagenesis of the GIIβ MRH domain (Y372A or Y372F). The numbering corresponds to the mature protein’s initial amino acid (i.e., without the first 23 residues corresponding to the signal peptide). The amplified mutant DNA containing both GIIβ and vector sequences was phosphorylated, religated, and electroporated into bacterial cells. Primers Y372F forward mutagenic primer 5′-TTCACATACAAGGTGGTG-3′ or Y372A forward mutagenic primer 5′-GCTACATACAAGGTGGTG-3′ and Y372 reverse primer 5′-GCCACCAATTTCACGTTTTG-3′ were used for amplification (mutagenic codons are underlined). After ligation and verification of the successful mutagenesis by sequencing, wild-type and mutant GIIβ DNA clones were transferred to the pREP1-ccdb2 Gateway-compatible S. pombe destination expression vector (RIKEN DNA Bank) by the LR clonase recombination reaction (Invitrogen). These constructs (pREP1-GIIβ Y372A and pREP1-GIIβ Y372F) were then electroporated into S. pombe competent ΔGIIβ cells. The pREP1-GIIβ W409A construct used in this study was described by Olson et al.,33 and the wild-type and MRH binding pocket mutant construct used in this study (pREP1-GIIβ and pREP1-GIIβ Y439F, respectively) were described by Stigliano et al. (please note that the numbering of this last mutant was changed from Y462F used in ref 18 as throughout this work the numbering corresponds to the mature protein sequence instead of starting at the initial Met).
GII Activity Assays.
GII activity was assayed in S. pombe microsomal fractions either (1) using the small substrate analogue p-nitrophenyl α-d-glucopyranoside (pNPG) or (2) using radioactively labeled monoglucosylated N-glycans as substrates as described in ref 18. Briefly, GII activity in ΔGIIβ mutants transformed with either wild-type or mutant GIIβ subunits was assayed using 5 mM pNPG as the substrate in 0.1 M HEPES buffer (pH 7.2) for 20 min at 37 °C, and the absorbance at 405 nm was measured, or with [14C-glucose]-Glc1Man9GlcNAc (obtained as described in ref 33) in 40 mM sodium phosphate buffer (pH 7.2) for 15 min at 30 °C. In the latter case, glucosidase activity was determined as the percentage of radioactive glucose released with respect to the total radioactivity.
Analysis of Glycans Produced in Vivo by S. pombe Cells.
To assess ER N-glycan composition in S. pombe cells expressing different GIIβs, we harvested S. pombe cells growing in the exponential growth phase, extensively washed them with 1% YNB medium without glucose, and resuspended them in 2 volumes (v/w) of the same medium. Cells (0.5 mL) were then preincubated for 5 min in 5 mM dithiothreitol and pulsed for 15 min with 5 mM glucose containing 150 μCi of [14C]glucose (300 Ci/mol). The labeling procedure and the preparation of whole cell endo-α-N-acetylglucosaminidase H-sensitive N-glycans have been previously described.41 Glycans were separated by paper chromatography (Whatman 1) in a 1-propanol/nitromethane/H2O mixture (5:2:4), and the peaks were identified by standards run in parallel. To improve the resolution, the identified glycans were eluted from the paper and resolved by high-performance liquid chromatography (HPLC) at room temperature using a TSK-GEL Amide-80 column (4.6 mm ϕ × 25 cm, Tosoh) with a H2O/CH3CN mobile phase with a linear gradient from 35:65 to 55:45 over 65 min at a flow rate of 0.75 mL/min. As slight variations in retention times among runs could be observed, the positions of the peaks in paper chromatography relative to known standards and not the retention times from HPLC were used to identify glycans.
Antibodies and Immunodetection.
Rat anti-GIIα polyclonal antiserum was obtained as described in ref 18 except that GIIα protein fragment expressed in bacteria and purified from inclusion bodies was injected in a Wistar male rat in four boosters of 50 μg protein each. Polyclonal antiserum thus obtained was tested by Western blotting using S. pombe wild-type microsomal fractions. Microsomes of ΔGIIα mutant cells were also used as a control to discard any unspecific binding of the obtained antiserum. Microsomal S. pombe proteins (125 μg) were resolved in an 8% sodium dodecyl sulfate (SDS)–polyacrylamide gel, electroblotted to Immobi-lonP membranes (Millipore), and incubated with rat anti-S. pombe GIIα (1:1000) or mouse anti-S. pombe GIIβ (1:5000) antibodies as described in ref 12. Secondary goat anti-mouse or goat anti-rat IgG conjugated to horseradish peroxidase was from Sigma. Immunodetection was conducted using enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate, Thermo Scientific) as described by the manufacturer.
Data Collection and Structure Determination by Heteronuclear NMR Spectroscopy.
NMR spectra were collected as previously described.32 Briefly, NMR spectra were collected at 25 °C on a Bruker 600 MHz spectrometer equipped with a triple-resonance CryoProbe. Data were processed using NMRPipe42 and visualized with XEASY.43 Complete 1H, 15N, and 13C resonance assignments for the W409A [0.7 mM protein, 10 mM deuterated imidazole (pH 7.1), and 150 mM NaCl] mutant of GIIβ MRH were determined by the transfer of assignments from the previously published wild-type structure collected under the same sample conditions33 and confirmed using three-dimensional HNCO, HCCH total correlation spectroscopy and 13C (aromatic)-edited NOESY-HSQC spectra. The structure was calculated using distance constraints derived from three-dimensional 15N-edited NOESY-HSQC and 13C-edited NOESY-HSQC spectra. Backbone ϕ and ψ dihedral angle constraints were generated using TALOS+44 and the secondary shifts of the 1H, 13Cα, 13Cβ, 13C′, and 15N nuclei. The NOEASSIGN module of CYANA45 was used to generate initial structures that subsequently went through iterative rounds of manual assignment in CYANA to remove constraint violations. The Xplor-NIH46 molecular dynamics protocol with explicit solvent47 was then used to further refine the top 20 CYANA conformers with the lowest target function. Final model statistics are listed in Table 2.
Table 2.
NMR and Refinement Statistics for the 20 MRH3 W409A Conformers
| experimental constraints | |
| distance constraints | |
| long-range (|i – j| > 5) | 598 |
| medium-range (1 < |i – j| ≤ 5) | 194 |
| sequential (|i – j| = 1) | 365 |
| intraresidue (i = j) | 297 |
| total | 1454 |
| dihedral angle constraints (ϕ and ψ) | 99 |
| average atomic rmsd from the mean structure (Å) (residues 365–409, 412–449) | |
| backbone (Cα, C′, N) | 0.47 ± 0.06 |
| heavy atoms | 0.89 ± 0.06 |
| deviations from idealized covalent geometry | |
| bond lengths (Å) | 0.019 |
| torsion angles (deg) | 1.4 |
| constraint violations | |
| no. of NOE distances >0.3 Å | 0.00 ± 0.00 |
| NOE distance rmsd (Å) | 0.016 ± 0.008 |
| no. of torsion angle violations >5° | 0.00 ± 0.00 |
| torsion angle violation rmsd (deg) | 0.588 ± 0.086 |
| WHATCHECK quality indicators | |
| Z-score | −0.37 ± 0.27 |
| rms Z-score | |
| bond lengths | 0.80 ± 0.03 |
| bond angles | 0.69 ± 0.02 |
| bumps | 0.00 ± 0.00 |
| Lennard-Jones energy (kJ mol−1) | −2,230 ± 48 |
| Ramachandran statistics (% of all residues) | |
| most favored | 76.2 ± 2.6 |
| additionally allowed | 18.0 ± 2.6 |
| generously allowed | 1.5 ± 1.2 |
| disallowed | 4.3 ± 1.5 |
RESULTS AND DISCUSSION
Crystal Structure of the GIIβ MRH Domain Bound to d-Mannose.
We previously reported the unbound structure determined by NMR at pH 7.2 (150 mM NaCl) of the isolated MRH domain of GIIβ.33 However, the low-affinity binding of the isolated GIIβ MRH domain for either mannose, Manα1,2-Man or Man-6-P, precluded our ability to determine the solution structure of the GIIβ MRH domain complexed to carbohydrate by NMR spectroscopy. We have now obtained crystals of the GIIβ MRH domain in complex with mannose. The final model (Figure 3A, PDB entry 4XQM) was refined to 1.6 Å resolution [Rwork = 16.7%, and Rfree = 19.7% (Table 1)] and shows that, as previously described,33 the MRH domain of GIIβ adopts the classical flattened β-barrel MRH fold comprised of a four-stranded antiparallel β-sheet orthogonal to a five-stranded antiparallel β-sheet, with strand 9 interjected between strands 7 and 8 (Figure 3B). In contrast to other MRH domains, the GIIβ MRH domain lacks disulfide bridges in the N-terminal, four-stranded β-sheet but does contain the two highly conserved disulfides in the C-terminal, five-stranded β-sheet (Figure 2B).
Figure 3.
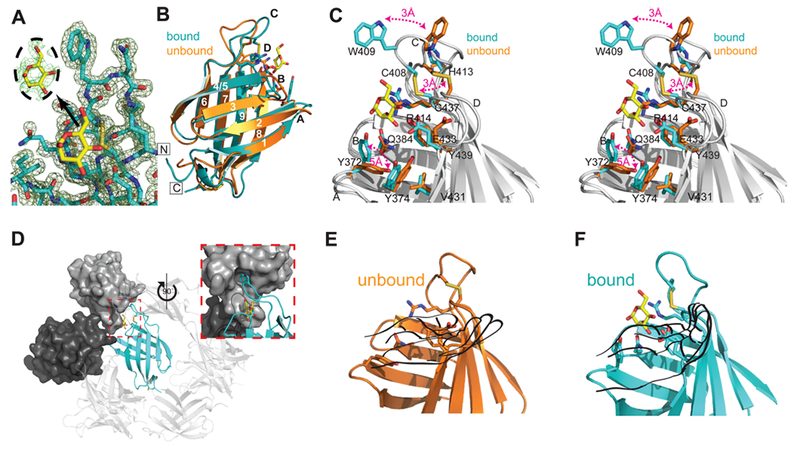
Crystal structure of the S. pombe GIIβ MRH domain bound to mannose. (A) Final 2Fo – Fc map contoured at 1σ with the Fo – Fc map for the ligand contoured at 2σ displayed in the inset. (B) Crystal structure of the MRH domain bound (PDB entry 4XQM) (cyan) to mannose (yellow) in comparison to the NMR structure of the unbound (orange) protein (PDB entry 2LVX). Loops A–D and β-strands 1–9 are labeled. The N- and C-termini are labeled and boxed. (C) Close-up stereoview of the binding pocket of the superimposed bound (cyan) and unbound (orange) structures with residues shown as sticks. Three major changes between the apo and bound structures are highlighted with red double arrows. Additionally, when ligand binds, the binding pocket narrows as the hydroxyl group of Y439 moves toward the N-terminal β-sheet by slightly more than 1 Å, and Tyr374 rotates ~45° so that the plane of its phenolic ring is oriented perpendicular to the side chain of V431. (D) Crystallographic packing of GIIβ MRH (cyan ribbon). The molecular surface of the cystallographic neighbor that contacts both W409 and the ligand is colored dark gray. The molecule that further occludes the binding region is colored light gray. The inset is rotated 90° with reference to a main panel. Ribbon diagram of GIIβ MRH domain in the absence (E) and presence (F) of mannose (yellow). Although in the unbound NMR structure previously reported33 the chemical shifts for these nitrogen atoms and their associated protons could not be determined, the energy-minimized structure consistently used the fully extended rotamer of R414. Figures were generated using PyMOL.51
The binding pocket is composed of a primary mannose binding site containing the four conserved residues (Q384, R414, E433, and Y439) located on both β-sheets and a more variable, extended site comprised of loops C (joining β-strands 6 and 7) and D (joining β-strands 8 and 9) that recognize the 6-OH group of mannose (Figure 3C). In the MPRs, this extended site recognizes moieties (phosphate or a phosphodiester, GlcNAc-1-phosphate) attached to the 6-OH of the mannose residue. Located within hydrogen bonding distance of the mannose are Y372 on β-strand 2 and Q384 on β-strand 3, forming the N-terminal side of the pocket, and residues on β-strands 7 (R414), 8 (E433), and 9 (Y439) that form the C-terminal side of the binding pocket.
Structural Comparison of GIIβ-MRH with and without Bound Mannose.
Ideally, we would have liked to make comparisons between apo and bound structures determined by the same biophysical technique. However, despite numerous attempts to crystallize the MRH domain of GIIβ in its unbound form, no suitable diffraction quality crystals were obtained. A comparison of apo and bound structures reveals that the MRH domain of GIIβ demonstrates no overall change in the fold with a rmsd of 1.4 Å over 89 Cα atoms (Figure 3B). This is unlike the dimeric CD-MPR that demonstrates an ~16 Å loop D movement that is also accompanied by changes in the dimer interface upon ligand binding.48 However, differences exist between the apo and bound states that are distributed over the entire binding pocket. Several major changes are highlighted in Figure 3C. One is the repositioning of residues in loop C: residues between C408 and H413 pivot toward the 6-OH of the mannose, moving the Cα atom of W409 3 Å (Figure 3C). In the bound structure, the W409 indole ring is stabilized by its insertion into a hydrophobic pocket of an adjacent crystallographic neighbor (Figure 3D).
To assist in visualizing the other major changes that occur upon ligand binding, the binding site can be thought of as a left hand with the thumb oriented along β-strand 4/5 and loop C being the two fingers adjacent to the thumb (Figure 3E,F). In the presence of ligand (Figure 3F), the guanidinium group of R414 on the middle finger is rotated upward ~90°, moving ~4.5 Å relative to the ligand-free structure. This repositioning of the R414 guanidinium group allows the C408–C437 disulfide bond to move 3 Å into the binding pocket (Figure 3C,E,F). The sulfur atoms are now positioned <4 Å from the εN atom of R414, allowing for a possible electrostatic interaction. The residues on the peripheral edge (i.e., the heel of the hand near the base of the thumb) of the binding site are also affected by the presence of ligand. The ~90° counter-clockwise rotation of the side chain of Y372 toward strand 3 is the most substantial, with the hydroxyl group being relocated by greater than 5 Å (Figure 3C,E,F). The movement of this side chain toward the ligand allows the hydroxyl group to come within hydrogen bonding distance of the 1-hydroxyl group of the ligand. Tyr374 is rotated ~45° in the direction of Y372. These movements together create a bowl in the palm of the hand surrounding the mannose ring (Figure 3C,E,F).
Comparison of GII-MRH to Other Ligand-Bound MRH Structures.
A comparison of the known MRH domain structures with bound ligands (CD-MPR, domains 1–3 of CI-MPR, OS-9, and GII-MRH) reveals the anticipated similarities: these MRH family members have the four essential mannose binding residues (glutamine, arginine, glutamate, and tyrosine) spatially conserved (Figure 4) and all within hydrogen bonding distance of their respective ligands (Figure 4A–D). However, this growing ensemble, now containing five bound MRH domain structures, allows for a more accurate structure-based sequence alignment leading to the ability to extract more information about how a MRH domain binding pocket is maintained. First, not surprisingly, there are a subset of residues that contribute to the hydrophobic core between the two β-sheets: L389, L400, V418, V431, and I441 (Figures 2B and 5A). These residues, with the exception of L389, fall on a line along the C-terminal sheet. Second, there are three absolutely conserved amino acids that may be important in maintaining the integrity of the binding pocket. The two absolutely conserved glycine residues (G390 and G405) are positioned at a critical junction in the structure: they tether the N-terminal side of loop C to β-strand 4/5 in a manner analogous to the function of the disulfide on the C-terminal side (Figure 5A). Together, these two structural elements appear to set the width of the 6-OH binding site. The other absolutely conserved residue among these five MRH domains is Y402 and appears to form the spacer between β-strand 3 and the C-terminal β-sheet by having its bulky side chain oriented perpendicular to the strands. Finally, Y374, which alters its position in the presence of ligand, is conserved in all but domain 5 (where it is a phenylalanine) and aligned in the same manner to allow for a favorable hydrophobic interaction with V431 in the GIIβ-MRH structure as well as that of OS-9 (Figure 5A).
Figure 4.
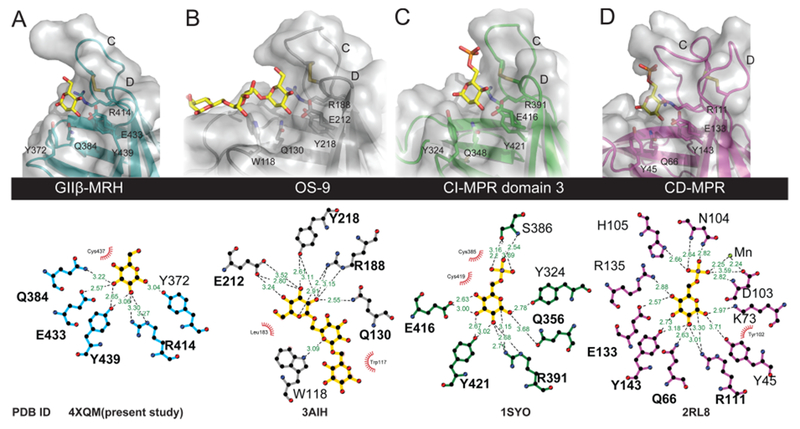
Comparison of ligand binding pockets of MRH domains. (A–D) Close-up view of the carbohydrate binding sites (top) of the MRH domains showing the four essential mannose binding residues along with the proposed glycosidic linkage-sensing tyrosine (Manα1,2Man) or tryptophan (Manα1,6Man). Ligands present in the determined structures are colored yellow. Molecular surfaces are shown over the ribbon models. The bottom panel was generated using LigPlot+52 and demonstrates the potential hydrogen bonding between the ligand and each of the MRH domains of the various proteins. Potential hydrogen bonding distances are colored green, and potential hydrophobic interactions are colored red. The PDB entries are listed for each structure (the bound structure of domain 5 was not included because the ligand is not refined).
Figure 5.
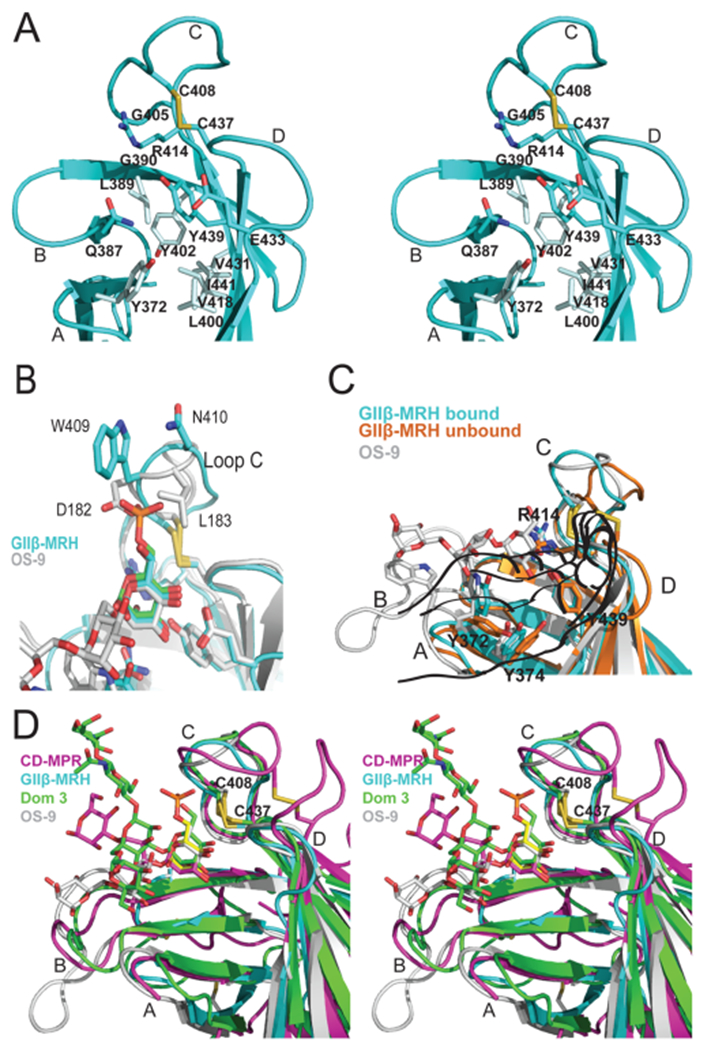
Specificity of the binding sites of MRH domains. (A) Ribbon diagram depicting a close-up stereoview of the binding pocket of the GIIβ MRH domain. Loops A–D along with the hydrophobic residues discussed in the text are labeled. The cysteine residues involved in a disulfide bridge (C408–C437, yellow) and side chains (sticks) of residues in the binding pocket are also shown. (B) Ribbon diagram of the loop C region of the MRH domains of GIIβ (cyan) and OS-9 (gray) with their respective ligands colored according to protein and the superimposed Man-6-P ligand colored green. Disulfide bridges are colored yellow. (C) Ribbon diagram of the GIIβ MRH domain in the absence (orange) and presence (cyan) of mannose. For comparison, the positioning of OS-9’s ligand (trisaccharide in the crystal structure) and the corresponding loop with W118 are colored gray. Disulfide bridges are colored yellow. (D) Ribbon diagram depicting a close-up stereoview of the binding pocket showing the superimposition of CD-MPR bound to pentamannosyl phosphate (magenta), domain 3 of CI-MPR (Dom3) bound to the asparagine-linked oligosaccharide of a crystallographic neighbor (green), OS-9 bound to a mannopentose (gray), and GIIβ (cyan) bound to mannose (yellow) MRH domains. Loops A–D are labeled, and the disulfide bridge is shown (C408–C437, yellow).
Although these four MRH domains are all specific for mannose, they differ in their specificity for modifications on the 6-OH as well as glycosidic linkage. A key to understanding the individual specificities lies in the evaluation of the differences between the four ligand-bound structures. The relative position of the binding pocket disulfide, along with the length and amino acid composition of loop C, is critical for specificity of ligand binding: this is the region that allows either a phosphate monoester or diester moiety to bind to the MPRs. As shown in Figure 2B, this region varies widely among MRH domains in amino acid composition, each reflecting the preference of that individual MRH domain. Focusing on the two ER resident protein MRH domains, OS-9 and GII, their loop C region should be similar because modifications to the 6-OH of mannose do not occur until after a protein’s exit from the ER. OS-9 bound to Manα1,6Man displays an occluded binding site at the 6-OH with D182 and L183 bracketing both sides of this hydroxyl (Figure 5B). We previously reported that the MRH domain from GIIβ is insensitive to modifications (phosphate or phosphate-GlcNAc) on the 6-OH group of mannose.33 From the structure presented here bound to mannose, it can now be seen how this lack of discrimination is possible. Although GIIβ MRH contains a tryptophan residue (W409) immediately C-terminal to the disulfide, this residue and its neighbors are oriented with the side chains positioned away from the mannose ring rather than toward mannose as observed in the other MRH domains. This positioning creates an open region, indifferent to the presence or absence of modifications on the 6-OH.
The heel of the hand harbors the extended binding site across which the remainder of the glycan, and the associated linkages, passes and is variable between the structures (Figure 5C,D). Inspection of the ligand-bound structures having two or more mannose rings bound illustrates that loop A is more highly conserved between these structures in length and position and harbors the proposed glycosidic linkage-sensing tyrosine (Y372) (see below) or WW motif, while loop B varies significantly in length and orientation as previously discussed. It should be noted that, with the exception of S. pombe, Arabidopsis thaliana, and Magnaporthe oryzae, the GIIβ MRH domain also possesses an extended loop B.
Role of a Conserved Tyrosine (Y372) among MRH Domains.
It has been proposed that Y372 and its counterpart in the other MRH domains serves as a glycosidic linkagesensing residue.17 As shown in Figure 2, Y372 is conserved in all MRH domains that recognize Manα1,2Man linkages, but tryptophan is in the corresponding position in OS-9 that recognizes Manα1,6Man linkages. In our previously published unbound structure of GIβ MRH,33 Y372 was highly constrained in position such that the side chain was perpendicular to the would-be-bound mannose ring. However, in the currently reported mannose-bound structure, the position of the phenolic ring has changed, rotating ~90° toward the mannose ring, and is within hydrogen bonding distance of the 1-OH of the terminal mannose residue (Figure 3C,E,F). This movement appears to be the result of a series of concerted movements of side chains that extends systematically throughout the binding site from Y439 to Y372. Residue Y372 is uniquely situated at the position of the glycosidic linkage and could serve to extend carbohydrate recognition out to the penultimate mannose. The residue directly preceding Y372 may also play an important role in the extended binding site. In the case of OS-9, W117 serves to ring stack with the third α1,6-linked mannose residue from the nonreducing end of the sugar [mannose c (Figure 1B)]. When ligand binds, the movements of R414, Y439, Y374, and Y372 together convey the presence of ligand from the most internal region of the carbohydrate binding site to the more external region of the binding site, a concerted rearrangement allowing for favorable, specific binding of ligand (Figure 4C).
To evaluate the role of this conserved residue, we studied the influence of Y372 of the GIIβ MRH domain on GII activity both in vitro and in vivo. Y372 was mutated (Y372A or Y372F) in full length GIIβ, and the wild type or mutated variants were expressed in a GIIβ deficient (ΔGIIβ) strain of S. pombe. Microsomal fractions from these strains were then assayed for GII activity toward both the small substrate analogue pNPG and the glycan [14C-Glc]Glc1Man9GlcNAc. We reported previously that GIIβ is not required to hydrolyze the small substrate analogue pNPG,18 and thus, microsomal GII activity using pNPG as a substrate reflects ER GIIα content, which appears to be the same for all constructs in these experiments (Figure 6A). Moreover, mutation of Y372 did not affect the GIIβ or GIIα content in the ER as shown by Western blotting (Figure 6B). Even though cells expressing mutant variants of GIIβ had normal GIIα ER levels, mutation of Y372 substantially decreased the in vitro activity of the enzyme toward glycans by approximately the same extent as the previously reported loop C W409A mutation, but not to the same degree as the MRH mannose hydroxyl binding mutant, Y439F (Figure 6A).
Figure 6.
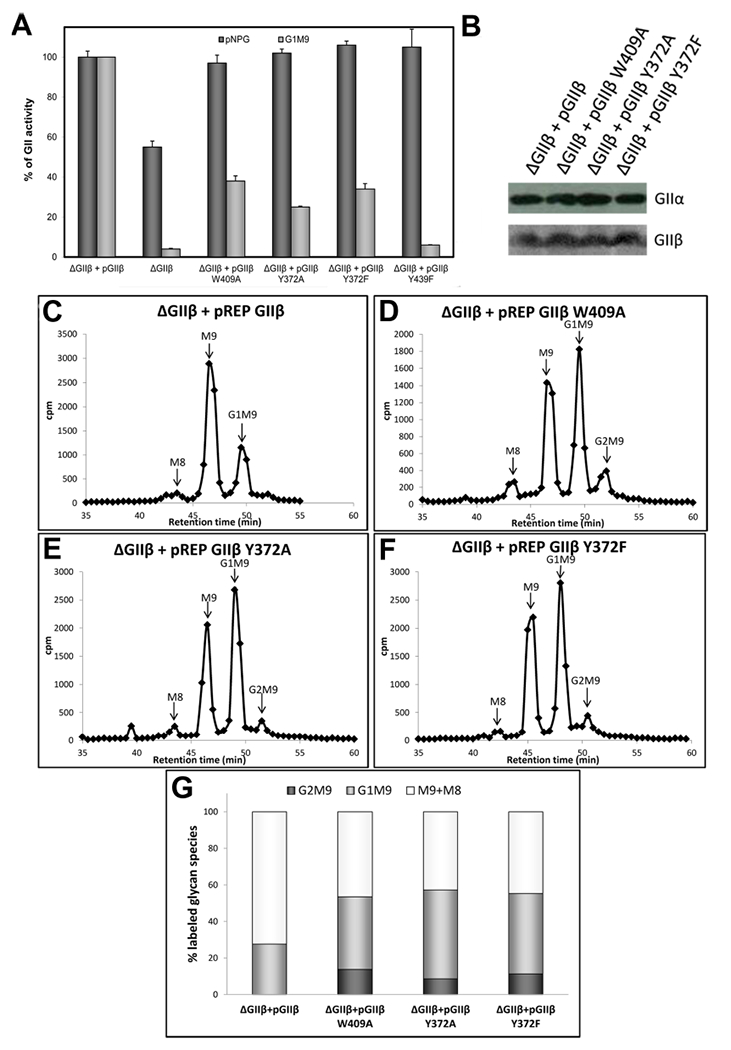
Influence of Y432 in GIIβ-mediated enhancement of GII activity toward N-glycans. (A) GII activity toward pNPG or Glc1Man9GlcNAc was measured using 125 μg of microsomes from S. pombe cells lacking endogenous GIIβ (ΔGIIβ) and expressing either GIIβ or the same subunit displaying the indicated mutations. The activity of ΔGIIβ cells transformed with wild-type GIIβ was taken as 100% for each substrate. Error bars represent the standard deviation. (B) Wild-type and mutant GIIβ expression levels and ER content of GIIα in the different S. pombe strains were evaluated by Western blotting. Microsomal proteins (125 μg) of S. pombe were resolved by 8% SDS-PAGE, transferred to a PVDF membrane, and blotted using rat polyclonal anti-GIIα (1:1000) or mouse polyclonal anti-GIIβ (1:5000). Goat antimouse IgG (1:5000) or goat antirat IgG (1:4000) conjugated to HRP were used as secondary antibodies. Reactions were detected by chemiluminescence. (C–F), glycan patterns synthesized in vivo by S. pombe ΔGIIβ mutant cells expressing GIIβ (C) or mutated GIIβ versions (D-F). Quantification of the relative amounts of the di-, mono-, and unglucosylated species from panels C–F is shown in panel (G). The label of Man8 species was added to that in Man9 species to account for unglucosylated glycans.
We then analyzed the N-glycan pattern produced in vivo by ΔGIIβ cells expressing GIIβ harboring either a Y372 mutation (Y372A, Y372F) or the previously characterized W409A mutation,33 thereby allowing for the evaluation of the effect of these amino acids on GII glucose trimming activity in vivo. S. pombe cells were incubated with [14C]Glc for 15 min in the presence of 5 mM dithiothreitol, which inhibits nascent glycoproteins from exiting the ER, thus preventing further extension of the newly synthesized N-glycans. Total cell high-mannose-type N-glycans were released with endo-β-N-acetylglucosaminidase H, which cleaves within the chitobiose core (Figure 1B), and the glycans were isolated and analyzed as described in Materials and Methods. The N-glycan patterns obtained from S. pombe cells expressing Y372 mutations in GIIβ revealed an altered pattern similar to that previously obtained for W409A: while deglucosylation of the transferred glycan Glc3Man9GlcNac2 was rapid and few glucose-containing glycans were detected in cells expressing wild-type GIIβ (Figure 6C), the amount of di- and monoglucosylated glycans was significantly higher in cells harboring W409 (Figure 6D) and Y372 (Figure 6E,F) mutations. Quantification of each dimono or unglucosylated glycan species in each panel is summarized in Figure 6G. Altogether, these results demonstrate that Y372 substitution in the context of the full length GIIβ has a moderate to strong inhibitory effect on GII activity; however, this residue does not have an effect as great as that of one of the four mannose hydroxyl-sensing residues (glutamine, arginine, glutamate, or tyrosine) previously tested. Future structural studies utilizing longer glycans and the GII heterodimer are needed to gain an understanding of how GII differentiates among the various mannose-containing ligands found in the ER to mediate its vital role in the glycoprotein folding pathway.
NMR Structure of the W409A Mutant.
We previously (and in this work) reported that a mutation of W409 in the MRH domain of GIIβ decreased the trimming efficiency of the holoenzyme both in vitro and in vivo.33 We have now determined the structure of the GIIβ MRH domain with a W409A mutation by NMR spectroscopy (Table 2). Mutation of this conserved tryptophan residue located near the tip of loop C causes no significant change in the overall fold of the molecule as this structure has an rmsd of 1.3 Å for 86 of 94 Cα atoms when compared to the ligand-free structure. The GIIβ MRH W409A domain yields a well-ordered ensemble of structures with the largest deviations from the mean in loop C (Figure 7A). Superimposition of the two unbound NMR structure ensembles (GIIβ MRH wild type and W409A) with the crystal structure determined in the presence of mannose (ligand-bound structure) reveals differences in loop regions (Figure 7B). Loop C of W409A is less constrained than in either of the other two proteins and displays an ~7 Å deviation between ensemble members for the positions of the Cα atom of N410, more than 2 times that deviation seen in this same residue in the wild-type protein ensemble. The residues in loop D are also displaced from the binding pocket by >1 Å, while residues in loop A and B are displaced >2 and 1 Å, respectively, when compared to those of the wild-type, unbound protein (Figure 7B). The absence of the tryptophan residue at position 409 in loop C appears to have global consequences on the residues in the mannose binding pocket. The four essential amino acids, as well as that of Y372, have altered their position from those of the wild-type, unbound protein to positions near or intermediate to that found in the bound structure even though no ligand was present. The substitution of this absolutely species-conserved tryptophan at position 409 with alanine somehow triggers a conformational response in the MRH domain similar to, although to a lesser extent than that seen in the presence of ligand.
Figure 7.
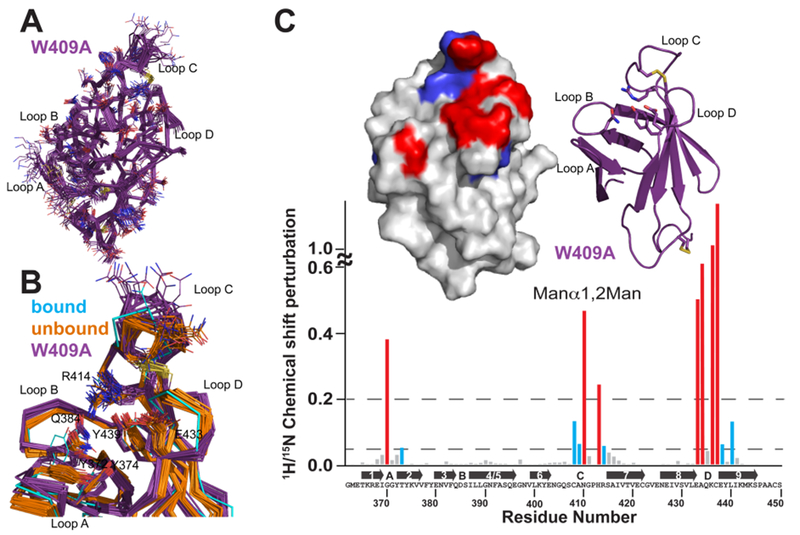
Solution structure of the S. pombe GIIβ MRH domain mutant W409A. (A) Superimposition of the polypeptide backbone heavy atoms of the 20 lowest-energy conformers of the ensemble representing the NMR structure of W409A. The side chains of the ensemble members are shown as lines. The chemical shifts for the first five N-terminal residues were not determined and for the sake of clarity are not shown. (B) Superimposition of the crystallographically bound model with the ensemble members of the S. pombe GIIβ MRH domain in the ligand-unbound state for the wild type (orange) and the W409A mutant (purple). Residues discussed in the text are represented by lines. A close-up view of the binding pocket is shown. (C) Spectrum of the GIIβ-MRH W409A mutant (0.1 mM) collected in the presence and absence of 50 mM Manβ1,2Man. 15N–1H chemical shift perturbations are plotted vs residue number for the protein. Secondary structural elements are listed above the sequence. Missing values correspond to proline residues or residues not observed in the 15N–1H HSQC spectra. The top left inset depicts chemical shift mapping on the structure surface. The surface is colored red for amino acids with the largest chemical shift perturbations (>0.2) and blue for those with more moderate (0.05) perturbations. The top right inset is a ribbon diagram for the molecule in the same orientation as the surface representation. The four essential amino acids are shown as sticks.
To further understand how substitution of W409 to alanine could trigger responses in the binding site similar to those after addition of ligand, we used our current chemical shift assignments to evaluate our previously reported interaction of the GIIβ MRH domain with Manα1,2Man.33 Chemical shift mapping reveals that the W409A mutation alters the interactions with residues on loop C, but not loop A or D (Figure 7C). In our previously reported chemical shift mapping experiments with the wild-type protein,33 loop C residues (406–408, 410, 413, 415, and 416) all showed chemical shift perturbations above background. The W409A mutation has the effect of decreasing the number of residues in loop C (408–410, 413, and 414) whose chemical shifts are altered in the presence of ligand.
Although positioning of W409 is influenced by crystal packing (Figure 3D), it strongly suggests that the W409 indole ring has a strong propensity for binding to a hydrophobic pocket of a nearby protein region or interacting with a hydrophobic face of the glycan ligand as shown in our model (Figure 8). However, the glycan is an unlikely binding partner because of the chemical shift perturbation results described above coupled with our previous biochemical studies that showed that GIIβ MRH domains harboring W409 mutations bound a glycoprotein bearing unmodified, phosphomonoester-modified, or phosphodiester-modified high-mannose-type glycans with affinities (within 3.5-fold) similar to or slightly lower than that of the wild type.33 Therefore, W409 is most likely bound to a hydrophobic region of the GIIβ or GIIα subunit in the intact heterodimer when bound to substrate. However, structural studies of the entire GIIβ subunit and/or the GII heterodimer are needed to validate this hypothesis. To further evaluate the interplay between W409 and the four conserved essential amino acids in the primary mannose binding pocket, the role of each of these four amino acids in the integrity of the protein was investigated.
Figure 8.
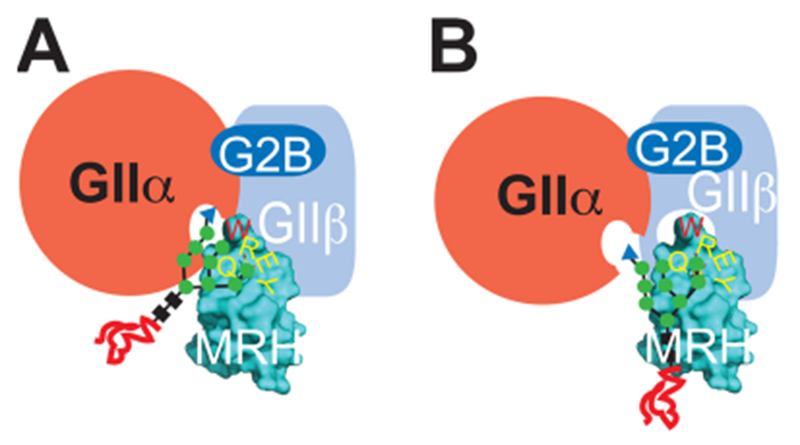
Models for the influence of W409 in GIIβ enhancement of GII activity.33 In both models, the catalytic GIIα subunit interacts with the GIIβ subunit (labeled in white) through the small G2B domain as well as through other yet to be defined contacts. In model A, the MRH domain enhances the catalytic activity of the GIIα subunit by W409 interacting with the B arm of the glycan. In model B, the W409 aromatic group interacts with a portion of the GIIβ subunit and enhances binding.
Effect of Single-Point Mutations on the Structural Integrity of the MRH Domain of GIIβ.
As previously discussed, substitution of any one of these four essential amino acids not only results in loss of MRH binding function but also results in loss of the ability of the heterodimeric GII to carry out glucose trimming in vivo: we showed that these same mutations in the context of the full length GIIβ domain resulted in a loss of GII activity in vivo without altering either its expression or its ability to form stable complexes with the GIIα subunit (as indicated by unperturbed levels of both α and β subunits in the ER).18 Despite this profound effect on protein function, no structural studies have been reported for any MRH domain containing a mutation of the conserved glutamine, arginine, glutamate, or tyrosine residues to date. To evaluate the consequence of these mutations on the structural integrity of the GIIβ MRH domain and to address the possibility that alterations in the MRH structure are conveyed to the rest of the heterodimer of GII, NMR spectroscopy was used to probe structural perturbations.
GIIβ MRH domain proteins containing a single-amino acid substitution in the primary binding pocket were isolated from inclusion bodies of E. coli grown in [15N]ammonium sulfate-containing medium and refolded as described in Materials and Methods. The conformation of each mutant was evaluated by standard 15N–1H HSQC experiments. Figure 9A shows that all four point mutants produce 15N–1H HSQC spectra that are indicative of a well-ordered and folded protein as the peaks are well-dispersed and exhibit similar intensity. Additionally, the total number of peaks is also similar to that produced by the wild-type protein under the same conditions. To verify that these point mutations render the MRH domain insensitive to the presence of ligand, 15N–1H HSQC spectra were collected in the presence of 200 mM Man-6-P (Figure 9C). These spectra show that in contrast to the wild-type protein (Figure 9B), significant chemical shift perturbations were not detected for any of the residues previously mapped to the primary binding site (Figure 9C), confirming the diminished ability of these mutants to bind ligand.
Figure 9.
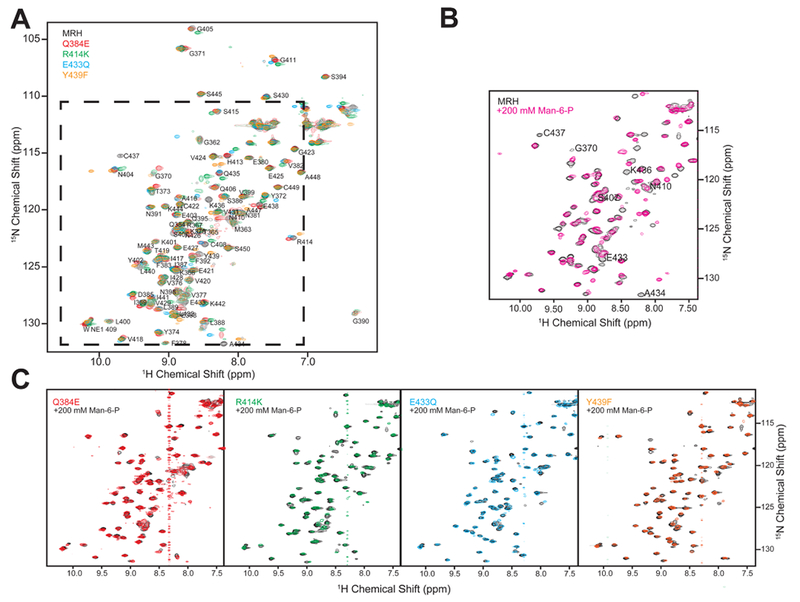
Influence of single-amino acid substitutions on the overall structure of the GIIβ MRH domain. Two-dimensional 15N–1H HSQC spectra of the wild type and four mutants containing a single-amino acid substitution in the binding pocket. (A) Overlay of four point mutants and wild-type protein in the absence of Man-6-P. (B) Overlay of spectra of the wild-type protein in the absence (black) and presence of 200 mM Man-6-P (pink). (C) Overlay of spectra of the GIIβ-MRH mutants in the absence and presence of 200 mM Man-6-P.
To further evaluate the effect of the Y439F mutation, as well as R414K, on the overall structure and to gain insight into how these mutations abrogate ligand binding, 15N-edited NOESY experiments were conducted. Inspection of these data localized changes in the spectra to residues in loop A (Q368–Y372) in which a loss or weakening of peak intensity of several long-range NOEs was observed (Figure 10) that may be suggestive of a general loosening of contacts between β-strand 6 (N391 NH) and loop C (Y402 NH). Thus, the increased mobility to loop C imparted by the R414K or Y439F substitution could destabilize the binding pocket. These results are consistent with the observation that in the presence of ligand, Y439 alters its position such that there is a concerted rearrangement of residues that becomes optimized for ligand binding (Figure 7B). Together, these data demonstrate that these point mutations do not exert their effects by forcing major structural alterations and suggest that their inhibitory effects on GII activity are due to either a reduction in the number of contacts to the glycan and/or a perturbation of the interaction between the MRH domain and the rest of the GII heterodimer (see Figure 8B).
Figure 10.
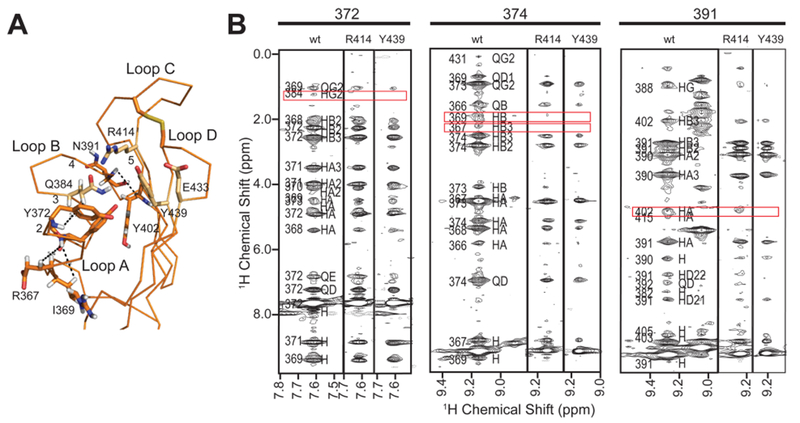
Influence of single-amino acid substitutions on 15N-edited NOESY-HSQC spectra of the GIIβ MRH domain. (A) Backbone trace of the GIIβ MRH domain in the ligand-unbound state (PDB entry 2LVX) with selected amino acids labeled along with loops A–D and β-strands 1–5. (B) Regions from 15N-edited NOESY-HSQC spectra of wild-type protein compared to the same region in the spectra collected for the two mutants, R414K and Y439F. Regions with significant differences are boxed in red.
CONCLUSIONS AND PERSPECTIVES
The importance of GII having a highly specialized domain such as an MRH domain stems from the complex nature of where this enzyme resides. The ER is a densely populated organelle containing a vast array of proteins, including other mannose binding proteins with which GII may compete for glycoprotein binding, as well as numerous substrates in the form of variable length glycans traversing through the protein folding and degradation pathways. The structure and/or composition of the glycan defines the fate of the glycoprotein that bears it. GII, an enzyme involved in quality control of glycoprotein folding, has a preference for glucosylated high-mannose (M9) ligands and has a lower deglucosylating activity as the mannose content drops.12,49 In contrast, the ERAD lectin OS-9 has a higher affinity for glycans with a lower mannose content15 that is reflective of their more terminal order in the folding–degradation pathway, as glycoproteins that have spent a significant amount of time in the ER folding pathway may eventually become substrates for ER mannosidase(s). This indicates that OS-9 and GII, and specifically their MRH domains, should have, in addition to their primary mannose binding pocket, an extended binding site that allows the proteins to sense the mannose content as well as linkages. The intriguing aspects of this proposed mechanism lie in the diversity of the mannose ligands (G1M9 to G1M7 and B arm vs C arm) and the spatial relationship between the MRH domain on the GIIβ subunit and the catalytic site located on the adjacent GIIα subunit, which is currently unknown and requires further structural studies.
Our studies demonstrate the importance of the absolutely conserved tryptophan residue in loop C on the function and structure of GII. Our previously presented model in which W409 could interact with either the glycan or an adjacent region of the heterodimer has been modified to reflect our inability to obtain evidence that this residue interacts with the glycan to enhance binding affinity. Crystallographic packing suggests that W409 favorably participates in a protein–protein interaction with the GIIβ subunit or the GIIα subunit. This interaction could be a permanent tethering of this residue away from the binding pocket, or it could be more transient and part of a conformational change that occurs upon ligand binding. Future studies will be directed at understanding the interplay between the MRH domain and the rest of the GIIβ subunit and, more globally, how the GIIα and GIIβ subunits interact to optimize the catalytic activity of GII.
ACKNOWLEDGMENTS
We thank Susana Raffo for synthesis of UDP-[14C]Glc and Marta Bravo for DNA sequencing.
Funding
This work was supported by National Institutes of Health Grant R01 DK042667 and Mizutani Foundation for Glycoscience Grant 10-0056 to N.M.D. This work was also supported by National Research Council (Argentina) Grant PIP-N824 and Agencia Nacional de Promoción Científica y Tecnológica (ANPCYT) Grant PICT2012-1504 to C.D. R.O. is a Doctoral Fellow of the National Research Council (Argentina), and A.J.P. and C.D. are Career Investigators of the National Research Council (Argentina).
ABBREVIATIONS
- CI-MPR
cation-independent mannose 6-phosphate receptor
- CD-MPR
cation-dependent mannose 6-phosphate receptor
- Man-6-P
mannose 6-phosphate
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- MRH
mannose 6-phosphate receptor homology
- GII
glucosidase II
- pNPG
p-nitrophenyl α-d-glucopyranoside
- pG3M9
Glc3Man9GlcNAc2
- G2M9
Glc2Man9GlcNAc2
- G1M9
Glc1Man9GlcNAc2
- M9
Man9GlcNAc2
- rmsd
root-mean-square deviation
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Yoshida Y, and Tanaka K (2010) Lectin-like ERAD players in ER and cytosol. Biochim. Biophys. Acta 1800, 172–180. [DOI] [PubMed] [Google Scholar]
- (2).Aebi M, Bernasconi R, Clerc S, and Molinari M (2010) N-glycan structures: Recognition and processing in the ER. Trends Biochem. Sci. 35, 74–82. [DOI] [PubMed] [Google Scholar]
- (3).D’Alessio C, Caramelo JJ, and Parodi AJ (2010) UDP-GlC: glycoprotein glucosyltransferase-glucosidase II, the ying-yang of the ER quality control. Semin. Cell Dev. Biol. 21, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).D’Alessio C, and Dahms NM (2015) Glucosidase II and MRH domain-containing proteins in the secretory pathway. Curr. Protein Pept. Sci. 16, 31–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Li E, Tabas I, and Kornfeld S (1978) The synthesis of complex-type oligosaccharides. I. Structure of the lipid-linked oligosaccharide precursor of the complex-type oligosaccharides of the vesicular stomatitis virus G protein. J. Biol. Chem. 253, 7762–7770. [PubMed] [Google Scholar]
- (6).Parodi AJ (2000) Protein glucosylation and its role in protein folding. Annu. Rev. Biochem. 69, 69–93. [DOI] [PubMed] [Google Scholar]
- (7).Aebi M (2013) N-linked protein glycosylation in the ER. Biochim. Biophys. Acta 1833, 2430–2437. [DOI] [PubMed] [Google Scholar]
- (8).Sousa M, and Parodi AJ (1995) The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J. 14, 4196–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hammond C, Braakman I, and Helenius A (1994) Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc. Natl. Acad. Sci. U.S.A. 91, 913–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Helenius A, and Aebi M (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049. [DOI] [PubMed] [Google Scholar]
- (11).Lederkremer GZ (2009) Glycoprotein folding, quality control and ER-associated degradation. Curr. Opin. Struct. Biol. 19, 515–523. [DOI] [PubMed] [Google Scholar]
- (12).Stigliano ID, Alculumbre SG, Labriola CA, Parodi AJ, and D’Alessio C (2011) Glucosidase II and N-glycan mannose content regulate the half-lives of monoglucosylated species in vivo. Mol. Biol. Cell 22, 1810–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Clerc S, Hirsch C, Oggier DM, Deprez P, Jakob C, Sommer T, and Aebi M (2009) Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J. Cell Biol. 184, 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hosokawa N, Kamiya Y, Kamiya D, Kato K, and Nagata K (2009) Human OS-9, a Lectin Required for Glycoprotein Endoplasmic Reticulum-associated Degradation, Recognizes Mannose-trimmed N-Glycans. J. Biol. Chem. 284, 17061–17068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Mikami K, Yamaguchi D, Tateno H, Hu D, Qin SY, Kawasaki N, Yamada M, Matsumoto N, Hirabayashi J, Ito Y, and Yamamoto K (2010) The sugar-binding ability of human OS-9 and its involvement in ER-associated degradation. Glycobiology 20, 310–321. [DOI] [PubMed] [Google Scholar]
- (16).Quan EM, Kamiya Y, Kamiya D, Denic V, Weibezahn J, Kato K, and Weissman JS (2008) Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol. Cell 32, 870–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Satoh T, Chen Y, Hu D, Hanashima S, Yamamoto K, and Yamaguchi Y (2010) Structural basis for oligosaccharide recognition of misfolded glycoproteins by OS-9 in ER-associated degradation. Mol. Cell 40, 905–916. [DOI] [PubMed] [Google Scholar]
- (18).Stigliano ID, Caramelo JJ, Labriola CA, Parodi AJ, and D’Alessio C (2009) Glucosidase II β Subunit Modulates N-Glycan Trimming in Fission Yeasts and Mammals. Mol. Biol. Cell 20, 3974–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Watanabe T, Totani K, Matsuo I, Maruyama J, Kitamoto K, and Ito Y (2009) Genetic analysis of glucosidase II β-subunit in trimming of high-mannose-type glycans. Glycobiology 19, 834–840. [DOI] [PubMed] [Google Scholar]
- (20).Takeda Y, Totani K, Matsuo I, and Ito Y (2009) Chemical approaches toward understanding glycan-mediated protein quality control. Curr. Opin. Chem. Biol. 13, 582–591. [DOI] [PubMed] [Google Scholar]
- (21).Hu D, Kamiya Y, Totani K, Kamiya D, Kawasaki N, Yamaguchi D, Matsuo I, Matsumoto N, Ito Y, Kato K, and Yamamoto K (2009) Sugar-binding activity of the MRH domain in the ER α-glucosidase II β subunit is important for efficient glucose trimming. Glycobiology 19, 1127–1135. [DOI] [PubMed] [Google Scholar]
- (22).Hofherr A, Wagner C, Fedeles S, Somlo S, and Kottgen M (2014) N-glycosylation determines the abundance of the transient receptor potential channel TRPP2. J. Biol. Chem. 289, 14854–14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Drenth JP, te Morsche RH, Smink R, Bonifacino JS, and Jansen JB (2003) Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat. Genet. 33, 345–347. [DOI] [PubMed] [Google Scholar]
- (24).Drenth JP, Martina JA, van de Kerkhof R, Bonifacino JS, and Jansen JB (2005) Polycystic liver disease is a disorder of cotranslational protein processing. Trends Mol. Med. 11, 37–42. [DOI] [PubMed] [Google Scholar]
- (25).Munro S (2001) The MRH domain suggests a shared ancestry for the mannose 6-phosphate receptors and other N-glycan-recognising proteins. Curr. Biol. 11, R499–R501. [DOI] [PubMed] [Google Scholar]
- (26.) Kornfeld S, and Sly WS (2001) I cell disease and pseudo-Hurler polydystrophy: Disorders of lysosomal enzyme phosphorylation and localization In Metabolic and Molecular Bases of Inherited Diseases (Scriver CR, Beaudet AL, Sly WS, and Valle D, Eds.) 8th ed., pp 3469–3482, McGraw Hill, New York. [Google Scholar]
- (27).Kim JJ, Olson LJ, and Dahms NM (2009) Carbohydrate recognition by the mannose-6-phosphate receptors. Curr. Opin. Struct. Biol. 19, 534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Roberts DL, Weix DJ, Dahms NM, and Kim J-JP (1998) Molecular basis of lysosomal enzyme recognition: Three-dimensional structure of the cation-dependent mannose 6-phosphate receptor. Cell 93, 639–648. [DOI] [PubMed] [Google Scholar]
- (29).Olson LJ, Zhang J, Lee YC, Dahms NM, and Kim J-JP (1999) Structural basis for recognition of phosphorylated high mannose oligosaccharides by the cation-dependent mannose 6-phosphate receptor. J. Biol. Chem. 274, 29889–29896. [DOI] [PubMed] [Google Scholar]
- (30).Olson LJ, Dahms NM, and Kim JJ (2004) The N-terminal carbohydrate recognition site of the cation-independent mannose 6-phosphate receptor. J. Biol. Chem. 279, 34000–34009. [DOI] [PubMed] [Google Scholar]
- (31).Olson LJ, Yammani RD, Dahms NM, and Kim JJ (2004) Structure of uPAR, plasminogen, and sugar-binding sites of the 300 kDa mannose 6-phosphate receptor. EMBO J. 23, 2019–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Olson LJ, Peterson FC, Castonguay A, Bohnsack RN, Kudo M, Gotschall RR, Canfield WM, Volkman BF, and Dahms NM (2010) Structural basis for recognition of phosphodiester-containing lysosomal enzymes by the cation-independent mannose 6-phosphate receptor. Proc. Natl. Acad. Sci. U.S.A. 107, 12493–12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Olson LJ, Orsi R, Alculumbre SG, Peterson FC, Stigliano ID, Parodi AJ, D’Alessio C, and Dahms NM (2013) Structure of the lectin mannose 6-phosphate receptor homology (MRH) domain of glucosidase II, an enzyme that regulates glycoprotein folding quality control in the endoplasmic reticulum. J. Biol. Chem. 288, 16460–16475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Moreno S, Klar A, and Nurse P (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194, 795–823. [DOI] [PubMed] [Google Scholar]
- (35).Soussilane P, D’Alessio C, Paccalet T, Fitchette AC, Parodi AJ, Williamson R, Plasson C, Faye L, and Gomord V (2009) N-glycan trimming by glucosidase II is essential for Arabidopsis development. Glycoconjugate J. 26, 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (37).Sheldrick GM (2010) Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr. D66, 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr. D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Matsuyama A, Arai R, Yashiroda Y, Shirai A, Kamata A, Sekido S, Kobayashi Y, Hashimoto A, Hamamoto M, Hiraoka Y, Horinouchi S, and Yoshida M (2006) ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 24, 841–847. [DOI] [PubMed] [Google Scholar]
- (41).Fernandez FS, Trombetta SE, Hellman U, and Parodi AJ (1994) Purification to homogeneity of UDP-glucose:glycoprotein glucosyltransferase from Schizosaccharomyces pombe and apparent absence of the enzyme from Saccharomyces cerevisiae. J. Biol. Chem 269, 30701–30706. [PubMed] [Google Scholar]
- (42).Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Vax A (1995) NMRPipe: A multidemensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (43).Bartels C, Xia TH, Billeter M, Guntert P, and Wuthrich K (1995) The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 6, 1–10. [DOI] [PubMed] [Google Scholar]
- (44).Shen Y, Delaglio F, Cornilescu G, and Bax A (2009) TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Herrmann T, Guntert P, and Wuthrich K (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227. [DOI] [PubMed] [Google Scholar]
- (46).Schwieters CD, Kuszewski JJ, Tjandra N, and Clore GM (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson 160, 65–73. [DOI] [PubMed] [Google Scholar]
- (47).Linge JP, Williams MA, Spronk CA, Bonvin AM, and Nilges M (2003) Refinement of protein structures in explicit solvent. Proteins 50, 496–506. [DOI] [PubMed] [Google Scholar]
- (48).Olson LJ, Zhang J, Dahms NM, and Kim J-JP (2002) Twists and turns of the CD-MPR: Ligand-bound versus ligand-free receptor. J. Biol. Chem 277, 10156–10161. [DOI] [PubMed] [Google Scholar]
- (49).Grinna LS, and Robbins PW (1980) Substrate specificities of rat liver microsomal glucosidases which process glycoproteins. J. Biol. Chem 255, 2255–2258. [PubMed] [Google Scholar]
- (50).Ashkenazy H, Erez E, Martz E, Pupko T, and Ben-Tal N (2010) ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51.) DeLano WL (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA. [Google Scholar]
- (52).Laskowski RA, and Swindells MB (2011) LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786. [DOI] [PubMed] [Google Scholar]