

Abstract
Systemic lupus erythematosus (SLE) is associated with an increased risk of vascular complications. Lupus nephritis is a major manifestation of SLE in the clinic. Lupus nephritis is elevated by T helper type 17 (Th17) cells, the major pro-inflammatory T-cell subset, leading to autoimmunity modulation. Therapeutic treatments targeting leukocyte recruitment may be useful in attenuating vascular complications linked to SLE progression. 3,7,3',4'-Tetrahydroxyflavone (fisetin) is a flavonol and a member of the flavonoid polyphenols. It is present in various fruits and vegetables, including persimmons, apples, kiwis, grapes, onions, strawberries and cucumbers. In the present study, the effects of fisetin against SLE induced by pristane (PRI) were evaluated in mice. Fisetin was indicated to reduce PRI-induced anti-double stranded DNA, anti- small nuclear ribonucleoprotein and the ratio of albumin to creatinine in urine. In addition, the chemokine (C-X-C motif) ligand (CXCL) signaling pathway was activated for PRI treatment, which was reversed by fisetin administration by reducing CXCL-1 and 2, chemokine (C-C motif) ligand 3, as well as CXC receptor 2 expression. In addition, the induction of inflammatory cytokines, including interleukin (IL)-6, tumor necrosis factor-α, IL-1β, as well as the chemokine interferon-γ, by PRI were downregulated by fisetin treatment in mice. Furthermore, Th17 cells and their associated cytokines were highly induced by PRI treatment, which was inhibited by fisetin administration. The present results indicated that fisetin may be an effective management for SLE by targeting the CXCL signaling pathway and regulating Th17 differentiation during lupus nephritis development.
Keywords: systemic lupus erythematosus, lupus nephritis, fisetin, CXCLs, Th17 differentiation
Introduction
Systemic lupus erythematosis (SLE) is an autoimmune disorder, which is linked to premature vascular disease (1,2). The enhanced macrovascular risk has not been fully investigated through exploring conventional risk factors or signals for vascular disease (3). Lupus nephritis is a major manifestation of SLE. Accordingly, SLE, as an autoimmune disease, is also characterized by autoantibody generation, elevated circulating inflammatory cytokines and injury to organs (4,5). Furthermore, SLE is a disease with various causes, including genetic and environmental factors (6). The pathogenesis of lupus nephritis includes an abnormal T-cell response, accelerating the generation of autoantibodies and immune complex deposits in renal or other organs (7). It has been indicated that T cells are essential for the pathogenesis of lupus nephritis, which involves the regulation of T helper (Th) cells (8). Th cells are central regulators of adaptive immune responses, which have an important role in the pathogenesis of SLE by regulating the interactions between other cells to regulate the generation of immunomodulatory cytokines (9). Once activated by antigens, the CD4+ T cells differentiate into various lineages of Th cells, including Th1, -9 and -17, which are associated with different types of cytokines (10). Th17 cells, which produce the pro-inflammatory cytokine interleukin (IL)-17, are of particular importance in the pathogenesis of SLE (11). According to previous studies, patients with SLE had higher circulating IL-17 levels in comparison with those in the controls (12). Chemokine (C-X-C motif) ligand (CXCL)-1 is a murine homolog to human CXCL-8 and a potent chemoattractant for neutrophils and IL-17; it mediates pro-inflammatory responses primarily by modulating the expression of other cytokines and chemokines, including IL-6, IL-1β, tumor necrosis factor (TNF)-α and monocyte chemoattractant protein (MCP)-1 (13,14).
3,7,3',4'-Tetrahydroxyflavone (fisetin), a naturally occurring bioactive flavonol, has been reported to improve the development of various diseases with its multiple bioactivities, including the reduction of oxidative stress, inflammatory response and apoptosis (15-17). In addition, fisetin was reported to ameliorate immune cell infiltration to improve organ injury (18). However, the crucial effects of fisetin in human SLE progression have remained elusive. The present study attempted to investigate whether fisetin has potential value to ameliorate SLE in mice induced by 2,6,10,14-tetramethylpen-tadecane [pristane (PRI)]. Previously, PRI has been suggested to induce nephritis in marine animals, characterized by mesangial expansion, glomerular proliferation and protein-uria, which are associated with elevated levels of anti-double stranded (ds)DNA antibodies (19). Of note, the present study indicated that fisetin reduces anti-autoimmune antibodies and pro-inflammatory cytokines release, and regulates Th17-cell differentiation through CXCL signaling. In addition, an in vitro experiment indicated that fisetin had no cytotoxic effects on treated cells. These results suggested that fisetin may be used as a promising candidate to attenuate SLE in the future.
Materials and methods
Animals and treatment
A total of 60 female C57BL/6 mice (weight, 18-20 g; age, 10 weeks), were purchased from Nanjing Medical University Animal Experiment Center (Nanjing, China). All experimental mice were housed under specific pathogen-free conditions in static microisolator cages with tap water ad libitum in a temperature-controlled room (25±2°C, 50±5% humidity) with a 12-h light/dark cycle and were fed a standard laboratory rodent diet with water ad libitum. The mice experiments were performed in a way to minimize animal suffering according to the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health from 1996. The Institutional Animal Care and Use Committee at Huai'an First People's Hospital, Nanjing Medical University (Huai'an, China) approved the animal study protocols. The animals were divided into 5 groups (n=10/group): Control (Con; without any treatment); PRI group; PRI+25 mg/kg fisetin; PRI+50 mg/kg fisetin and PRI+100 mg/kg fisetin (20,21). Fisetin (purity, >98%) was from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and was administered orally by gavage once a day. In order to induce lupus in mice, the animals received an intraperitoneal (i.p.) injection of 0.5 ml PRI (Sigma-Aldrich; Merck KGaA), as previously described (22). PBS was administered to the control group. At 30 weeks following treatment with PRI or PBS, blood samples were collected through retroorbital bleeding with capillary tubes to obtain the serum. Next, the mice were sacrificed, and renal and spleen tissue samples were immediately isolated and stored at −80°C for the further experiments described below. Proteinuria was measured through urine albumin-to-creatinine ratio determined using the Albumin assay kit (A028-1; Nanjing Jiancheng Bioengineering Institute, Nanjing, China) and Creatinine Assay kit (C011-2; Nanjing Jiancheng Bioengineering Institute).
Cells and culture
The RAW264.7 murine macrophage cell line and the 293FT cell line were purchased from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1% (v/v) penicillin/streptomycin in an incubator at 37°C in a humidified atmosphere containing 5% CO2. For the experiments, cells were pre-treated with 100 ng/ml lipopolysaccharide (LPS; Sigma-Aldrich; Merck KGaA) for 2 h, followed by incubation with fisetin (15 and 30 µM) for another 24 h. Subsequently, all cells were harvested for reverse transcription-quantitative polymerase chain reaction (RT-qPCR), western blot and flow cytometric analysis.
MTT assay
In order to measure the cytotoxicity of fisetin on normal cells, 293FT and RAW264.7 cells were seeded into 96-well plates at 1×103 cells/well in complete growth medium. On the following day, the cells were treated with different concentrations of fisetin ranging from 0-160 µM at 37°C for 24, 48 or 72 h. The cell viability was determined by an MTT assay with the absorbance read at 570 nm (23).
Analysis of serum samples
The serum concentrations of anti-dsDNA antibodies (5110; Alpha Diagnostic, San Antonio, TX, USA), anti-small nuclear ribonucleoprotein antibody (AB-23240-A; anti-snRNP; Alpha Diagnostic), IL-1β (MLB00C), IL-6 (M6000B), TNF-α (MTA00B), interferon-γ (IFN-γ) (DY485), CXCL-1 (DY453), MCP-1/CCL-2 (MJE00), chemokine (C-C motif) ligand 3 (CCL-3) (MMA00) and CXCL-2 (MM200; all from R&D Systems, Minneapolis, MN, USA) were measured with corresponding ELISA kits following the manufacturers' protocols.
Histological analysis
To histologically evaluate the renal tissue samples after various treatments, kidney tissue specimens were fixed in 10% formalin at room temperature for 48 h and embedded in paraffin. A series of 4-µm sections containing the hilum of the renal tissue samples were stained with a periodic acid-Schiff (PAS) Stain Kit (Abcam, Cambridge, MA, USA) following the manufacturer's protocols. In brief, a semi-quantitative scoring system was applied to calculate 10 various parameters, including mesangial deposits, mesangial hypercellularity, mesangial sclerosis, endocapillary sclerosis, endocapillary cellular infiltrate, endocapillary organized crescents, endocapillary cellular crescents, tubular atrophy, interstitial inflammation and interstitial fibrosis: 0, no involvement; 0.5, minimal involvement (<10%); 1, mild involvement (10-30%); 2, moderate involvement (31-60%); 3, severe involvement (60%). An activity and chronicity index was determined through compiling scores from groups of associated parameters (as for the activity: Mesangial deposits, mesangial hypercellularity and endocapillary cell infiltration; and as for the chronicity: Tubular atrophy, endocapillary organized crescents, endocapillary sclerosis and interstitial fibrosis). Spleen tissue samples were soaked in 4% formalin overnight at room temperature, embedded in paraffin and sectioned at 4 µm thickness. Sections were stained with hematoxylin and eosin (H&E) and visualized by light microscopy at a magnification of x400.
Flow cytometric analysis
The spleen tissue was disaggregated into single cells as described previously (24) and stained for 30 min in dark at room temperature for surface markers with the following antibodies: CD3 (554832; 1:100), CD4 (553651; 1:100) and CD8 (557085; 1:100) purchased from BD Biosciences (Franklin Lakes, NJ, USA). Spleen cells were stimulated and then stained for intracellular cytokines. RAW264.7 cells were treated as indicated above and then harvested for CD3 conjugation. The following fluorochrome-conjugated antibodies were used: CD4 (563933; 1:100 dilution, GK1.5), CD3 (561042; 1:100 dilution, 145-2C11), IL-17 (eBio17B7; 1:100 dilution) and RAR-related orphan receptor (ROR)γt (12-6988-82; 1:100 dilution, AFKJ-9), and all were purchased from BD Biosciences or eBioscience (Thermo Fisher Scientific, Inc.). The antibodies were stained in dark for 30 min Finally, the labeled cells were quantified with a FACSCanto instrument (BD Biosciences).
T-cell differentiation in vitro
CD4+CD25− T cells were isolated from spleens of 6-month-old mice as described previously (25). Cells were purified using a MACS magnetic column (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) with a CD4+ T cell enrichment kit (11-0041-82; eBioscience; Thermo Fisher Scientific, Inc.). The cells were then stimulated with a plate-bound anti-CD3e antibody (5 µg/ml; 11-0038-42, eBioscience; Thermo Fisher Scientific, Inc.) and anti-CD28 antibody (2 µg/ml; 16-0281-82, eBioscience; Thermo Fisher Scientific, Inc.). For Th17-cell polarization, the following exogenous cytokines and antibodies were added: Transforming growth factor-β (5 ng/ml, R&D Systems); anti-IL-4 antibody (46-7041-82; 10 µg/ml; eBioscience; Thermo Fisher Scientific, Inc.); IL-6 (MAB406; 30 ng/ml; R&D Systems); anti-IFN-γ antibody (MAB485; 10 µg/ml; eBioscience; Thermo Fisher Scientific, Inc.) and IL-1β (AF-401-NA; 20 ng/ml; R&D Systems). After 5 days of culture, the proportion of Th17 cells among all cells was determined by flow cytometry.
RT-qPCR
Total RNA extraction of cells and spleen tissue samples was performed using TRIzol reagent (Life Technologies; Thermo Fisher Scientific, Inc.). Total RNA (1 µg) was reverse transcribed using the Moloney murine leukemiavirus RT system (Promega Corp., Madison, WI, USA). A mixture of 2 µl RT product with 7.3 µl nuclease-free H2O, 0.4 µl ROX I (50X), 10 µl TaqMan PCR Mixture (2X) and 0.3 µl TaqMan MiRNA Assay (20X) primer was prepared. The reaction was performed at 42°C for 1 h and terminated by deactivation of the enzyme at 70°C for 10 min. PCR was performed using SYBR-Green (Bio-Rad Laboratories, Inc., Hercules, CA, USA) in an ABI PRISM 7900HT detection systems (Applied Biosystems; Thermo Fisher Scientific, Inc.). Equal amounts of cDNA were diluted and amplified through real-time PCR using All-in-One qPCR Mix (GeneCopoeia, Maryland, USA) in a 20-µl reaction volume containing 10 µl of 2X All-in-One qPCR Mix (GeneCopoeia, Maryland, USA), 1 µl of 2 µM forward primer, 1 µl of 2 µM reverse primer, 1 µl of cDNA and 6 µl of nuclease-free water. All of the primers were from Invitrogen (Thermo Fisher Scientific, Inc.). Amplification of pre-denatured products was performed at 94°C for 60 sec followed by 45 cycles at 95°C for 30 sec, 58°C for 30 sec and 72°C for 30 sec; this was followed by 95°C for 10 sec, 65°C for 45 sec and 40°C for 60 sec. Fold induction values were calculated according to the 2−ΔΔCq method, where ΔCt represents the differences in cycle threshold number between the target gene and GAPDH, and ΔΔCt represents the relative change in the differences between control and treatment groups (26). The following primers were used: IL-6 forward, 5'-CAA GCA ACA ATG GAC TTT AGG-3' and reverse, 5'-GAG CCA TTA ACT ATG GAA CCA-3'; IL-1β forward, 5'-AGA CTC CAC ATT ACA GGC AAT GCC-3' and reverse, 5'-AAT CGC TAC GGA TCT CCA GA-3'; MCP-1 forward, 5'-AGA TCA GGC GAT GCA TGT AGA CG-3' and reverse, 5'-CAA TAC CAG TGG ACA AAC GAC-3'; IFN-γ forward, 5'-ACA AGC TCA AGT ACC CAA GGT ACA-3' and reverse, 5'-GTG CTA CTG GTG TGA TCA-3'; TNF-α forward, 5'-ATG TCC GTC CTC ATC GGT AGC-3' and reverse, 5'-CCG GAG GAC ACA GTC CAC C-3'; IL-17 forward, 5'-CCC ATG AGA TAC ACG GGT A-3' and reverse, 5'-CCG ATA CTG CAA GGT GAA C-3'; GAPDH forward, 5'-TCA CAT GAA CCG GAC AGA GG-3' and reverse, 5'-CCA ATC CTA CGA CAC CGA CTA AC-3'.
Western blot analysis
The cells and spleen tissue samples were homogenized with hypotonic buffer [25 mM Tris-HCl (pH 8.0), 1 mM EDTA, 5 µg/ml leupeptin, 1 mM Pefabloc SC, 50 µg/ml aprotinin, 5 µg/ml soybean trypsin inhibitor and 4 mM benzamidine] at 10% (wt/vol) to yield a homogenate. The final supernatants were the obtained by centrifugation at 12,000 x g for 20 min at 4°C. The protein concentration was determined with a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Inc.) with bovine serum albumin as a standard. Subsequently, equal amounts of total protein (20-40 µg) were subjected to 10% SDS-PAGE and electrophoretically transferred onto polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were then blocked with 5% skimmed milk Tris buffered saline with 0.1% Tween 20 (TBST), washed, and then incubated with the following primary antibodies at 1:1,000 dilution overnight at 4°C: Rabbit anti-IL-17 (13838), rabbit anti-CXCL-1 (PA1-29220), rabbit anti-CD3 (4443), rabbit anti-CXCL-2 (701126), rabbit anti-CCL-3 (MA5-24364), rabbit anti-CXCR-2 (PA5-38620) and rabbit anti-GAPDH (2118; Cell Signaling Technology, Inc., Danvers, MA, USA), followed by incubation with HRP-conjugated goat anti rabbit secondary antibody (1:5,000; ab6721; Abcam) for 2 h at room temperature. Western blot bands were observed using the GE Healthcare ECL Western Blotting Analysis System (GE Healthcare, Little Chalfont, UK) and exposed to X-ray film (Eastman Kodak, Rochester, NY, USA). Protein expression levels were quantified by determining the grey value, standardized to the housekeeping gene (GAPDH) and expressed as a fold of the control.
Statistical analysis
Values are expressed as the mean ± standard error of the mean. Statistical analyses were performed using GraphPad PRISM (version 6.0; GraphPad Inc., La Jolla, CA, USA). Analysis of variance with Dunnet's least-significant difference post-hoc test was performed for comparison between groups. P<0.05 was considered to indicate a statistically significant difference.
Results
Fisetin treatment reduces auto-antibodies and nephritis in PRI-induced mice
Compared with the Con group, anti-dsDNA antibodies were significantly increased in PRI-induced mice (Fig. 1A). Of note, fisetin administration significantly reduced PRI-induced anti-dsDNA antibody levels in a dose-dependent manner. In addition, the levels of anti-snRNPs were markedly increased in PRI-treated mice, but were reduced by fisetin in a dose-dependent manner (Fig. 1B). Finally, the ratio of urine albumin to creatinine was evaluated to assess lupus nephritis after PRI administration. As presented in Fig. 1C, PRI treatment significantly enhanced the ratio of albumin to creatinine, suggesting that it caused injury in mice, which was ameliorated by fisetin treatment. These results indicated that fisetin may improve various autoimmune features of lupus in mice induced by PRI.
Figure 1.

Effect of fisetin treatment on auto-antibodies and nephritis in PRI-induced mice. (A) Circulating levels of anti-dsDNA antibody in PRI-induced mice. (B) Circulating levels of anti-snRNP antibody in PRI-induced mice. (C) Ratio of urine albumin/creatinine of mice. Values are expressed as the mean ± standard error of the mean (n=10 in each group). ***P<0.001 vs. the Con group; +P<0.05, ++P<0.01 and +++P<0.001 vs. the PRI group. Con, control; PRI, pristine; dsDNA, double stranded DNA; snRNP, small nuclear ribonucleoprotein; F25/50/100, treatment with 25/50/100 mg/kg fisetin.
Fisetin reduces characteristics of nephritis in a murine model of SLE
In order to determine the effect of fisetin on PRI-induced nephritis in mice, the renal sections were histologically examined by PAS staining, which was evaluated using indexes of activity and chronicity. As presented in Fig. 2A and B, the activity index was markedly enhanced by PRI induction, which was comparable to that in the control group. Fisetin administration significantly reduced the activity index, indicating that lupus nephritis was attenuated by fisetin. Consistently, the PRI-induced elevation in the chronicity index was also reduced by fisetin treatment (Fig. 2A and C). In addition, serum BUN and creatinine levels were highly induced by PRI, which was inhibited by fisetin, indicating renal injury induced by PRI and that fisetin had the capacity to attenuate nephritis (Fig. 2D and E). Furthermore, infiltration of CD3+ cells was assessed to calculate the effects of fisetin on SLE. The immunohistochemical analysis demonstrated that PRI treatment led to increases of CD3+ cells throughout the kidney tissue compared with the control group. Of note, fisetin-treated mice exhibited significantly reduced infiltration of CD3+ cells compared with that in the PRI group (Fig. 3A). Western blot analysis further indicated that PRI treatment increased CD3 levels, which was inhibited by fisetin (Fig. 3B). Taken together, the above results demonstrated similar results to those of other PRI-induced animal models of lupus, and various features associated with lupus nephritis induced by PRI administration were dose-dependently reduced by fisetin treatment.
Figure 2.

Effects of fisetin on nephritis in SLE models. (A) Histological PAS staining of glomeruli in PRI-induced mice. Representative images are displayed. Scale bar=50 µm. (B) The activity index for each group was determined from the PAS-stained tissue sections. (C) The chronicity index of PAS-stained renal tissue sections was measured. (D) Serum BUN and (E) creatinine were examined. Values are expressed as the mean ± standard error of the mean (n=10 in each group). ***P<0.001 vs. the Con group; +P<0.05, ++P<0.01 and +++P<0.001 vs. the PRI group. Con, control; PRI, pristine; PAS, periodic acid-Schiff; BUN, blood urea nitrogen; F50/100, treatment with 50/100 mg/kg fisetin.
Figure 3.

Fisetin treatment reduces renal immune cell infiltration in PRI-induced mice. (A) Immunohistochemical analysis was performed to determine the amount of CD3-positive cells in renal sections isolated from mice subjected to the indicated treatments. Scale bar=50 µm. (B) Renal protein expression of CD3 was examined using by western blot analysis. Values are expressed as the mean ± standard error of the mean (n=10 in each group). ***P<0.001 vs. the Con group; ++P<0.01 and +++P<0.001 vs. the PRI group. Con, control; PRI, pristine; F50/100, treatment with 50/100 mg/kg fisetin.
Fisetin reduces the circulating levels of inflammatory signaling factors in mice with PRI-induced SLE
In order to assess the role of fisetin in circulating inflammatory biomarkers associated with leukocyte recruitment, the serum levels of CXCL-1, CXCL-2, CCL-3, CXCR-2, IL-6, IFN-γ, TNF-α, IL-1β and MCP-1 were evaluated in PRI-induced mice treated with or without fisetin. In the pathogenesis of lupus, leukocyte extravasation has an important role (27). The results of the present study indicated that serum CXCL-1 (Fig. 4A), CXCL-2 (Fig. 4B), CCL-3 (Fig. 4C) and CXCR-2 (Fig. 4D) were highly increased in PRI-induced mice compared with those in the Con group. However, after fisetin treatment, reduced levels of these signals were observed. Next, the inflammatory cytokines IL-6, IFN-γ, TNF-α and IL-1β, and the chemokines MCP-1 were assessed in the plasma of mice after various treatments (Fig. 4E-I). The results indicated that PRI treatment enhanced the levels of these factors, which were reduced by administration of fisetin in a concentration-dependent manner. Thus, fisetin may attenuate SLE due to reducing chemokines and cytokines in PRI-induced mice.
Figure 4.

Fisetin reduces the levels of inflammatory signaling proteins in mice with PRI-induced systemic lupus erythematosus. The circulating levels of (A) CXCL-1, (B) CXCL-2, (C) CCL-3, (D) CXCR-2, (E) IL-6, (F) IFN-γ, (G) TNF-α, (H) IL-1β and (I) MCP-1 levels were determined by ELISA. Values are expressed as the mean ± standard error of the mean (n=10 in each group). ***P<0.001 vs. the Con group; +P<0.05, ++P<0.01 and +++P<0.001 vs. the PRI group. Con, control; PRI, pristine; IL, interleukin; TNF, tumor necrosis factor; MCP, monocyte chemoattractant protein; CXC, chemokine (C-X-C) motif; CXCL, CXC ligand; CXCR, CXC receptor; CCL-3, chemokine (C-C motif) ligand 3; F25/50/100, treatment with 25/50/100 mg/kg fisetin.
Fisetin inhibits PRI-induced changes the composition of lymphocytes in the spleen of mice
According to previous studies, splenomegaly is a characteristic of mice with SLE (27). As presented in Fig. 5A, the spleen size of mice induced with PRI was larger than that of mice without any treatment. By contrast, fisetin administration reduced the spleen size in PRI-induced mice. In addition, the total number of lymphocytes in the spleen tissue samples was markedly increased in PRI-treated mice, which was reduced by fisetin treatment (Fig. 5B). Subsequently, the composition of splenic lymphocytes was explored by flow cytometric analysis. The results presented in Fig. 5C indicated that the proportion of CD3+CD4+ T cells was highly augmented in PRI-induced mice, which was inhibited by fisetin administration. The ratio of CD4+ vs. CD8+ T cells among the total splenic lymphocytes was significantly increased in the PRI-treated mice, which was attenuated by fisetin administration (Fig. 5D). Of note, a reduced CD4+/CD8+ ratio was observed after fisetin administration compared with that in mice treated with PRI only. Furthermore, the B-cell number was also increased in the PRI-induced mice compared with that in the control group (Fig. 5E). Finally, the H&E staining indicated that PRI induced histological changes compared with control group, which was attenuated by fisetin administration (Fig. 5F). These results demonstrated that PRI treatment caused an abundance of different lymphocyte subsets in mice, which was reduced by fisetin administration, indicating its role in improving SLE induced by PRI in mice.
Figure 5.

Effects of fisetin on the composition of lymphocytes in the spleen of mice induced by PRI. (A) Representative images of spleens obtained from PRI-induced mice after fisetin administration. The weight of the spleen was determined at the end of the experiment. Scale bar=50 µm. (B) Total number of lymphocytes in PRI-induced mice treated with different concentrations of fisetin. (C) The proportion of CD3+ and CD4+ T cells was evaluated in PRI-induced mice after fisetin administration. (D) The proportion of CD4+ to CD8+ cells among the splenic lymphocytes was calculated. (E) The B-cell number was determined in mice under various treatments. Scale bar=50 µm. (F) Histological analysis of spleens obtained from mice induced by PRI with or without fisetin treatment. Values are expressed as the mean ± standard error of the mean (n=10 in each group). ***P<0.001 vs. the Con group; +P<0.05, ++P<0.01 and +++P<0.001 vs. the PRI group. Con, control; PRI, pristine; F25/50/100, treatment with 25/50/100 mg/kg fisetin.
Fisetin inhibits SLE-associated increases in splenic Th17 cells and pro-inflammatory cytokine expression
The present study attempted to evaluate the proportion of Th17 cells among the splenic CD3+CD4+ T cells through flow cytometric analysis. As presented in Fig. 6A-C, the percentage of RORγ+ cells among CD3+CD4+ T cells was significantly increased in PRI-induced mice, as well as the percentage of IL-17-generating CD3+CD4+ T cells, which were reduced by fisetin treatment in a dose-dependent manner. In addition, the pro-inflammatory cytokines in splenic cells were determined by RT-qPCR analysis. The mRNA expression of pro-inflammatory cytokines and MCP-1 was highly induced by PRI, which was in line with the circulating levels. Of note, fisetin administration markedly reduced the mRNA levels of these cytokines and MCP-1, indicating its role in suppressing the inflammatory response in mice with SLE (Fig. 6D). Finally, in order to further confirm the capacity of fisetin to ameliorate SLE induced by PRI in mice, the mRNA and protein levels of IL-17 were assessed in the splenic cells through RT-qPCR, western blotting and ELISA. As presented in Fig. 6E, PRI induced IL-17, which was inhibited by fisetin.
Figure 6.

Splenic Th17 cells and pro-inflammatory cytokine expression. The lymphocytes were isolated from mice subjected to various treatments. (A) Flow cytometry was used to evaluate RORγt+CD3+CD4+ Th17 cells among splenic CD4+ cells obtained from mice. Quantification of (B) RORγ and (C) IL-17 from the flow cytometry results. (D) The mRNA levels of the cytokines IL-6, IFN-γ, TNF-α, IL-1β, and MCP-1β in the spleen tissue samples were assessed. (E) IL-17 mRNA and protein levels were evaluated by reverse transcription-quantitative polymerase chain reaction, western blot and ELISA. Values are expressed as the mean ± standard error of the mean (n=10 in each group). ***P<0.001 vs. the Con group; +P<0.05, ++P<0.01 and +++P<0.001 vs. the PRI group. Con, control; PRI, pristine; F25/50/100, treatment with 25/50/100 mg/kg fisetin; Th17, T-helper type 17; IL, interleukin; TNF, tumor necrosis factor; MCP, monocyte chemoattractant protein; IFN, interferon; ROR, RAR-related orphan receptor.
Fisetin regulates CXCL signaling in PRI-induced mice with SLE
The present study further investigated how CXCLs signaling was altered in PRI-induced mice with or without fisetin treatment. Western blot analysis indicated that the CXCL signaling pathway was activated by PRI in mice with SLE, supported by an elevation in CXCL-1, CXCL-2, CCL-3 and CXCR-2 protein expression levels. Of note, fisetin administration significantly reduced the abundance of CXCL-1, CXCL-2, CCL-3 and CXCR-2 protein, which may be involved in the mechanisms by which fisetin attenuates SLE induced by PRI treatment (Fig. 7A and B).
Figure 7.

Role of fisetin in regulating the CXCLs signaling pathway in mice with PRI-induced systemic lupus erythematosus. (A) Western blot analysis was used to determine the expression levels of CXCL-1, CXCL-2, CCL-3 and CXCR-2 in the spleen tissue samples obtained from mice subjected to various treatments. Representative images are provided. (B) Quantification of CXCL-1, CXCL-2, CCL-3 and CXCR-2 protein levels from the western blots. Values are expressed as the mean ± standard error of the mean (n=10 in each group). ***P<0.001 vs. the Con group; +P<0.05, ++P<0.01 and +++P<0.001 vs. the PRI group. Con, control; PRI, pristine; CXC, chemokine (C-X-C) motif; CXCL, CXC ligand; CXCR, CXC receptor; CCL-3, chemokine (C-C motif) ligand 3; F25/50/100, treatment with 25/50/100 mg/kg fisetin.
In order to further confirm the effects of fisetin on SLE in vitro, RAW264.7 cells were included
According to the in vivo experiment, induction of inflammation was a key molecular mechanism by which SLE progressed. Hence, RAW264.7 cells were pre-treated with 100 ng/ml LPS for 2 h, followed by fisetin administration for another 24 h. As presented in Fig. 8A and B, the LPS-induced increases in CD3 expression levels were significantly reduced by fisetin. Furthermore, the mRNA levels of the cytokines IL-6, IFN-γ, TNF-α and IL-1β, and the chemokine MCP-1 were all increased by LPS, which was inhibited by fisetin treatment (Fig. 8C). Consistently, the protein expression of CXCL-1, CXCL-2, CCL-3 and CXCR-2 was also enhanced by LPS, which was markedly reduced by fisetin treatment (Fig. 8D). In conclusion, the above results indicated that in vitro, fisetin reduced inflammation and CXCL pathway activation triggered by LPS as a possible molecular mechanism by which it also attenuates SLE in vivo. Finally, to assess whether fisetin had any possible cytotoxic effects, the cell line 293FT and the RAW264.7 mouse macrophage cell line of were treated with fisetin at 0, 5, 10, 20, 40, 80 or 160 µM for 24, 48 or 72 h. Subsequently an MTT assay was used to measure the cell viability. The results presented in Fig. 9A and B indicated no significant difference in the number of viable 293FT and RAW264.7 cells after fisetin treatment for 24, 48 and even 72 h, which suggested that fisetin was safe for SLE treatment under the above conditions.
Figure 8.

Fisetin reduces inflammation and expression of CXCLs in LPS-induced RAW 264.7 cells. RAW 264.7 cells were pre-treated with LPS (100 ng/ml) for 2 h, followed by treatment with fisetin (15 or 30 µM) for another 24 h. (A) Flow cytometric analysis was used to quantify CD3-expressing cells. (B) CD3 expression levels were measured using western blot analysis. (C) Reverse transcription-quantitative polymerase chain reaction analysis of pro-inflammatory cytokines as indicated. (D) CXCL-1, CXCL-2, CCL-3 and CXCR-2 expression levels were assessed using western blot analysis. Values are expressed as the mean ± standard error of the mean (n=8 in each group). ***P<0.001 vs. the Con group; +P<0.05, ++P<0.01 and +++P<0.001 vs. the LPS group. Con, control; LPS, lipopolysaccharide; CXC, chemokine (C-X-C) motif; CXCL, CXC ligand; CXCR, CXC receptor; CCL-3, chemokine (C-C motif) ligand 3; F15/30, treatment with 15/30 µm fisetin.
Figure 9.
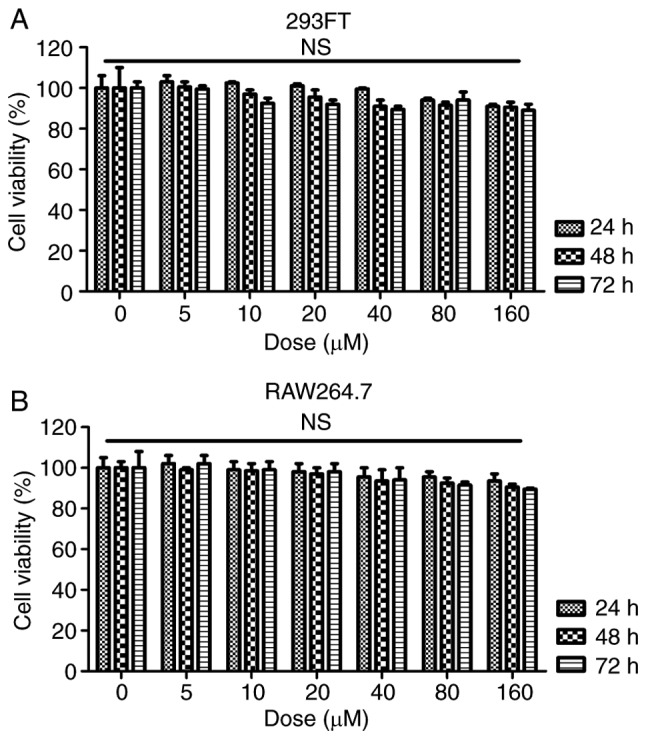
Analysis of the effects of fisetin in vitro. (A) The 293FT and (B) the RAW264.7 mouse macrophage cell line were treated with different concentrations of fisetin (0, 5, 10, 20, 40, 80 and 160 µM) for 24, 48 or 72 h. The MTT assay was used to determine the cell viability. n.s., no significant difference.
Discussion
SLE has been reported to be the most common autoimmune disease among women aged 40-49 years (28,29). The course of SLE exhibits a great variation among individuals, from mild to rapidly progressive disease and eventually even a fatal outcome (30). Patients with SLE usually present with high levels of auto-antibodies, pro-inflammatory cytokines and nephritis (31). The underlying pathogenesis of SLE is complex due to a variety of potential disease mechanisms among individuals (32). Effective therapeutic strategies to treat SLE are urgently required. Fisetin, which has been reported to prevent pro-inflammatory cytokine expression and to have immune-regulatory effects, was assessed in the present study as a potential strategy to treat SLE induced by PRI in mice.
In the present study, pristane was used to induce SLE in mice, which was characterized by elevated anti-dsDNA antibodies, anti-snRNP antibodies and the ratio of albumin to creatinine, indicating renal injury in mice. The renal architecture was significantly preserved, as fisetin treatment prevented PRI-induced histological alterations, indicating that nephritis was caused in this model of lupus. Of note, fisetin administration significantly reduced the levels of these antibodies, as well as the activity and chronicity indexes determined from PAS staining analysis.
Of note, in the SLE model, alterations in the proportion of Th17 cells were identified among splenic CD4+ cells and the expression of Th17-associated genes, including IL-17. It is well known that Th17 cells are closely associated with the pathogenesis of lupus nephritis via multiple mechanisms (33). Th17 cells are considered as a major type of pro-inflammatory T cell (34). In addition, a variety of cytokines and transcription factors regulate the signaling pathways that promote Th17 cell differentiation and enhance Th17 immune responses (35). RORγt is an essential transcription factor, which regulates the development and progression of Th17 cells (36). In the present study, PRI induced high expression of RORγ, contributing to Th17-cell differentiation, which was reduced by fisetin administration. Furthermore, IL-17, another important cytokine, indicates the extent of Th17-cell differentiation, which is associated with certain immune conditions (37,38). The present results indicated that IL-17 was markedly upregulated at the gene and protein level in PRI-induced mice in comparison to the control group, which was inhibited by fisetin.
Cytokines are also considered to have a critical role in SLE progression, which is associated with Th17-cell differentiation (39). Accordingly, IL-6, TNF-α, IL-1β, IFN-γ and MCP-1 drive the differentiation of Th17 cells, and IL-17 promotes the expansion of Th17 cells (40). Lupus-prone mice universally display high IFN levels, thus providing excellent animal models to investigate this matter (41). In the present study, the levels of IL-6, TNF-α, IL-1β, IFN-γ and MCP-1 in the serum and in the splenic lymphocytes were significantly increased in PRI-treated mice, which may be another factor to enhance Th17 function and lupus nephritis development. The CXCR-2-binding chemokines, CXCL-1 and -2, are potential chemoattractants, which are associated with the immune response (42). Inflammatory molecules, including TNF-α, IL-1β, IFN-γ, MCP-1 and leukotrienes, have been reported to be important for CXCL-1/2 recruitment (43). According to previous studies, pro-inflammatory cytokines, including TNF-α, may enhance CXCL-1 and -2 expression in various diseases (44). Macrophages have an important role in the progression of SLE (45,46). In addition, the two pro-inflammatory cytokines CXCL-1 and -2 have been reported to be linked to the progression of various diseases, including breast cancer, lung injury and prostate cancer, via their upstream signaling pathways, including nuclear factor-κB, which is associated with the secretion of TNF-α and IL-1β (47). Similarly, in the present study, CXCL-1, CXCL-2 and CXCR-2 were highly induced by PRI induction, which was markedly reduced by fisetin administration. Furthermore, CCL-3 was also enhanced in PRI-treated mice, and fisetin exerted an inhibitory effect by regulating CCL-3 expression, which was in line with CXCL-1/2 alterations in mice with SLE. Macrophages are the central effector cells and regulatory cells in inflammation (48,49). Therefore, RAW264.7 macrophages were selected as an in vitro cell model in the present study. Incubation with fisetin reduced the LPS-induced inflammatory response and expression of CXCLs in RAW264.7 cells, contributing to the attenuation of the immune response, which may be a potential molecular mechanism by which SLE was alleviated.
In conclusion, the present study indicated that PRI induced SLE in mice, which was inhibited by fisetin treatment in a dose-dependent manner. Specifically, the elevated levels of Th17 cells were reduced by fisetin treatment, which was associated with the suppression of pro-inflammatory cytokines and its downstream signaling pathway of CXCLs, contributing to the disruption of Th17 cell differentiation in mice with SLE. Our present study suggested that fisetin is a potential therapeutic strategy for SLE.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
SX performed the experiments and YL supervised the experiments and wrote the paper.
Ethics approval and consent to participate
The animal study protocols were approved by the Institutional Animal Care and Use Committee at Huai'an First People's Hospital, Nanjing Medical University (Huai'an, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that no conflicts of interest exist.
References
- 1.Mikdashi JA, Wozniak M, Ashraf U, Regenold W. Patterns of vascular brain injury in systemic lupus e2rythematosus patients with ischemic strokes: Impact on neuropsychological, neurobehavioral and physical function outcome. Arthritis Rheumatol. 2015;67:3568–3569. [Google Scholar]
- 2.Ajeganova S, Gustafsson T, Jogestrand T, Frostegård J, Hafström I. Bone mineral density and carotid atherosclerosis in systemic lupus erythematosus: A controlled cross-sectional study. Arthritis Res Ther. 2015;17:84. doi: 10.1186/s13075-015-0595-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischer K, Sawicki M, Chamiak-Ciemińska K, Stolarczyk J, Winikajtis-Burzyńska A, Milchert M, Ostanek L, Bobrowska-Snarska D, Kapłon Ł, Przepiera-Będzak H, et al. A5.07 The role of immunologic and inflammatory factors in the risk of microvascular and macrovascular impairment development in systemic lupus erythematosus-preliminary data. Ann Rheum Dis. 2016;75:A44–A44. doi: 10.1136/annrheumdis-2016-209124.105. [DOI] [Google Scholar]
- 4.Abou-Raya A, Abou-Raya S, Helmii M. The effect of vitamin D supplementation on inflammatory and hemostatic markers and disease activity in patients with systemic lupus erythematosus: A randomized placebo-controlled trial. J Rheumatol. 2013;40:265–272. doi: 10.3899/jrheum.111594. [DOI] [PubMed] [Google Scholar]
- 5.Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, Bengtsson AA, Blom AM. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. 2012;188:3522–3531. doi: 10.4049/jimmunol.1102404. [DOI] [PubMed] [Google Scholar]
- 6.Choi J, Kim ST, Craft J. The pathogenesis of systemic lupus erythematosusan update. Curr Opin Immunol. 24:651–657. doi: 10.1016/j.coi.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madhok R. Systemic lupus erythematosus: Lupus nephritis. BMJ Clin Evidence. 2015;1123:2015. [PMC free article] [PubMed] [Google Scholar]
- 8.Alunno A, Bartoloni E, Bistoni O, Nocentini G, Ronchetti S, Caterbi S, Valentini V, Riccardi C, Gerli R. Balance between regulatory T and Th17 cells in systemic lupus erythematosus: The old and the new. Clin Dev Immunol. 2012;2012;823085 doi: 10.1155/2012/823085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wahren-Herlenius M, Dörner T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet. 2013;382:819–831. doi: 10.1016/S0140-6736(13)60954-X. [DOI] [PubMed] [Google Scholar]
- 10.Shirota Y, Yarboro C, Fischer R, Pham TH, Lipsky P, Illei GG. Impact of anti-interleukin-6 receptor blockade on circulating T and B cell subsets in patients with systemic lupus erythematosus. Ann Rheum Dis. 2013;72:118–128. doi: 10.1136/annrheumdis-2012-201310. [DOI] [PubMed] [Google Scholar]
- 11.Hundorfean G, Neurath MF, Mudter J. Functional relevance of T helper 17 (Th17) cells and the IL-17 cytokine family in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:180–186. doi: 10.1002/ibd.21677. [DOI] [PubMed] [Google Scholar]
- 12.Zhang W, Cai Y, Xu W, Yin Z, Gao X, Xiong S. AIM2 facilitates the apoptotic DNA-induced systemic lupus erythematosus via arbitrating macrophage functional maturation. J Clin Immunol. 2013;33:925–937. doi: 10.1007/s10875-013-9881-6. [DOI] [PubMed] [Google Scholar]
- 13.Sun W, Jiao Y, Cui B, Gao X, Xia Y, Zhao Y. Immune complexes activate human endothelium involving the cell-signaling HMGB1-RAGE axis in the pathogenesis of lupus vasculitis. Lab Invest. 2013;93:626–638. doi: 10.1038/labinvest.2013.61. [DOI] [PubMed] [Google Scholar]
- 14.Adhami VM, Syed DN, Khan N, Mukhtar H. Dietary flavonoid fisetin: A novel dual inhibitor of PI3K/Akt and mTOR for prostate cancer management. Biochem Pharmacol. 2012;84:1277–1281. doi: 10.1016/j.bcp.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ying TH, Yang SF, Tsai SJ, Hsieh SC, Huang YC, Bau DT, Hsieh YH. Fisetin induces apoptosis in human cervical cancer HeLa cells through ERK1/2-mediated activation of caspase-8-/caspase-3-dependent pathway. Arch Toxicol. 2012;86:263–273. doi: 10.1007/s00204-011-0754-6. [DOI] [PubMed] [Google Scholar]
- 16.Currais A, Prior M, Dargusch R, Armando A, Ehren J, Schubert D, Quehenberger O, Maher P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer's disease transgenic mice. Aging Cell. 2014;13:379–390. doi: 10.1111/acel.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitra S, Biswas S, Sinha A, Jana NR, Banerjee EM. Therapeutic use of fisetin and fisetin loaded on mesoporous carbon nanoparticle (MCN) in thioglycollate-induced peritonitis. J Nanomed Nanotechnol. 2015;6:332. doi: 10.4172/2157-7439.1000332. [DOI] [Google Scholar]
- 18.Rabb H, Noel S, Martina-Lingua MN, Racusen LC, Hamad AR, Bandapalle S. Compositions and methods for the study and treatment of acute kidney injury. 14/930,883. US Patent. Filed November 3, 2015 issued May 5, 2016.
- 19.Summers SA, Odobasic D, Khouri MB, Steinmetz OM, Yang Y, Holdsworth SR, Kitching AR. Endogenous interleukin (IL)-17A promotes pristane-induced systemic autoimmunity and lupus nephritis induced by pristane. Clin Exp Immunol. 2014;176:341–350. doi: 10.1111/cei.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Léotoing L, Davicco MJ, Lebecque P, Wittrant Y, Coxam V. The flavonoid fisetin promotes osteoblasts differentiation through Runx2 transcriptional activity. Mol Nutr Food Res. 2014;58:1239–1248. doi: 10.1002/mnfr.201300836. [DOI] [PubMed] [Google Scholar]
- 21.Lee JD, Huh JE, Jeon G, Yang HR, Woo HS, Choi DY, Park DS. Flavonol-rich RVHxR from rhus verniciflua stokes and its major compound fisetin inhibits inflammation-related cytokines and angiogenic factor in rheumatoid arthritic fibroblast-like synovial cells and in vivo models. Int Immunopharmacol. 2009;9:268–276. doi: 10.1016/j.intimp.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 22.Feng D, Yang L, Bi X, Stone RC, Patel P, Barnes BJ. Irf5-deficient mice are protected from pristane-induced lupus via increased Th2 cytokines and altered IgG class switching. Eur J Immunol. 2012;42:1477–1487. doi: 10.1002/eji.201141642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Angius F, Floris A. Liposomes and MTT cell viability assay: An incompatible affair. Toxicology In Vitro. 2015;29:314–319. doi: 10.1016/j.tiv.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Donnenberg VS, Donnenberg A. Flow cytometry on disaggregated solid tissues. Int Drug Discov. 2011;6:14–18. [Google Scholar]
- 25.Yuan JS, Wang D, Stewart CN. Statistical methods for efficiency adjusted real-time PCR quantification. Biotechnol J. 2008;3:112–123. doi: 10.1002/biot.200700169. [DOI] [PubMed] [Google Scholar]
- 26.Ding Q, Zhao M, Yu B, Bai C, Huang Z. Identification of tetraazacyclic compounds as novel potent inhibitors antagonizing RORγt activity and suppressing Th17 cell differentiation. PloS One. 2015;10:e0137711. doi: 10.1371/journal.pone.0137711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janda J, Plattet P, Torsteinsdottir S, Jonsdottir S, Zurbriggen A, Marti E. Generation of equine TSLP-specific antibodies and their use for detection of TSLP produced by equine keratinocytes and leukocytes. Vet Immunol Immunopathol. 2012;147:180–186. doi: 10.1016/j.vetimm.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 28.Clowse MEB, Chakravarty E, Costenbader KH, Chambers C, Michaud K. Effects of infertility, pregnancy loss, and patient concerns on family size of women with rheumatoid arthritis and systemic lupus erythematosus. Arthritis Care Res. 2012;64:668–674. doi: 10.1002/acr.21593. [DOI] [PubMed] [Google Scholar]
- 29.Chung SA, Brown EE, Williams AH, Ramos PS, Berthier CC, Bhangale T, Alarcon-Riquelme ME, Behrens TW, Criswell LA, Graham DC, et al. Lupus nephritis susceptibility loci in women with systemic lupus erythematosus. J Am Soc Nephrol. 2014;25:2859–2870. doi: 10.1681/ASN.2013050446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Askanase A, Shum K, Mitnick H. Systemic lupus erythematosus: An overview. Soc Work Health Care. 2012;51:576–586. doi: 10.1080/00981389.2012.683369. [DOI] [PubMed] [Google Scholar]
- 31.Llanos C, Mackern-Oberti JP, Vega F, Jacobelli SH, Kalergis KM. Tolerogenic dendritic cells as a therapy for treating lupus. Clin Immunol. 2013;148:237–245. doi: 10.1016/j.clim.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 32.Shen N, Liang D, Tang Y, Tak PP. MicroRNAs-novel regulators of systemic lupus erythematosus pathogenesis. Nat Rev Rheumatol. 2012;8:701–709. doi: 10.1038/nrrheum.2012.142. [DOI] [PubMed] [Google Scholar]
- 33.Talaat RM, Mohamed SF, Bassyouni IH. Th1/Th2/Th17/Treg cytokine imbalance in systemic lupus erythematosus (SLE) patients: Correlation with disease activity. Cytokine. 2015;72:146–153. doi: 10.1016/j.cyto.2014.12.027. [DOI] [PubMed] [Google Scholar]
- 34.Xing Q, Wang B, Su H, Cui J, Li J. Elevated Th17 cells are accompanied by FoxP3+ Treg cells decrease in patients with lupus nephritis. Rheumatol Int. 2012;32:949–958. doi: 10.1007/s00296-010-1771-0. [DOI] [PubMed] [Google Scholar]
- 35.Chatterjee M, Rauen T, Kis-Toth K, Kyttaris VC, Hedrich CM, Terhorst C, Tsokos GC. Increased expression of SLAM receptors SLAMF3 and SLAMF6 in systemic lupus erythematosus T lymphocytes promotes Th17 differentiation. J Immunol. 2012;188:1206–1212. doi: 10.4049/jimmunol.1102773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu Y, Liu Y, Shi FD, Zou H, Matarese G, La Cava A. Cutting edge: Leptin-induced RORγt expression in CD4+ T cells promotes Th17 responses in systemic lupus erythematosus. J Immunol. 2013;190:3054–3058. doi: 10.4049/jimmunol.1203275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30:455–464. doi: 10.1016/j.it.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hägerbrand K, Marsal J, Gudjonsson S, Håkansson U, Reizis B, Kotarsky K, et al. IRF4 transcription-factor-dependent CD103+CD11b+ dendritic cells drive mucosal T helper 17 cell differentiation. Immunity. 2013;38:958–969. doi: 10.1016/j.immuni.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 39.Chen DY, Chen YM, Wen MC, Hsieh TY, Hung WT, Lan JL. The potential role of Th17 cells and Th17-related cytokines in the pathogenesis of lupus nephritis. Lupus. 2012;21:1385–1396. doi: 10.1177/0961203312457718. [DOI] [PubMed] [Google Scholar]
- 40.Barbado J, Martin D, Vega L, Almansa R, Gonçalves L, Nocito M, Jimeno A, Ortiz de Lejarazu R, Bermejo-Martin JF. MCP-1 in urine as biomarker of disease activity in Systemic Lupus Erythematosus. Cytokine. 2012;60:583–586. doi: 10.1016/j.cyto.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 41.Petri M, Wallace DJ, Spindler A, Chindalore V, Kalunian K, Mysler E, Neuwelt CM, Robbie G, White WI, Higgs BW, et al. Sifalimumab, a human anti-interferon-α monoclonal antibody, in systemic lupus erythematosus: A phase I randomized, controlled, dose-escalation study. Arthritis Rheum. 2013;65:1011–1021. doi: 10.1002/art.37824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lisi S, Sisto M, Lofrumento DD, D'Amore M, De Lucro R, Ribatti D. A potential role of the GRO-α/CXCR2 system in Sjögren's syndrome: Regulatory effects of pro-inflammatory cytokines. Histochem Cell Biol. 2013;139:371–379. doi: 10.1007/s00418-012-1035-z. [DOI] [PubMed] [Google Scholar]
- 43.Zhao PW, Jiang WG, Wang L, Jiang ZY, Shan YX, Jiang YF. Plasma levels of IL-37 and correlation with TNF-α, IL-17A, and disease activity during DMARD treatment of rheumatoid arthritis. PLoS One. 2014;9:e95346. doi: 10.1371/journal.pone.0095346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moles A, Murphy L, Wilson CL, Chakraborty JB, Fox C, Park EJ, Mann J, Oakley F, Howarth R, Brain J, et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J Hepatol. 2014;60:782–791. doi: 10.1016/j.jhep.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tung YT, Chua MT, Wang SY, Chang ST. Anti-inflammation activities of essential oil and its constituents from indigenous cinnamon (Cinnamomum osmophloeum) twigs. Bioresour Technol. 2008;99:3908–3913. doi: 10.1016/j.biortech.2007.07.050. [DOI] [PubMed] [Google Scholar]
- 46.Zughaier S, Aneja R, Stephens DS. Noscapine and analogs and methods related thereto. 8,841,317. US Patent. Filed August 24, 2011 issued September 23, 2014.
- 47.Smiljanovic B, Grün JR, Biesen R, Schulte-Wrede U, Baumgrass R, Stuhlmüller B, Maslinski W, Hiepe F, Burmester GR, Radbruch A, et al. The multifaceted balance of TNF-α and type I/II interferon responses in SLE and RA: How monocytes manage the impact of cytokines. J Mol Med. 2012;90:1295–1309. doi: 10.1007/s00109-012-0907-y. [DOI] [PubMed] [Google Scholar]
- 48.Schiffer L, Bethunaickan R, Ramanujam M, Huang W, Schiffer M, Tao H, Madaio MP, Bottinger EP, Davidson A. Activated renal macrophages are markers of disease onset and disease remission in lupus nephritis. J Immunol. 2008;180:1938–1947. doi: 10.4049/jimmunol.180.3.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuroiwa T, Lee EG. Cellular interactions in the pathogenesis of lupus nephritis: The role of T cells and macrophages in the amplification of the inflammatory process in the kidney. Lupus. 1998;7:597–603. doi: 10.1191/096120398678920712. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.