

Abstract
A large body of evidence has demonstrated that one mechanism by which cholecystokinin (CCK) inhibits food intake through activation of CCK1 receptors (CCK1R) on vagal afferent neurons that innervate the gastrointestinal tract and project to the hindbrain. OLETF rats, which carry a spontaneous null mutation of the CCK1R, are hyperphagic, obese, and predisposed to type 2 diabetes. Recently, by introgressing the OLETF-derived, CCK1R-null gene onto a Fischer 344 genetic background, we have been able to generate a CCK1R-deficient, congenic rat strain, F344.Cck1r−/−, that in contrast to OLETF rats, possesses a lean and normoglycemic phenotype. In the present study, the behavioral and neurobiological phenotype of this rat strain was characterized more fully. As expected, intraperitoneal injections of CCK-8 inhibited intake of chow and Ensure Plus and induced Fos responses in the area postrema and the gelatinosus, commissural and medial subdivisions of the nucleus tractus solitarius of F344.Cck1r+/+ rats, whereas CCK-8 was without effect on food intake or Fos induction in the F344.Cck1r−/− rats. F344.Cck1r−/− and F344.Cck1r+/+ rats did not differ in body weight and showed comparable weight gain when maintained on Ensure Plus for 2 weeks. Also, no difference was found in 24-h food intake, and dark-phase meal frequency or meal size between F344.Cck1r+/+ and F344.Cck1r−/− rats. As expected, blockade of endogenous CCK action at CCK1R increased food intake and blocked the effects of peripheral CCK-8 in control F344.Cck1r+/+ rats. These results confirm that in rats with a F344 background, CCK-1R mediates CCK-8-induced inhibition of food intake and Fos activation in the hindbrain and demonstrate that selective genetic ablation of CCK1R is not associated with altered meal patterns, hyperphagia, or excessive weight gain on a palatable diet.
Keywords: satiety, meal size, cholecystokinin, CCK1 receptors
1. Introduction
Postprandial release of the anorexigenic peptide, cholecystokinin (CCK), generates a well characterized neural signal from the gastrointestinal tract to the hindbrain. Increasing evidence indicates that CCK inhibits food intake by activating intestinal vagal afferent axons that project to the hindbrain nucleus tractus solitarius (NTS), which in turn, activates hindbrain mechanisms that control meal size [11,39,46,48,49,51,65]. Sulfated cholecystokinin octapeptide (CCK-8) binds with high affinity to both CCK1 and CCK2 receptors (CCK1R/CCK2R) [20]. Peripheral administration of CCK-8 induces Fos (a marker of neuronal activation) in the commissural (cNTS), gelatinosus (gNTS), caudal-medial subdivisions of the NTS (cmNTS) [5,9,11,16,35,44,53,63] and the area postrema (AP) [11,35,44,52,63]. These hindbrain areas are important in the regulation of food intake because they receive vagal afferent information from the gut including signals from gastric distension, CCK, and other satiety peptides [55], and they integrate these meal-related cues to control feeding [16,44,55,67].
A large body of literature supports the hypothesis that CCK’s acute effects on neuronal activation in the hindbrain and inhibition of food intake are mediated through the CCK1R [39,51]. Peripheral administration of selective CCK1R agonists and CCK1R antagonists inhibit and stimulate food intake, respectively, [39,51,65] and peripheral administration of the selective CCK1R antagonist, devazepide, blocks CCK-8-induced stimulation of Fos [15,35,63] or elevation of c-fos mRNA [11] in the AP and NTS, as well as CCK-8-induced inhibition of food intake [39,50,65]. In addition, devazepide blocks the effects of food intake inhibition by intestinally infused nutrients [8,48,68]. Both devazepide and another CCK1R antagonist, lorglumide, block the effects of nutrient-induced Fos activation in the AP and NTS [19,28]. Together, these findings indicate that both endogenous and exogenous CCK acutely stimulate neuronal activity in the AP and NTS and inhibit feeding through activation of the CCK1R. Previous studies using the Otsuka Long-Evans Tokushima (OLETF) rats and transgenic mice have reconfirmed that both exogenous and endogenous CCK acts via CCK1R to inhibit food intake [24,34].
Whether chronic ablation of CCK1R signaling contributes to hyperphagia, obesity and impaired glycemic regulation, remains an unresolved issue, as illustrated by the divergent phenotypes of the OLETF rat and CCK1R null mice (CCK1R−/−). The OLETF rat is a well characterized genetic model of type 2 diabetes mellitus (Type 2 DM) [23] that lacks CCK1R [17] because of mutation in the region of the CCK1R gene [58], and this mutation is associated with an inability of CCK-8 to reduce food intake [40] and induce Fos in the hindbrain [19]. These animals also become both hyperphagic and obese relative to their wild-type controls, the Long-Evans Tokushima Otsuka rats (LETO) [34,40]. The Long-Evans strain is reported to be mildly obese with higher basal plasma glucose levels and impaired glucose tolerance to compared to Sprague-Dawley and Wistar strain rats [22,26]. The obesity and hyperphagia of the OLETF rat strain is thought to result from the lack of CCK1R signaling, overexpression of the orexigenic neuropeptide Y (NPY) in the dorsomedial hypothalamus (DMH) [3], as well as the presence of several quantitative trait loci (QTL’s) associated with obesity and type 2 diabetes mellitus (DM) that are intrinsic to the classical strain F344 and Brown Norway rats [21,37,42,43,64,66]. While several factors appear to contribute to the obesity, hyperphagia, and impaired glycemic regulation in the OLETF rat, the CCK1R−/− mouse model has been used as an alternative strategy to characterize the role of endogenous CCK in the control of food intake and body weight.
CCK1R−/− mice, a second rodent model with genetically ablated CCK1R signaling, similar to OLETF rats, show no reduction in food intake after CCK-8 administration, and have meal patterns characterized by increased meal size compared to wild-type control animals [6,25]. However, in contrast to OLETF rats, CCK1R−/− mice are normoglycemic [25], and do not develop hyperphagia and obesity when maintained on regular chow [4,24]. Both CCK1R−/− mice and wild-type mice lack CCK1R in the DMH [4] and recent studies suggest that the regulation of NPY in the DMH is independent of CCK1R signaling in CCK1R−/−mice [2,4], but not in OLETF rats [2,4]. Taken together, these findings indicate that the absence of CCK1R specifically increases meal size in chow fed CCK1R−/− mice, whereas dysregulation of NPY signaling in the DMH in the OLETF rat is one of multiple factors that may explain the obesity and hyperphagia of the OLETF rat.
In order to address if the QTL of the OLETF rats contribute, in part, to their obese phenotype, we recently generated a congenic Fischer 344 rat strain, F344.Cck1r−/−, in which the OLETF-derived CCK1R gene identified by the QTL (Niddm10/of) on chromosome 14 was introgressed into the normoglycemic and nonobese Fischer 344 rat genetic background. As we reported earlier, F344.Cck1r−/− is a unique, CCK1R-deficient Fischer 344 rat strain with a lean and normoglycemic phenotype [36,37], that offers a new way to assess the behavioral and neurobiological consequences of selective CCK1R ablation.
The present study was conducted to further dissect the behavioral and neurobiological phenotype of the lean, normoglycemic F344.Cck1r−/− rat strain. We hypothesized that F344.Cck1r−/− rats, relative to their Fischer 344 wild-type counterparts, would not respond to the suppressive or stimulatory effects of CCK-8 on food intake and Fos induction in the hindbrain, respectively. Further, we hypothesized that the mutant F344.Cck1r−/− rats, relative to F344 wild-type counterparts would not show hyperphagia, excessive weight gain or altered meal patterns during exposure to a highly palatable diet. Specifically, we compared F344.Cck1r−/− rats with their Fischer 344 wild-type counterparts on the following behaviors: 1) chow intake or intake of a palatable diet following CCK-8 vs. vehicle administration; 2) Fos induction in several hindbrain areas (AP, cNTS, cmNTS, and gNTS) that commonly respond to CCK-8 in wild-type rats; 3) meal patterns and body weight gain during a 2-week exposure to a palatable diet, Ensure Plus Vanilla.
2. Results
Characteristics of the Congenic F344.Cck1r rat.
The congenic line was developed by K. Matsumoto from the University of Tokushima, Japan, as a collaborative project with D.H. Moralejo from the University of Washington, USA. The line has been maintained for sixteen inter-cross matings of sister-brother littermates at the University of Washington in Seattle. The OLETF DNA recombinant fragment has been kept as the original line that contains the locus Niddm10/of previously identified by quantitative trait loci (QTL’s) analyzes [25]. When new STS markers became available, we were able to define the DNA fragment introgressed within 37 Mb. The Cck1r gene is co-localized within the locus Niddm10 on rat chromosome 14 between D14Wox5 at 42.60 Mb and D14Rat45 at 70.23 Mb, this critical interval from OLETF rat introgressed onto the F344 rat is shown in Figure 1.
Figure 1. Genotyping of Chromosome 14. STS/SSLP marker map of chromosome 14.
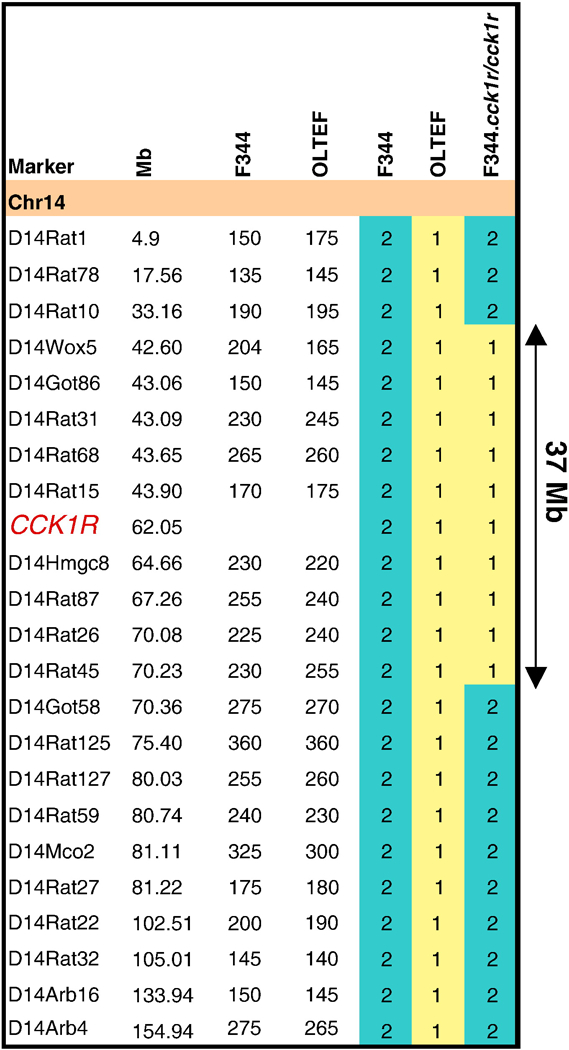
The new availability of high density markers from the Rat Genome Database expanding the size of chromosomes allow us to define more precisely the fragment introgressed onto F344 rat to 37-Mb. The UCSC map marker location and the size of the polymorphic marker between the two parental strains OLETF and F344 are shown. The numerical color code is: yellow (1) OLETF, blue (2) F344, and yellow-blue (2–1-2) F344.Cck1r congenic line carrying a DNA fragment of 37 Mb from OLETF rat.
CCK1R, CCK2R, and GAPDH Gene Expression in Mutant F344.CCK1r−/− and Wild-type F344.CCK1r+/+ rats.
Our aim was to confirm the presence and absence of CCK1R in pancreatic tissue because the pancreas is a known site of CCK1R expression [12], and this tissue was chosen in order to confirm the presence of the CCK1R deletion from F344.CCK1r+/+ and F344.CCK1r−/− rats, respectively. There was no significant difference in age (days) between groups at the start of the experiment: (F344.Cck1r−/− (92.2 ± 27.7 days) vs. F344.Cck1r+/+ (99.2 ± 32.6 days)) (F(1,8)=0.027, p=NS). As expected, CCK1R expression was virtually absent in the pancreas of F344.CCK1r−/− rats (n=5/group), with a copy number of 0.02 ± 0.02. CCK1R was expressed in the pancreas of F344.CCK1r+/+ rats, with a copy number of 469.8 ± 104.9, and this was significantly different from CCK1R expression in pancreas in F344.CCK1r−/− rats (F(1,8)=20.06, P<0.01). CCK2R expression levels in pancreas were equal in both strains of rats, with a copy number of 0 ± 0 in the F344.CCK1r−/− and 0 ± 0 in the F344.CCK1r+/+ rats (p=NS). The housekeeping gene GAPDH was also expressed in equal amounts in the pancreas from the two strains of rats, with copy numbers of 0.22 ± 0.4 and 0.18 ± 0.02 in the F344.CCK1r−/− and F344.CCK1r+/+ rats, respectively (P=NS). As a positive control for CCK2R, brain tissue, known to contain CCK2R [41], was also collected and screened for CCK2R expression. There was also a significant difference in age (3.6 days) between groups (n=5/group) at the start of the experiment: (F344.Cck1r−/− (57.2 ± 0.7 days) vs. F344.Cck1r+/+ (60.8 ± 0.7 days)) (F(1,8)=12.0, p<0.05). CCK2R levels were approximately equal in both strains of rats, with a copy number of 191.2 ± 19.4 in the F344.CCK1r−/− and 183.4 ± 22.9 in the F344.CCK1r+/+ rats (p=NS). The housekeeping gene GAPDH was expressed in equal amounts in the two strains of rats, with copy numbers of 1461±90.7 and 1554.2±148.2 in the F344.CCK1r−/− and F344.CCK1r+/+ rats, respectively (P=NS).
Study 1: Effects of CCK-8 on Food Intake in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
Study 1A: Effects of CCK-8 on Chow Diet.
Our aim was to investigate the effects of CCK-8 on solid chow intake in F344.Cck1r−/− and F344.Cck1r+/+ rats (n=7/group) in a crossover design with each animal serving as its own control. There was no significant difference in body weight between groups at the start of the experiment: (F344.Cck1r−/− (347 ± 2.8 vs. F344.Cck1r+/+ (359 ± 6.1 g)) (F(1,12)=3.62, p=NS). There was also no significant difference in age (days) between groups at the start of the experiment: (F344.Cck1r−/− (135 ± 0 days) vs. F344.Cck1r+/+ (151 ± 8.3 days)) (F(1,12)=3.93, p=NS).
CCK-8 (2 nmol/kg) “Low Dose”
The effects of two doses of CCK-8 (2 and 8 nmol/kg) were examined in both in mutant F344.Cck1r−/− and wild-type F344.Cck1r+/+ rats in order determine the food intake response to different doses of CCK-8. There was no significant difference in body weight between groups at the start of the experiment: (F344.Cck1r−/− (347 ± 2.8 g) vs. F344.Cck1r+/+ (359 ± 6.1 g)) (F(1,12)=3.62, p=NS).
CCK-8 (2 nmol/kg) alone had no effect in the F344.Cck1r−/− rats, but inhibited 30-minute intake in the F344.Cck1r+/+ rats by 87% (Figure 2A). There was a significant main effect of CCK-8 (F(1,12) = 10.49, p<0.01), strain (F(1,12) = 24.68, p<0.01), and a significant interaction of CCK-8 and strain (F (1,12) = 8.83, p<0.05), indicating that strain influenced the ability of CCK-8 to inhibit intake of chow.
Figure 2A-B. Effects of CCK-8 on Chow Intake in Mutant F344.CCK1r−/− and Wild-type F344.CCK1r+/+ Rats :
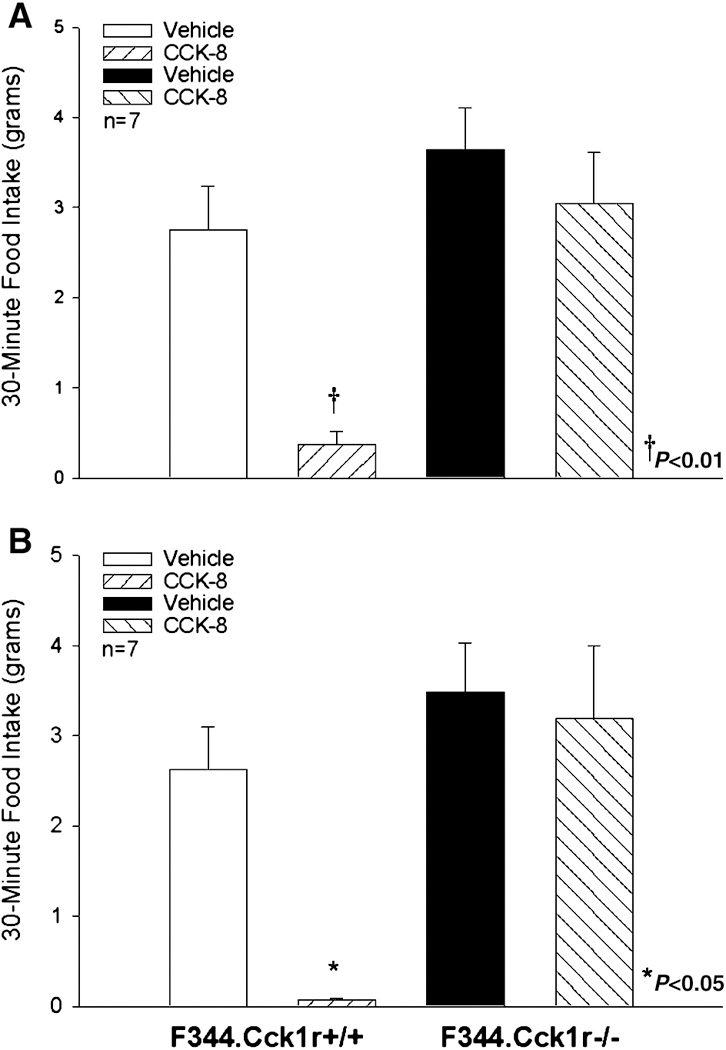
CCK-8 (2 nmol/kg), as expected, inhibited food intake at 30 min by 87% in F344.Cck1r+/+ (Figure 2A), whereas CCK-8 at this dose was ineffective at inhibiting food intake in F344.Cck1r−/− rats (Figure 2A). Similarly, the higher dose of CCK-8 (8 nmol/kg) inhibited food intake at 30 min by 97% in F344.Cck1r+/+ whereas in F344.Cck1r−/− rats, CCK-8 did not inhibit food intake (Figure 2B).
CCK-8 (8 nmol/kg) “High Dose”
CCK-8 (8 nmol/kg) alone had no effect in the F344.Cck1r−/− rats, but inhibited 30-minute intake in the F344.Cck1r+/+ rats by 97% (Figure 2B). There was a significant main effect of CCK-8 (F(1,12) = 10.58, p<0.01), strain (F(1,12) 9.54, p<0.01), and a significant interaction of CCK-8 and strain (F (1,12) = 5.95, p<0.05), indicating that strain influenced the ability of CCK-8 to inhibit intake of chow.
Study 1B: Effects of CCK-8 on Palatable Diet (Ensure Plus Vanilla)
Our aim was to investigate the effects of CCK-8 on a palatable diet intake in F344.Cck1r−/− and F344.Cck1r+/+ rats (n=7/genotype) in a crossover design with each animal serving as its own control. There was no significant difference in body weight between groups at the start of the experiment: (F344.Cck1r−/− (339 ± 4.4 g) vs. F344.Cck1r+/+ (336 ± 4.3 g)) (F(1,14)=0.256, p=NS). There was a significant difference in age of approximately 5.5 weeks between groups, also used in Study 3A, 3B, and 4 at the start of the experiment: (F344.Cck1r−/− (123 ± 1.7 days) vs. F344.Cck1r+/+ (162 ± 15.2 days)) (F(1,14)=5.165, p=0.04). CCK-8 (8 nmol/kg) alone had no effect on food intake in the F344.Cck1r−/− rats, but did inhibit 30-minute food intake by 87% in F344.Cck1r+/+ rats (Figure 3). There was no significant main effect of CCK-8 (F(1,14)=1.92, p=NS), strain (F(1,14) = 3.27, p=0.09), but there was a significant interaction of CCK-8 and strain (F (1,14) = 14.62, p<0.01), indicating that strain influenced the ability of CCK-8 to inhibit intake of Ensure Plus Vanilla.
Figure 3. Effects of CCK-8 on 30-minute intake of Ensure Plus Vanilla in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
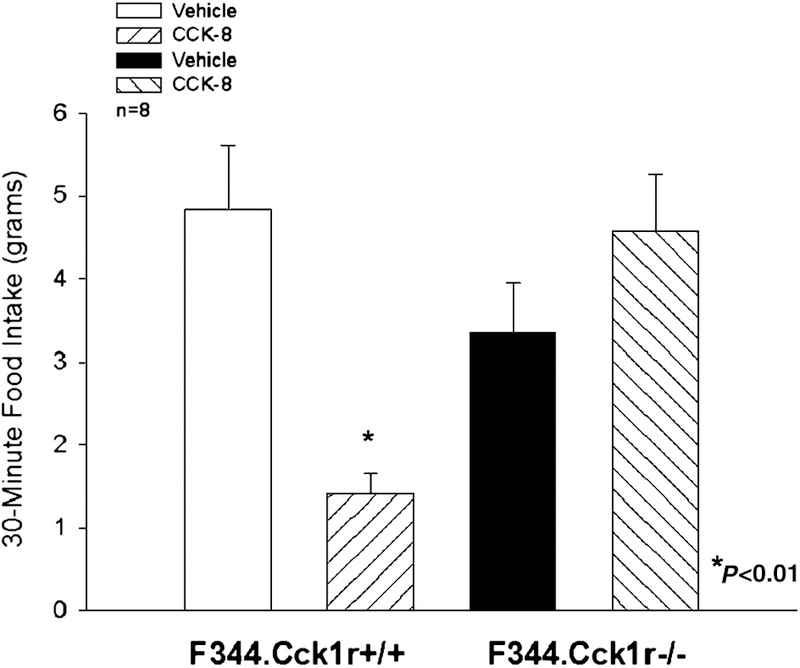
CCK-8 (8 nmol/kg) inhibited Ensure intake at 30 min by 87% in F344.Cck1r+/+ rats, but failed to alter food intake F344.Cck1r−/− rats.
Study 2. Effects of CCK-8 on Fos Induction in the Hindbrain in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
Our aim was to examine the effects of CCK-8 (8 nmol/kg) to induce Fos in the area postrema (AP) and nucleus tractus solitarius (NTS)) in F344.Cck1r−/− and F344.Cck1r+/+ rats. There was no significant difference in body weight between groups injected with saline at the start of the experiment: (F344.Cck1r−/− (355 ± 4.4 g) vs. F344.Cck1r+/+ (357 ± 8.7 g)) (F(1,12)=0.086, p=NS). There was no significant difference in age between groups injected with saline at the start of the experiment: (F344.Cck1r−/− (142 ± 6.6 days) vs. F344.Cck1r+/+ (140 ± 8.6 days)) (F(1,12)=0.029, p=NS). In animals to be injected with CCK-8, there was an average difference in age of 2 weeks, which also resulted in a difference in body weight between groups at the start of the experiment: (F344.Cck1r−/− (374 ± 2.6 g) vs. F344.Cck1r+/+ (347 ± 6.3 g)) (F(1,18)=15.652, p<0.05). There was no significant difference in age between groups injected with CCK-8 at the start of the experiment: (F344.Cck1r−/− (165 ± 12.1 days) vs. F344.Cck1r+/+ (146 ± 8.4 days)) (F(1,18)=1.768, p=NS).
Area Postrema (AP)
CCK-8 alone had no effect in the F344.Cck1r−/− rats, but CCK-8 stimulated Fos induction in the AP in the F344.Cck1r−/− rats by 5237% relative to vehicle-injected animals (Figure 4A-D, Figure 4A-B). There was a significant main effect of CCK-8 (F(1,29) = 78.3, p<0.01), strain (F(1,29) = 78.6, p<0.01), and a significant interaction of CCK-8 and strain (F (1,29) = 84.5, p<0.01), indicating that strain influenced the ability of CCK-8 to induce Fos in the AP.
Figure 4A-D. Effects of CCK-8 on Fos induction in the Area Postrema in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
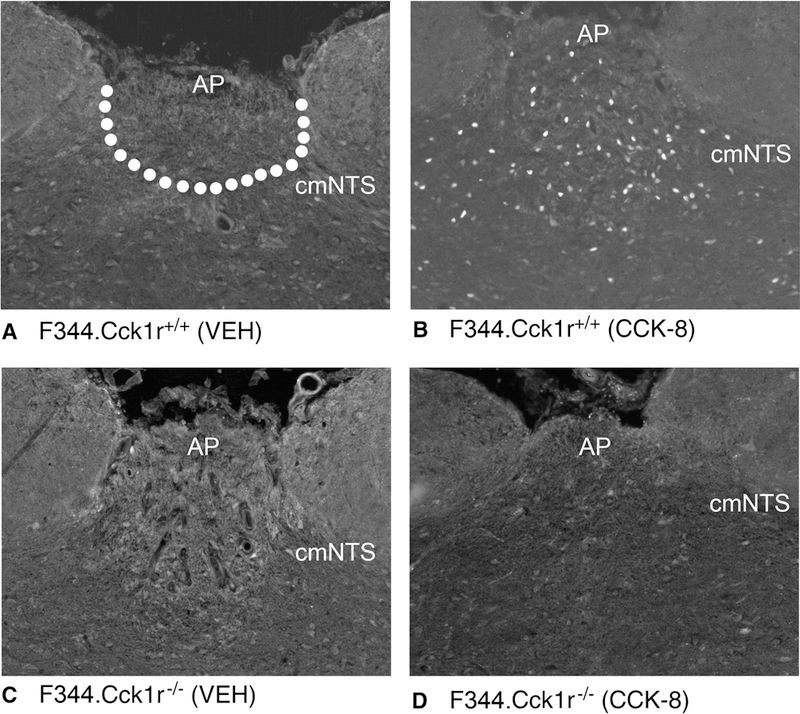
Fos activation is revealed by concentration of immunoreactive Fos in cell nuclei (Fos (+) cells) in the AP. Fos-immunostaining was done by Cy3 fluorescence. Images were taken from the whole AP. Figure 4A: Peripheral injection of vehicle induced little or no Fos (+) neurons in the AP of F344.Cck1r+/+ rats. Figure 4B: Peripheral injection of CCK-8 (8 nmol/kg) induced numerous Fos (+) neurons in the AP of F344.Cck1r+/+ rats. Figure 4C: Peripheral injection of vehicle induced little or no Fos (+) neurons in the AP of F344.Cck1r−/− rats. Figure 4D: Peripheral injection of CCK-8 (8 nmol/kg) induced little or no Fos (+) neurons in the AP of F344.Cck1r−/− rats. A-D all visualized at 20X magnification.
Caudal-medial NTS (cmNTS)
CCK-8 alone had no effect in the F344.Cck1r−/− rats, but CCK-8 stimulated Fos induction in the cmNTS by 1931% relative to vehicle injected animals (Figure 4A-B, Figure 8A-D). There was a significant main effect of CCK (F(1,29) = 79.0, p<0.01), strain (F(1,29) = 83.8, p<0.01), and a significant interaction of CCK-8 and strain (F (1,29) = 87.3, p<0.01), indicating that strain influenced the ability of CCK-8 to induce Fos in the cmNTS.
Figure 8. Body weight gain following 2-week exposure to Ensure Plus Vanilla in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
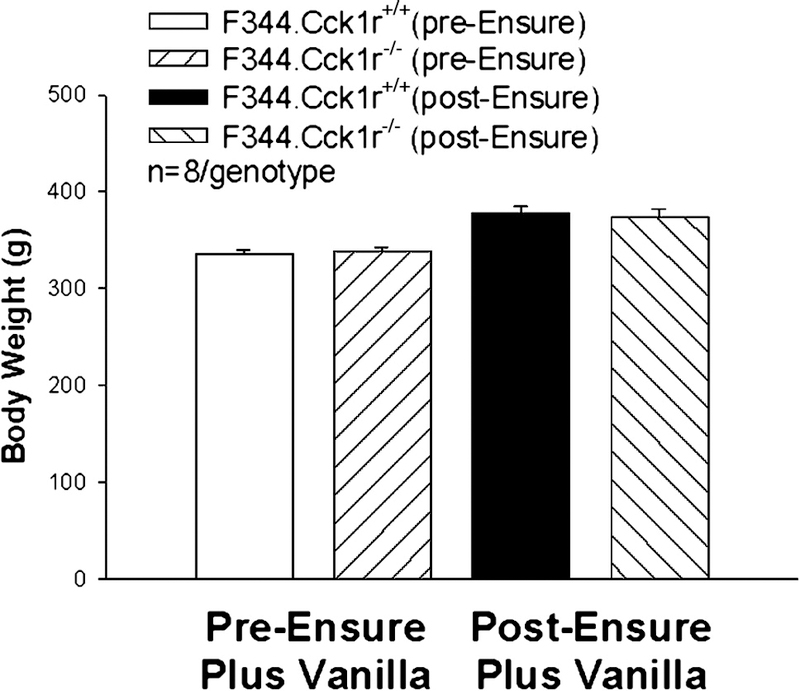
Body weight in animals (n=8/genotype) was monitored before and after a 2-week exposure to Ensure Plus Vanilla. Both genotypes gained a comparable amount of weight: (F344.Cck1r−/− (65.5 ± 2.6 g) vs. F344.Cck1r+/+ (61.8 ± 4.5 g)). There also was no difference in body weights between groups at the start or completion of exposure to Ensure Plus Vanilla.
Commissural NTS (cNTS)
CCK-8 alone had no effect in the F344.Cck1r−/− rats, but CCK-8 stimulated Fos induction in the cNTS by 13757% relative to vehicle injected animals (Figure 7A-B). There was a significant main effect of CCK-8 (F(1,29) = 42.3, p<0.01), strain (F(1,29) = 40.9, p<0.01) and a significant interaction of CCK-8 and strain (F (1,29) = 46.1, p<0.01), indicating that strain influenced the ability of CCK-8 to induce Fos in the cNTS.
Figure 7A-B. Meal number and meal size in 6 h fasted and ad libitum fed Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
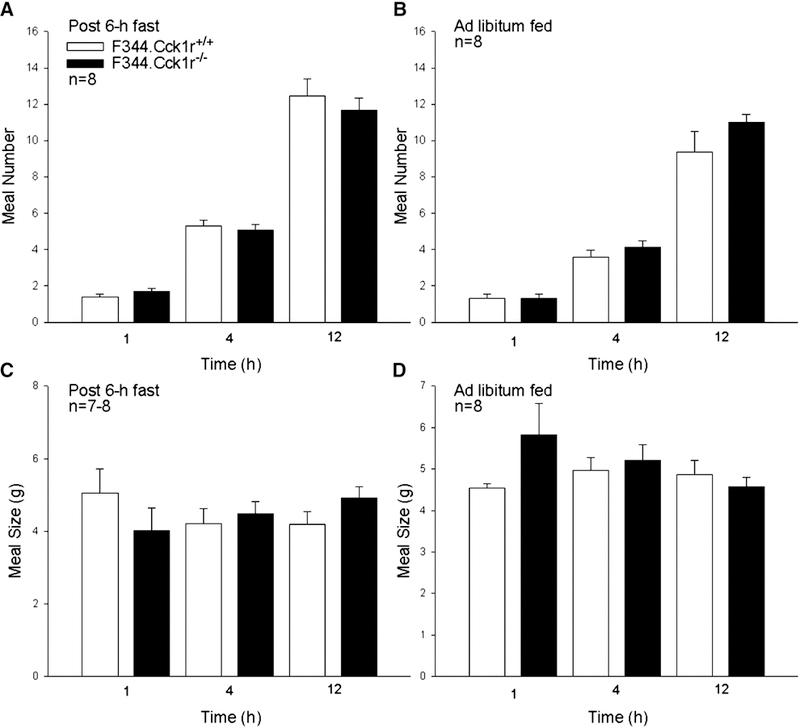
Meal number was not different at 1, 4, or 12 hours after provision of food between F344.Cck1r+/+ and F344.Cck1r−/− rats following a 6-h fast or when fed Ensure Plus ad libitum (Figure 7A). Meal size was not different at 1, 4, or 12 hours after provision of food between F344.Cck1r+/+ and F344.Cck1r−/− rats following a 6-h fast or when fed Ensure Plus ad libitum (Figure 7B).
Gelatinosus NTS (gNTS)
CCK-8 alone had no effect in the F344.Cck1r−/− rats, but CCK-8 stimulated Fos induction in the gNTS by 5471% relative to vehicle injected animals (Figure 7A-B). There was a significant main effect of CCK-8 (F(1,29) = 7.7, p<0.01), strain (F(1,29) = 8.6, p<0.01), and a significant interaction of CCK-8 and strain (F (1,29) = 31.3, p<0.01), indicating that strain influenced the ability of CCK-8 to induce Fos in the cNTS.
Study 3. Meal Patterns During Access to Palatable Diet in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
There was no significant difference in body weight between groups used in Study 3A or 3B at the start of the experiment: (F344.Cck1r−/− (338 ± 4.6 g) vs. F344.Cck1r+/+ (336 ± 4.4 g)) (F(1,14)=0.124, p=NS). There was a significant difference in age of approximately 5.5 weeks between groups in Study 3A, 3B, and 4 at the start of the experiment: (F344.Cck1r−/− (123 ± 1.7 days) vs. F344.Cck1r+/+ (162 ± 15.2 days)) (F(1,14)=5.165, p=0.04).
Study 3A: Response to CCK-8 on Wild-type Meal Size in F344.Cck1r+/+ and Mutant F344.Cck1r−/− Rats.
Our aim was to investigate the effect of CCK-8 on 60-min meal patterns in F344.Cck1r−/− and F344.Cck1r+/+ rats (n=8/genotype) fed a liquid diet (Ensure Plus Vanilla) in a crossover design in which each animal served as its own control. CCK-8 (8 nmol/kg) inhibited 60-min meal size in animals fed Ensure Plus Vanilla by 37% in F344.Cck1r+/+ rats (F(1,7)=11.372, p<0.05) (5.63 ± 0.82 g veh vs. 3.53 ± 0.55 g CCK-8), but was without effect in F344.Cck1r−/− rats (F(1,7)=0.563, p=NS) (5.66 ± 1.20 g veh vs. 7.35 ± 1.67 g CCK-8). CCK-8, as expected, had no effect on 60-min meal frequency in either the F344.Cck1r+/+ (F(1,7)=0.636, p=NS) (1.88 ± 0.23 meals veh vs. 1.63 ± 0.26 meals CCK-8) or the F344.Cck1r−/− rats (F(1,7)=1.00, p=NS) (1.88 ± 0.23 meals veh vs. 1.88 ± 0.30 meals CCK-8).
Study 3B: Meal Number and Meal Size in Mutant F344.Cck1r−/− F344.Cck1r+/+ and Wild-type Rats.
Our aim was to investigate the meal patterns in F344.Cck1r−/− and F344.Cck1r+/+ rats (n=8/genotype) fed a palatable diet (Ensure Plus Vanilla) when fasted 6-h or ad libitum fed.
6-h Fasted Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
Meal number was not different at 1, 4, or 12 h from the start of the dark cycle between F344.Cck1r+/+ and F344.Cck1r−/− rats (P=NS at all time points, Figure 7A). There was also no significant difference in meal size between F344.Cck1r+/+ and F344.Cck1r−/− rats at 1, 4, or 12 h from the start of the dark cycle (P=NS at all time points, Figure 7C). Cumulative 18-h intake was not different in F344.Cck1r+/+ (55.3±6.3 g) and F344.Cck1r−/− rats (52.1±1.8 g) (P=NS).
Ad Libitum Fed Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
Meal number was not different at 1, 4, or 12 hours between F344.Cck1r+/+ and F344.Cck1r−/− rats (p=NS at all time points, Figure 7B). There was also no significant effect on meal size between F344.Cck1r+/+ and F344.Cck1r−/− rats at 1, 4, or 12 h (P=NS at all time points, Figure 7D). Cumulative 24-h intake was not different in F344.Cck1r+/+ (56.9±2.1 g) andF344.Cck1r−/− rats (58.8±1.8 g) (P=NS).
Study 3C: Food intake in Chow-fed Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
There was a significant difference in body weight between groups at the start of the experiment: (F344.Cck1r−/− (336 ± 3.1 g) vs. F344.Cck1r+/+ (320 ± 3.1 g)) (F(1,19)=0.13.553, p<0.05). There was no significant difference in age between groups in Study 3C at the start of the experiment: (F344.Cck1r−/− (115.5 1.1 days) vs. F344.Cck1r+/+ (115.4 ± 3.0 days)) (F(1,19)=0.000, p=NS). Our aim was to investigate the feeding effects of chow-fed F344.Cck1r−/− and F344.Cck1r+/+ rats. Food intake was measured at 0.5 and 18 h after food access was restored, starting at the onset of the dark cycle. F344.Cck1r−/− (n=11) and the F344.Cck1r+/+ (n=10) rats (each assessed on 3 separate days), which did not receive any injection, consumed similar amounts of chow at 0.5 h (2.1±0.3g F344.Cck1r−/− vs. 2.0±0.3g F344.Cck1r+/+) (P=NS) and 18 h (17.8±0.7g F344.Cck1r−/− vs. 17.0±0.5g F344.Cck1r+/+) (P=NS).
Study 4. Body Weight Gain During 2-week Access to Palatable Diet (Ensure Plus Vanilla) in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+.
Our aim was to investigate the body weight gain in F344.Cck1r−/− and F344.Cck1r+/+ rats (n=8/genotype) fed a palatable diet (Ensure Plus Vanilla) after a 2-week exposure. Body weight was monitored before and after a 2-week exposure to Ensure Plus Vanilla. Both genotypes gained a comparable amount of weight: (F344.Cck1r−/− (36 ± 4.7 g) vs. F344.Cck1r+/+ (42 ± 4.2 g)) (p=NS). There was a significant difference in age of approximately 5.5 weeks between groups at the start of the experiment: (F344.Cck1r−/− (123 ± 1.7 days) vs. F344.Cck1r+/+ (162 ± 15.2 days)) (F(1,14)=5.165, p=0.04). There also was no difference in body weights between groups at the start (F344.Cck1r−/− (338 ± 4.7 vs. F344.Cck1r+/+ (336 ± 4.4 g)) or completion of exposure (F344.Cck1r−/− (374 ± 7.8 g) vs. F344.Cck1r+/+ (378 ± 7.4 g)) to Ensure Plus Vanilla (p=NS) (Figure 8).
Study 5. Response of F344.Cck1r+/+ Rats to Endogenous CCK1R Blockade.
Our aim was to determine if blockade of endogenous CCK1R in Fischer 344 rats would result in a stimulation of food intake. Animals weighed approximately 285 ± 1.7 g and were 110–123 days old at the start of the experiments. Devazepide (1 mg/kg) alone stimulated 2, 3, and 4 h food intake in F344.Cck1r−/− rats by 47, 25 and 25% (Figure 9A). Devazepide also stimulated 24 h food intake by 20% (data not shown). Significant main effect of devazepide were obtained at 2 h (F(1,13) = 29.428, p<0.01), 3 h (F(1,13) = 10.364, p<0.01), 4 h (F(1,13) = 14.250, p<0.01), and 24 h (F(1,13) = 32.587, p<0.01).
Figure 9. Response of Wild-type F344.Cck1r+/+ Rats to Endogenous CCK1R Blockade.
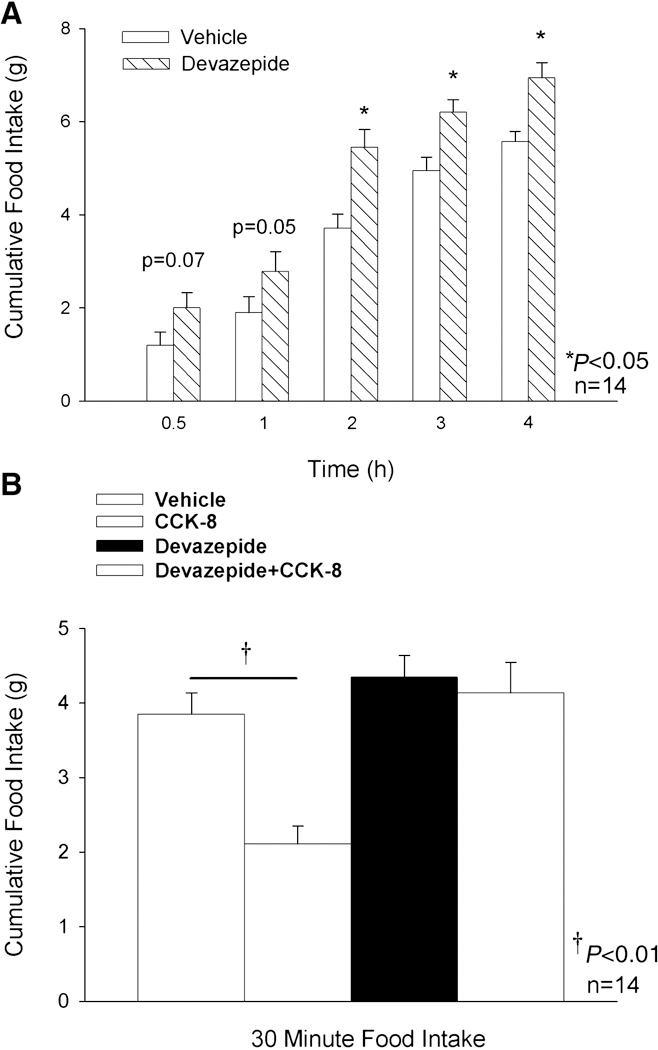
Figure 9A. Devazepide (1 mg/kg) or vehicle (10% DMSO, 10% Tween 80, 80% saline) was administered to 30-min. fasted animals at 30 min. prior to the start of the dark cycle and access to food. Figure 9B. Devazepide (1 mg/kg) or vehicle was administered to 6-h fasted animals 30 min. prior to CCK-8 (8 nmol/kg) or vehicle (0.1% BSA, saline). CCK-8 was administered immediately prior to the dark cycle and access to food. Food intake was measured at 0.5, 1, 2, 3, 4, and 24 h after access to food and the start of the dark cycle for studies described in Figure 9A and 9B.
Study 6. Effect of Endogenous CCK1R Blockade on the Ability of F344.Cck1r+/+ Rats to Respond to CCK-8.
Our aim was to determine if blockade of endogenous CCK1R in Fischer 344 rats would impair the ability of CCK-8 to inhibit food intake. Animals weighed approximately 334 ± 2.6 g and were 126–139 days old at the start of the experiments. Devazepide (1 mg/kg) alone had no effect in 6-h fasted F344.Cck1r−/− rats, and CCK-8 (8 nmol/kg) alone inhibited 30-minute intake in F344.Cck1r+/+ rats by 45% (Figure 9B). Devazepide blocked the ability of CCK-8 to inhibit 30-min food intake. There was a significant main effect of devazepide (F(1,39) = 20.173, p<0.01), CCK-8 (F(1,39) = 13.309, p<0.01), and a significant interaction of devazepide and CCK-8 (F (1,39) = 7.484, p<0.01).
3. Discussion.
In the present study, we dissected the feeding behavior and neurobiology of a novel, CCK1R-deficient rat strain (Fischer 344.Cck1r−/−), whose lean and normoglycemic phenotype contrasts sharply with that of the OLETF rat, a well-established model of CCK1R-deficiency. With this unique animal model, we hypothesized that F344.Cck1r−/− rats, relative to their Fischer 344 wild-type counterparts, would not respond to the suppressive or stimulatory effects of CCK-8 on food intake and Fos induction, respectively. As expected, CCK-8 inhibited chow and Ensure Plus intake and induced Fos in the hindbrain in F344.Cck1r+/+ rats but not in F344.Cck1r−/− rats. We further hypothesized that F344.Cck1r−/− rats would not show hyperphagia, excessive weight gain or altered meal patterns compared to F344.Cck1r+/+ rats, during exposure to a highly palatable diet. We found that there was no difference between the F344.Cck1r+/+ and F344.Cck1r−/− animals in meal size during the dark cycle or any differences in body weight gain between the F344.Cck1r−/− and F344.Cck1r+/+ animals, following 2 weeks on a highly palatable Ensure diet. We found that blockade of endogenous CCK at CCK1R stimulates food intake in ad libitum fed F344.Cck1r+/+ rats and blocks the effects of CCK-8 to inhibit food intake in 6-h fasted F344.Cck1r+/+ rats confirming that control F344.Cck1r+/+ rats with intact CCK1R signaling maintain the ability to respond to endogenous CCK1R blockade. Together, these results confirm the CCK1R gene deletion, but we find that in rats with a F344 background, genetic ablation of CCK1R is not associated with obesity, hyperphagia, or altered meal patterning.
The present results are the first description of a rat model lacking CCK1Rs that is lean, non-hyperphagic, and without obvious aberrations in meal frequency or meal size. Body weight development of the congenic F344.Cck1r−/− and F344.Cck1r+/+ animals were equivalent to one another on both regular chow diet or during a 2-week diet of highly palatable Ensure Vanilla diet. Aside from an inability to respond to CCK-8 [23,38], these findings contradict observations from OLETF rats, which show hyperphagia [40], increased meal size [40], decreased meal frequency [40], overconsumption on high-fat diets [2] and exacerbation of their obesity [2,54] compared to their LETO littermates.
The question remains why the OLETF rat is obesity prone and hyperglycemic whereas the congenic F344.Cck1r−/− is lean and normoglycemic. The congenic F344.Cck1r−/− rats may be resistant to the obesity and diabetes phenotype because it lacks the genes that favor type 2 DM which are found in the OLETF animals, as demonstrated by QTL analyses [36,37,43,55,57,66]. Another possibility is that the Niddm10/of QTL may not cause type 2 DM in rats on a Fischer 344 background because this genetic background might offer some protection against type 2 DM, and introgression of the Niddm10/of QTL alone might be insufficient to cause type 2 DM in the Fischer 344 rats [25].
It is not clear what common denominator if any that is shared between the CCK1R−/− mice, the CCK null (CCK−/−) mice, and the F344.Cck1r−/− rats which explains the lack the obesity and hyperphagia in all three animals models fed a chow diet [4,24,29]. It is possible that the lean phenotype in all three animal models may be explained by upregulation and increased activity of satiety signals other than CCK (ie glucagon-like peptide 1 [61], bombesin-related peptides [27,32], PYY [1], oxyntomodulin [10], or islet amyloid polypeptide [30,47]). Identifying any abnormalities in neuropeptide gene expression in these animal models certainly merits further investigation.
Differences between studies that have examined CCK1R−/− and CCK−/−mice have reported marked differences in hyperphagia based on diet. CCK1R−/−mice have been shown to be either hyperphagic and obese [13] or not hyperphagic [2,29] depending on diet. It would be useful for future studies to examine effects of identical diets on meal patterns in F344.Cck1r−/− rats, CCK1R−/− and CCK−/− mice in order to further delineate the role of CCK1R in these animal models.
The present study demonstrates for the first time a rat model with selective genetic ablation of CCK1R is not associated with altered meal patterns, hyperphagia, or excessive weight gain on a palatable diet. As expected, CCK-8 requires CCK1R to inhibit food intake and to activate neurons in hindbrain satiety centers. In addition, control rats on a Fischer 344 background were found to exhibit the expected increased food intake in response to the CCK1R antagonist devazepide and as expected, devazepide also blocked the effects of CCK-8 to inhibit food intake. Together, these results confirm the CCK1R gene deletion, but we find that in rats with a F344 background that is not predisposed to Type 2 DM or obesity, genetic ablation of CCK1R is not associated with obesity, hyperphagia, or altered meal patterning.
4. Experimental Procedure
Experimental Animals.
Experimental protocols were approved by the Institutional Animal Care & Use Committee (IACUC) from the University of Washington and the Seattle VA Puget Sound Medical Center. Adult male Fischer rats (weight range 283–373 g) were used in all studies. The animals were housed individually in Plexiglass cages in a temperature-controlled room (21–23 º under a 12/12 h light-dark cycle. Animals were adapted to lights-off at 1800 h and were fed a standard rat chow diet (Harlan Teklad, Madison, WI) with the exception of Study 1, in which Ensure Plus was substituted for chow diet. All animals used in all studies with the exception of Study 5 and 6 were obtained from a breeding colony at the University of Washington, originally derived from Charles River. Adult male Fischer 344 rats were purchased from Charles River for Study 5 and 6 due to the lack of availability of animals from the University of Washington. All animals were age-matched and weight-matched unless otherwise indicated. De-ionized water was freely available.
Development of congenic line F344.Cck1r−/−.
The congenic line was constructed by the speed congenic method [31,62]. The Fischer 344.Cck1r−/− rats were developed by an initial cross between male OLETF and female Fischer 344 rats, followed by 5 back-crossings between male progenies and female Fischer 344 rats [25], and we have subsequently bred over 16 generations by inter-crossings at the University of Washington. At each generation, microsatellite-based genotyping was performed to select the best male that harbored OLETF Cck1r gene donor genome segment. Heterozygous sisters and brothers were mated to obtain rats homozygous for the introgressed strain. To verify the purity of the strain, a genome-wide analysis with 100 markers spaced at an average of 22 cM was performed. No DNA OLETF-derived alleles outside the target region were found [36]. All animals used in these studies are progeny of 16 inter-crosses at the University of Washington.
Genotyping.
The congenic line F344.Cck1r−/− was genotyped to assure the integrity of the DNA fragment carrying the CCKR gene from OLETF rat onto the F344 rat. The DNA transferred fragment from OLETF included the locus F.O-Nidd10/of, as recently published [25]. Between 25–30 days of age, 5-mm tail snips were obtained and DNA isolated using a phenol-chloroform extraction protocol. The tail snips were digested overnight with 12.5 µl Proteinase K (Promega, Corp., Madison, WI) in 500 µl SET (1% SDS in 150 mmol/L NaCl, 5 mmol/L EDTA and 50 mmol/L Tris, pH 8.0) and then diluted in 500 µl phenolchloroform. The samples were vortexed for 1 min and the mixture centrifuged at 20,800xg for 15 min; the aqueous layer was transferred to 500 µl cold isopropanol. The samples were again centrifuged at 20,800 x g for 5 min, the isopropanol discarded and the DNA pellet washed by centrifugation in cold 70% EtOH. The pellet was air dried for 15 min and resuspended in TE (10 mmol/L Tris, 1 mmol/L EDTA, pH 7.4) at 55 oC for 2–4 hours and then at 4 oC overnight. The samples were diluted to 25 ng/µl in TE and 2 µl of this genomic DNA solution was used per one of the following 10 µL reactions.
Cck1r region primers:
1µLof 10X reaction buffer (Promega), 0.8µl MgCl (Promega), 0.2µl 10mmol/L dNTP’s (New England BioLabs), 0.5µLof 1µmol/L M13 labeled forward primer (Qiagen), 0.5µl of 20 µmol/L unlabeled reverse primer (Qiagen), 0.1µl Taq DNA Polymerase (Promega), 1 µL M13–700 (LiCor Biosciences) and 3.9 µL ddH2O. All samples were then amplified using the following standard PCR protocol: 95 oC 5 min, 95 oC 20 sec, 60 oC 20 sec, 72 oC 30 sec, steps 2–4 repeated 30 times, 72 oC 3 min. Samples were kept at 4oC until use. PCR products were diluted to 25% with STOP solution (LiCor Biosciences) and analyzed using a NEN Global IR2 DNA Analyzer System (Model 4200S-2) with a 6.5% gel matrix (LiCor Biosciences). Simple sequence length polymorphism (SSLP’s) markers were found using the UCSC rat genome browser http://genome.ucsc.edu/index.html.
RNA Isolation.
Tissue was homogenized with a Kinematica Polytron PT 10/35 (Brinkmann, Westbury, NY) in RNA lysis solution (Stratagene, La Jolla, CA or Qiagen, Valencia, CA) immediately after dissection. Total RNA was isolated using a RNeasy (Qiagen) or Absolutely RNA Miniprep Kit (Stratagene) with DNAse treatment. PolyA+ RNA was isolated with Oligotex Direct mRNA Midi/Maxi Kit, (Qiagen). cDNA synthesis was performed using SuperScript II Reverse Transcriptase (Invitrogen) according to manufacturer’s recommendations.
Quantitative PCR.
Quantitative PCR was performed on an Mx4000® Multiplex Quantitative PCR System (Stratagene) in duplex reactions with rat glyceraldehyde phosphate dehydrogenase (GAPDH) (NM_017008) and samples loaded in triplicate using 80 ng of total RNA. Quantitative PCR was run in a 20 μL reaction using a Brilliant® Single-Step QRT-PCR Kit (2 μL of 10x core RT-PCR buffer, 5 mM MgCl2, 400 nM each primer, 200 nM each probe, 800 µM of dNTP mix, 75 nM passive reference dye, 1 units of AffinityScript RT, 1 unit of SureStart TaqDNA-polymerase) with PCR cycling conditions of 45°C for 30 min, 95°C for 10 min, and 40 cycles of 95°C for 30 sec, 60°C for 1 min. The positions of the probes in the genes are all in the 3’ regions of the transcripts, at the end of the coding regions, where the sequences are more variable. The primers and probes used for each gene are listed in Table 1. Primers were from Integrated DNA Technologies (Skokie, IL) or Qiagen Operon (Valencia, CA). Fluorescent labeled probes were from Integrated DNA Technologies or Applied Biosystems, Inc. (Foster City, CA). The standard curves, multiplexed with GAPDH, showed reaction efficiencies as follows: Cck1r: 90.6 %, Cck2r: 86.9 %, and Gapdh: 96.7 To compare values from wild-type rats of different litters, common threshold values were chosen and the Cts were converted to copy number and normalized to the amount of RNA in each well as determined by RiboGreen quantitation (Molecular Probes, Inc.).
Table 1.
– Probe and primers used in quantitative PCR
| Primer name | Accession | Primer sequence 5′ to 3′ |
|---|---|---|
| rCck1r-Qf | NM_012688 | TGCCAAGTGACGCTATGCA |
| rCck1r-Qr | GCCACCACCATCACAATCC | |
| rCck1r-Qp | 6FAM-AGAAGAAAGAGGATGAGTAGCAGGAATGTTTGCC-BHQ1 | |
| rCck2r-Qf | NM_013165 | CATAGCCCTGGAGCGATACAG |
| rCck2r-Qr | CGTGGGAGCGTGTTTGC | |
| rCck2r-Qp | 6FAM-ATCTGCCGACCACTGCAAGCACG-BHQ1 | |
| rGapdh-Qf | NM_017008 | CGGCCTCGTCTCATAGACAAG |
| rGapdh-Qr | ACCAGGCGGCCAATACG | |
| rGapdh-Qp | HEX-AAATCCGTTGACACCGACCTTCACCA-BHQ1 |
Bioinformatics.
The following resources were used:
University of California Santa Cruz (UCSC) Rat Genome Browser: http://genome.ucsc.edu/index.html
Rat Genome Database (RGD): http://rgd.mcw.edu/
National Center for Biotechnology Information (NCBI): http://www.ncbi.nlm.nih.gov/
Primer3 (MIT): http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi.
CCK-8 and devazepide injections.
Intraperitoneal (IP) injections were administered to rats via a 1.0 ml syringe with a 25-gauge needle. CCK-8 (Bachem/Peninsula, Belmont, CA) was dissolved in saline with 0.1% BSA and given as an IP injection (1 ml/kg injection volume) immediately prior to the start of the dark cycle , i.e., at a time when the animals normally begin eating and when CCK-8 has a potent effect on reducing food intake. Devazepide (Tocris, Ellisville, MO) was dissolved in 10% Tween 80, and 80% saline and given as an IP injection (1 mg/kg injection volume) 30-min prior to the start of the dark cycle.
Feeding studies.
Study 1. Effects of CCK-8 on Chow Intake in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
Study 1A: Effects of CCK-8 on Chow Diet.
Our aim was to investigate the effects of CCK-8 on solid chow intake in F344.Cck1r−/− and F344.Cck1r+/+ rats (n=7/group) in a crossover design with each animal serving as its own control. Briefly, rats were adapted to handling and to a 6-h fast for 1 week prior to the experiment. After a 6-h fast, the two groups of rats received injections of a high dose of CCK-8 (8 nmol/kg) or saline in randomized order with injections separated by 48 h. Preweighed amounts of chow pellets were placed onto the top of each cage immediately after each animal received an injection of vehicle or CCK-8. Food intake was measured manually by collecting chow pellets placed at the top of each cage at 30 min. and 24 h after animals received injections of CCK-8 or vehicle. Differences in amount of food weighed at 30 min and 24 h from the preweighed amount of food reflected the amount of food consumed. This particular feeding paradigm has been used previously [5,7], and the results obtained from CCK-8 injections at doses used in these studies (2, 8 nmol/kg, IP) are similar to what we have reported previously (4.8 nmol/kg, IP) in a food intake model whereby spillage was minimized [6]. One week later, these studies were repeated in chow-fed animals using a crossover design in which each animal received injections of a lower dose of CCK-8 (2 nmol/kg) or saline over a 48 h interval. Visual examination of the cage bottom revealed negligible spillage in all chow-fed animals, and there were no apparent differences in spillage within or between treatment groups in the chow fed animals.
Study 1B: Effects of CCK-8 on Palatable Diet (Ensure Plus Vanilla).
Our aim was to investigate the effects of CCK-8 on palatable diet intake in F344.Cck1r−/− and F344.Cck1r+/+ rats (n=7/group) in a crossover design with each animal serving as its own control. Briefly, rats were adapted to handling and to a 6-h fast for 1 week prior to the experiment. After a 6-h fast, the two groups of rats received injections of a high dose of CCK-8 (8 nmol/kg) or saline in randomized order with injections separated by 48 h. Preweighed volumetric flasks containing Ensure Plus Vanilla were placed in the front of each cage immediately after each animal received an injection of vehicle or CCK-8. Food intake was measured manually by weighing each volumetric flask at 30 min and 18 h after animals received injections of CCK-8 or vehicle.
Study 2. Effects of CCK-8 on Fos Induction in the Hindbrain in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
Our aim was to examine the effects of CCK-8 (8 nmol/kg) to induce Fos in the area postrema (AP) and nucleus tractus solitarius (NTS)) in F344.Cck1r−/− and F344.Cck1r+/+ rats. Briefly, rats were adapted to handling and to a 6-h fast for 1 week prior to the experiment. After 6-h food deprivation, rats received injections of saline or CCK-8 (8 nmol/kg) (n= 8–10 per group). All animals were returned to their cages immediately following injections. Food was made unavailable to prevent feeding-induced stimulation of hindbrain neurons through gastric distension [14,60]. At the conclusion of each study (1.5 h following CCK-8 or vehicle injections and 1.5 h from the start of the dark cycle), rats were anesthetized with a lethal dose of pentobarbital (Nembutal) (50 mg/kg, IP) and transcardially exsanguinated by perfusion with saline followed by 4% paraformaldehyde in 10 mM phosphate buffer (PBS), pH 7.2. Brains were removed and stored overnight in fresh fixative at 4°C and then transferred to 10 mM phosphate buffer containing 25% sucrose for 48 h. Brains were frozen by submersion in chilled isopentane for 10–15 seconds and placed under crushed dry ice. Coronal cryostat sections (14 m) through the hindbrain were mounted on slides and stored at –80ºC.
Immunocytochemical staining.
Slides were washed with 10 mM PBS at room temperature followed by blocking buffer (5% normal goat serum in 10 mM PBS) for 90 min and additional buffer washes. The primary antibody was rabbit polyclonal anti-Fos, 1:5,000 dilution (Ab-5, Oncogene, San Diego, CA ) [33,59]. The Fos antibody was diluted in 10 mM PBS. Following an overnight incubation in the primary antibody at 4° C, slides were washed in 10 mM PBS, and then incubated for 1 h in goat anti-rabbit IgG-Alexa 488 (Molecular Probes, Eugene, OR) diluted 1:200 in 10 mM PBS. Slides were washed in 10mM PBS and coverslipped using an anti-fading glycerol-based mounting media. The immunostaining specificity control included replacement of the primary antibody with normal rabbit serum at the same dilution as the primary antibody. Under conditions used in the immunocytochemical staining protocol, immunoreactive Fos protein was concentrated in the nuclei of labeled cells.
Immunocytochemical data analysis.
Slides were analyzed with a Zeiss Axioplan fluorescence microscope and all measurements were made with a 20X objective lens. Identification of anatomic landmarks was assisted by staining cell nuclei with Hoechst 33258 (Sigma, St. Louis, MO), which was added to the mounting medium and observed with a conventional DAPI filter set. Digital RGB images of the fluorescent preparations were acquired with a Nikon Eclipse E-800 (Melville, NY) with a QImaging Retiga 1300i Fast 1394 high performance digital CCD camera (Burnaby, B.C., Canada) plus the Image-Pro Express imaging system (Media Cybernetics, Inc., Bethesda, MD) and were exported to Photoshop (Adobe, Tucson, AZ). Measurements of Fos expression in the cmNTS were made on four sections separated by 240 m between NTS levels 4.24 and 5.08 mm posterior to the interaural line [45]. In each NTS section analyzed, the number of neurons that had Fos-positive immunofluorescence in the nucleus was recorded bilaterally. The total number of cmNTS Fos (+) neurons across the 4 anatomically matched sections was analyzed across the treatment groups. Unlike the cmNTS, the data for the AP were sampled and pooled from only two sections because the AP was found on only two of the four sections sampled for analysis of the cmNTS (4.8 and 5.08 mm posterior to interaural line) [45]. The number of Fos (+) cells in the cmNTS or the AP was derived from the cumulative number of Fos (+) cells between the treatments across 4 sections for the cmNTS or 2 sections for the AP, respectively.
Study 3. Meal patterns During Access to Palatable Diet in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
Measurement of Meal Patterns:
Briefly, an 8-channel lickometer (Dilog, Tallahassee, Fl) was used to record the number and timing of licks from the individual animals [18]. Custom-made software written in Visual Basic (Microsoft, Redmond, WA) was used off-line to convert raw lick data into meal-pattern parameters. Trains of more than 50 licks with > 90% of the interlick intervals between 80–250 milliseconds [56] were considered as meals. The criterion for meal termination was an absence of ingestion for at least 10 min. The weight of food ingested during meals and 1, 4 and 12-h time intervals was estimated from the number of recorded licks. For each rat, the conversion factor used was equal to the quotient of the total number of licks and weight of the food ingested during the day of observation.
Study 3A: Response to CCK-8 on Meal Size in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
Our aim was to investigate the effect of CCK-8 on 60-min meal patterns in F344.Cck1r−/− and F344.Cck1r+/+ rats (n=8/genotype) fed a liquid diet (Ensure Plus Vanilla) in addition to using a lickometer in a crossover design in which each animal served as its own control. F344.Cck1r−/−and F344.Cck1r+/+ rats were adapted to a 6-h fast and to handling for 1 week prior to the experiment. After a 6-h fast, rats received injections of saline or CCK-8 (8 nmol/kg) in randomized order in a cross-over design with injections separated by 48 h. In animals injected with CCK-8 or vehicle, food intake was measured manually at 0.5 and 18 h coupled with computerized recordings of meal patterns for 1 h from the start of the dark cycle and access to food.
Study 3B: Meal Number and Meal Size in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
Our aim was to investigate the meal patterns in F344.Cck1r−/− and F344.Cck1r+/+ rats (n=8/genotype) fed a palatable diet (Ensure Plus Vanilla);In addition, meal size and meal frequency data were collected in these animals in the absence of any injections and determined from data collected at 1, 4, 12, and 18 or 24 h from the start of the dark cycle and this data was averaged across 2 consecutive days in either 6-h food deprived animals or ad libitum fed animals.
Study 3C: Food Intake in Chow-fed Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
Our aim was to investigate the feeding effects of chow-fed F344.Cck1r−/− and F344.Cck1r+/+ rats. Daily cumulative 0.5-h and 18-h chow intake was recorded in 6 h fasted F344.Cck1r−/− (n=11) and F344.Cck1r+/+ (n=10) rats that did not receive any injections and averaged over a 3 day period.
Study 4. Body Weight Gain During 2-week Access to Palatable Diet (Ensure Plus Vanilla) in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
Our aim was to investigate the body weight gain in F344.Cck1r−/− and F344.Cck1r+/+ rats (n=8/genotype) fed a palatable diet (Ensure Plus Vanilla) after a 2-week exposure. Body weight was monitored on a daily basis before and after a 2-week exposure to Ensure Plus Vanilla.
Study 5. Response of F344.Cck1r+/+ Rats to Endogenous CCK1R Blockade
Our aim was to determine if blockade of endogenous CCK1R in Fischer 344 rats would result in a stimulation of food intake. Briefly, rats were adapted to handling and to a 30-min fast for 1 week prior to the experiment. Devazepide (1 mg/kg, IP) or vehicle (1 ml/kg injection volume) was administered into 30-min. fasted animals (n=14) approximately 30 min. prior to the start of the dark cycle and access to food. Food intake was measured at 0.5, 1, 2, 3, 4, and 24 h following the start of the dark cycle and access to food.
Study 6. Effect of Endogenous CCK1R Blockade on the Ability of F344.Cck1r+/+ Rats to Respond to CCK-8.
Our aim was to determine if blockade of endogenous CCK1R in Fischer 344 rats would impair the ability of CCK-8 to inhibit food intake. Briefly, rats were adapted to handling and to a 6-h fast for 1 week prior to the experiment. Devazepide (1 mg/kg, IP) or vehicle (1 ml/kg injection volume) was administered into 30-min. fasted animals (n=14) approximately 30 min. prior to administration of CCK-8 (8 nmol/kg, IP) or vehicle (0.1% BSA, salilne). CCK-8 was administered immediately prior to the the start of the dark cycle and access to food. Food intake was measured at 0.5, 1, 2, 3, 4, and 24 h following the start of the dark cycle and access to food.
Statistical Analysis.
Mean values ± SE are shown as indicated. All results are expressed as means ± SEM. Comparisons in the responsiveness to CCK-8 on food intake between groups in a within-subjects design were made using a two-way repeated-measures analysis of variance (ANOVA) followed by Tukey’s Honestly Significantly Different (HSD) test as a post hoc test. Comparisons in the responsiveness to CCK-8 on Fos induction between groups as a between-subjects design were made using a two-way ANOVA followed by Tukey’s HSD test as a post hoc test. Comparisons between groups (body weight, meal patterns) were made using a one-tailed Student’s t-test. Analyses were performed using the statistical program SYSTAT version 11.0 (Systat Software Inc., San Jose, CA), and Statistica 4.1 (StatSoft Inc., Tulsa, OK). Differences were considered significant if P<0.05.
Figure 5A-B. Quantitative Effects of CCK-8 on Fos induction in the AP, cmNTS, cNTS, and gNTS in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
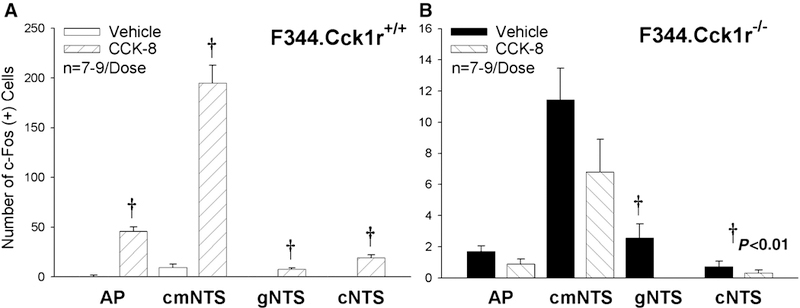
CCK-8 (8 nmol/kg), as expected, stimulated Fos induction in the AP, cmNTS, cNTS, and gNTS (AP, cmNTS, cNTS, and gNTS noted on Y axis labels) by 5237, 1931, 13757, and 5471% in F344.Cck1r+/+ (Figure 4A), but was without effect in F344.Cck1r−/− (Figure 5B). As can be observed by the scale on the Y axis between Figures 5A-B, the absolute magnitude of CCK-8-elicited Fos induction in the hindbrain was nearly 20–50-fold greater in F344.Cck1r+/+ vs. F344.Cck1r−/− rats.
Figure 6A-D. Effects of CCK-8 on Fos induction in the cmNTS in Mutant F344.Cck1r−/− and Wild-type F344.Cck1r+/+ Rats.
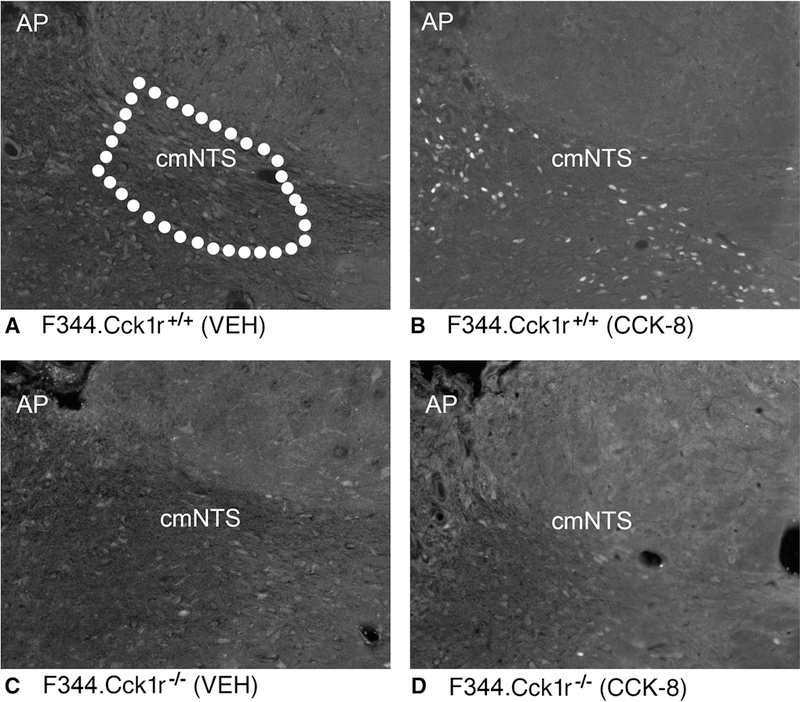
Fos activation is revealed by concentration of immunoreactive Fos in cell nuclei (Fos (+) cells) in the cmNTS. Fos-immunostaining was done by Cy3 fluorescence. Images were taken from the right side of the cmNTS with the AP shown in the upper left. Figure 6A: Peripheral injection of vehicle induced little or no Fos (+) neurons in the cmNTS of F344.Cck1r+/+ rats. Figure 6B: Peripheral injection of CCK-8 (8 nmol/kg) induced numerous Fos (+) neurons in the cmNTS of F344.Cck1r+/+ rats. Figure 6C: Peripheral injection of vehicle induced little or no Fos (+) neurons in the cmNTS of F344.Cck1r−/− rats. Figure 6D: Peripheral injection of CCK-8 (8 nmol/kg) induced little or no Fos (+) neurons in the cmNTS of F344.Cck1r−/− rats. A-C all visualized at 20X magnification.
Acknowledgements
This material is based upon work supported by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs (VA), the biomedical research core programs of the National Institutes of Health (NIH) Diabetes Endocrinology Research Center and the Clinical Nutrition Research Unit (CNRU) at the University of Washington, and the American Diabetes Association. The research in our laboratory has been supported by the Department of VA Merit Review Research Program, NIH Grants DK-17047, RO1DK-61516, P30DK017047-31, P30DK017047-31689, P30DK035816, and by the Junior Faculty Award 1-05-JF-32 by the American Diabetes Association. The authors would like to thank the University of Washington CNRU biostatistician Brian Fish and Drs. Diana Williams and Tami Wolden-Hanson for their input on statistics.
Literature references
- [1].Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD and Bloom SR, Gut hormone PYY(3–36) physiologically inhibits food intake, Nature, 418 (2002) 650–4. [DOI] [PubMed] [Google Scholar]
- [2].Bi S, Chen J, Behles RR, Hyun J, Kopin AS and Moran TH, Differential body weight and feeding responses to high-fat diets in rats and mice lacking cholecystokinin 1 receptors, Am J Physiol Regul Integr Comp Physiol, 293 (2007) R55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bi S, Ladenheim EE, Schwartz GJ and Moran TH, A role for NPY overexpression in the dorsomedial hypothalamus in hyperphagia and obesity of OLETF rats, Am J Physiol Regul Integr Comp Physiol, 281 (2001) R254–60. [DOI] [PubMed] [Google Scholar]
- [4].Bi S, Scott KA, Kopin AS and Moran TH, Differential roles for cholecystokinin a receptors in energy balance in rats and mice, Endocrinology, 145 (2004) 3873–80. [DOI] [PubMed] [Google Scholar]
- [5].Blevins JE, Eakin TJ, Murphy JA, Schwartz MW and Baskin DG, Oxytocin innervation of caudal brainstem nuclei activated by cholecystokinin, Brain Res, 993 (2003) 30–41. [DOI] [PubMed] [Google Scholar]
- [6].Blevins JE, Hamel FG, Fairbairn E, Stanley BG and Reidelberger RD, Effects of paraventricular nucleus injection of CCK-8 on plasma CCK-8 levels in rats, Brain Res, 860 (2000) 11–20. [DOI] [PubMed] [Google Scholar]
- [7].Blevins JE, Schwartz MW and Baskin DG, Evidence that paraventricular nucleus oxytocin neurons link hypothalamic leptin action to caudal brain stem nuclei controlling meal size, Am J Physiol Regul Integr Comp Physiol, 287 (2004) R87–96. [DOI] [PubMed] [Google Scholar]
- [8].Brenner LA and Ritter RC, Type A CCK receptors mediate satiety effects of intestinal nutrients, Pharmacol Biochem Behav, 54 (1996) 625–31. [DOI] [PubMed] [Google Scholar]
- [9].Chen DY, Deutsch JA, Gonzalez MF and Gu Y, The induction and suppression of c-fos expression in the rat brain by cholecystokinin and its antagonist L364,718, Neurosci Lett, 149 (1993) 91–4. [DOI] [PubMed] [Google Scholar]
- [10].Dakin CL, Gunn I, Small CJ, Edwards CM, Hay DL, Smith DM, Ghatei MA and Bloom SR, Oxyntomodulin inhibits food intake in the rat, Endocrinology, 142 (2001) 4244–50. [DOI] [PubMed] [Google Scholar]
- [11].Day HE, McKnight AT, Poat JA and Hughes J, Evidence that cholecystokinin induces immediate early gene expression in the brainstem, hypothalamus and amygdala of the rat by a CCKA receptor mechanism, Neuropharmacology, 33 (1994) 719–27. [DOI] [PubMed] [Google Scholar]
- [12].De Weerth A, Pisegna JR and Wank SA, Guinea pig gallbladder and pancreas possess identical CCK-A receptor subtypes: receptor cloning and expression, Am J Physiol, 265 (1993) G1116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Donovan MJ, Paulino G and Raybould HE, CCK(1) receptor is essential for normal meal patterning in mice fed high fat diet, Physiol Behav, 92 (2007) 969–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Emond M, Ladenheim EE, Schwartz GJ and Moran TH, Leptin amplifies the feeding inhibition and neural activation arising from a gastric nutrient preload, Physiol Behav, 72 (2001) 123–8. [DOI] [PubMed] [Google Scholar]
- [15].Fraser KA and Davison JS, Meal-induced c-fos expression in brain stem is not dependent on cholecystokinin release, Am J Physiol, 265 (1993) R235–9. [DOI] [PubMed] [Google Scholar]
- [16].Fraser KA, Raizada E and Davison JS, Oral-pharyngeal-esophageal and gastric cues contribute to meal-induced c-fos expression, Am J Physiol, 268 (1995) R223–30. [DOI] [PubMed] [Google Scholar]
- [17].Funakoshi A, Miyasaka K, Jimi A, Kawanai T, Takata Y and Kono A, Little or no expression of the cholecystokinin-A receptor gene in the pancreas of diabetic rats (Otsuka Long-Evans Tokushima Fatty = OLETF rats), Biochem Biophys Res Commun, 199 (1994) 482–8. [DOI] [PubMed] [Google Scholar]
- [18].Gannon KS, Smith JC, Henderson R and Hendrick P, A system for studying the microstructure of ingestive behavior in mice, Physiol Behav, 51 (1992) 515–21. [DOI] [PubMed] [Google Scholar]
- [19].Glatzle J, Kreis ME, Kawano K, Raybould HE and Zittel TT, Postprandial neuronal activation in the nucleus of the solitary tract is partly mediated by CCK-A receptors, Am J Physiol Regul Integr Comp Physiol, 281 (2001) R222–9. [DOI] [PubMed] [Google Scholar]
- [20].Hill DR and Woodruff GN, Differentiation of central cholecystokinin receptor binding sites using the non-peptide antagonists MK-329 and L-365,260, Brain Res, 526 (1990) 276–83. [DOI] [PubMed] [Google Scholar]
- [21].Kanemoto N, Hishigaki H, Miyakita A, Oga K, Okuno S, Tsuji A, Takagi T, Takahashi E, Nakamura Y and Watanabe TK, Genetic dissection of “OLETF”, a rat model for non-insulin-dependent diabetes mellitus, Mamm Genome, 9 (1998) 419–25. [DOI] [PubMed] [Google Scholar]
- [22].Kawano K, HHirashima T, Mori S, Man ZW and Natori T, Establishment of the OLETF rat. In Shima K (Ed.), Obesity and NIDDM: Lessons from the OLETF rat, Elsevier, 1999. [Google Scholar]
- [23].Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M and Natori T, Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain, Diabetes, 41 (1992) 1422–8. [DOI] [PubMed] [Google Scholar]
- [24].Kopin AS, Mathes WF, McBride EW, Nguyen M, Al-Haider W, Schmitz F, Bonner-Weir S, Kanarek R and Beinborn M, The cholecystokinin-A receptor mediates inhibition of food intake yet is not essential for the maintenance of body weight, J Clin Invest, 103 (1999) 383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kose H, Moralejo DH, Ogino T, Mizuno A, Yamada T and Matsumoto K, Examination of OLETF-derived non-insulin-dependent diabetes mellitus QTL by construction of a series of congenic rats, Mamm Genome, 13 (2002) 558–62. [DOI] [PubMed] [Google Scholar]
- [26].Kozma C, Cummins LM and Tekeli S, The use of Long-Evans rats in long term toxicity studies. In Spiegal A (Ed.), The Laboratory Animal in Drug Testing, Hannover, 1972, pp. 283–301. [Google Scholar]
- [27].Ladenheim EE, Wirth KE and Moran TH, Receptor subtype mediation of feeding suppression by bombesin-like peptides, Pharmacol Biochem Behav, 54 (1996) 705–11. [DOI] [PubMed] [Google Scholar]
- [28].Lo CM, Ma L, Zhang DM, Lee R, Qin A, Liu M, Woods SC, Sakai RR, Raybould HE and Tso P, Mechanism of the induction of brain c-Fos-positive neurons by lipid absorption, Am J Physiol Regul Integr Comp Physiol, 292 (2007) R268–73. [DOI] [PubMed] [Google Scholar]
- [29].Lo CM, Samuelson LC, Chambers JB, King A, Heiman J, Jandacek RJ, Sakai RR, Benoit SC, Raybould HE, Woods SC and Tso P, Characterization of mice lacking the gene for cholecystokinin, Am J Physiol Regul Integr Comp Physiol, 294 (2008) R803–10. [DOI] [PubMed] [Google Scholar]
- [30].Lutz TA, Geary N, Szabady MM, Del Prete E and Scharrer E, Amylin decreases meal size in rats, Physiol Behav, 58 (1995) 1197–202. [DOI] [PubMed] [Google Scholar]
- [31].Markel P, Shu P, Ebeling C, Carlson GA, Nagle DL, Smutko JS and Moore KJ, Theoretical and empirical issues for marker-assisted breeding of congenic mouse strains, Nat Genet, 17 (1997) 280–4. [DOI] [PubMed] [Google Scholar]
- [32].Martin CF and Gibbs J, Bombesin elicits satiety in sham feeding rats, Peptides, 1 (1980) 131–4. [DOI] [PubMed] [Google Scholar]
- [33].McMinn JE, Sindelar DK, Havel PJ and Schwartz MW, Leptin deficiency induced by fasting impairs the satiety response to cholecystokinin, Endocrinology, 141 (2000) 4442–8. [DOI] [PubMed] [Google Scholar]
- [34].Miyasaka K, Kanai S, Ohta M, Kawanami T, Kono A and Funakoshi A, Lack of satiety effect of cholecystokinin (CCK) in a new rat model not expressing the CCK-A receptor gene, Neurosci Lett, 180 (1994) 143–6. [DOI] [PubMed] [Google Scholar]
- [35].Monnikes H, Lauer G and Arnold R, Peripheral administration of cholecystokinin activates c-fos expression in the locus coeruleus/subcoeruleus nucleus, dorsal vagal complex and paraventricular nucleus via capsaicin-sensitive vagal afferents and CCK-A receptors in the rat, Brain Res, 770 (1997) 277–88. [DOI] [PubMed] [Google Scholar]
- [36].Moralejo DH, Ogino T, Kose H, Yamada T and Matsumoto K, Genetic verification of the role of CCK-AR in pancreatic proliferation and blood glucose and insulin regulation using a congenic rat carrying CCK-AR null allele, Res Commun Mol Pathol Pharmacol, 109 (2001) 259–74. [PubMed] [Google Scholar]
- [37].Moralejo DH, Wei S, Wei K, Weksler-Zangen S, Koike G, Jacob HJ, Hirashima T, Kawano K, Sugiura K, Sasaki Y, Ogino T, Yamada T and Matsumoto K, Identification of quantitative trait loci for non-insulin-dependent diabetes mellitus that interact with body weight in the Otsuka Long-Evans Tokushima Fatty rat, Proc Assoc Am Physicians, 110 (1998) 545–58. [PubMed] [Google Scholar]
- [38].Moran TH, Unraveling the obesity of OLETF rats, Physiol Behav (2007). [DOI] [PMC free article] [PubMed]
- [39].Moran TH, Ameglio PJ, Schwartz GJ and McHugh PR, Blockade of type A, not type B, CCK receptors attenuates satiety actions of exogenous and endogenous CCK, Am J Physiol, 262 (1992) R46–50. [DOI] [PubMed] [Google Scholar]
- [40].Moran TH, Katz LF, Plata-Salaman CR and Schwartz GJ, Disordered food intake and obesity in rats lacking cholecystokinin A receptors, Am J Physiol, 274 (1998) R618–625. [DOI] [PubMed] [Google Scholar]
- [41].Moran TH, Robinson PH, Goldrich MS and McHugh PR, Two brain cholecystokinin receptors: implications for behavioral actions, Brain Res, 362 (1986) 175–9. [DOI] [PubMed] [Google Scholar]
- [42].Mountjoy KG, Mortrud MT, Low MJ, Simerly RB and Cone RD, Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain, Mol Endocrinol, 8 (1994) 1298–308. [DOI] [PubMed] [Google Scholar]
- [43].Ogino T, Wei S, Wei K, Moralejo DH, Kose H, Mizuno A, Shima K, Sasaki Y, Yamada T and Matsumoto K, Genetic evidence for obesity loci involved in the regulation of body fat distribution in obese type 2 diabetes rat, OLETF, Genomics, 70 (2000) 19–25. [DOI] [PubMed] [Google Scholar]
- [44].Olson BR, Freilino M, Hoffman GE, Stricker EM, Sved AF and Verbalis JG, c-Fos expression in rat brain and brainstem nuclei in response to treatments that alter food intake and gastric motility, Mol. Cell. Neurosci, 4 (1993) 93–106. [DOI] [PubMed] [Google Scholar]
- [45].Paxinos G, Carrive P, Wang H and Wang P-Y, Chemo Architectonic Atlas of the Rat Brainstem Academic Press, Orlando, 1999. [Google Scholar]
- [46].Reidelberger RD, Abdominal vagal mediation of the satietiy effects of exogenous and endogenous cholecystokinin in rats, Am J Physiol, 263 (1992) R1–R5. [DOI] [PubMed] [Google Scholar]
- [47].Reidelberger RD, Haver AC, Arnelo U, Smith DD, Schaffert CS and Permert J, Amylin receptor blockade stimulates food intake in rats, Am J Physiol Regul Integr Comp Physiol, 287 (2004) R568–74. [DOI] [PubMed] [Google Scholar]
- [48].Reidelberger RD, Heimann D, Kelsey L and Hulce M, Effects of peripheral CCK receptor blockade on feeding responses to duodenal nutrient infusions in rats, Am J Physiol Regul Integr Comp Physiol, 284 (2003) R389–98. [DOI] [PubMed] [Google Scholar]
- [49].Reidelberger RD, Hernandez J, Fritzsch B and Hulce M, Abdominal vagal mediation of the satiety effects of CCK in rats, Am J Physiol Regul Integr Comp Physiol, 286 (2004) R1005–12. [DOI] [PubMed] [Google Scholar]
- [50].Reidelberger RD and O-Rourke MF, Potent cholecystokinin antagonist L-364,718 stimulates food intake in rats, Am J Physiol, 257 (1989) R1512–R1518. [DOI] [PubMed] [Google Scholar]
- [51].Reidelberger RD, Varga G and Solomon TE, Effects of selective cholecystokinin antagonists L364,718 and L365,260 on food intake in rats, Peptides, 12 (1991) 1215–21. [DOI] [PubMed] [Google Scholar]
- [52].Rinaman L, Oxytocinergic inputs to the nucleus of the solitary tract and dorsal motor nucleus of the vagus in neonatal rats, J Comp Neurol, 399 (1998) 101–9. [DOI] [PubMed] [Google Scholar]
- [53].Rinaman L, Verbalis JG, Stricker EM and Hoffman GE, Distribution and neurochemical phenotypes of caudal medullary neurons activated to express cFos following peripheral administration of cholecystokinin, Journal of Comparative Neurology, 338 (1993) 475–490. [DOI] [PubMed] [Google Scholar]
- [54].Schwartz GJ, Whitney A, Skoglund C, Castonguay TW and Moran TH, Decreased responsiveness to dietary fat in Otsuka Long-Evans Tokushima fatty rats lacking CCK-A receptors, Am J Physiol, 277 (1999) R1144–51. [DOI] [PubMed] [Google Scholar]
- [55].Shapiro RE and Miselis RR, The central organization of the vagus nerve innervating the stomach of the rat, J Comp Neurol, 238 (1985) 473–88. [DOI] [PubMed] [Google Scholar]
- [56].Smith GP, John Davis and the meanings of licking, Appetite, 36 (2001) 84–92. [DOI] [PubMed] [Google Scholar]
- [57].Sugiura K, Miyake T, Taniguchi Y, Yamada T, Moralejo DH, Wei S, Wei K, Sasaki Y and Matsumoto K, Identification of novel non-insulin-dependent diabetes mellitus susceptibility loci in the Otsuka Long-Evans Tokushima fatty rat by MQM-mapping method, Mamm Genome, 10 (1999) 1126–31. [DOI] [PubMed] [Google Scholar]
- [58].Takiguchi S, Takata Y, Funakoshi A, Miyasaka K, Kataoka K, Fujimura Y, Goto T and Kono A, Disrupted cholecystokinin type-A receptor (CCKAR) gene in OLETF rats, Gene, 197 (1997) 169–75. [DOI] [PubMed] [Google Scholar]
- [59].Thiele TE, van Dijk G, Yagaloff K, Fisher SL, Schwartz MW, Burn P and Seeley RJ, Central infusion of melanocortin agonist MTII in rats: assessment of c-Fos expression and taste aversion, Am J Physiol, 274 (1998) R248–254. [DOI] [PubMed] [Google Scholar]
- [60].Traub RJ, Sengupta JN and Gebhart GF, Differential c-fos expression in the nucleus of the solitary tract and spinal cord following noxious gastric distention in the rat, Neuroscience, 74 (1996) 873–84. [DOI] [PubMed] [Google Scholar]
- [61].Turton MD, O’Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, Choi SJ, Taylor GM, Heath MM, Lambert PD, Wilding JP, Smith DM, Ghatei MA, Herbert J and Bloom SR, A role for glucagon-like peptide-1 in the central regulation of feeding, Nature, 379 (1996) 69–72. [DOI] [PubMed] [Google Scholar]
- [62].Wakeland E, Morel L, Achey K, Yui M and Longmate J, Speed congenics: a classic technique in the fast lane (relatively speaking), Immunol Today, 18 (1997) 472–7. [DOI] [PubMed] [Google Scholar]
- [63].Wang L, Martinez V, Barrachina MD and Tache Y, Fos expression in the brain induced by peripheral injection of CCK or leptin plus CCK in fasted lean mice, Brain Res, 791 (1998) 157–66. [DOI] [PubMed] [Google Scholar]
- [64].Watanabe TK, Okuno S, Oga K, Mizoguchi-Miyakita A, Tsuji A, Yamasaki Y, Hishigaki H, Kanemoto N, Takagi T, Takahashi E, Irie Y, Nakamura Y and Tanigami A, Genetic dissection of “OLETF,” a rat model for non-insulin-dependent diabetes mellitus: quantitative trait locus analysis of (OLETF x BN) x OLETF, Genomics, 58 (1999) 233–9. [DOI] [PubMed] [Google Scholar]
- [65].Weatherford SC, Chiruzzo FY and Laughton WB, Satiety induced by endogenous and exogenous cholecystokinin is mediated by CCK-A receptors in mice, Am J Physiol, 262 (1992) R574–8. [DOI] [PubMed] [Google Scholar]
- [66].Wei S, Wei K, Moralejo DH, Ogino T, Koike G, Jacob HJ, Sugiura K, Sasaki Y, Yamada T and Matsumoto K, Mapping and characterization of quantitative trait loci for non-insulin-dependent diabetes mellitus with an improved genetic map in the Otsuka Long-Evans Tokushima fatty rat, Mamm Genome, 10 (1999) 249–58. [DOI] [PubMed] [Google Scholar]
- [67].Willing AE and Berthoud HR, Gastric distension-induced c-fos expression in catecholaminergic neurons of rat dorsal vagal complex, Am J Physiol, 272 (1997) R59–67. [DOI] [PubMed] [Google Scholar]
- [68].Yox DP, Brenner L and Ritter RC, CCK-receptor antagonists attenuate suppression of sham feeding by intestinal nutrients, Am J Physiol, 262 (1992) R554–61. [DOI] [PubMed] [Google Scholar]