

Abstract
More than 40% of patients with luminal breast cancer treated with endocrine therapy agent tamoxifen demonstrate resistance. Emerging evidence suggests tumor initiating cells (TICs) and aberrant activation of Src and Akt signaling drive tamoxifen resistance and relapse. We previously demonstrated that aryl hydrocarbon receptor (AhR) ligand Aminoflavone (AF) inhibits the expression of TIC gene α6-integrin and disrupts mammospheres derived from tamoxifen-sensitive breast cancer cells. In the current study, we hypothesize that tamoxifen resistant (TamR) cells exhibit higher levels of α6-integrin than tamoxifen sensitive cells and that AF inhibits the growth of TamR cells by suppressing α6-integrin-Src-Akt signaling. In support of our hypothesis, TamR cells and associated mammospheres were found to exhibit elevated α6-integrin expression compared to their tamoxifen sensitive counterparts. Furthermore, tumor sections from patients who relapsed on tamoxifen showed enhanced α6-integrin expression. Gene expression profiling from the TCGA database further revealed that basal-like breast cancer samples, known to be largely unresponsive to tamoxifen, demonstrated higher α6-integrin levels than luminal breast cancer samples. Importantly, AF reduced TamR cell viability and disrupted TamR mammospheres while concomitantly reducing α6-integrin mRNA and protein levels. In addition, AF and siRNA against α6-integrin blocked tamoxifen-stimulated proliferation of TamR MCF-7 cells and further sensitized these cells to tamoxifen. Moreover, AF reduced Src and Akt signaling activation in TamR MCF-7 cells. Our findings suggest elevated α6-integrin expression is associated with tamoxifen resistance and AF suppresses α6-integrin-Src-Akt signaling activation to confer activity against TamR breast cancer.
Keywords: breast cancer, α6-integrin, Src-Akt signaling, AF; tamoxifen resistance
INTRODUCTION
Breast cancer is the most commonly diagnosed cancer in women worldwide. Resistance to therapies often results in metastasis which leads to recurrence and breast cancer mortality (Ahmad, 2013). Estrogen receptor positive (ER+) breast cancer is the most frequently diagnosed breast cancer subtype. Tamoxifen is widely used to treat ER+ breast cancer although the emergence of resistance significantly diminishes its clinical efficacy (Tanic et al., 2012). Tumor initiating cells (TICs) are key contributors to tamoxifen resistance owing to their ability to evade treatment and self-renew to produce recurrent tumors (Ojo et al., 2015). Tamoxifen treatment itself has been shown to select for cells with self-renewal capacity (Raffo et al., 2013). As such, elimination of TICs is crucial to circumvent tamoxifen resistance and confer long-term clinical benefit (Gruber et al., 2017; Ricci-Vitiani et al., 2008).
Integrins have been identified as important regulators of tumor initiation or cancer stemness and drug resistance (Seguin et al., 2015). In particular, α6-integrin is important for TIC maintenance and function (Lathia et al., 2010). Indeed, elevated α6-integrin expression in breast tumor tissues has been associated with poor overall survival among patients (Friedrichs et al., 1995). We recently demonstrated that, contrary to tamoxifen, aryl hydrocarbon receptor (AhR) ligand Aminoflavone (AF) inhibits α6-integrin expression to suppress TIC proliferation in ER+ breast cancer models and though α6-integrin often partners with β1 and β4 integrins, AF did not markedly alter the expression of these integrins (Brantley et al., 2016). Although another non-toxic AhR ligand Tranilast has been shown to synergize with Tamoxifen in vitro (Darakhshan et al., 2013) and inhibit TIC proliferation (Prud’homme et al., 2010), our recent study was the first to link α6-integrin with AhR ligand-mediated suppression of TIC proliferation (Brantley et al., 2016). Thus far, factors that contribute to TIC survival in TamR cancers have not been fully elucidated.
Though endocrine therapy resistance has been associated with elevated expression of AhR target genes cytochrome P450s 1A1 and 1B1, elevated expression of these genes did not mediate resistance to endocrine therapy agent fulvestrant (Brockdorff et al., 2000). Interestingly, fulvestrant induces AhR signaling to suggest cross-talk interactions occur between ER and AhR signaling pathways. McDonnell and colleagues previously demonstrated the ability of 4-hydroxy-tamoxifen (4OHTam), an active tamoxifen metabolite, to induce AhR target genes in the absence of estrogen (DuSell et al., 2010). Safe and colleagues previously reported that AhR agonists, in certain contexts, block estradiol-mediated mammary tumor growth via AhR-ER crosstalk mechanisms (Safe and McDougal, 2002). In addition, small molecules that activate AhR signaling were found to inhibit cancer cell invasion and metastases in breast cancer cells including basal-like subtypes known to resist endocrine therapy (Hall et al., 2010; Jin et al., 2014). Moreover, AhR ligand Aminoflavone demonstrates the potential to activate AhR signaling yet demonstrates potent and selective anticancer activity in certain breast cancer cell lines and corresponding tumors (Loaiza-Pérez et al., 2004).
The purpose of this study is to examine an association between α6-integrin expression and tamoxifen resistance and to determine whether AF demonstrates anticancer activity in TamR cells by targeting the α6-integrin-Src-Akt signaling axis. AF has undergone extensive preclinical development and has been evaluated in clinical trials for efficacy against solid tumors. However, the ability for AF to demonstrate efficacy in TamR cells of varying molecular subtypes and the potential mechanism(s) of such anticancer actions has not been fully explored. A better understanding of the molecular targets, such as α6-integrin, that contribute to tamoxifen resistance provides an avenue to identify biomarkers useful in recognizing patients less likely to benefit from endocrine therapy.
MATERIALS AND METHODS
Cell Culture and Reagents.
Human MCF-7 and T47D Parental (Par MCF-7, Par T47D) and MCF-7 and T47D Tamoxifen resistant (TamR MCF-7 and TamR T47D) cells are of the luminal A breast cancer subtype and were developed and maintained as previously described (Fu et al., 2016; Morrison et al., 2014). Parental MCF-7 cells were originally obtained from Dr. Marc Lippman (National Cancer Institute, Bethesda, MD) while the parental T47D (ATCC cat# HTB-133, RRID:CVCL_0553) cells were originally obtained from the American Type Culture Collection (ATCC). Luminal B ZR-75–30 (ATCC cat# CRL-1504, RRID:CVCL_1661) cells were a kind gift from Dr. Daisy De Leon (Loma Linda University Health School of Medicine, Loma Linda, CA). Luminal B BT-474 (ATCC cat# HTB-20, RRID:CVCL_0179) cells were obtained from the American Type Culture Collection (ATCC). All cell lines were either authenticated once Tamoxifen resistance was established or using STR DNA profiling. ZR-75–30 breast cancer cells were cultured in RPMI-1640 medium containing 10% FBS (Hyclone, Logan, UT), supplemented with 2 mM glutamine and penicillin and streptomycin antibiotics (Mediatech, Herndon, VA). BT-474 cells were cultured in ATCC Hybri-Care Medium, reconstituted in 1 L cell-culture-grade water and supplemented with 1.5 g/L sodium bicarbonate, 10% FBS and 2 mM glutamine and penicillin and streptomycin antibiotics. The α6-integrin blocking antibody GoH3 (clone NKI-GoH3) was obtained from Millipore (cat# MAB1378; Temecula, CA, RRID:AB_1121–794). 5-amino-2-(4-amino-3-fluorophenyl)-6,8-difluoro-7-methyl-4H-1-benzopyran-4-one (Aminoflavone, AF) was obtained from the “The NCI/DTP Open Chemical Repository” (http://dtp.cancer.gov, Frederick, MD) at the Frederick National Laboratory for Cancer Research. 4-hydroxy-tamoxifen (4OHTam) was obtained from Sigma-Aldrich (St. Louis, MO). Stock solutions of AF and 4OHTam were dissolved in dimethyl sulfoxide (DMSO). All stocks were stored protected from light at −20°C until use.
Determination of Cancer Cell Viability.
We evaluated the ability of AF to inhibit the growth of breast cancer cells with varying degrees of sensitivity to tamoxifen. Briefly, MCF-7 cells and T47D cells (Par and TamR), as well as BT-474 and ZR-75–30 cells were cultured in their respective media as mentioned above and plated in 96 well plates. Approximately 24 h later, cells were treated with AF (0.1 nM-10,000 nM), 4OHTam or 0.1% DMSO for 72 h for all cell lines except BT-474 cells which received treatment for 120 h. Cytotoxicity was determined using the Alamar Blue™ assay as previously described elsewhere (McLean et al., 2008). Otherwise, cells were grown in suspension as mammospheres as described in accordance with the Mammosphere assay, exposed to AF or 4OHTam followed by harvesting and disruption in trypsin by thorough mixing. The resulting individual cell suspensions were transferred to a 96 well plate and the Alamar Blue Assay™ was performed as previously described (Brantley et al., 2016). To determine whether α6-integrin mediates responsiveness of 4OHTam in TamR cells, TamR monolayers were exposed to blocking antibody GoH3 (1 or 10 μg/ml) for 3 d (TamR MCF-7) for 5 d (BT-474 cells) alone or in combination with either 4OHTam or AF. Cells were otherwise transfected with a pool of siRNAs against α6-integrin as described below. Cell viability was then determined as described above.
siRNA Transfection.
siRNA and transfection reagents were obtained from GE Dharmacon (Lafayette, Colorado, US). Positive control siRNA (ON-TARGETplus Cyclophilin B Control Pool (Human), cat#D-001820–10-05), negative control siRNA (ON-TARGETplus Non-targeting Pool, cat# D-001810–10-05), test siRNA (ON-TARGETplus Human ITGA6 (3655) siRNA - SMARTpool, cat#L-007214–00-0005) were resuspended in RNase free water and aliquoted for short-term storage at −200C prior to use. TamR MCF-7 cells were diluted in antibiotic-free complete medium to achieve a plating density of 60–80% confluency in either 96 or 6 well plates followed by incubation at 37°C with 5% CO2 overnight. Transfection medium was prepared according to the manufacturer’s instructions. Cells were transfected with 25nM control siRNAs or 10nM ITGA6 siRNA for 24 h followed by an additional 24 h incubation in complete media. Transfection efficiency was verified using qPCR and western blotting analyses. Conditions that showed target mRNA knockdown of > 80% as well as > 80 % cell viability were used in subsequent studies.
Mammosphere Assay.
Cells were cultured in suspension as mammospheres using the MammoCult™ Human Medium Kit (Stem Cell Technologies, Vancouver, BC, Canada). Mammospheres were cultured for 5 days in Falcon 6-well non-treated polystyrene plates (product# 351146) before being exposed to respective treatments. Mammospheres were visualized using an IX-71 Olympus microscope (relief contrast mode) and pictures taken before and after treatment. Additionally, mammospheres were collected and prepared for Alamar Blue™, semi-quantitative or qPCR analyses as described previously (Brantley et al., 2016).
RNA extraction, semi-quantitative RT-PCR, and quantitative PCR analyses.
Total RNA was isolated from Par MCF-7, TamR MCF-7, BT-474, and ZR-75–30 cells (grown in monolayers) or as Par MCF-7, TamR MCF-7, ZR-75–30 and BT-474 mammospheres using either the Quick-RNA MiniPrep Kit (Zymo Research, Irvine, CA, USA) or miRNeasy Mini Kit (Qiagen, Germantown, MD, USA) in accordance with the manufacturers’ instructions. cDNA was prepared using an iScript Advanced cDNA synthesis kit (BioRad, Richmond, CA). Semi-quantitative PCR was conducted as detailed elsewhere (van Riggelen et al., 2005) to determine the relative expression of α6-integrin variant A (875 bp) and variant B (745 bp) in mammospheres. Primers used for semi-quantitative PCR have been described elsewhere and were as follows: α6-integrin- Forward: 5’-CTA ACG GAG TCT CAC AAC TC-3’, Reverse: 5’-AGT TAA AAC TGT AGG TTC G-3’ and GAPDH: 460 bp, Forward: 5’-TGG ATA TTG TTG CCA TCA ATG ACC-3’ and Reverse: 5’-GAT GGC ATG GAC TGT GGT CAT G-3’ (Dydensborg et al., 2009). Quantitative real-time PCR analysis was also performed using a CFX-96 PCR instrument (Bio-Rad, Hercules, CA). PCR products were obtained using the following primers from Qiagen (Germantown, MD): human ITGA6, human BAX, human GAPDH, and human RPLP0.
Western Blot Analysis.
Cells were seeded at 3–4×106 cells per plate (100 mm) and allowed to attach. Cells were then serum starved for approximately 24 h before treatment with 1 μM AF or 0.1% DMSO for 8, 24 or 48h. In some instances, cells were treated with GoH3 blocking antibody. Following treatment, the cells were harvested on ice by scraping, washed twice with cold PBS before adding CelLytic™ M lysis buffer (Sigma, St. Louis, MO) supplemented with protease and phosphatase inhibitors. Protein concentration was determined using the BCA™ Protein Assay Kit (Prod#23250, ThermoScientific, Rockford, IL), according to the manufacturer’s instructions. For Western blot analysis, proteins were resolved on 4–12% NuPage® Bis-Tris Mini Gels at a constant voltage of 200V. Gels were then blotted onto PVDF membranes using the iBlot® 7-Minute Blotting System (ThermoScientific, Rockford, IL). The membranes were blocked for 1h in blocking buffer consisting of 5% non-fat dry milk in 1X TBST at room temperature. The membranes were then incubated with primary antibody overnight at 40C with gentle rocking. The primary antibodies used were phospho-Src (Tyr527)(Cell Signaling Technology [CST] cat#2105, RRID:AB_10829463), phospho-Akt (Ser473) (CST cat#9271, RRID:AB_329825), phospho-Akt (Thr308) (CST cat#9275, RRID:AB_32928), Integrin α6 (CST cat#3750, RRID:AB_2249263), total Akt (CST cat#9272, RRID:AB_329827), Src (36D10) Rabbit mAb (CST cat#2109, RRID:AB_2106059) purchased from Cell Signaling Technology (Danvers, MA). Monoclonal anti-β-actin antibody (cat#A2228, RRID:AB_476697) was purchased from Sigma-Aldrich. Membranes were incubated with an ant-rabbit IgG, HRP-linked secondary antibody (CST cat#7074, RRID:AB_2099233) from Cell Signaling Technology or goat anti-mouse IgG-HRP (cat# sc-2005) from Santa Cruz Biotechnology (Dallas, Texas) for 1 h at room temperature. Protein detection was then done using the SuperSignal West Dura Extended Duration Substrate enhanced chemiluminescence detection system (ThermoFisher Scientific, Rockford, IL).
Tumor Specimens & Immunohistochemistry.
Fourteen breast tumor specimens were retrieved from patients who relapsed on endocrine therapy in accordance with an IRB approved protocol from the Loma Linda University ethics committee. Three of the patients experienced relapse following treatment with tamoxifen. All patients provided informed consent. Formalin-fixed paraffin embedded (FFPE) tissues were cut into 4μm sections and α6-integrin expression was detected using an EXPOSE Mouse and Rabbit-specific HRP/DAB detection IHC kit (Abcam, Cambridge, MA) in accordance with manufacturer’s recommendations. FFPE cancer tissue sections were deparaffinized by baking overnight at 560C, followed by xylene treatment. Tissue sections were then immediately rehydrated in graded concentrations (100% to 70%) of ethanol. Antigen retrieval was then performed via microwaving in citrate buffer (6.0 pH) for 10 minutes. Endogenous peroxidase activity was blocked via the application of a Hydrogen Peroxide Block. Non-specific staining was also blocked using a Protein Block. This was followed by overnight incubation with a rabbit polyclonal antibody to α6-integrin (ab133386, Abcam; Cambridge, MA). Thereafter, the sections were exposed to a Mouse Specifying Reagent and a Goat anti-rabbit HRP conjugate for 15 minutes and 1h respectively. Tissue sections were then stained using a DAB Chromogen and Substrate mixture, followed by counterstaining with hematoxylin. Positive and negative controls included normal lymph node tissue sections (ab4350, Abcam) and thyroid carcinoma tissue sections, known to express our target α6-integrin, incubated with or without primary antibody respectively (data not shown). Stained tissue sections were visualized via light microscopy. A pathologist (LD) blinded to the sample identity manually quantified all stains. Stains were scored as 1 (weak), 2 (moderate) or 3 (strong) to describe relative α6-integrin expression.
Molecular and histological assessment of tumor subtypes.
Using RNA sequencing data derived from The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas, 2012)[RRID:SCR_003193], we evaluated α6-integrin expression in patient tumors stratified based on molecular subtypes, which were determined by the Pam 50 gene set. The molecular subtypes include: basal-like, luminal A, luminal B and Her2 enriched. In brief, these subtypes are defined based upon the expression levels of specific hormone receptors (Estrogen Receptor (ER), Progesterone Receptor (PR) and v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (ERBB2 or HER2). The presence of ER defines the Luminal subtypes and the absence of HER2 amplification distinguishes Luminal A from Luminal B. The absence of all three receptors in tumors further characterized with EGFR and ck5/6 expression, are selected as ‘Basal-like’.
Statistical analysis.
Differences between groups were analyzed using one-way ANOVA with Tukey’s test or the Tukey–Kramer multiple comparison tests for evaluating three or more groups. To compare two groups, the unpaired Student’s t-test with Welch’s correction was used. Statistical analysis was performed using GraphPad Prism 4.0, Graph Pad software, Inc. San Diego, California, USA, www.graphpad.com. Differences were considered significant at p < 0.05.
RESULTS
Elevated levels of α6-integrin are found in cells and patient tumors that are TamR.
Overexpression of α6-integrin has been shown to promote breast cancer resistance to radiotherapy (Hu et al., 2016). To determine whether α6-integrin expression is associated with tamoxifen resistance in ER+ breast cancer, we measured the expression of α6-integrin in a panel of tamoxifen resistant breast cancer cells including TamR MCF-7, BT-474 and ZR-75–30 cells in comparison to Par MCF-7 cells. We found that basal α6-integrin mRNA levels were significantly elevated in these cells compared to Par MCF-7 cells (Figure 1A). Furthermore, α6-integrin expression levels were higher in TamR MCF-7 and BT-474 mammospheres compared to Par MCF-7 mammospheres (Figure 1B). We also found elevated α6-integrin protein expression levels among TamR MCF-7, BT-474 and ZR-75–30 breast cancer cells compared to Par MCF-7 cells (Figure 1C). Immunohistochemistry data from a representative patient revealed that treatment naïve tumor tissue sections stained positive for α6-integrin expression. However, once patients relapse on Tamoxifen, α6-integrin expression intensifies (Figure 1D). Positive staining was also evident among tissue sections taken from bone metastases (data not shown). Furthermore, α6-integrin expression levels were significantly higher in tumor samples of the basal-like molecular subtype than the luminal A, luminal B or Her2 enriched subtypes (Figure 1E) and basal-like tumors are known to exhibit resistance to tamoxifen. Taken together, our data suggest that α6-integrin overexpression is associated with Tamoxifen resistance.
Figure 1. α6-integrin expression in tamoxifen-resistant breast cancer cells and breast tumor tissues.
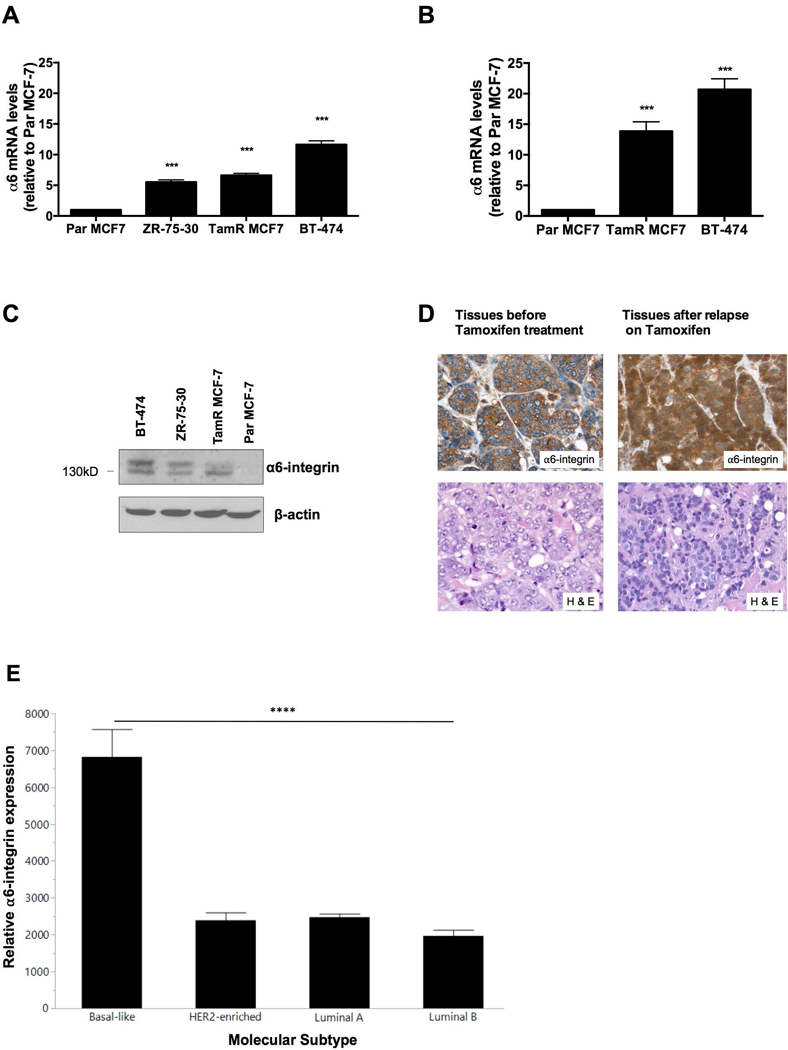
(A) Endogenous α6-integrin mRNA expression was evaluated in Par MCF-7, TamR MCF-7, ZR-75–30 and BT-474 cells and (B) in Par MCF-7, TamR MCF-7 and BT-474 mammospheres. Data represent the mean of at least 3 independent experiments. Bars, SEM. Significantly different at ***P < 0.001 in comparison to Par MCF-7 cells or mammospheres. (C) Western blot revealing relative α6-integrin protein expression in Par MCF-7, TamR MCF-7, ZR-75–30 and BT-474 cells. (D) Representative α6-integrin IHC stains for treatment naïve patient tumor tissues (left) and patient tumor tissues following relapse on tamoxifen (right). Magnification 40X. (E) Bar graph depicting α6-integrin mRNA expression levels (Pam50 gene set) from different breast tumor types derived from the TCGA database. Bars, SD. Significantly different as denoted **** P < 0.0001 when comparing basal-like subtypes with luminal A, luminal B and Her2-amplified subtypes.
Aminoflavone inhibits ER+ TamR cell proliferation and disrupts ER+ TamR mammospheres.
We previously showed that AFP464 (AF pro-drug) and AF disrupt mammospheres derived from in vitro and in vivo models via α6-integrin suppression (Brantley et al., 2016). Therefore, we sought to determine whether AF inhibits the proliferation of TamR cells and disrupts TamR mammospheres. Interestingly, the luminal A T47D cells (both Par and TamR) and to a lesser extent luminal A, MCF-7 cells (both Par and TamR) exhibited a biphasic dose response following treatment with AF while this effect was not apparent in the luminal B ZR-75–30 or BT-474 cells (Figure 2A). With the exception of the TamR T47D cells (IC50 ∼ 1 μM), all cells demonstrated responsiveness to AF at sub-micromolar concentrations, with TamR MCF-7 cells showing the most sensitivity to AF (Figure 2A). In support of other studies indicating the tendency for Her2/neu enriched cells to resist tamoxifen (Chen et al., 2008), we found that BT-474 and ZR-75–30 cells were unresponsive to tamoxifen (data not shown). Notably, TamR MCF-7 cells were not only insensitive to tamoxifen but demonstrated an increase in viability following tamoxifen exposure, while AF treatment prevented tamoxifen-induced TamR cell proliferation as seen by increased cell viability (Figure 2B). In keeping with our observations, it has been reported that ER+ tumors that have acquired resistance to tamoxifen often demonstrate tamoxifen-stimulated proliferation while retaining ER expression (Chang and Fan, 2013). AF helped to restore sensitivity to tamoxifen in TamR MCF-7 and BT-474 cells (Figure 1B,C). We previously demonstrated that AF impedes mammosphere formation in MCF-7 cells sensitive to tamoxifen (Brantley et al., 2016). In the current study, we found that AF disrupted mammospheres derived from TamR MCF-7, BT-474 and ZR-75–30 cells (Figure 2D). AF was also able to reduce the number of mammospheres formed by the TamR MCF-7 cells (Figure 2E). Due to size differences between untreated mammospheres and fragmented, AF exposed mammospheres, manual count appeared to show an increase in the number of AF exposed BT-474 mammospheres compared to control (data not shown). An accurate count on ZR-75–30 mammospheres was not readily achievable as these cells, at best, formed very loose mammospheres and were completely disrupted following AF treatment. Thus, determining actual mammosphere number was not readily feasible. However, using the Alamar Blue™ assay, we found AF reduced cell viability of TamR MCF-7, ZR-75–30 and BT-474 mammospheres (Figure 2F). Our data suggest that AF inhibits TamR cell viability, impedes tamoxifen-induced TamR MCF-7 cell proliferation and disrupts TamR mammospheres.
Figure 2. Determination of AF-mediated anticancer activity in Tamoxifen-resistant breast cancer cells and mammospheres.
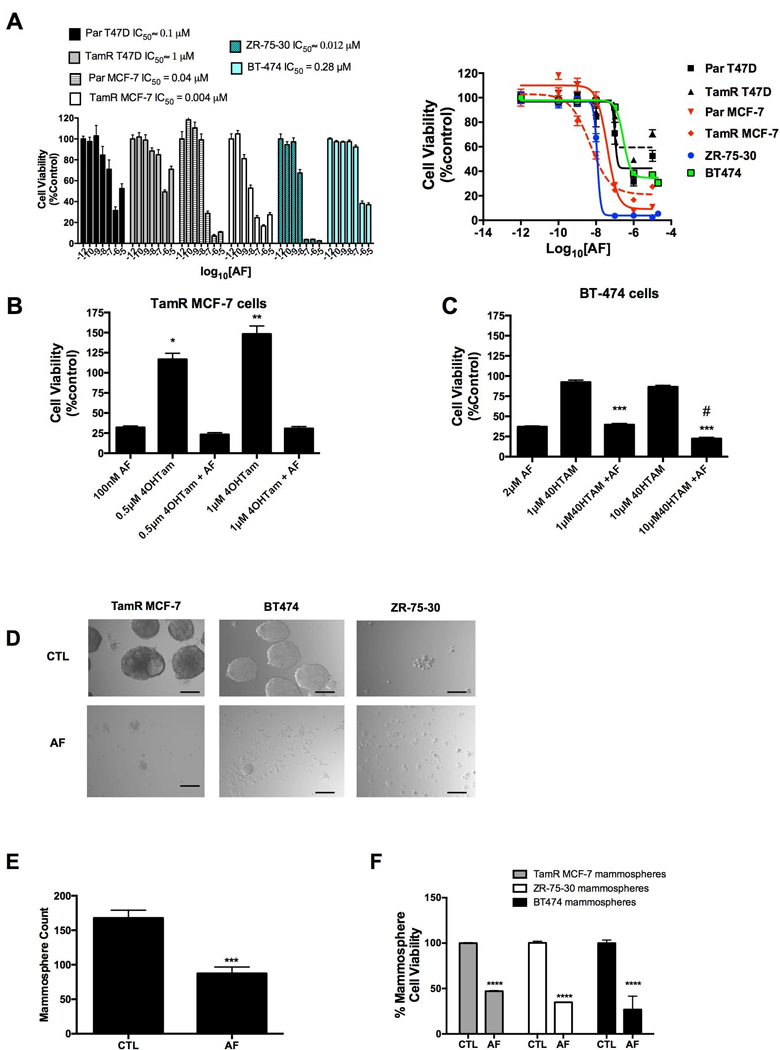
(A) Parental MCF-7 (Par MCF-7), Parental T47D (Par T47D), TamR MCF-7, TamR T47D, BT-474 and ZR-75–30 cells were exposed to AF (0.1–10000 nM) up to 5 d before analysis via the Alamar Blue™ assay in accordance with Materials and Methods. Data represent the mean of at least 4 independent experiments using at least quadruplicate samples for each concentration (B) TamR MCF-7 cells were exposed to AF, 4-hydroxytamoxifen (4OHTam) or AF and 4OHTam in combination before using the Alamar Blue™ assay as described in detail in Materials and Methods. Statistically significant at *P < 0.05 or **P < 0.01 in comparison to control (0.1% DMSO). (C) BT-474 cells were exposed to AF, 4OHTam or AF and 4OHTam in combination before using the Alamar Blue™ assay as described in detail in Materials and Methods. Statistically significant at ***P < 0.001 in comparison to cells treated with 4OHTam alone. (D) Mammospheres derived from TamR MCF-7, BT-474 and ZR-75–30 cells were treated with 0.1% DMSO (control, CTL) or AF in accordance with Materials and Methods before imaging using relief contrast microscopy. Scale bar = 50 μm. (E) TamR MCF-7 mammospheres were treated with CTL or AF (1 μM, 48h) and then counted in accordance with Materials and Methods. (F) The cell viability of mammospheres derived from TamR cells was determined following treatment with CTL or AF (2 μM for BT-474 cells, 1 μM for TamR MCF-7 cells and 100 nM for ZR-75–30 cells) for 48 h. Viability was determined in accordance with Materials and Methods.
Blocking α6-integrin expression and function inhibits 4OHTam-induced TamR cell proliferation and enhances the anticancer efficacy of AF.
We previously revealed that cells that substantially overexpress α6-integrin are rescued from the cytotoxic effects of AF (Brantley et al., 2016). To determine whether α6-integrin contributes to driving the resistance phenotype in TamR cells, we used a functional blocking antibody in select studies. In addition, we used a pool of siRNAs against α6-integrin. We used 100 nM AF rather than 1 μM due to the longer incubations times and to better determine whether AF in combination with other treatments would lead to an enhancement in anticancer activity as compared to AF alone. Blocking antibody GoH3 enhanced the anticancer activity of tamoxifen and AF in Par MCF-7 cells and in TamR cells (Figure 3A-C). Suppressing α6-integrin’s function or silencing α6-integrin reduced the cell viability of TamR cells, prevented the 4OHTam-induced proliferation, and enhanced responsiveness of these cells to 4OHTam (Figure 3B,D). As expected, the effects on cell proliferation were a bit more pronounced with AF and siRNA against α6-integrin as compared to the blocking antibody since the blocking antibody is unable to negate the downstream effects (e.g., cell proliferation) while AF and α6-integrin siRNA are able to. Furthermore, blocking both the function and expression of α6-integrin enhanced the cytotoxic effects of AF against TamR cells (Figure 3B,D). Notably, the TamR MCF-7 cells were more responsive to the GoH3 treatment alone compared to the Par MCF-7 cells suggesting greater reliance on α6-integrin by these resistant cells for survival. These data suggest α6-integrin is important in the survival of TamR cells, particularly tamoxifen-induced cell proliferation, and contributes to AF-mediated anticancer actions.
Figure 3. Impact of AF and α6-integrin suppression on the responsiveness of breast cancer cells to tamoxifen.
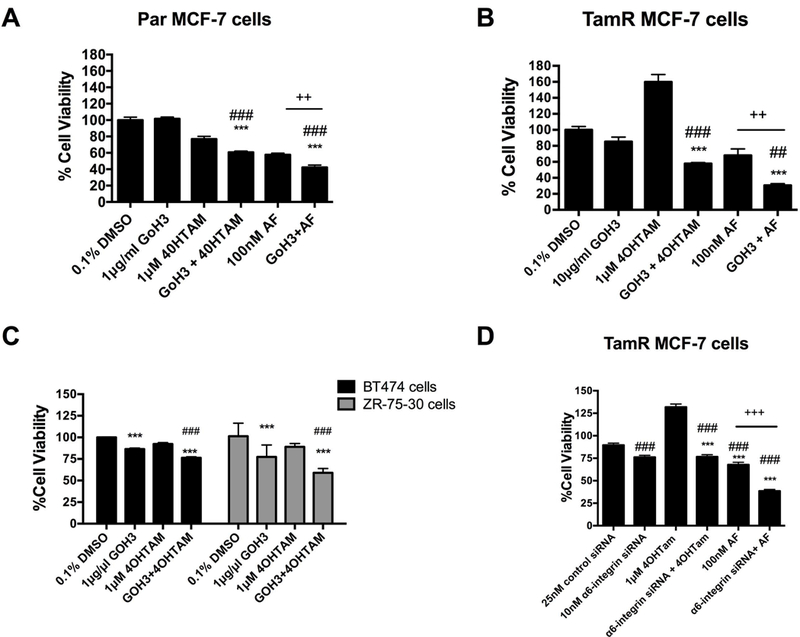
(A) Par MCF-7 cells were treated with DMSO (control), 4OHTam, α6-integrin blocking antibody GoH3, AF or GoH3 in combination with 4OHTam or AF before cell viability was assessed using the Alamar Blue™ assay as described in Materials and Methods. Statistically significant at ###P < 0.001 in comparison to DMSO (control) exposed. Statistically significant at ***P < 0.001 in comparison to 4OHTam alone and statistically significant at ++ P = 0.002, where indicated. (B) TamR MCF-7 cells were exposed to 4OHTam, α6-integrin blocking antibody GoH3, AF or GoH3 in combination with 4OHTam or AF before cell viability was assessed using the Alamar Blue™ assay as described in Materials and Methods. Statistically significant at ###P < 0.001 or ##P < 0.01 in comparison to 0.1% DMSO (control) exposed. Statistically significant at ***P < 0.001 in comparison to 4OHTam alone. Statistically significant at ++ P = 0.01, where indicated. (C) BT-474 and ZR-75–30 cells were exposed to GoH3, 4OHTam or the combination for up to 5 days before the Alamar Blue™ assay was used in accordance with Materials and Methods. Statistically significant at ***P < 0.001 in comparison to DMSO (control) exposed cells. Bars, SEM. Statistically significant at ### P < 0.001 in comparison to 4OHTam alone. (D) TamR MCF-7 cells were transfected with a pool of siRNAs against α6-integrin or non-targeting siRNAs. Transfected cells were exposed to 4OHTam or AF alone. Cell viability was determined using the Alamar Blue™ assay as described in the Materials and Methods. Statistically significant at ###P < 0.001 in comparison to DMSO (control) exposed. Statistically significant at ***P < 0.001 in comparison to 4OHTam alone. Statistically significant at +++P < 0.001, where indicated.
Aminoflavone inhibits α6-integrin expression, α6-integrin-Src-Akt signaling activation and induces BAX expression in TamR cells.
We found that AF reduced the expression of both cytoplasmic variants of α6-integrin (α6A and α6B) in TamR MCF-7 mammospheres (Figure 4A). AF also reduced α6-integrin gene expression in TamR MCF-7 mammospheres (Figure 4B). AF treatment was also found to significantly reduce α6-integrin expression in TamR MCF-7 and BT-474 cells (Figure 4C). However, AF was unable to inhibit α6-integrin expression in ZR-75–30 cells, despite their sensitivity to this agent (data not shown) which suggests that ZR-75–30 cells demonstrate sensitivity to AF via α6-integrin-independent mechanisms. It is interesting to note that ZR-75–30 cells lack progesterone receptor (PR) expression while BT-474 cells express the PR and this may account for some of the differences seen in viability and α6-integrin expression inhibition in these cells following AF treatment. AF decreased α6-integrin protein expression in TamR MCF-7 breast cancer cells (Figure 4D).
Figure 4. AF suppresses α6-integrin expression in tamoxifen-resistant breast cancer cells.
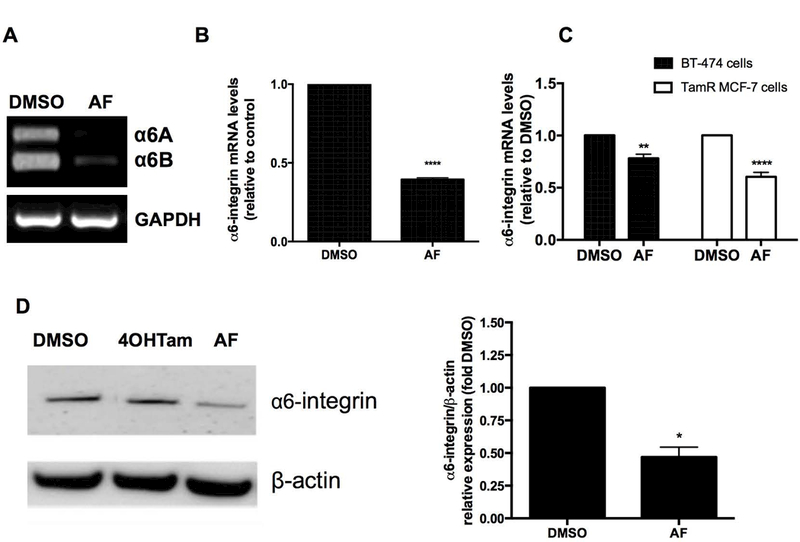
(A) semi-quantitative PCR analysis was performed to evaluate the expression of A and B isoform variants of α6-integrin in TamR MCF-7 mammospheres exposed to CTL (0.1% DMSO) or 1 μM AF for 48h. (B) Tam MCF-7 mammospheres were treated with 0.1% DMSO or 1 μM AF for 48h before qPCR analyses were performed to evaluate α6-integrin expression. Data represent the mean of at least 5 independent experiments performed in quadruplicate. Bars, SEM. Statistically significant at ****P < 0.0001 in comparison to 0.1% DMSO. (C) BT-474 and TamR MCF-7 cells were exposed to 0.1% DMSO or 2 μM AF for 120h and CTL 0.1% DMSO or 1 μM AF for 48h respectively before qPCR analyses were performed to evaluate α6-integrin expression. Data represent the mean of at least 5 independent experiments performed in quadruplicate. Bars, SEM. Statistically significant at **P < 0.01 or ****P < 0.0001 in comparison to 0.1% DMSO. (D) TamR MCF-7 cells were treated with 0.1% DMSO, 1 μM 4OHTam or 1 μM AF for 48 h before cells were lysed and analyzed for α6-integrin protein expression using Western blotting in accordance with Materials and Methods. Bars, SEM. Statistically significant at *P < 0.05 in comparison to DMSO.
α6-integrin signaling events that are crucial in cancer progression include α6-FAK/Src activation of the PI3K-Akt pathway (Kim et al., 2009). To assess whether down-regulation of α6-integrin lead to a reduction in Src and Akt signaling, we assessed levels of phosphorylated Src (p-Src) and Akt (p-AKT). AF caused an increase in pAkt (ser 473) that was inhibited by the α6-integrin blocking antibody GoH3 (Figure 5A). We observed a more pronounced increase in pAkt (ser 473) expression in Par MCF-7 cells following AF treatment (data not shown) that is consistent with a previous study using MCF-7 cells (Meng et al., 2007). GoH3 treatment caused no appreciable change in pAkt (ser 473) phosphorylation at either time point in TamR MCF-7 cells (Figure 5A). Both AF and GoH3 reduced pAkt (thr 308) levels in TamR MCF-7 cells at both time points while GoH3 enhanced the ability of AF to reduce pAkt (thr 308) activation after 24 h of treatment (Figure 5B). AF and GoH3 increased phosphorylation at the Src inactivation site, Tyr527, in TamR MCF-7 cells as early as 8h (Figure 5C). This phosphorylation was sustained up to 24 h of treatment (Figure 5C), though GoH3 was unable to enhance AF-mediated inhibition of Src signaling at either time point causing a paradoxical decrease after 24 h of combined treatment (Figure 5C). Taken together, AF caused a net decrease in Akt and Src signaling activation.
Figure 5. AF modulates Akt and Src signaling in Tamoxifen-resistant breast cancer cells.
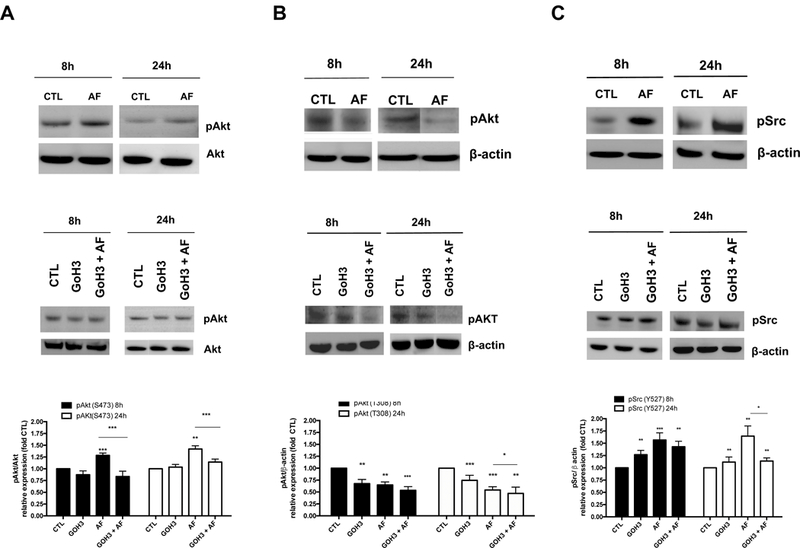
(A-B) TamR MCF-7 cells were exposed to media only or media containing 0.01% DMSO (CTL), 1 μM AF, 1μg/ml GoH3, or AF + GoH3 in combination for 8 and 24 h before Akt phosphorylation was assessed using Western blot analyses in accordance with Materials and Methods. (C) TamR MCF-7 cells were exposed to media only or media containing 0.01% DMSO (CTL), 1 μM AF, 1μg/ml GoH3, or AF + GoH3 in combination for 8 and 24 h before Src phosphorylation was assessed using Western blot analyses in accordance with Materials and Methods. Data represent the mean of at least 3 independent experiments. Bars, SEM. Statistically significant at *P < 0.05, **P < 0.01 or ***P < 0.001 in comparison to CTL or where indicated.
Integrin-mediated cell survival has been linked to the regulation of the pro-apoptotic gene BAX and integrin signaling appears to block BAX-induced apoptosis by preventing BAX translocation to the mitochondria (Gilmore et al., 2000).We previously demonstrated the ability of AF to induce apoptosis in sensitive breast cancer cells as evidenced by PARP cleavage and caspase 9 activation (McLean et al., 2008). We therefore evaluated the expression of BAX following AF treatment in Par and TamR MCF-7 cells. We found that AF significantly increased BAX expression in both Par and TamR MCF-7 cells (Figure 6A-B). Our data suggest that AF inhibits Src and Akt signaling activation to initiate TamR cell death via BAX induction and to suppress TamR cell proliferation (Figure 7).
Figure 6. AF induces the expression of pro-apoptotic gene BAX in Tamoxifen-sensitive and Tamoxifen-resistant breast cancer cells.
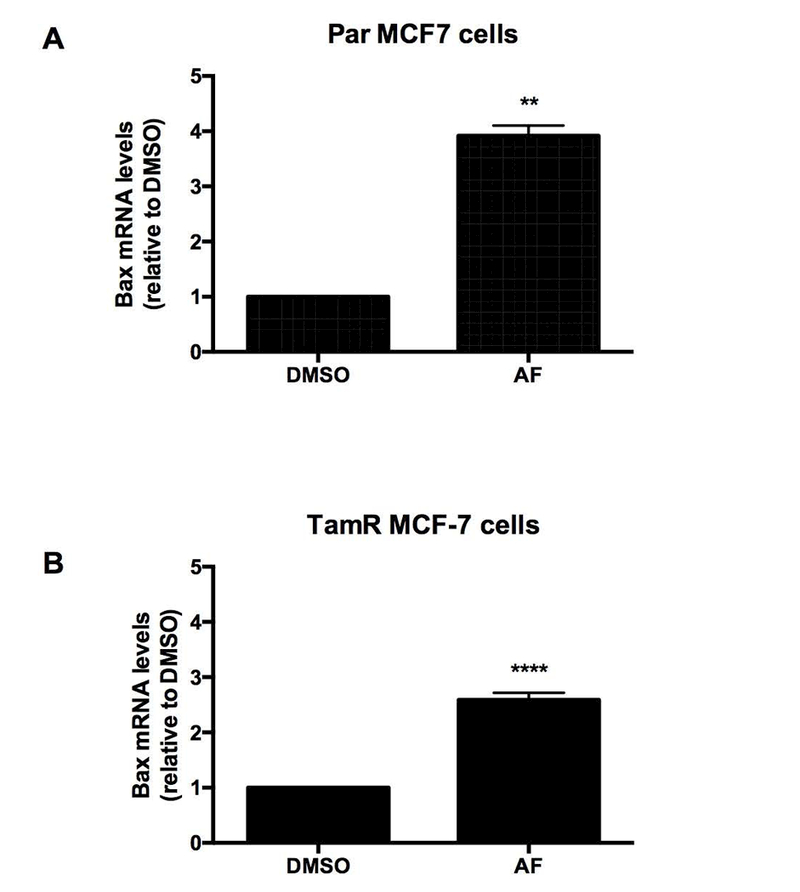
(A) Par and (B) TamR MCF-7 cells were exposed to CTL or 1 μM AF for 48 h before qPCR analysis was employed to detect BAX mRNA expression. Data represent the mean of at least 3 independent experiments. Bars, SEM. Statistically significant at **P < 0.01 or ****P < 0.0001 in comparison to DMSO.
Figure 7. Schematic depiction of proposal mechanism by which AF confers anticancer actions in TamR breast cancer cells.
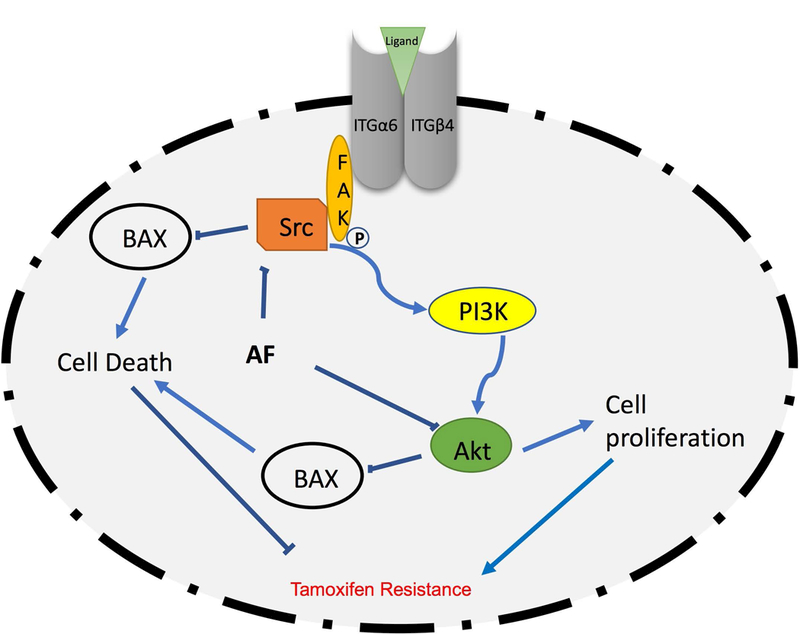
Ligands such as laminin bind to the α6/β4 integrin heterodimer to stimulate FAK/Src activation. This activation in turn stimulates cell-survival pathways such as the PI3K/Akt pathway, which increases cell proliferation and inhibits cell death to promote Tamoxifen resistance. On the contrary, AF inhibits α6-integrin/Src/Akt signaling to overcome resistance.
DISCUSSION
Tamoxifen resistance frequently leads to relapse, metastases and death. It is therefore imperative to develop effective therapeutic agents to combat tamoxifen resistance. In this study, we discovered that AhR ligand AF inhibits the proliferation of TamR cells at least in part by reducing α6-integrin expression and inhibiting activation of down-stream Src and Akt signaling pathways. Our findings and that of others also suggest that elevated α6-integrin expression is linked to tamoxifen resistance.
Although AhR signaling activation has been shown to promote tumorigenesis, emerging evidence indicates that certain AhR agonists exhibit anti-invasive and anti-metastatic actions (Hall et al., 2010; Hanieh et al., 2016; Prud’homme et al., 2010). AF selectively and potently inhibits the growth of cancer cells and tumors with no appreciable toxicity to non-malignant cells (Loaiza-Pérez et al., 2004; McLean et al., 2008). Non-toxic AhR agonists such as AF and Tranilast behave like partial AhR agonists which often oppose the tumor promoting actions of toxic, full AhR agonists similar to AhR antagonists. Small molecule AhR antagonists have been shown to inhibit the progenitor population within TamR cells in vitro and in vivo (Dubrovska et al., 2012).
Cells with higher levels of α6-integrin expression such as the BT-474 cells were less sensitive to the cytotoxic actions of AF and this supports our earlier observation that breast cancer cells with very high α6-integrin expression resist the cytotoxic actions of AF (Figure. 2) (Brantley et al., 2016). There is likely a threshold of α6-integrin expression that when exceeded, renders cells resistant to AF (Brantley et al., 2016). In the current study, TamR cells also demonstrated varying levels of sensitivity to AF due to differences in their molecular makeup. Synergism has been reported between AF and fulvestrant, in ER+ breast cancer cells (Shelton et al., 2007). Importantly, fulvestrant is a standard of care agent used to treat patients who have relapsed on tamoxifen.
The ability of α6-integrin blockade to enhance AF efficacy in TamR cells suggests further benefit is plausible from combining α6-integrin blocking agents with anti-cancer AhR agonists to treat TamR breast cancer. Furthermore, tamoxifen in combination with other AhR agonists such as the selective aryl hydrocarbon receptor modulator, 6-methyl-1,3,8-trichlorodibenzofuran (6-MCDF) has previously shown remarkable efficacy in mouse models of breast cancer that show responsiveness to tamoxifen (McDougal et al., 2001). Interestingly, 6-MCDF decreased levels of ERα through proteasomal degradation. Thus, AhR ligands have potential to demonstrate efficacy in the treatment of breast cancer including subtypes that are resistant to endocrine therapy.
ER expression does not entirely define the anticancer efficacy of AF. For instance, certain basal-like breast cancer cells such as MDA-MB-468 are highly sensitive to AF (Brinkman et al., 2014), yet treatment with histone deacetylase inhibitor vorinostat is necessary to sensitize basal-like MDA-MB-231 breast cancer cells to AF via ER reactivation (Stark et al., 2013). Responsiveness to AF appears to rely in part on the ability of this small molecule to induce AhR-mediated signaling activation and to suppress α6-integrin-mediated signaling pathways.
Our data suggest that elevated α6-integrin expression is linked to tamoxifen resistance and sustains the proliferation and survival of tamoxifen resistant cells. Notably, AF reduced the expression of both cytoplasmic splice variants of α6-integrin (α6A and α6B) in TamR MCF-7 mammospheres (Figure 4A). Importantly, α6B expression defines the mesenchymal population in breast cancer that is necessary for TIC function (Goel et al., 2014). Our findings are consistent with previous reports that revealed elevated α6-integrin expression of more than 3-fold in patient-derived ER+ breast cancer xenografts with acquired resistance to tamoxifen (Cottu et al., 2014). Furthermore, α6-integrin expression was comparatively higher in mammosphere-derived cells than cells from 2D cell culture (monolayers). This finding is consistent with what we found previously (Brantley et al., 2016). Indeed, mammospheres are known to enrich for TICs (Saadin et al., 2013). Though our patient sample size was small in the IHC study (Fig. 1), the trend toward elevated α6-integrin expression in patients who relapsed on tamoxifen was further demonstrated in basal-like tumors (tamoxifen unresponsive) in comparison to other tumor types from the TCGA database involving a much larger cohort of patients. Nonetheless, the above-mentioned findings suggest that elevated levels of α6-integrin are associated with tamoxifen resistance and α6-integrin may be valuable as a predictive biomarker of tamoxifen responsiveness.
TICs have been shown to play a key role in the development of resistance to tamoxifen (Bostner et al., 2013). In fact, tamoxifen treatment itself has been shown to select for cells with self-renewal capacity and promote mammosphere formation (Raffo et al., 2013). A recent study showed that α6-integrin ligand laminin conferred resistance to tamoxifen in an estrogen-dependent, tamoxifen-sensitive LM05-E breast cancer cell line via α6-integrin (Berardi et al., 2016). These observations support the hypothesis that tamoxifen may promote its own resistance by up-regulating α6-integrin levels and other TIC-related pathways and genes. Tamoxifen can also act as an ER agonist in breast cancer cells to promote Tamoxifen resistance. In keeping with our observations, it has been reported that ER+ tumors that have acquired resistance to tamoxifen may either be unresponsive to this agent or demonstrate tamoxifen stimulated growth while retaining ER expression (Chang, 2012). Reduced expression of co-repressors observed in tamoxifen resistance, results in stabilization of the agonist confirmation of the ERα, thereby allowing ERα activation by tamoxifen. (Chakraborty and Biswas, 2014). This may explain why tamoxifen stimulates proliferation in certain resistant cells.
Integrins have been shown to activate cell survival pathways such as PI3K to promote cancer cell proliferation and cell death via downstream FAK/Src signaling activation (Kim et al., 2009). In particular, α6-integrin primarily activates PI3K signaling to promote cancer cell migration, invasion, and survival (Lipscomb and Mercurio, 2005). In the current study, we found that increased α6-integrin expression correlated with an overall increase in Src-Akt signaling since we found TamR cells exhibited not only increased α6-integrin expression, but elevations in Akt phosphorylation (Fig. 1C, supplementary Fig 2). Additionally, AF effectively suppressed α6-integrin expression and this lead to an overall decrease Src-Akt signaling. Thus, Src-Akt signaling is decreased after α6-integrin expression is suppressed.
AF phosphorylated Src at Tyr527 in TamR MCF-7 cells as early as 8h and this phosphorylation was sustained for at least 24h (Figure 5C). GoH3 also promoted this phosphorylation as well, though GoH3 combined with AF did not enhance this effect (Fig. 5C). Phosphorylation of p-Src(Tyr527) results in Src inactivation through interaction with the SH2 domain and protein folding which makes Src inaccessible to substrates (Frame, 2002). Interestingly, acquired tamoxifen resistance leads to integrin-induced FAK/Src activation; inhibition of integrin-mediated FAK/Src/Akt activation was found to produce small yet significant sensitization to tamoxifen (Cowell et al., 2006). Taken together, our findings indicate AF suppresses Src activation in TamR MCF-7 cells.
AF increased pAkt( ser473) in Par MCF-7 cells (data not shown) consistent with a previous report which showed that sub-micromolar concentrations of AF caused S phase arrest when these cells were treated up to 8 h (Meng et al., 2007). AF increased Akt activation in Par MCF-7 cells to a greater extent than TamR MCF-7 cells and interestingly the α6 integrin blocking antibody GoH3 inhibited AF-mediated increases in Akt activation in TamR MCF-7 cells (Fig. 5A). We concur with Pommier and colleagues that our findings suggest that activation of Akt might reflect a cellular defense mechanism to AF-mediated DNA damage. It is, therefore, possible that this switch from Akt inactivation to activation with 1μM AF used in the current study may represent an initial apoptotic response followed by cell cycle arrest in response to DNA damage caused by more prolonged exposure. Indeed, AF induces oxidative DNA damage and S-phase arrest in triple negative MDA-MB-468 cells (McLean et al., 2008).
Phosphorylation of Thr308 in the activation loop of the kinase domain and Ser473 in the C-terminal regulatory domain is needed for full activation of Akt, with Thr308 phosphorylation playing the dominant role in Akt activation (Song et al., 2005; Vincent et al., 2011). Furthermore, Akt phosphorylation at these two sites occurs independently of each other (Alessi et al., 1996) with PDK1 phosphorylating Akt at Thr308 and mTORC2 phosphorylating Akt at Ser473. Therefore, since AF significantly reduced Thr308 phosphorylation, we can conclude that this AhR ligand decreased overall Akt kinase activity in TamR MCF-7 cells, an effect that was enhanced by GoH3 following 24 h of co-treatment (Fig. 5B). AF has targets other than α6-integrin that may contribute to its ability to inhibit Src-Akt signaling activation. For instance, β-naphthoflavone, another AhR agonist with in vivo anti-tumor activity, was found to inhibit PI3K/Akt signaling in MCF-7cells in an AhR-dependent manner (Wang et al., 2014). On the other hand, GoH3 specifically blocks the function of α6-integrin and thus AF and GoH3 have the potential to inhibit Src-Akt signaling by related as well as distinct mechanisms.
Activated Akt and Src resulting from integrin signaling and concomitant inhibition of pro-apoptotic BAX activity opposes cell death (Bouchard et al., 2008; Shishido et al., 2014). These observations support our findings that AF inhibits α6-integrin/Src/Akt signaling and induces BAX expression to promote TamR MCF-7 cell death. Additionally, AF suppresses the proliferation of TamR MCF-7 cells by suppressing Thr308 Akt phosphorylation. In our study, both Par and TamR MCF-7 cells showed increased α6-integrin/Src/Akt signaling though TamR cells exhibited this enhanced signaling to a greater extent (Fig. 1C and supplementary Fig 2). Thus, Src-Akt inhibition in TamR and Par MCF-7 cells likely occurs via similar means and the greater level of BAX induction observed in Par MCF-7 cells compared to TamR MCF-7 cells concurs with the enhanced ability of AF to suppress α6-integrin expression in these cells. It is quite plausible that when these cells are untreated, BAX translocation to the mitochondria is suppressed. We speculate that following AF treatment, α6-integrin/Src/Akt signaling becomes inhibited to enable BAX translocation irrespective of tamoxifen responsiveness. This may explain why BAX induction was observed in both cell lines after AF treatment. Taken together, our data suggest that BAX translocation is readily restored following AF-mediated α6-integrin/Src/Akt signaling blockade.
In conclusion, our data suggest AF inhibits α6-integrin-Src-Akt signaling to induce apoptosis, reduce cell proliferation and counteract tamoxifen resistance in ER+ breast cancer cells. More in-depth studies are needed to conclusively determine whether α6-integrin plays a causal role in tamoxifen resistance as has been recently determined for TIC genes OCT-4 and SOX-9 (Bhatt et al., 2016; Jeselsohn et al., 2017). Our findings do suggest that AhR ligands such as AF have the potential to help combat tamoxifen resistance to ultimately improve clinical outcomes for patients who have relapsed on tamoxifen. Other AhR ligands such as anti-allergy agent Tranilast disrupt mammospheres (Prud’homme et al., 2010). We recently determined that related AhR ligand, 5F 203 suppresses α6-integrin expression and disrupts mammospheres (data not shown). To the best of our knowledge, our report is the first to demonstrate the ability of AhR ligands to reverse tamoxifen resistance by attenuating α6-integrin-Src-Akt signaling. Our study provides a rationale for evaluating α6-integrin as a potential biomarker for tamoxifen resistance and to more appropriately stratify luminal breast cancer patients that would ultimately benefit from endocrine therapy in combination with AhR ligands such as AF.
Supplementary Material
Confirmation of α6-integrin silencing.
Basal phosphorylation of Akt in Par and TamR MCF-7 breast cancer cells.
ACKNOWLEDGEMENTS
This research was supported in part from Grants to Promote Collaborative and Translational Research (GCAT), Grants for Research and School Partnerships (GRASP), grants from the National Institute of General Medical Sciences for enhancing minority access to research careers (MARC U*STAR 2T34 GM008663) and the National Institute of Health Disparities and Minority Health of the National Institutes of Health (NIH) under award number P20MD006988. The content is solely the responsibility of the authors and does not necessarily represent views of NIH.
Contract grant sponsors: Grants to Promote Collaborative and Translational Research (GCAT), Grants for Research and School Partnerships (GRASP), National Institute of General Medical Sciences for enhancing minority access to research careers and the National Institute of Health Disparities and Minority Health of the National Institutes of Health.
Contract grant numbers: MARC U*STAR 2T34 GM008663; P20MD006988
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- Ahmad A 2013. Pathways to breast cancer recurrence. ISRN oncology 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. 1996. Mechanism of activation of protein kinase B by insulin and IGF-1. The EMBO Journal 15(23):6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Berardi DE, Raffo D, Todaro LB, Simian M. 2016. Laminin Modulates the Stem Cell Population in LM05-E Murine Breast Cancer Cells Through the Activation of the MAPK/ERK Pathway. Cancer research and treatment : official journal of Korean Cancer Association. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Stender JD, Joshi S, Wu G, Katzenellenbogen BS. 2016. OCT-4: a novel estrogen receptor-alpha collaborator that promotes tamoxifen resistance in breast cancer cells. Oncogene 35(44):5722–5734. [DOI] [PubMed] [Google Scholar]
- Bostner J, Karlsson E, Pandiyan MJ, Westman H, Skoog L, Fornander T, Nordenskjold B, Stal O. 2013. Activation of Akt, mTOR, and the estrogen receptor as a signature to predict tamoxifen treatment benefit. Breast cancer research and treatment 137(2):397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard V, Harnois C, Demers MJ, Thibodeau S, Laquerre V, Gauthier R, Vezina A, Noel D, Fujita N, Tsuruo T, Arguin M, Vachon PH. 2008. B1 integrin/Fak/Src signaling in intestinal epithelial crypt cell survival: integration of complex regulatory mechanisms. Apoptosis 13(4):531–542. [DOI] [PubMed] [Google Scholar]
- Brantley E, Callero MA, Berardi DE, Campbell P, Rowland L, Zylstra D, Amis L, Yee M, Simian M, Todaro L, Loaiza-Perez AI, Soto U. 2016. AhR ligand Aminoflavone inhibits alpha6-integrin expression and breast cancer sphere-initiating capacity. Cancer letters 376(1):53–61. [DOI] [PubMed] [Google Scholar]
- Brinkman AM, Wu J, Ersland K, Xu W. 2014. Estrogen receptor alpha and aryl hydrocarbon receptor independent growth inhibitory effects of aminoflavone in breast cancer cells. BMC cancer 14:344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockdorff BL, Skouv J, Reiter BE, Lykkesfeldt AE. 2000. Increased expression of cytochrome p450 1A1 and 1B1 genes in anti-estrogen-resistant human breast cancer cell lines. International journal of cancer Journal international du cancer 88(6):902–906. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. 2012. Comprehensive molecular portraits of human breast tumours. Nature 490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Biswas PK. 2014. Structural insights into selective agonist actions of tamoxifen on human estrogen receptor alpha. J Mol Model 20(8):2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Fan W. 2013. Endocrine therapy resistance: current status, possible mechanisms and overcoming strategies. Anti-cancer agents in medicinal chemistry 13(3):464–475. [PubMed] [Google Scholar]
- Chang M 2012. Tamoxifen Resistance in Breast Cancer. Biomolecules & Therapeutics 20(3):256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Wang Y, Kane SE, Chen S. 2008. Improvement of sensitivity to tamoxifen in estrogen receptor-positive and Herceptin-resistant breast cancer cells. J Mol Endocrinol 41(5):367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottu P, Bieche I, Assayag F, El Botty R, Chateau-Joubert S, Thuleau A, Bagarre T, Albaud B, Rapinat A, Gentien D, de la Grange P, Sibut V, Vacher S, Hatem R, Servely JL, Fontaine JJ, Decaudin D, Pierga JY, Roman-Roman S, Marangoni E. 2014. Acquired resistance to endocrine treatments is associated with tumor-specific molecular changes in patient-derived luminal breast cancer xenografts. Clinical cancer research : an official journal of the American Association for Cancer Research 20(16):4314–4325. [DOI] [PubMed] [Google Scholar]
- Cowell LN, Graham JD, Bouton AH, Clarke CL, O’Neill GM. 2006. Tamoxifen treatment promotes phosphorylation of the adhesion molecules, p130Cas//BCAR1, FAK and Src, via an adhesion-dependent pathway. Oncogene 25(58):7597–7607. [DOI] [PubMed] [Google Scholar]
- Darakhshan S, Bidmeshkipour A, Khazaei M, Rabzia A, Ghanbari A. 2013. Synergistic effects of tamoxifen and tranilast on VEGF and MMP-9 regulation in cultured human breast cancer cells. Asian Pacific journal of cancer prevention : APJCP 14(11):6869–6874. [DOI] [PubMed] [Google Scholar]
- Dubrovska A, Hartung A, Bouchez LC, Walker JR, Reddy VA, Cho CY, Schultz PG. 2012. CXCR4 activation maintains a stem cell population in tamoxifen-resistant breast cancer cells through AhR signalling. British journal of cancer 107(1):43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuSell CD, Nelson ER, Wittmann BM, Fretz JA, Kazmin D, Thomas RS, Pike JW, McDonnell DP. 2010. Regulation of aryl hydrocarbon receptor function by selective estrogen receptor modulators. Mol Endocrinol 24(1):33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dydensborg AB, Teller IC, Groulx JF, Basora N, Pare F, Herring E, Gauthier R, Jean D, Beaulieu JF. 2009. Integrin alpha6Bbeta4 inhibits colon cancer cell proliferation and c-Myc activity. BMC cancer 9:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame MC. 2002. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta 1602(2):114–130. [DOI] [PubMed] [Google Scholar]
- Friedrichs K, Ruiz P, Franke F, Gille I, Terpe HJ, Imhof BA. 1995. High expression level of alpha 6 integrin in human breast carcinoma is correlated with reduced survival. Cancer research 55(4):901–906. [PubMed] [Google Scholar]
- Fu X, Jeselsohn R, Pereira R, Hollingsworth EF, Creighton CJ, Li F, Shea M, Nardone A, De Angelis C, Heiser LM, Anur P, Wang N, Grasso CS, Spellman PT, Griffith OL, Tsimelzon A, Gutierrez C, Huang S, Edwards DP, Trivedi MV, Rimawi MF, Lopez-Terrada D, Hilsenbeck SG, Gray JW, Brown M, Osborne CK, Schiff R. 2016. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proceedings of the National Academy of Sciences of the United States of America 113(43):E6600–E6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore AP, Metcalfe AD, Romer LH, Streuli CH. 2000. Integrin-Mediated Survival Signals Regulate the Apoptotic Function of Bax through Its Conformation and Subcellular Localization. The Journal of cell biology 149(2):431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel HL, Gritsko T, Pursell B, Chang C, Shultz LD, Greiner DL, Norum JH, Toftgard R, Shaw LM, Mercurio AM. 2014. Regulated splicing of the alpha6 integrin cytoplasmic domain determines the fate of breast cancer stem cells. Cell reports 7(3):747–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber W, Scheidt T, Aberger F, Huber CG. 2017. Understanding cell signaling in cancer stem cells for targeted therapy - can phosphoproteomics help to reveal the secrets? Cell communication and signaling : CCS 15(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JM, Barhoover MA, Kazmin D, McDonnell DP, Greenlee WF, Thomas RS. 2010. Activation of the aryl-hydrocarbon receptor inhibits invasive and metastatic features of human breast cancer cells and promotes breast cancer cell differentiation. Molecular endocrinology (Baltimore, Md) 24(2):359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanieh H, Mohafez O, Hairul-Islam VI, Alzahrani A, Bani Ismail M, Thirugnanasambantham K. 2016. Novel Aryl Hydrocarbon Receptor Agonist Suppresses Migration and Invasion of Breast Cancer Cells. PloS one 11(12):e0167650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T, Zhou R, Zhao Y, Wu G. 2016. Integrin α6/Akt/Erk signaling is essential for human breast cancer resistance to radiotherapy. Scientific reports 6:33376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeselsohn R, Cornwell M, Pun M, Buchwalter G, Nguyen M, Bango C, Huang Y, Kuang Y, Paweletz C, Fu X, Nardone A, De Angelis C, Detre S, Dodson A, Mohammed H, Carroll JS, Bowden M, Rao P, Long HW, Li F, Dowsett M, Schiff R, Brown M. 2017. Embryonic transcription factor SOX9 drives breast cancer endocrine resistance. Proceedings of the National Academy of Sciences of the United States of America 114(22):E4482–E4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin UH, Lee SO, Pfent C, Safe S. 2014. The aryl hydrocarbon receptor ligand omeprazole inhibits breast cancer cell invasion and metastasis. BMC cancer 14:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MR, Choi HS, Yang JW, Park BC, Kim JA, Kang KW. 2009. Enhancement of vascular endothelial growth factor-mediated angiogenesis in tamoxifen-resistant breast cancer cells: role of Pin1 overexpression. Molecular cancer therapeutics 8(8):2163–2171. [DOI] [PubMed] [Google Scholar]
- Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, MacSwords J, Wu Q, Vasanji A, McLendon RE, Hjelmeland AB. 2010. Integrin alpha 6 regulates glioblastoma stem cells. Cell stem cell 6(5):421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscomb EA, Mercurio AM. 2005. Mobilization and activation of a signaling competent alpha6beta4integrin underlies its contribution to carcinoma progression. Cancer Metastasis Rev 24(3):413–423. [DOI] [PubMed] [Google Scholar]
- Loaiza-Pérez AI, Kenney S, Boswell J, Hollingshead M, Alley MC, Hose C, Ciolino HP, Yeh GC, Trepel JB, Vistica DT, Sausville EA. 2004. Aryl hydrocarbon receptor activation of an antitumor aminoflavone: basis of selective toxicity for MCF-7 breast tumor cells. Molecular cancer therapeutics 3. [PubMed] [Google Scholar]
- McDougal A, Wormke M, Calvin J, Safe S. 2001. Tamoxifen-induced antitumorigenic/antiestrogenic action synergized by a selective aryl hydrocarbon receptor modulator. Cancer research 61(10):3902–3907. [PubMed] [Google Scholar]
- McLean L, Soto U, Agama K, Francis J, Jimenez R, Pommier Y, Sowers L, Brantley E. 2008. Aminoflavone induces oxidative DNA damage and reactive oxidative species-mediated apoptosis in breast cancer cells. Int J Cancer 122(7):1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lh Meng, Kohn KW Pommier Y. 2007. Dose-response transition from cell cycle arrest to apoptosis with selective degradation of Mdm2 and p21WAF1//CIP1 in response to the novel anticancer agent, aminoflavone (NSC 686288). Oncogene 26(33):4806–4816. [DOI] [PubMed] [Google Scholar]
- Morrison G, Fu X, Shea M, Nanda S, Giuliano M, Wang T, Klinowska T, Osborne CK, Rimawi MF, Schiff R. 2014. Therapeutic potential of the dual EGFR/HER2 inhibitor AZD8931 in circumventing endocrine resistance. Breast cancer research and treatment 144(2):263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo D, Wei F, Liu Y, Wang E, Zhang H, Lin X, Wong N, Bane A, Tang D. 2015. Factors promoting tamoxifen resistance in breast cancer via stimulating breast cancer stem cell expansion. Current medicinal chemistry. [DOI] [PubMed] [Google Scholar]
- Prud’homme GJ, Glinka Y, Toulina A, Ace O, Subramaniam V, Jothy S. 2010. Breast cancer stem-like cells are inhibited by a non-toxic aryl hydrocarbon receptor agonist. PLoS One 5(11):e13831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffo D, Berardi DE, Pontiggia O, Todaro L, de Kier Joffe EB, Simian M. 2013. Tamoxifen selects for breast cancer cells with mammosphere forming capacity and increased growth rate. Breast cancer research and treatment 142(3):537–548. [DOI] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Pagliuca A, Palio E, Zeuner A, De Maria R. 2008. Colon cancer stem cells. Gut 57(4):538–548. [DOI] [PubMed] [Google Scholar]
- Saadin K, Burke JM, Patel NP, Zubajlo RE, White IM. 2013. Enrichment of tumor-initiating breast cancer cells within a mammosphere-culture microdevice. Biomedical Microdevices 15(4):645–655. [DOI] [PubMed] [Google Scholar]
- Safe S, McDougal A. 2002. Mechanism of action and development of selective aryl hydrocarbon receptor modulators for treatment of hormone-dependent cancers (Review). International journal of oncology 20(6):1123–1128. [PubMed] [Google Scholar]
- Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. 2015. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol 25(4):234–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton P, Sausville E, Nakanishi T, Brodie A, Belosay A, LoRusso P, Njar V, Burger A. 2007. Breast cancer cells resistant to anti-hormone treatments retain sensitivity to Aminoflavone (NSC 686288). Molecular cancer therapeutics 6(11 Supplement):PR-4–PR-4. [Google Scholar]
- Shishido S, Bonig H, Kim YM. 2014. Role of integrin alpha4 in drug resistance of leukemia. Front Oncol 4:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song G, Ouyang G, Bao S. 2005. The activation of Akt/PKB signaling pathway and cell survival. Journal of cellular and molecular medicine 9(1):59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark K, Burger A, Wu J, Shelton P, Polin L, Li J. 2013. Reactivation of estrogen receptor alpha by vorinostat sensitizes mesenchymal-like triple-negative breast cancer to aminoflavone, a ligand of the aryl hydrocarbon receptor. PLoS One 8(9):e74525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanic N, Milovanovic Z, Tanic N, Dzodic R, Juranic Z, Susnjar S, Plesinac-Karapandzic V, Tatic S, Dramicanin T, Davidovic R, Dimitrijevic B. 2012. The impact of PTEN tumor suppressor gene on acquiring resistance to tamoxifen treatment in breast cancer patients. Cancer biology & therapy 13(12):1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Riggelen J, Buchwalter G, Soto U, De-Castro Arce J, zur Hausen H, Wasylyk B, Rosl F. 2005. Loss of net as repressor leads to constitutive increased c-fos transcription in cervical cancer cells. The Journal of biological chemistry 280(5):3286–3294. [DOI] [PubMed] [Google Scholar]
- Vincent EE, Elder DJE, Thomas EC, Phillips L, Morgan C, Pawade J, Sohail M, May MT, Hetzel MR, Tavaré JM. 2011. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. British journal of cancer 104(11):1755–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Xu CX, Bu Y, Bottum KM, Tischkau SA. 2014. Beta-naphthoflavone (DB06732) mediates estrogen receptor-positive breast cancer cell cycle arrest through AhR-dependent regulation of PI3K/AKT and MAPK/ERK signaling. Carcinogenesis 35(3):703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Confirmation of α6-integrin silencing.
Basal phosphorylation of Akt in Par and TamR MCF-7 breast cancer cells.