

Abstract
Tumor cell motility is the essential step in cancer metastasis. Previously we showed that oxytocin and EGF effects on cell migration in prostate cancer cells require Giα2 protein. In the present study, we investigated the interactions among GPCR, Giα2, PI3-kinase and Rac1 activation in the induction of migratory and invasive behavior by diverse stimuli. Knockdown and knockout of endogenous Giα2 in PC3 cells resulted in attenuation of TGFβ1, oxytocin, SDF-1α and EGF effects on cell migration and invasion. In addition, knock-down of Giα2 in E006AA cells attenuated cell migration and over-expression of Giα2 in LNCaP cells caused significant increase in basal and EGF stimulated cell migration. Pretreatment of PC3 cells with Pertussis toxin (PTX) resulted in attenuation of TGFβ1 and oxytocin induced migratory behavior and PI3-kinase activation without affecting EGF induced PI3-kinase activation and cell migration. Basal and EGF induced activation of Rac1 in PC3 and DU145 cells were not affected in cells after Giα2 knockdown. On the other hand, Giα2 knockdown abolished the migratory capability of PC3 cells over-expressing constitutively active Rac1. The knockdown or knockout of Giα2 resulted in impaired formation of lamellipodia at the leading edge of the migrating cells. We conclude that Giα2 protein acts at two different levels which are both dependent and independent of GPCR signaling to induce cell migration and invasion in prostate cancer cells and its action is downstream of PI3-kinase/AKT/Rac1 axis.
Keywords: Migration, G-protein coupled receptors, Epidermal growth factor, F-actin, lamellipodia
Introduction
The process of metastasis starts with the dissemination of cancer cells from the primary tumor and ends with the formation of detectable macrometastases at distant sites (Vanharanta and Massague, 2013). Metastasis of epithelial cancer cells is a multi-stage process involving epithelial-mesenchymal transition (EMT), degradation of extracellular matrix (ECM), cell migration and invasion of the neighboring tissues, intravasation into blood or lymph vessels, transportation and extravasation, mesenchymal-epithelial transition (MET), and growth of metastatic tumors at distant locations (Berx et al., 2007; Rycaj and Tang, 2017; Shibue and Weinberg, 2011). In prostate, early stage of cancer is treatable by surgery and radiation therapy and the prognosis in these patients is very good. However, the prostate cancers in later stages of disease metastasize to other tissues and bone and pose a significant problem for treatments (DeSantis et al., 2014).
Tumor cell motility is the initial step in the process of invasion and metastasis and it is an essential component of dissemination of tumor cells from the primary tumor to local and distant sites. Although the movement of cancer cells can be either random or in a definite direction, most metastatic cancer cells exhibit directed migration (Roussos et al., 2011). Directed cell migration is a complex process which is regulated by numerous intracellular proteins; most important proteins regulating cell motility belong to Rho family of GTPases (Raftopoulou and Hall, 2004). Rho-subfamily members, including RhoA, Rac1 and Cdc42, play important roles in the regulation of cell migration (Rathinam et al., 2011). During cell migration, actin-driven protrusions at the leading edge are driven by Rac1 activation, whereas actomyosin contractility at the cell body and rear are coordinated by active Rho A (Raftopoulou and Hall, 2004), while Cdc42 regulates the establishment of cell polarity (Fortin Ensign et al., 2013; Srinivasan et al., 2003).
Several growth factors, chemokines, hormones and prostaglandins, acting through different types of membrane receptors, have been shown to induce migratory and invasive behavior in prostate cancer cells (Maxwell et al., 2014; Vo et al., 2013). Several of these ligands bind and activate G-protein coupled receptors (GPCRs), which signal via heterotrimeric G-proteins (classified into Gs, Gi, Gq, G12) in the form of activated Gα-GTP and Gβγ subunits (Luo et al., 2017; Salazar et al., 2013; Scarlett et al., 2018). Several studies have implicated different members of Gi family of proteins in cell migration in cancer cells (Ghosh et al., 2008; Li et al., 2013; Ward and Dhanasekaran, 2012). It has also been shown that Gβγ released from Gi-protein promotes migration and invasion of metastatic breast cancer cells (Xie et al., 2013). On the other hand, growth factors such as EGF and VEGF bind to their specific receptor tyrosine kinases which signal through their intracellular substrates to induce cell migration in cancer cells including prostate cancer cells (Festuccia et al., 2005; Ghosh et al., 2012; Sweeney et al., 2002). The activation of GPCRs or tyrosine kinase receptors by chemokines and growth factors, respectively also leads to the activation of phosphoinositide 3-kinase (PI3-kinase)/AKT/mammalian target of rapamycin (mTOR) pathway which plays an essential role in induction of migratory and invasive behavior (Liang et al., 2015; Xue and Hemmings, 2013). PI3-kinase is the enzyme responsible for generation of 3-phosphorylated phosphoinositides and activation of the protein kinase B/AKT leading to downstream activation of mTOR (Fruman and Rommel, 2014; Khwaja et al., 1997; Luo et al., 2003; O’Reilly et al., 2006). mTOR is a serine/threonine kinase that exists as two distinct protein complexes (mTORC1 and mTORC2) which differ in their biological effects (Francipane and Lagasse, 2014). Several studies have shown that inhibition of PI3-kinase/AKT/mTOR pathway results in attenuation of cell migration induced by chemokines and growth factors (Britschgi et al., 2012; Xue and Hemmings, 2013). Recently, both mTORC1 and mTORC2 were found to be critical for migration and invasion and activation of Rac1 and RhoA in several prostate cancer cell lines (Chen et al., 2014).
Previous studies from our laboratory have established the role of oxytocin through Gi-coupled receptor induced cell migration in prostate cancer cells and that Giα2 protein is essential for these effects (Zhong et al., 2010). Interestingly, EGF effects on cell migration in PC3 and DU145 prostate cancer cells were also attenuated in the absence of Giα2 (Zhong et al., 2012). While these studies indicate essential roles of Giα2 and PI3-kinase/AKT/mTOR in the migratory behavior of prostate cancer cells, the exact role of Giα2 and its interaction with the PI3-kinase pathway remains unknown. The present study was carried out to investigate the interactions among GPCR, Giα2, PI3-kinase and Rac1 activation in the induction of migratory and invasive behavior by diverse stimuli in prostate cancer cell lines. Specifically we attempted to seek answers to the following questions: 1) if activation of Giα2 is essential for cell migration and invasion in response to diverse extracellular stimuli, 2) if Giα2 acts upstream or downstream of PI3-kinase and/or Rac1 activation and, 3) how the Gβγ subunit may influence the migratory capability of prostate cancer cell lines.
Materials and Methods
Chemicals and Reagents
Human oxytocin (OXT), Pertussis toxin (PTX), mouse anti-β-actin antibody, mouse anti-α-tubulin and bovine serum albumin (BSA) were obtained from Sigma Chemicals (St. Louis, MO). The specific inhibitor of Gβγ subunit (M119K) was obtained from Developmental Therapeutics Program, National Cancer Institute (Bathesda, MD) and HA-tagged RGS2 plasmid was obtained from Dr. Yaping Tu (Cao et al., 2006). Recombinant human TGFβ1 and recombinant human SDF-1α (CXCL12) were purchased from PeproTech (Rocky Hill, NJ). Rat tail collagen, Matrigel and transwell inserts were obtained from BD Biosciences (San Jose, CA). Antibodies against phospho-AKTser473, AKT, phospho-EGFR and EGFR were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Control (sc-37007) and Giα2-targeting (sc-41752) siRNAs, transfection reagent (sc-29528), rabbit polyclonal anti-Giα2 antibody (sc-7276) and mouse β-actin antibody were purchased from Santa Cruz Biotechnology (Dallas, TX). Alexa Fluor® 488 goat anti-rabbit IgG (H+L) antibody and epidermal growth factor (EGF) were obtained from Life Technologies (Grand Island, NY). The anti-rabbit and anti-mouse immunoglobulins coupled with horseradish peroxidase (IgG-HRP), the FuGENE® 6 Transfection reagent and Wizard® Genomic DNA Purification Kit were obtained from Promega (Madison, WI). Rhodamine phalloidin (PHDR1) and GLisa Rac1 activation assay kit were purchased from Cytoskeleton, Inc. (Denver, CO). Rabbit RGS2 antibody was purchased from Proteintech, Chicago, IL. Mouse HA antibody was purchased from Covance, Princeton, NJ. Lipofectamine 3000 Transfection Reagent and DAPI were obtained from Thermo Fisher Scientific Inc. (Waltham, MA). Cell culture reagents were obtained from Mediatech, Inc. (Manassas, VA).
Cell culture
Prostate cancer cell lines (LNCaP, DU145 and PC3) were obtained from American Type Culture Collection (ATCC) (Rockville, MD). LNCaP is a low aggressive, androgen sensitive, prostate cancer cell line; DU145 and PC3 are androgen independent cell lines, derived from brain and bone metastatic sites, respectively. They were maintained in a 5% CO2 environment at 37°C, as previously described (Vo and Khan, 2011; Vo et al., 2013; Zhong et al., 2012). E006AA cells are derived from a localized prostate cancer in a patient of African American descent and were kindly provided by Dr. Shahriar Koochekpour (Roswell Park Cancer Institute, Buffalo, NY). These cells were maintained and cultured as described previously (Elliott et al., 2018; Koochekpour et al., 2004).
Transient transfection with Giα2 siRNAs and RGS2 plasmid
PC3 and E006AA cells were seeded in 6-well plates at a density of 1.5 × 105 cells per well and transfected with control or Giα2 specific siRNAs as previously described (Zhong et al., 2012) according to the manufacture’s protocol. Briefly, media with no antibiotics (200 µl/well) containing 30 nmol/L of siRNA was mixed with the transfection reagent (6 µl/well) and, after incubation, the siRNA mixtures were added drop by drop on the cells. The transfection media were replaced with the regular culture media (2 ml/well) the next day. Cells were harvested 72 hours after transfection and used for different assays.
PC3 cells in suspension (2 × 105) were transfected with 5.0 µg of either pcDNA3.1 control vector or vector encoding HA-tagged RGS2 (Cao et al., 2006) by Lipofectamine 3000, according to the manufacture’s protocol. Cells were seeded on 6-well plates and harvested 48 h later for Western blot analysis of RGS2 expression and migration assay.
Knockout of Giα2 using CRISPR/Cas9 gene editing
The plasmid “all in one-WT-Cas9” was ligated with three different sgRNAs, immediately after the U6 promoter according to previous protocol (Vo et al., 2017). In brief, sgRNAs were designed to target three different exons of Giα2 gene. PC3 cells were transfected using Lipofectamine 3000 reagent, according the manufacturer’s protocol. The transfected cells were sorted for GFP-tag 72 hours after transfection, using a BD FACSJAZZ cell sorter (BD Biosciences, San Jose, CA) and 500 positive cells were selected and maintained in MEM+10% FBS. Genomic DNA was extracted using Wizard® Genomic DNA Purification Kit, and a PCR was conducted to amplify the region of interest, flanking the sgRNA targeted sequence (Forward primer: 5’-TGGCTATCAGGATCATCCACG-3’; Reverse primer: 5’- CTCCGGTCCAGCCTTTAGTC-3’). Sanger sequencing was used to sequence the PCR product using the following Forward primer: 5’-TGGCTATCAGGATCATCCACG-3’. To confirm the knockout of the Giα2 protein, Western blot analyses were performed, using antibodies against Giα2.
Overexpression of Giα2 full length in PC3-Giα2 and LNCaP Cells
Giα2 knockout PC3 (PC3-Giα2) and LNCaP cells were seeded in 6-well plates at a density of 2.5 × 105 cells per well. The next day, cells were transfected with pcDNA3.1 empty vector and with the overexpression plasmid of the full length (FL), wild type (WT) of Giα2 gene (pcDNA3.1-Giα2-FL-WT; cDNA Resource Center, Bloomsberg, PA), using the Lipofectamine 3000 Transfection reagent, following the manufacturer’s protocol. Briefly, 2.5 μg of plasmid DNA were diluted with MEM for PC3-Giα2 and RPMI for LNCaP without antibiotics and were incubated for 15 minutes in the presence of transfection reagent. After the incubation time the complex DNA/Transfection reagent (ratio 1:3) was added to the cells. After 72 hours the cells were selected for 2 to 3 weeks in growth medium containing 400 μg/ml for PC3-empty vector, 800 μg/ml for the PC3-Giα2+Giα2-FL-WT, and 200 μg/ml for both LNCaP-empty vector and LNCaP+ Giα2-FL-WT of G418, and maintained under the same condition for further studies. Migration assays and Western blots were performed to confirm the re-expression and overexpression of the protein and the migratory capability of the cells.
Stable overexpression of Rac1Q61L in PC3 cells
Bacterial Stab containing pcDNA3-EGFP (empty vector) and pcDNA3-EGFP-Rac1Q61L (Rac1 constitutively active) plasmids were purchased from Addgene (plasmids #13031 and #12981, gifts from Doug Golenbock and Gary Bokoch, respectively) (Subauste MC, 2000). Plasmids were isolated and purified, according to the manufacturer’s protocol, using ZYMOPURE™ plasmid Maxiprep kit (Zymo Research, Irvin, CA). Purified plasmids (2 μg) were transfected into PC3 cells, using Lipofectamine 3000 transfection reagent for 24 hours. Cells were sorted based on EGFP expression using BD FACSJAZZ Cell Sorter (BD Bioscience, San Jose, CA). The enriched populations were grown in MEM, 10% FBS and different concentrations of the selective antibiotic G418 (400 μg/ml for PC3-empty vector and 800 μg/ml for PC3-Rac1Q61L) and then used for further experiments.
Western blot analysis
Western blot analyses were performed as described previously (Elliott et al., 2018; Vo et al., 2013; Zhong et al., 2012). Briefly, protein samples (30–35 µg proteins) were separated on 10% SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore Corp., Bedford, Massachusetts). After blocking, the membranes were incubated with appropriate dilutions of specific primary antibodies (1:1000 for p-AKT, AKT, p-EGFR, EGFR,; 1:500 for Giα2; 1:5000 for β-actin: 1:3000 for α-tubulin) overnight at 4°C. After washing, the blots were incubated with appropriate secondary antibodies and developed in ECL mixture. The blots were then exposed to an X-ray film and visualized by autoradiography. The density of specific protein bands was determined by ImageJ software (NIH, Bethesda, MD) and normalized using density of β-actin or α-tubulin bands which were used as loading controls. For RGS2, protein samples (40 µg) were loaded on 15% SDS-PAGE gel, electrophoresed, and transferred to an Immobilon-FL membrane (Millipore). Primary antibodies were used to identify RGS2, HA tag and loading control β-actin. IRdye700- or IRdye800-labeled secondary antibodies were used for protein band detection. The images were captured with a LI-COR Odyssey infrared imaging system (LI-COR Biosciences) at wavelengths of 700 or 800 nm.
Cell migration and invasion assays
In vitro cell migration and invasion assays were conducted using 24-well transwell inserts (8 μm) as described previously (Elliott et al., 2018; Vo et al., 2013; Zhong et al., 2012). Briefly, transwell inserts were coated with rat tail collagen (50 mg/ml), for migration assay, and with 50 μl of a 1:4 Matrigel/Coating buffer solution for invasion assay. Cells were treated with different chemoattractant solutions. For the migration assay the ligands used were OXT (100 nmol/L), TGFβ1 (5 ng/ml), SDF-1α (100ng/ml), EGF (10 ng/ml). For the invasion assay TGFβ1 (5 ng/ml), SDF-1α (100ng/ml), EGF (10 ng/mL) and 5% FBS as a positive control were used as treatments. The plates were incubated at 37°C for 5 hours (DU145 and PC3), and 24 hours (LNCaP and E006AA) for migration assays, and 48 hours for invasion assays. After fixation the cells were stained with 3 ng/ml of DAPI and images of five non-overlapping fields were captured using Axiovert 200M, Carl Zeiss (Göttingen, Germany) microscope, and the number of stained nuclei were determined with automatic counting using image analysis software (ZEN 2012; Carl Zeiss). Results were expressed as migration and invasion index defined as: the average number of cells per field for test substance/the average number of cells per field for the medium control.
Immunofluorescence and actin staining
Cells grown (0.5 × 105 cells/ml) on coverslips for 72 hours were fixed with 4% paraformaldehyde in phosphate buffered saline (PBS) for 15 minutes and washed with PBS three times. Cells were permeabilized with 0.1% Triton X-100 in PBS for 10 minutes and incubated with 10% normal goat serum for 1 hour to block nonspecific antibody binding. Then the cells were incubated with anti-Giα2 antibody (1:200) overnight at 4°C. After washing, the cells were incubated with secondary antibody, Alexa Fluor 488-conjugated anti-rabbit immunoglobulins (1:1000) for 45 minutes. To validate the specificity of the antibodies, parallel cell preparations were incubated with either primary or secondary antibodies alone and processed as negative controls. The cells were washed with PBS and incubated with Rhodamine-phalloidin for 30 minutes to detect F-actin filaments and DAPI for 10 minutes to detect the nuclei, and mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Images were captured using Zeiss LSM 700 Confocal Microscope with a 40× magnification objective.
RAC1 activation assay
PC3 and DU145 cells were seeded in 6-well plates at a density of 1.5 × 105 cells per well. The next day, cells were transfected with control siRNA or the Giα2-targeting siRNA using siRNA transfection reagent as described above. After 48 hours, cells were serum starved for 24 hours and then treated with EGF (100 ng/ml) for 3 minutes. Rac1 activity was then measured in cell lysate proteins (0.1–0.2 mg/ml) with GLISA (colorimetric format, Cytoskeleton, Denver, CO) according to the manufacturer’s protocol.
Statistical analysis
All experiments were repeated at least three times using different cell preparations. The results are presented as mean ± SEM of three independent experiments and images from a single representative experiment are presented. ANOVA and Duncan’s modified multiple range tests were employed to assess the significance of differences among various treatment groups (p < 0.05).
Results
Giα2 is essential for cell migration and invasion in prostate cancer cells
Previously, we found that endogenous Giα2 is essential for cell migration in prostate cancer cells, in response to both oxytocin and EGF, acting via GPCR and PTKR, respectively (Zhong et al., 2012). To determine whether Giα2 is required for cell migration in response to additional diverse stimuli, we knocked down endogenous Giα2 by over 80% (Fig. 1A) using Giα2 siRNA and then carried out cell migration assays in PC3 cells transfected with control siRNA or Giα2 siRNA in the presence of TGFβ1, SDF-1α and EGF. All ligands caused significant increase (p<0.05) in migration in cells transfected with control siRNA whereas TGFβ1, SDF-1α and EGF did not induce cell migration in Giα2 deficient cells (Fig. 1B). In parallel experiments, we also determined the effects of Giα2 knockdown in invasive behavior of PC3 cells. As shown in Fig. 1C, TGFβ1, SDF-1α and EGF effects on invasion were also significantly reduced in Giα2 deficient cells. In addition, FBS (5%) was used as a general positive control in the invasion assays and the knockdown of Giα2 resulted in a significant reduction in the number of the invading cells in FBS treated cells as well (Fig. 1C).
Fig. 1. Knockdown of endogenous Giα2 and RGS2 overexpression negatively regulate migration and invasion of PC3 cells in response to diverse stimuli.
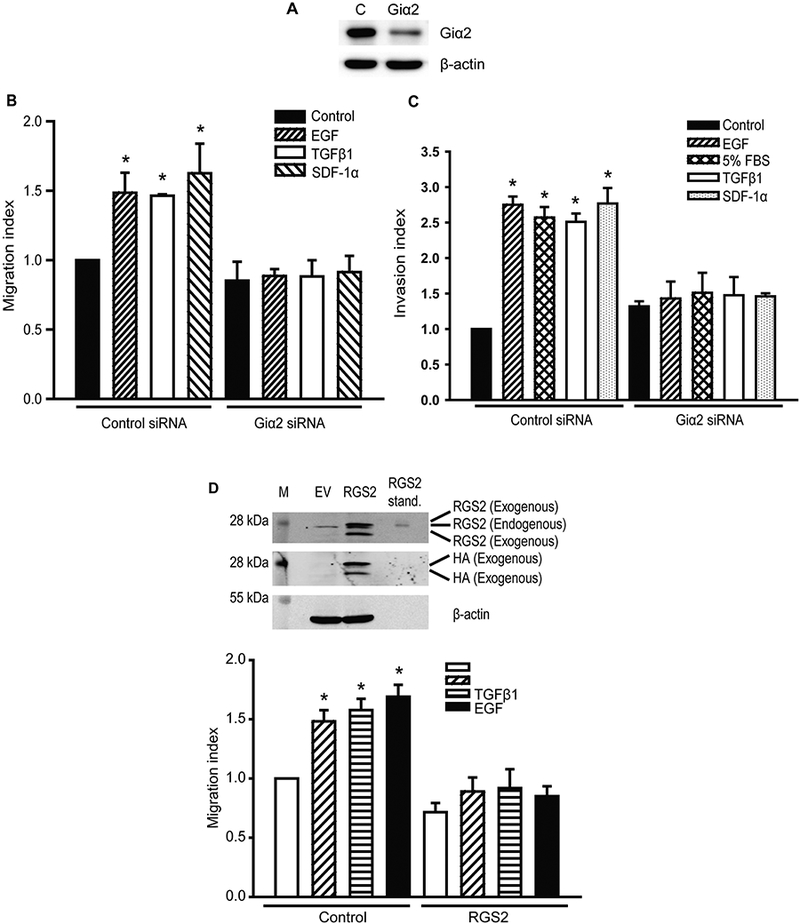
A. Western blot analysis of Giα2 in PC3 cells lysates to validate the knockdown of endogenous Giα2 protein using siRNA. β-actin was used as an internal control. B. Cell migration of PC3 cells after transfection with control siRNA or Giα2 siRNA in response to SDF-1α (100 nM), TGFβ1 (5 ng/ml) or EGF (10 ng/ml). Results are expressed as migration index defined as the average number of cells per field for the ligand tested/the average number of cells per field for the vehicle control. Each bar represents mean ± SEM (n=3). Significant differences (P<0.05) among different groups are represented with different lowercase letters. C. Invasive behavior of PC3 cells transfected with control siRNA and Giα2 siRNA in response to SDF-1α (100 nM), TGFβ1 (5 ng/ml), or EGF (10 ng/ml). Results are expressed as invasion index defined as average number of cells per field for the ligand tested/the average number of cells per field for the vehicle control. Each bar represents mean ± SEM (n=3). Significant differences (P<0.05) among different groups are represented with different lowercase letters. 5% FBS was used as a positive control. D. The upper panel shows Western blot analysis of HA-tag and RGS2 in cell lysates to validate the over-expression of RGS2 protein. β-actin was used as an internal control. Cell migration in PC3 cells transfected with control or RGS2 over-expression plasmids in response to stimulation with OXT (100 nmol/L), TGFβ1 (5 ng/ml) or EGF (10 ng/ml). Results are expressed as migration index. Each bar represents mean ± SEM (n=3). *Significantly different (P<0.05) when compared with appropriate controls.
To investigate whether Giα2 effects on cell migration induced by different ligands require its activation, we overexpressed RGS2 (Regulator of G-protein signaling 2) which inactivates Gαi and Gq subunits by increasing GTPase activity (Cao et al., 2006; Wolff et al., 2012) in PC3 cells. As shown in Fig. 1D (upper panel), PC3 cells express very low levels of endogenous RGS2 protein and RGS2 levels were significantly increased after overexpression of exogenous RGS2. Overexpression of exogenous RGS2 resulted in complete attenuation of cell migration in PC3 cells in response to OXT, TGFβ1 and EGF (Fig. 1D, lower panel). To conclusively establish a specific role of Giα2 in cell migration and invasion, we also generated Giα2 knockout prostate cancer cell lines (PC3-Giα2) using CRISPR/Cas9 gene editing approach. Clones 1, 2 and 3 have complete knockout of Giα2 protein (Fig. 2A). Specific knockout of endogenous Giα2 led to significant reduction of migratory and invasive behavior in the PC3-Giα2 cells (clones 1 and 2) in the presence or absence of exogenous stimuli (Fig. 2B, 2C). Re-expression of Giα2 protein in these cell lines restored their migratory behavior (Fig. 2D).
Fig. 2. Knockout, knockdown and overexpression of Giα2 have differential effects on migration and invasion in various prostate cancer cell lines.
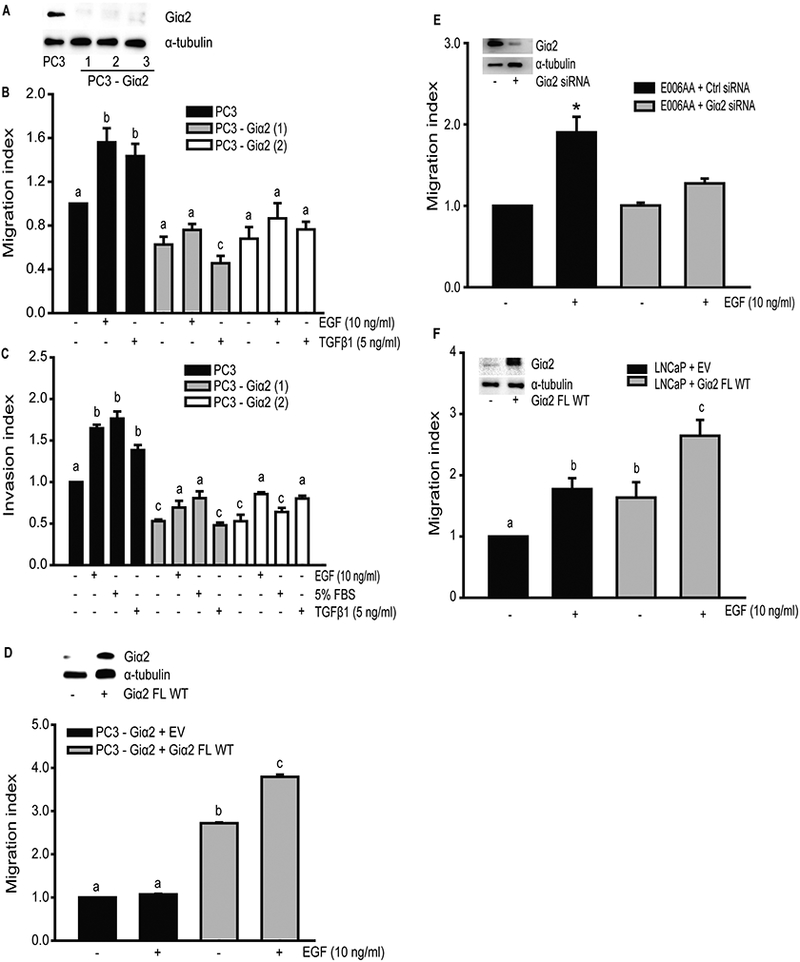
A. Western blot analysis of Giα2 in lysates from PC3 cells and PC3-Giα2-null cell lines after knockout of Giα2, using CRISPR/Cas9 gene editing. α-tubulin was used as an internal control. B. Cell migration in PC3 and PC3-Giα2-null cells in response to TGFβ1 (5 ng/ml) or EGF (10 ng/ml). Results are expressed as migration index. Each bar represents mean ± SEM (n=3). Significant differences (P<0.05) among different groups are represented with different lowercase letters. C. Invasive behavior of PC3-Giα2-null cells in response to TGFβ1 (5 ng/ml) or EGF (10 ng/ml). Results are expressed as invasion index. Each bar represents mean ± SEM (n=3). Significant differences (P<0.05) among different groups are represented with different lowercase letters. 5% FBS was used as a positive control. D. Upper panel shows Western blot analysis of Giα2 in cell lysates, to validate the re-expression of the protein. (EV= empty vector). α-tubulin was used as an internal control. Cell migration in PC3-Giα2-null cells after re-expression of full length (FL) wild-type (WT) Giα2, was significantly increase in both absence and in response to EGF (10 ng/ml) stimulation. Results are expressed as migration index. Each bar represents mean ± SEM (n=3). Significant differences (P<0.05) among different groups are represented with different lowercase letters. E. Upper panel shows western blot analysis of Giα2 in cell lysates, to validate the knockdown of the protein. α-tubulin was used as an internal control. Cell migration in E006AA cells is decreased after Giα2 knockdown, in response to EGF (10 ng/ml) stimulation. Results are expressed as migration index. Each bar represents mean ± SEM (n=3). * Significantly different (P<0.05) from appropriate controls. F. Upper panel shows western blot analysis of Giα2 in cell lysates, to validate the over-expression of the protein. α-tubulin was used as an internal control. Cell migration in LNCaP cells overexpressing Giα2 is increased, in absence and presence of EGF (10 ng/ml), compared to the control. Results are expressed as migration index. Each bar represents mean ± SEM (n=3). Significant differences (P<0.05) among different groups are represented with different lowercase letters.
We also knocked down endogenous Giα2 protein in E006AA cells. The knockdown of Giα2 significantly decreased (p<0.05) migratory capability of E006AA cells in response to EGF (Fig. 2E). On the other hand, LNCaP cells have a low expression of endogenous Giα2 protein and lower capacity to migrate. Overexpression of Giα2 caused a significant increase (p<0.05) in basal and EGF stimulated cell migration in LNCaP cells (Fig. 2F). These results showed that activated Giα2 subunit is essential for both cell migration and invasion in prostate cancer cell lines in response to diverse stimuli, acting through a variety of signaling pathways.
Role of Giα2 in the activation of PI3-kinase/AKT signaling pathway in prostate cancer cells
Our previous studies have shown that activation of PI3-kinase/AKT pathway is essential for cell migration and invasion in prostate cancer cells (Vo et al., 2013; Walker et al., 2013). Therefore, we determined a possible role of Giα2 in the activation of this pathway and whether or not this action involved its activation via GPCR. In the first set of experiments, we determined the activation of PI3-kinase pathway in response to various ligands after pre-incubation of PC3 cells with Pertussis toxin (PTX). PTX pretreatment inhibits activation of Gi/Go proteins via GPCR. As shown in Fig. 3A and 3B, OXT, TGFβ1 and EGF caused significant increase in p-AKT levels in PC3 cells. Pretreatment with PTX blocked the effects of OXT and TGFβ1 on p-AKT levels but had no effects on EGF induced p-AKT levels. The effects of various ligands on cell migration correlated with their effects on activation of PI3-kinase pathway (Fig. 3C). These results suggested that effects of both TGFβ1 and OXT on activation of PI3-kinase and cell migration are mediated via Gi/Go-coupled receptors while EGF effects do not involve activation of Gi/Go coupled GPCR.
Fig. 3. PTX pretreatment and inhibition of Gβγ subunit have differential effects on PI3-kinase activation and migration in response to different ligands.
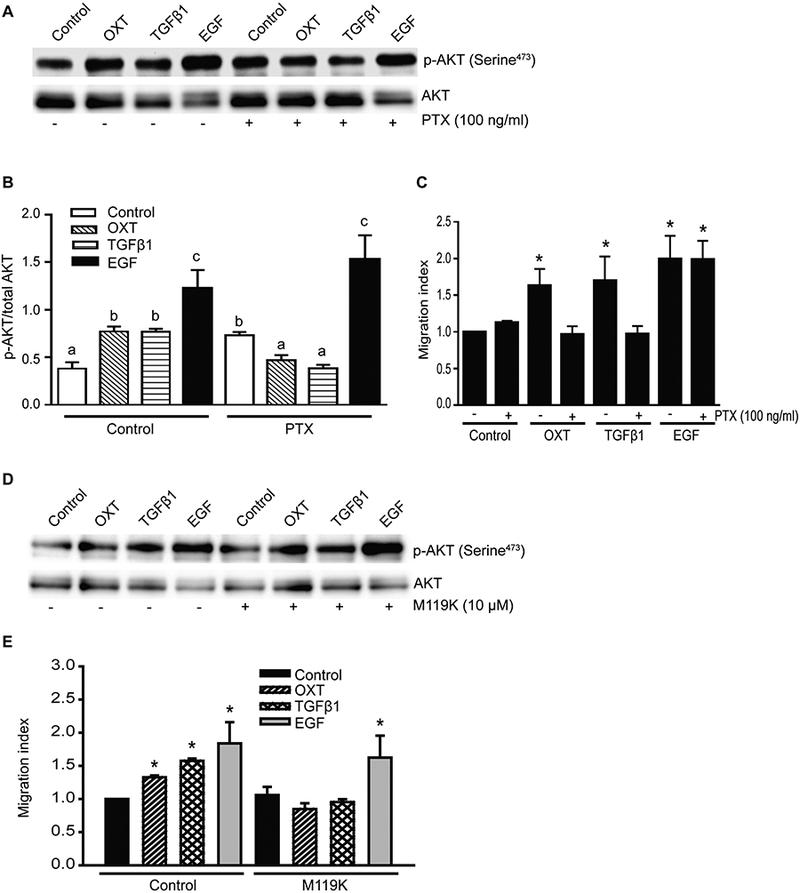
A. Western blot analysis of p-AKT in total cell lysates from PC3 cells after pretreatment with PTX for 1 hour, followed by OXT (100 nmol/L), TGFβ1 (5 ng/ml), or EGF (10 ng/ml) treatment for 30 minutes. Total AKT served as a loading and normalization control. B. Quantitative differences in levels of p-AKT in different groups. Data are expressed as mean ± SEM (n=3) and analyzed by ANOVA and Duncan’s modified range tests. Significant differences between groups in a given category (P<0.05) are designated with different lowercase letters. C. PC3 cells were incubated with or without PTX (100 ng/ml) overnight and then subjected to transwell migration assay in the presence of OXT (100 nmol/L), TGFβ1 (5 ng/ml) or EGF (10 ng/ml). Results are expressed as migration index. Each bar represents mean ± SEM (n=3). * Significantly Different (P<0.05) when compared with appropriate controls. D. Western blot analysis of p-AKT in cell lysates from PC3 cells after pretreatment with M119K inhibitor (10 μM) followed by OXT (100 nmol/L), TGFβ1 (5 ng/ml) or EGF (3 ng/ml) treatment for 30 minutes. AKT blots served as loading controls. E. PC3 cells were pretreated with or without M119K (10 µM) for 15 minutes and then subjected to transwell migration assay in the presence of OXT (100 nmol/L), TGFβ1 (5 ng/ml) or EGF (10 ng/ml). Results are expressed as migration index. Each bar represents mean ± SEM (n=3). *Significantly different (P<0.05) when compared with appropriate controls.
To determine whether the effects on cell migration and PI3-kinase activation were due to activated Gβγ subunits, PC3 cells were treated with TGFβ1, OXT and EGF in the presence or absence of specific Gβγ inhibitor (M119K). The Gβγ inhibitor blocked the stimulatory effects of the TGFβ1 and OXT on activation of PI3-kinase pathway (Fig. 3D) and cell migration (Fig. 3E). On the other hand, incubation with Gβγ inhibitor had no effect on EGF-induced activation of PI3-kinase activation and cell migration under identical conditions (Fig. 3D, 3E).
Giα2 is essential for activation of PI3-kinase pathway in response to TGFβ1 and OXT but is not required for EGF induced activation of PI3-kinase pathway
Next, we determined whether PTX inhibition of PI3-kinase activation and cell migration was due to specific effects on Giα2 activation and whether Giα2 protein plays a role in activation of PI3-kinase pathways downstream of GPCR activation in EGF treated cells. We investigated the effects of various ligands on activation of PI3-kinase signaling and cell migration after selective knockdown of endogenous Giα2 using specific siRNAs. As shown in Fig. 4A (upper panel), all three ligands increased levels of p-AKT in PC3 cells transfected with control siRNA. In the cells transfected with Giα2 siRNA, the basal levels of p-AKT were slightly higher than the control cells; however, OXT and TGFβ1 failed to cause any increase in p-AKT levels in these cells (Fig. 4A, 4B). On the other hand, the induction of PI3-kinase activity and increased p-AKT levels induced by EGF were not affected by the absence of Giα2 protein in PC3 cells (Fig. 4A, 4B). In spite of divergent effects of different ligands on activation of PI3-kinase pathway, all ligands including EGF failed to induce cell migration in the absence of endogenous Giα2 protein (Fig. 4C). It should be noted that the decrease in cell migration in Giα2 deficient cells in response to EGF is not due to decreased activation of EGFR because EGF-increased levels of p-EGFR were similar in cells transfected with control siRNA or Giα2 siRNA (Fig. 4A, lower panel). Together, these results suggest that essential effects of Giα2 in EGF induced cell migration are exerted downstream of activation of EGFR and PI3-kinase pathway.
Fig. 4. Knockdown of endogenous Giα2 has differential effects on PI3-kinase activation and cell migration in response to diverse ligands.
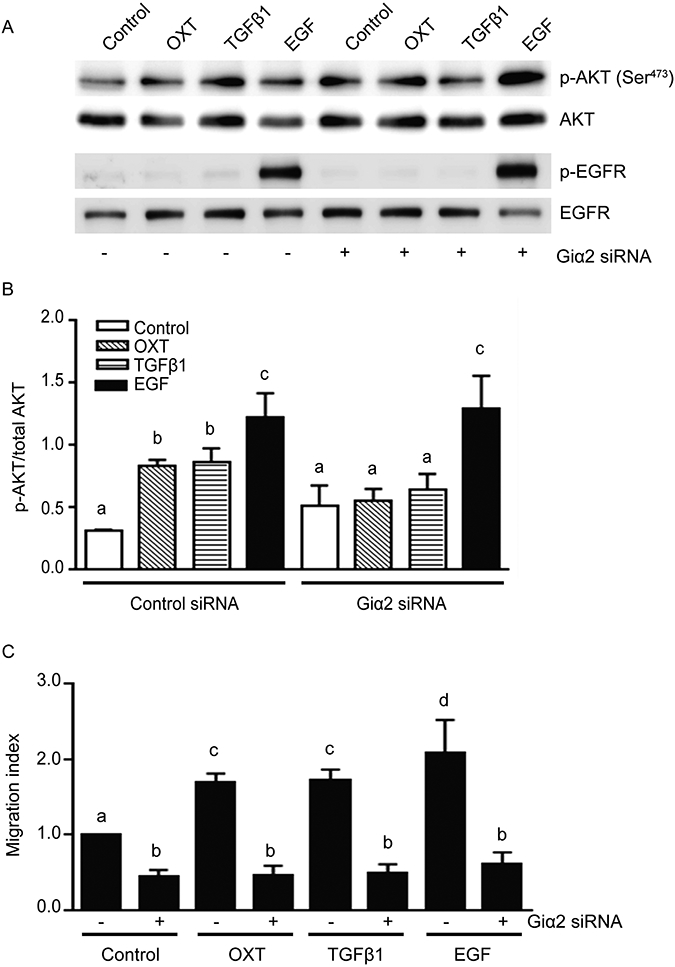
A. Western blot analysis of p-AKT and p-EGFR in PC3 cells lysates, transfected with control siRNA or Giα2 siRNA in response to OXT (100 nmol/L), TGFβ1 (5 ng/ml) or EGF (3 ng/ml). Total AKT and EGFR served as loading and normalization controls. B. Quantitative analysis of p-AKT activation in PC3 cells. Data is presented as mean ± SEM (n=3) and analyzed by ANOVA and Duncan’s modified range tests. Significant differences between groups in a given category (P<0.05) are designated with different lowercase letters. C. Cell migration in PC3 cells after transfection with control siRNA or Giα2 siRNA transfected cells in response to OXT (100 nmol/L), TGFβ1 (5 ng/ml) or EGF (10 ng/ml). Results are expressed as migration index. Each bar represents mean ± SEM (n=3). Significant differences (P<0.05) among different groups are represented with different lowercase letters.
Giα2 requirement for EGF-induced cell migration is independent and down-stream of Rac1 activation
Rac1 is a Rho GTPase family member which when activated induces the formation of actin-rich lamellipodia at the leading edge of motile cells (Spiering and Hodgson, 2011; Vega and Ridley, 2008). Therefore, we determined Rac1 activity in PC3 and DU145 cell lysates after knockdown of endogenous Giα2 and treatment with EGF. As shown in Fig. 5A, the basal activity of Rac1 was not decreased after knockdown of endogenous Giα2 in PC3 and DU145 cells and the EGF induced activation of Rac1 was slightly decreased in both cell types which was not statistically significant.
Fig. 5. Giα2 acts independent of Rac1 activation in EGF-stimulated cell migration and it is required for actin polymerization and lamellipodia formation.
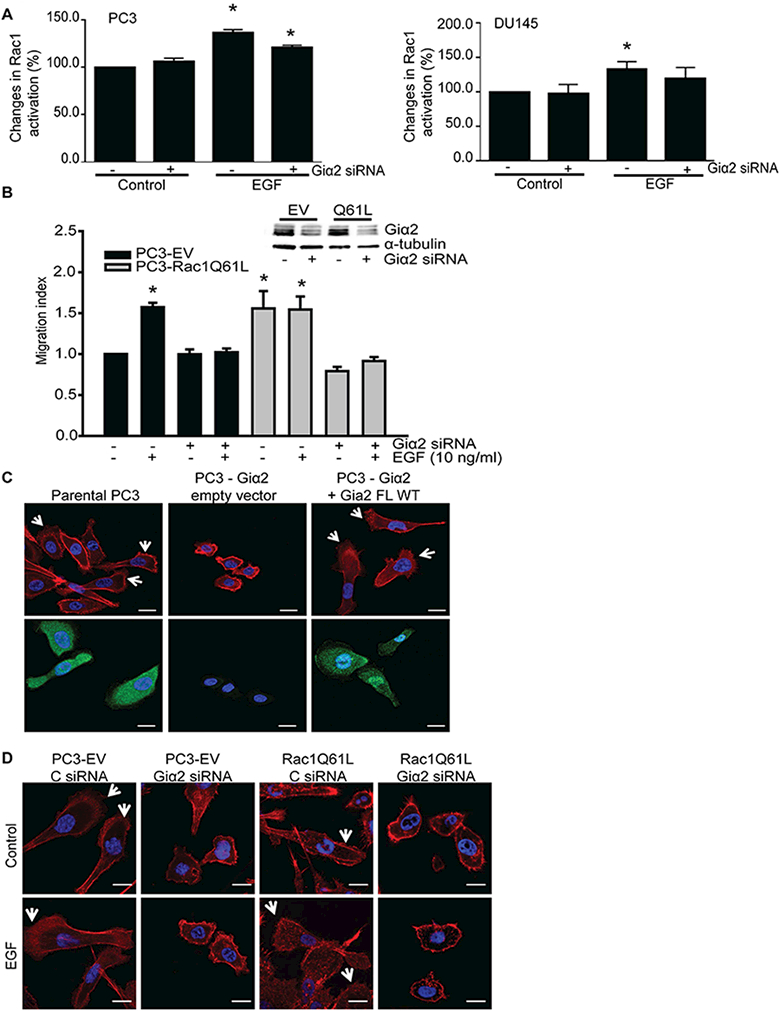
A. Activation of Rac1, after knockdown of endogenous Giα2. PC3 and DU145 cells were transfected with control siRNA or Giα2 siRNA for 48 hours; cells were serum starved overnight and stimulated with EGF (100 ng/ml) for 3 minutes and Rac1 activity was then measured with G-LISA kit. Data were expressed as mean ± SEM (n=3) and analyzed by ANOVA and Duncan’s modified range tests. * Significantly different (P<0.05) compared to controls. B. Upper panel shows Western blot analysis of Giα2 in cell lysates, to validate the knockdown of the protein. Cell migration in parental PC3-EV and PC3-Rac1Q61L cells was performed after knockdown of endogenous Giα2, in response to EGF (10 ng/ml). Results are expressed as migration index. Each bar represents mean ± SEM (n=3). * Significantly different (P<0.05) compared to controls. C. F-actin staining (red color, upper panel) and immunofluorescence (green color, lower panel) of Giα2, in parental PC3, PC3-Giα2 and PC3-Giα2+Giα2 FL WT cells. The arrows indicate the lamellipodia structures present in normal PC3 and in the restored PC3-Giα2+Giα2 WT. Cells were visualized with 40× objective. The scale bars represent 20 μm. D. PC3-EV cells and PC3-Rac1Q61L cells were transfected with control siRNA and Giα2 siRNA and incubated without (control) or with EGF (10 ng/ml) for 30 minutes. Cells were stained for F-actin (red) and DAPI (blue). Arrows indicate lamellipodia at cell edges. Cells were visualized with 40× objective. The scale = 20 μm.
Next, we overexpressed constitutively active Rac1 (Rac1Q61L) in PC3 cells and determined the role of Giα2 in cell migration in these cells. As shown in Fig. 5B, overexpression of Rac1Q61L in PC3 cells led to significant increase in cell migration which was not further increased in the presence of EGF compared to cell transfected with empty vectors (PC3-EV). Knockdown of endogenous Giα2 by specific siRNAs resulted in attenuation of basal and EGF stimulated cell migration in both control and Rac1Q61L overexpressing PC3 cells. These results suggest that EGF and constitutively active Rac1 act on same migratory pathway that is required for the function of Giα2.
Giα2 is required for lamellipodia formation in prostate cancer cells
Cell migration involves Rac-dependent formation of lamellipodia at the leading edge of migrating cells, which is a key structure for cancer cell migration and invasion (Yamaguchi and Condeelis, 2007). We determined the role of Giα2 in lamellipodia formation after knockout of endogenous Giα2 in PC3 cells. Absence of endogenous Giα2 resulted in a significant decrease in cell size and lack of lamellipodia formation in PC3 cells (Fig. 5C). These effects were reversed after re-expression of Giα2 in PC3-Giα2 cells (Fig. 5C). Furthermore, transfection of Giα2 siRNA in parental PC3 and PC3-Rac1Q61L cell lines resulted in decrease in cell size and formation of lamellipodia extensions compared with control siRNA transfected cells (Fig. 5D). These effects of knockdown of Giα2 on lamellipodia formation were similar in control and EGF treated cells (Fig. 5D).
Discussion
The novel findings reported in this study show that Giα2 is essential for cell migration and invasion in prostate cancer cells in response to diverse extracellular stimuli acting via both GPCRs and protein tyrosine kinase receptors. We also show that Giα2 acts at two distinct levels to exert its effects on cell migration. First, its activation through specific GPCRs is required for induction of migration and invasion in response to extracellular stimuli such as chemokines, TGFβ1 and OXT. This effect is PTX sensitive and is upstream of the activation of PI3-kinase pathway and Rac1. However, this activation of Giα2 via GPCR is not required for cell migration induced by ligands such as EGF acting via protein tyrosine kinase receptors. Second, Giα2 protein is required for accumulation of F-actin and formation of lamellipodia at the leading edge of migrating cells. This novel effect of Giα2 is PTX insensitive, does not require activation by GPCRs, and is independent of PI3-kinase and Rac1 activation. In addition, this effect of Giα2 is required for migratory behavior induced by all stimuli including EGF.
The essential role of Giα2 in migration and invasive behavior of prostate cancer cells in response to a diverse group of ligands was evident after both selective knockdown by specific siRNAs and by complete knockout of Giα2 expression using CRISPR/Cas9 technology. These effects are specific to Giα2 since re-expression of this protein restored migratory behavior in Giα2-null PC3 cells. A wide variety of ligands acting through distinct membrane receptors have been implicated in the induction of migratory and invasive behavior in prostate and other cancer cells (Roussos et al., 2011). These effects are critical for the development of metastatic disease which poses significant problems for treatment of these cancers and is the major cause of mortality associated with these cancers. Majority of ligands such as chemokines (such as CXCR12 or SDF-1α), OXT and PGE2 exert their effects on cell motility and invasion through the activation of Giα proteins via their specific GPCRs (Roussos et al., 2011; Vo et al., 2013; Zhong et al., 2010). The role of Giα proteins is supported by the fact that the effects of these ligands are attenuated by pretreatment of the cells with PTX which inhibits the activation of Go/Giα proteins (Roussos et al., 2011; Vo et al., 2013; Zhong et al., 2010). Previous studies from our laboratory have shown that OXT induces migratory behavior in PC3 prostate cancer cell line and these effects were mediated via a GPCR which is sensitive to PTX inhibition (Zhong et al., 2010). Further studies showed that these effects of OXT were exerted specifically through the activation of Giα2 protein since a) the over-expression of PTX-insensitive Giα2 restored OXT effects on cell migration in cells pretreated with PTX, b) specific knockdown of Giα2 by siRNA resulted in attenuation of effects of OXT in the presence of other Giα family members (Giα1 and Giα3), and c) the effects of OXT were restored by expression of siRNA-insensitive Giα2 in these cells (Zhong et al., 2010; Zhong et al., 2012). The present results confirm our previous studies with OXT and extend the essential role of this protein in cell migration and invasion in response to SDF-1α and TGFβ1. We have previously shown that TGFβ1 induces migratory and invasive behavior in prostate cancer (PC3) cells and these effects are mediated by PGE2 activation of its EP4 receptors (Vo et al., 2013). Human EP4 receptor has previously been shown to couple to Gαi and activate PI3-kinase/AKT pathway (Fujino and Regan, 2006; Yoshida et al., 2013). Our current findings show that Giα2 is essential for induction of migration in prostate cancer cells by TGFβ1 presumably via its activation of PGE2 receptor (Vo et al., 2013). This notion is supported by the inhibition of TGFβ1 effects on cell migration by PTX pretreatment or by selective knockdown of Giα2 protein.
Several growth factors such as EGF, VEGF and IGF-1 have also been shown to induce cell migration in many different types of cancer cells including prostate cancer cells (Roussos et al., 2011; Zhong et al., 2012). These growth factors act through specific cell membrane receptor tyrosine-kinases and do not involve activation of GPCRs to exert their effects on cell migration. In our previous studies, we have shown that EGF induces migratory behavior in prostate cancer cells (PC3 and DU145) and these effects of EGF are not inhibited by pretreatment of cells by PTX (Zhong et al., 2010; Zhong et al., 2012) indicating that activation of Giα2 by GPCR is not required for the effects of EGF on cell migration. The present study confirmed these results. However, we made an interesting finding in the previous study on the role of Giα2 in cell migration in prostate cancer cells; PTX pretreatment inhibited effects of OXT but had no effects on EGF induced cell migration. On the other hand, specific knockdown of Giα2 by siRNA resulted in attenuation of both OXT and EGF induced cell migration (Zhong et al., 2012). Our present findings using specific knockdown or silencing of Giα2 expression confirm an essential role of this protein in the effects of EGF on cell migration and invasion. Indeed, absence of Giα2 attenuated FBS effects on cell invasion suggesting its essential role in these processes irrespective of the ligands. Inhibition of cell migration induced by all ligands in RGS2 over-expressing cells suggests that the effects of Giα2 are exerted by its activated GTP-bound form.
It is well established that activation of PI3-kinase/AKT signaling is required for chemotaxis and cell motility in a wide variety of normal and cancer cells (Van Haastert and Devreotes, 2004). We have previously shown that TGFβ1, PGE2 and EGF on cell migration in prostate cancer cells require the activation of PI3-kinase/AKT pathway (Vo et al., 2013). The activation of PI3-kinase pathways by GPCR is assumed to be primarily dependent on activated Gβγ which is dissociated from active Gα subunit after the activation of GPCR (Van Haastert and Devreotes, 2004). In accordance with this notion, Gβγ has been shown to be required for induction of migration in breast cancer cells (Kirui et al., 2010).
In attempts to understand the interaction between the PI3-kinase pathway and the role of Giα2 protein during the induction of cell migration by various ligands, we determined the levels of p-AKT under various experimental conditions. Our data indicated that PTX pretreatment resulted in a slight but significant increase in p-AKT in PC3 cells but it attenuated OXT and TGFβ1 induced p-AKT levels and cell migration. Similar lack of increase in p-AKT levels in response to OXT, TGFβ1 were observed in PC3 cell after selective knockdown of endogenous Giα2 protein. These data suggest that Giα2 activation via GPCR is required for OXT and TGFβ1 induced activation of PI3-kinase pathway and cell migration. Our data also showed that OXT and TGFβ1 stimulation of PI3-kinase activity is dependent on the activity of Gβγ subunit; however, the critical need for Giα2 for this activation indicates that Gβγ subunit is generated by the activation of GPCR coupled with Giα2 and Gβγ subunits. Interestingly, activation of Giα2 or Gβγ by GPCRs was not required for EGF induced activation of PI3-kinase pathway and induction of cell migration since PTX pretreatment or inhibition of Gβγ did not affect EGF-induced p-AKT and cell migration (Kue and Daaka, 2000). On the other hand, knockdown of Giα2 resulted in inhibition of EGF-induced cell migration and invasion without affecting its induction of PI3-kinase activation. These results indicate that EGF effects on activation of PI3-kinase pathway is not dependent on the presence or activation of Giα2 protein and the critical effects of Giα2 in EGF-induced cell migration and invasion must be exerted down-stream of activation of PI3-kinase pathway.
Rac1, a member of Rho family of GTPases, plays a central role in cell motility and its activation is required for actin polymerization and branching and formation of lamellipodia at the leading edge of the migrating cells (Raftopoulou and Hall, 2004; Rathinam et al., 2011). Our results show that the essential role of Giα2 downstream of activation of PI3-kinase in EGF treated cells does not involve activation of Rac1. First, knockdown of Giα2 did not cause any reduction in basal Rac1 activity in both PC3 and DU145 cells and had only marginal effects on EGF-induced increase in Rac1 activity. Second, overexpression of constitutively active Rac1 in PC3 resulted in significantly increased cell migration; knockdown of endogenous Giα2 completely blocked basal and EGF induced cell migration in these cells. These results suggest that Giα2 is involved in the regulation of cell motility and invasion at a step which is independent or downstream of Rac1 activation.
The knockdown of endogenous Giα2 by siRNAs or silencing of expression of Giα2 by CRISPR/Cas9 technology was associated with distinct morphological changes in the PC3 cells, such as reduction in the cell size, reduced actin polymerization and reduction in lamellipodia formation in both stimulated and non-stimulated cells. Similar morphological effects were observed after knockdown of Giα2 in cell overexpressing constitutively active Rac1. These results suggest a fundamental role of Giα2 in actin polymerization and lamellipodia formation in prostate cancer cells which is independent of Rac1 activity. Previous studies in different cell types have indicated a similar role of Giα family members (Giα2 and Giα3) in cell migration (Li et al., 2013) and especially in the leading edge of migrating cells (Ghosh et al., 2008; Ward et al., 2015). Recent studies have also reported specific interactions of Giα subunit with proteins involved in chemotaxis such as GIV (Ghosh et al., 2008), ELMO1/Dock180 (Li et al., 2013), AGS3/mInsc (Kamakura et al., 2013), Src and β-pix (Ward et al., 2015).
Giα2 is expressed ubiquitously and represents the quantitatively predominant Giα isoform (Gohla et al., 2007). Our previous study determined the relative expression of three Giα isoforms (Giα1, Giα2 and Giα3) in PC3 cells and found a very high expression of Giα2 in these cells (Zhong et al., 2010). Immunohistochemical localization studies in prostate cancer patient tissue microarrays showed higher expression of Giα2 in tissue samples from advanced and metastatic stages of prostate cancers than those from normal tissue and early stage cancer tissues (Caggia et al. unpublished data). This data indicate an increase in the levels of this protein in accordance of its role in more invasive and metastatic prostate cancers.
On the basis of the results reported in the current study we conclude that Giα2 acts at two different levels which are both dependent and independent of GPCR signaling to induce migration in prostate cancer cells. These results also indicate that reduction in the synthesis or activation of this protein may provide a therapeutic target for advanced metastatic stages of prostate cancer. Inhibition of Giα2 at the level of its interaction with other proteins involved in lamellipodia formation at the leading edge of the migrating cells may provide specific therapeutic target without interfering with its activation by GPCR and other biological effects (Fig. 6).
Fig. 6. Hypothetical mechanism of action of Giα2 protein during migration and lamellipodia formation in prostate cancer cells.
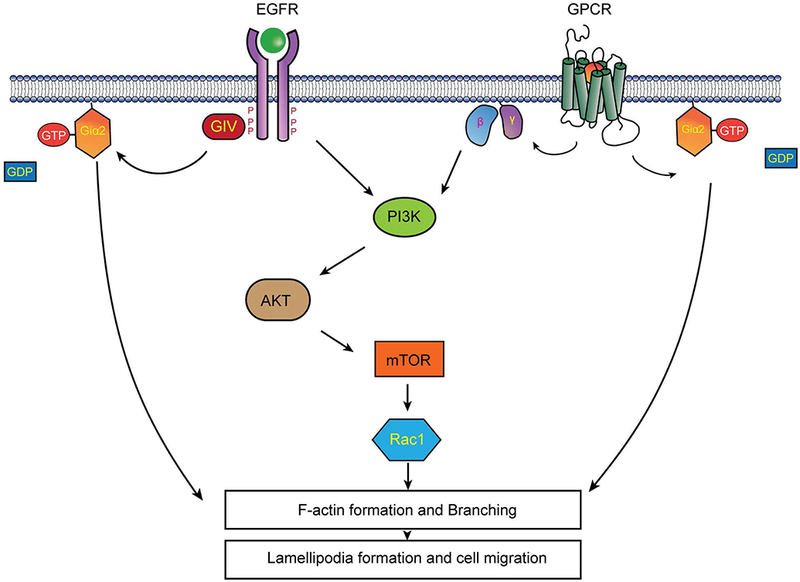
Giα2 protein acts at two different levels which are both dependent and independent of GPCR signaling to induce cell migration, invasion and lamellipodia formation in prostate cancer cells. Its effects on lamellipodia formation are exerted downstream of PI3-kinase/AKT/Rac1 axis.
Acknowledgments
This work was supported by NIH (NIMHD/RCMI G12MD007590 and NIMHD/P20MD002285) and Georgia Research Alliance. The authors wish to thank Dr. Peri Nagappan for his assistance in immunostaining, Dr. Jin Zou for his help in cell sorting and Ms. Ma Vittoria Andrea Ordonio for her technical support in Bioinformatics analysis.
References
- Berx G, Raspe E, Christofori G, Thiery JP, Sleeman JP. 2007. Pre-EMTing metastasis? Recapitulation of morphogenetic processes in cancer. Clin Exp Metastasis 24(8):587–597. [DOI] [PubMed] [Google Scholar]
- Britschgi A, Andraos R, Brinkhaus H, Klebba I, Romanet V, Muller U, Murakami M, Radimerski T, Bentires-Alj M. 2012. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell 22(6):796–811. [DOI] [PubMed] [Google Scholar]
- Cao X, Qin J, Xie Y, Khan O, Dowd F, Scofield M, Lin MF, Tu Y. 2006. Regulator of G-protein signaling 2 (RGS2) inhibits androgen-independent activation of androgen receptor in prostate cancer cells. Oncogene 25(26):3719–3734. [DOI] [PubMed] [Google Scholar]
- Chen X, Cheng H, Pan T, Liu Y, Su Y, Ren C, Huang D, Zha X, Liang C. 2014. mTOR regulate EMT through RhoA and Rac1 pathway in prostate cancer. Mol Carcinog. [DOI] [PubMed] [Google Scholar]
- DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, Alteri R, Robbins AS, Jemal A. 2014. Cancer treatment and survivorship statistics, 2014. CA: a cancer journal for clinicians 64(4):252–271. [DOI] [PubMed] [Google Scholar]
- Elliott B, Zackery DL, Eaton VA, Jones RT, Abebe F, Ragin CC, Khan SA. 2018. Ethnic Differences in TGFbeta Signaling Pathway May Contribute to Prostate Cancer Health Disparity. Carcinogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festuccia C, Angelucci A, Gravina GL, Biordi L, Millimaggi D, Muzi P, Vicentini C, Bologna M. 2005. Epidermal growth factor modulates prostate cancer cell invasiveness regulating urokinase-type plasminogen activator activity. EGF-receptor inhibition may prevent tumor cell dissemination. Thromb Haemost 93(5):964–975. [DOI] [PubMed] [Google Scholar]
- Fortin Ensign SP, Mathews IT, Symons MH, Berens ME, Tran NL. 2013. Implications of Rho GTPase Signaling in Glioma Cell Invasion and Tumor Progression. Front Oncol 3:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francipane MG, Lagasse E. 2014. mTOR pathway in colorectal cancer: an update. Oncotarget 5(1):49–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman DA, Rommel C. 2014. PI3K and Cancer: Lessons, Challenges and Opportunities. Nat Rev Drug Discov 13(2):140–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino H, Regan JW. 2006. EP(4) prostanoid receptor coupling to a pertussis toxin-sensitive inhibitory G protein. Mol Pharmacol 69(1):5–10. [DOI] [PubMed] [Google Scholar]
- Ghosh P, Garcia-Marcos M, Bornheimer SJ, Farquhar MG. 2008. Activation of Galphai3 triggers cell migration via regulation of GIV. J Cell Biol 182(2):381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Basu M, Roy SS. 2012. ETS-1 protein regulates vascular endothelial growth factor-induced matrix metalloproteinase-9 and matrix metalloproteinase-13 expression in human ovarian carcinoma cell line SKOV-3. J Biol Chem 287(18):15001–15015. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gohla A, Klement K, Piekorz RP, Pexa K, vom Dahl S, Spicher K, Dreval V, Haussinger D, Birnbaumer L, Nurnberg B. 2007. An obligatory requirement for the heterotrimeric G protein Gi3 in the antiautophagic action of insulin in the liver. Proc Natl Acad Sci U S A 104(8):3003–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamakura S, Nomura M, Hayase J, Iwakiri Y, Nishikimi A, Takayanagi R, Fukui Y, Sumimoto H. 2013. The cell polarity protein mInsc regulates neutrophil chemotaxis via a noncanonical G protein signaling pathway. Dev Cell 26(3):292–302. [DOI] [PubMed] [Google Scholar]
- Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J. 1997. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J 16(10):2783–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirui JK, Xie Y, Wolff DW, Jiang H, Abel PW, Tu Y. 2010. Gbetagamma signaling promotes breast cancer cell migration and invasion. J Pharmacol Exp Ther 333(2):393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koochekpour S, Maresh GA, Katner A, Parker-Johnson K, Lee TJ, Hebert FE, Kao YS, Skinner J, Rayford W. 2004. Establishment and characterization of a primary androgen-responsive African-American prostate cancer cell line, E006AA. Prostate 60(2):141–152. [DOI] [PubMed] [Google Scholar]
- Kue PF, Daaka Y. 2000. Essential role for G proteins in prostate cancer cell growth and signaling. J Urol 164(6):2162–2167. [PubMed] [Google Scholar]
- Li H, Yang L, Fu H, Yan J, Wang Y, Guo H, Hao X, Xu X, Jin T, Zhang N. 2013. Association between Galphai2 and ELMO1/Dock180 connects chemokine signalling with Rac activation and metastasis. Nat Commun 4:1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Yue J, Wang J, Zhang L, Fan R, Zhang H, Zhang Q. 2015. GPCR48/LGR4 promotes tumorigenesis of prostate cancer via PI3K/Akt signaling pathway. Med Oncol 32(3):49. [DOI] [PubMed] [Google Scholar]
- Luo J, Manning BD, Cantley LC. 2003. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 4(4):257–262. [DOI] [PubMed] [Google Scholar]
- Luo W, Tan P, Rodriguez M, He L, Tan K, Zeng L, Siwko S, Liu M. 2017. Leucine-rich repeat-containing G protein-coupled receptor 4 (Lgr4) is necessary for prostate cancer metastasis via epithelial-mesenchymal transition. The Journal of biological chemistry 292(37):15525–15537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PJ, Neisen J, Messenger J, Waugh DJ. 2014. Tumor-derived CXCL8 signaling augments stroma-derived CCL2-promoted proliferation and CXCL12-mediated invasion of PTEN-deficient prostate cancer cells. Oncotarget 5(13):4895–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. 2006. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66(3):1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A. 2004. Cell migration: Rho GTPases lead the way. Dev Biol 265(1):23–32. [DOI] [PubMed] [Google Scholar]
- Rathinam R, Berrier A, Alahari SK. 2011. Role of Rho GTPases and their regulators in cancer progression. Front Biosci (Landmark Ed) 16:2561–2571. [DOI] [PubMed] [Google Scholar]
- Roussos ET, Condeelis JS, Patsialou A. 2011. Chemotaxis in cancer. Nat Rev Cancer 11(8):573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rycaj K, Tang DG. 2017. Molecular determinants of prostate cancer metastasis. Oncotarget 8(50):88211–88231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar N, Castellan M, Shirodkar SS, Lokeshwar BL. 2013. Chemokines and chemokine receptors as promoters of prostate cancer growth and progression. Crit Rev Eukaryot Gene Expr 23(1):77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlett KA, White EZ, Coke CJ, Carter JR, Bryant LK, Hinton CV. 2018. Agonist-induced CXCR4 and CB2 Heterodimerization Inhibits Galpha13/RhoA-mediated Migration. Molecular cancer research : MCR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibue T, Weinberg RA. 2011. Metastatic colonization: settlement, adaptation and propagation of tumor cells in a foreign tissue environment. Semin Cancer Biol 21(2):99–106. [DOI] [PubMed] [Google Scholar]
- Spiering D, Hodgson L. 2011. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh Migr 5(2):170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan S, Wang F, Glavas S, Ott A, Hofmann F, Aktories K, Kalman D, Bourne HR. 2003. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J Cell Biol 160(3):375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subauste MC VH M, Benard V, Chamberlain CE, Chuang TH, Chu K, Bokoch GM, Hahn KM. . 2000. Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas. J Biol Chem 275(13):9725–9733. [DOI] [PubMed] [Google Scholar]
- Sweeney P, Karashima T, Kim SJ, Kedar D, Mian B, Huang S, Baker C, Fan Z, Hicklin DJ, Pettaway CA, Dinney CP. 2002. Anti-vascular endothelial growth factor receptor 2 antibody reduces tumorigenicity and metastasis in orthotopic prostate cancer xenografts via induction of endothelial cell apoptosis and reduction of endothelial cell matrix metalloproteinase type 9 production. Clin Cancer Res 8(8):2714–2724. [PubMed] [Google Scholar]
- Van Haastert PJ, Devreotes PN. 2004. Chemotaxis: signalling the way forward. Nat Rev Mol Cell Biol 5(8):626–634. [DOI] [PubMed] [Google Scholar]
- Vanharanta S, Massague J. 2013. Origins of metastatic traits. Cancer Cell 24(4):410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega FM, Ridley AJ. 2008. Rho GTPases in cancer cell biology. FEBS Lett 582(14):2093–2101. [DOI] [PubMed] [Google Scholar]
- Vo BT, Khan SA. 2011. Expression of nodal and nodal receptors in prostate stem cells and prostate cancer cells: autocrine effects on cell proliferation and migration. Prostate 71(10):1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo BT, Li C, Morgan MA, Theurillat I, Finkelstein D, Wright S, Hyle J, Smith SMC, Fan Y, Wang YD, Wu G, Orr BA, Northcott PA, Shilatifard A, Sherr CJ, Roussel MF. 2017. Inactivation of Ezh2 Upregulates Gfi1 and Drives Aggressive Myc-Driven Group 3 Medulloblastoma. Cell Rep 18(12):2907–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo BT, Morton D Jr., Komaragiri S, Millena AC, Leath C, Khan SA. 2013. TGF-beta effects on prostate cancer cell migration and invasion are mediated by PGE2 through activation of PI3K/AKT/mTOR pathway. Endocrinology 154(5):1768–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L, Millena AC, Strong N, Khan SA. 2013. Expression of TGFbeta3 and its effects on migratory and invasive behavior of prostate cancer cells: involvement of PI3-kinase/AKT signaling pathway. Clin Exp Metastasis 30(1):13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JD, Dhanasekaran DN. 2012. LPA Stimulates the Phosphorylation of p130Cas via Galphai2 in Ovarian Cancer Cells. Genes Cancer 3(9–10):578–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JD, Ha JH, Jayaraman M, Dhanasekaran DN. 2015. LPA-mediated migration of ovarian cancer cells involves translocalization of Galphai2 to invadopodia and association with Src and beta-pix. Cancer Lett 356(2 Pt B):382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff DW, Xie Y, Deng C, Gatalica Z, Yang M, Wang B, Wang J, Lin MF, Abel PW, Tu Y. 2012. Epigenetic repression of regulator of G-protein signaling 2 promotes androgen-independent prostate cancer cell growth. Int J Cancer 130(7):1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Abel PW, Kirui JK, Deng C, Sharma P, Wolff DW, Toews ML, Tu Y. 2013. Identification of upregulated phosphoinositide 3-kinase gamma as a target to suppress breast cancer cell migration and invasion. Biochem Pharmacol 85(10):1454–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue G, Hemmings BA. 2013. PKB/Akt-dependent regulation of cell motility. J Natl Cancer Inst 105(6):393–404. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Condeelis J. 2007. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta 1773(5):642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Fujino H, Otake S, Seira N, Regan JW, Murayama T. 2013. Induction of cyclooxygenase-2 expression by prostaglandin E2 stimulation of the prostanoid EP4 receptor via coupling to Galphai and transactivation of the epidermal growth factor receptor in HCA-7 human colon cancer cells. Eur J Pharmacol 718(1–3):408–417. [DOI] [PubMed] [Google Scholar]
- Zhong M, Boseman ML, Millena AC, Khan SA. 2010. Oxytocin induces the migration of prostate cancer cells: involvement of the Gi-coupled signaling pathway. Mol Cancer Res 8(8):1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Clarke S, Vo BT, Khan SA. 2012. The essential role of Gialpha2 in prostate cancer cell migration. Mol Cancer Res 10(10):1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]