

Abstract
Background:
Injury to the blood brain barrier exposes endothelium rich in von Willebrand factor (vWF) which may play a role in altered platelet aggregation following traumatic brain injury (TBI). Ristocetin is an antimicrobial substance which induces vWF-mediated aggregation of platelets. We examined these mechanisms in injured patients by measuring the aggregation response of platelets to stimulating agonists (including ristocetin) via whole blood multiple electrode platelet aggregometry. We hypothesized that patients with TBI have an altered platelet aggregation response to ristocetin stimulation compared to patients without TBI.
Methods:
Blood was collected from 233 trauma patients without thrombocytopenia. Platelet aggregation was assessed using multiple electrode platelet aggregometry (Multiplate®). Platelet aggregation response to stimulating agonists collagen, thrombin receptor-activating peptide-6, adenosine diphosphate, arachidonic acid, and ristocetin were measured. Factor activity was measured.
Results:
Of the 233 patients, 23% had TBI. There were no differences in platelet aggregation responses to any agonists between TBI and non-TBI patients except ristocetin. Platelet aggregation response to ristocetin stimulation was significantly lower in TBI patients (p=0.03). TBI patients also had higher factor VIII activity (215% vs. 156%, p=0.01). In multivariate analysis, there was a significant independent association of impaired platelet aggregation response to ristocetin stimulation with TBI (OR 3.05, p=0.04).
Conclusions:
Given the importance of platelets in hemostasis, understanding the mechanisms of impaired platelet aggregation following injury is critical. The impaired platelet aggregation response to ristocetin stimulation and corresponding increase in factor VIII activity in TBI patients may be secondary to a TBI-induced effect on vWF quantity (due to injury driven consumption of vWF) or vWF function with resultant increase in circulating factor VIII activity (due to impaired carrying capacity of vWF). Given there are multiple known therapies for vWF deficits including desmopressin, purified and recombinant vWF, and estrogens, these lines of investigation are particularly compelling in patients with TBI and hemorrhage.
Keywords: ristocetin, von Willebrand factor, platelet, trauma
BACKGROUND
Trauma is a leading killer worldwide with five million deaths a year (1–3). Hemorrhage accounts for 60% of deaths in patients with potentially salvageable injuries (3–5). One-third of these hemorrhaging patients suffer from trauma-induced coagulopathy, a multi-factorial disorder of inflammation and coagulation characterized by impaired clot formation from a distinct response to tissue injury and shock (6, 7). More recently, investigations have begun to uncover the importance of the role of both platelets and endothelium in hemorrhage, organ failure, and mortality after injury (8–15).
Platelets have a pivotal role in hemostasis via two key pathways: facilitation of pro-coagulant events and regulation of endothelial integrity (16). However, the role of platelets in trauma-induced coagulopathy is not well understood (13, 17–22). This hinders our ability to understand and appropriately provide platelet-specific targeted treatments to hemorrhaging trauma patients. It is known that post-injury thrombocytopenia is related to bleeding, progression of brain injury, and mortality (20, 23), but accumulating evidence suggests that impaired platelet aggregation (independent of platelet count) following injury is common and associated with worse outcomes (13, 17–22). In fact, nearly half of all injured patients demonstrate evidence of impaired platelet aggregation despite having normal platelet counts (13). Identifying the biology of this impaired platelet aggregation is critical given the association with worse outcomes and higher mortality in these patients (13, 14, 24–26). Post-injury platelet aggregation has been primarily quantified using multiple electrode platelet aggregometery, which assays the aggregation response of platelets to various stimulating agonists via measurement of increasing electrical impedance in a sample of whole blood over a 6-minute time period.
Injuries characterized by significant tissue and endothelial damage have the strongest associations with impaired platelet aggregation. The most severely impaired platelet aggregation has been identified in patients with traumatic brain injury (TBI), high injury severity scores (ISS), presence of shock, and those who ultimately do not survive; these patients have increased platelet activation but impaired platelet aggregation (13, 14, 27, 28). Together, these findings suggest that damage to endothelium, such as the blood-brain barrier which is known to be rich in von Willebrand factor (vWF) (29), may lead to excessive platelet activation but impaired aggregation (14, 27, 28, 30).
Ristocetin is an antimicrobial substance which forms complexes with vWF causing conformational change in vWF and inducing the aggregation of platelets (Figure 1, B1). This relies on the binding of vWF to the platelet GPIb receptor. Impaired platelet aggregation response to ristocetin stimulation is therefore consistent with either an absence of GPIb receptors (Figure 1, B2) or with a decrease in the amount or function of circulating vWF (Figure 1, B3/B4). In addition, it has been demonstrated that a decrease in vWF, the carrier protein of factor VIII, results in a change in the circulating amount of factor VIII (31) (Figure 1, B5). Given these relationships, we examined these mechanisms as they relate to TBI by performing multiple electrode platelet aggregometry, hypothesizing that patients with TBI have an altered platelet aggregation response to ristocetin stimulation compared to patients without TBI consistent with disruption of the vWF rich blood-brain barrier.
Figure 1. Normal and Impaired Platelet Aggregation Responses to Ristocetin Stimulation Measured by Multiple Electrode Platelet Aggregometry.
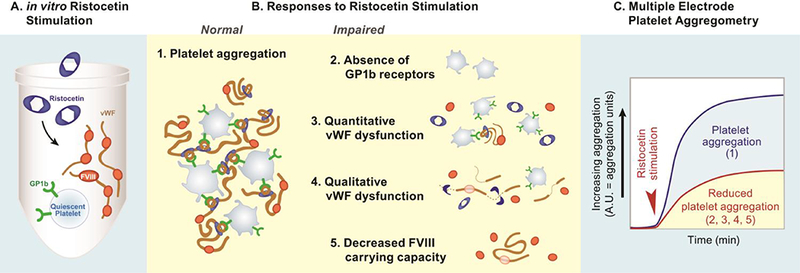
A. in vitro Ristocetin Stimulation
B. Responses to Ristocetin Stimulation
Normal
B1. Platelet aggregation. This illustration represents a normal response to ristocetin stimulation. Ristocetin binds to vWF resulting in a conformational change to von Willebrand factor (vWF). The ristocetin-vWF complex then binds to platelet GP1b receptors leading to ristocetin induced aggregation.
Impaired
B2. Absence of GP1b receptors. This illustration represents an absence of GP1b receptors on platelets (Bernard-Soulier syndrome). Due to the absence of GP1b receptors, the ristocetin-vWF complex is unable to bind to platelets to induce aggregation. This disease state is rare and unlikely to be an explanation of the findings.
B3. Quantitative vWF dysfunction. This illustration represents a quantitative dysfunction of vWF as an explanation of impaired platelet aggregation response to ristocetin stimulation. It is plausible that injury causes the majority of the circulating vWF to be consumed in the process of binding to platelets to form clot. This may lead to a lack of circulating vWF left to complex with the ristocetin and therefore a downstream decrease in platelet aggregation via Gp1b receptors.
B4. Qualitative vWF dysfunction. This illustration represents a qualitative dysfunction of vWF as an explanation of impaired platelet aggregation response to ristocetin stimulation. It is plausible that a TBI-induced injurious effect on vWF function leads to an inability of ristocetin to bind and cause conformational change to vWF and therefore a downstream impairment in platelet aggregation via Gp1b receptors.
B5. Decreased FVIII carrying capacity. This illustration represents an explanation of the downstream FVIII effects of both quantitative (B3) and qualitative vWF dysfunction (B4). It is plausible that the elevated factor VIII activity in the patients with TBI may be explained by the decreased amount (B3) or function (B4) of circulating vWF due to impaired factor VIII carrying capacity.
C. Multiple Electrode Platelet Aggregometry. Platelet aggregation can be quantified using multiple electrode platelet aggregometery which assays the aggregation response of platelets to various stimulating agonists (ristocetin) via measurement of increasing electrical impedance in a sample of whole blood. Platelet aggregation responses to the various agonists are reported as area under the aggregation curve in units (AUC) over a 6-minute measurement period. Therefore, reduced platelet aggregation is measured as a lower AUC.
METHODS
Blood samples were collected and multiple electrode platelet aggregometry was performed on whole blood from 233 injured trauma patients on arrival to a Level I urban trauma emergency department (ED) as part of a longitudinal study examining perturbations in coagulation and inflammation after trauma (2010–2016). Exclusions included patients who were pediatric, pregnant, in-custody, had burns >20% body surface area, were transferred from another facility, did not require ICU level care, or were atraumatic. Patients with thrombocytopenia (platelet count 150–400 × 109/L), on anti-platelet or anti-coagulant medications were excluded to control for confounding effects on platelet aggregometry. Comprehensive injury, demographic, clinical, and outcome data were collected under a waiver of consent approved by the University of California Institutional Review Board.
TBI was defined by clinical notes and chart review for imaging identified intracranial injury and confirmed with AIS (Abbreviated Injury Score) head score ≥3. Isolated TBI (iTBI) was defined as AIS head score ≥3 and AIS all other categories <3. TBI + Polytrauma (pTBI) was defined as AIS head score ≥3 and any other AIS categories ≥3. Multi-organ failure (MOF) was defined using the Denver Post-Injury Multiple Organ Failure Score (32). Acute respiratory distress syndrome (ARDS) was defined by Berlin criteria, as described previously (33). Coagulopathy was defined as international normalized ratio (INR) of 1.3 or higher. During the study period, there were no explicit changes to transfusion practices or protocols and all blood product transfusions occurred following the blood draw.
Admission samples were collected via initial placement of a 16G or larger peripheral IV as described previously (13, 21, 34) in vacuum-sealed tubes containing 3.2% (0.109M) sodium citrate. Platelet aggregation response to various stimulating agonists was assessed using multiple electrode platelet aggregometry (Multiplate®, Verum Diagnostica GmbH; Munich, Germany) immediately after sample collection (13). Briefly, 0.3mL of whole blood was diluted in warmed normal saline containing 3mM CaCl2 and incubated for 3 minutes at 37°C with continuous stirring in a Multiplate® test cell. Each test cell contains two sets of 3mm silver-coated copper wires, across which electrical resistance is measured at 0.57 second intervals. Platelet aggregation was stimulated by adenosine diphosphate (ADP, final concentration 6.5μM; via P2 receptors), thrombin receptor activating peptide-6 (TRAP, final concentration 32μM; via PAR receptors), arachidonic acid (AA, final concentration 0.5mM; via the cyclooxygenase pathway), collagen (final concentration 3.2μg/mL; via GpIa/IIa and GpVI receptors), and ristocetin (final concentration 0.77 mg/mL; via vWF complex to Gp1b receptors). Platelet adhesion to the electrodes was detected as increasing electrical impedance, measured by duplicate sets of sensor wires in each test cell. Platelet aggregation responses to the various stimulating agonists are reported as area under the aggregation curve in units (AUC) over a 6-minute measurement period. Reference ranges for citrated whole blood were provided by the manufacturer. Samples were immediately centrifuged, and plasma extracted and stored at −80°C. Fibrinogen; factors II, V, VII, VIII, IX, and X were measured with a Stago Compact Coagulation Analyzer (Diagnostica Stago Inc, Parsippany, NJ) in accordance with manufacturer instructions.
Post-hoc retrospective analysis of prospectively collected data was performed. The primary outcomes examined were platelet aggregation responses to stimulating agonists measured by multiple electrode platelet aggregometry in TBI vs. non-TBI cohorts. Data are presented as mean (± standard deviation), median (interquartile range [IQR]), or percentage; univariate comparisons were made using Student’s t test for normally distributed data, Wilcoxon rank sum or Kruskal-Wallis testing for nonparametric data, and Fisher’s exact test for proportions. Multivariate logistic regression was performed to examine the association of impaired platelet aggregation with TBI, controlling for confounding variables including extra-cranial injury severity (AIS in each category), gender, mechanism, and platelet count. An [alpha] < 0.05 was considered significant. Bonferroni corrections was used for multiple comparisons. All analysis was performed using Stata version 14 (StataCorp, College Station, TX).
RESULTS
Overall Cohort
The 233 patients were a relatively standard trauma population with respect to age (median 34, IQR 26–48) and gender (88% male). They were moderately injured with a median ISS of 9 (IQR 12–15), and a mean base deficit of 2.2 ±5.5. Five percent of the cohort was coagulopathic on presentation (INR ≥1.3), 25% required transfusion of products in the first 24 hours, and there was a 7% mortality at discharge (Table 1). The overall cohort had a normal median platelet count of 263 × 109/L (IQR 222–308; Table 1). The percent of patients with impaired platelet aggregation response to any stimulating agonist stimulation was 36% (10% ADP, 4% collagen, 17% TRAP, 18% AA, and 13% ristocetin; Table 2). Despite this, the median AUCs (and IQRs) for each agonist were within the normal manufacturer ranges (Table 2).
TABLE 1.
Patient Demographics/Outcomes
| N=233 | |
|---|---|
| Age (years) | 34 (26–48) |
| Male | 88% |
| BMI (kg/m2) | 27 +/− 5 |
| Blunt mechanism | 47% |
| Injury severity score | 9 (1–18) |
| Admit GCS | 15 (12–15) |
| TBI | 23% |
| Pre-hospital crystalloid volume (mL) | 0 (0–200) |
| Admit temperature (°) | 36.6 +/− 0.7 |
| Admit pH | 7.3 +/− 0.1 |
| Admit base deficit | 2.2 +/− 5.5 |
| Admit INR ≥1.3 | 5% |
| Admit INR | 1.1 +/− 0.1 |
| Admit PTT (sec) | 28 +/− 3.8 |
| Admit platelets (x 109/L) | 263 (222–308) |
| Admit fibrinogen (mg/dL) | 218 (177–279) |
| Transfused in 24 hours | 25% |
| Transfused platelets in 24 hours | 7% |
| Total hospital days | 4 (2–9) |
| Total ICU days (to 28 days) | 1 (0–3) |
| Ventilator free days (to 28 days) | 28 (26–28) |
| ARDS | 5% |
| Multi-organ failure | 5% |
| Mortality at 24h | 2% |
| Mortality at discharge | 7% |
Patient demographics for the 233 patients. Data are mean +/− SD, median (inter-quartile range), or percentage. Data for skewed variables reported as median with inter-quartile ranges. Ventilator free days are counted for the first 28 days of hospitalization. Patients who expired received 0 ventilator free days.
TABLE 2.
Admit Multiple Electrode Platelet Aggregometry
| N=233 | Manufacturer Normal Ranges | |
|---|---|---|
| ADP AUC (U) | 60 (47–71) | 36–101 |
| Low ADP Response (%) | 10% | n/a |
| Collagen AUC (U) | 49 (39–61) | 24–79 |
| Low Collagen Response (%) | 4% | n/a |
| TRAP AUC (U) | 97 (82–112) | 75–137 |
| Low TRAP Response (%) | 17% | n/a |
| AA AUC (U) | 61 (48–71) | 42–100 |
| Low AA Response (%) | 18% | n/a |
| Ristocetin AUC (U) | 66 (42–87) | 27–124 |
| Low Ristocetin Response (%) | 13% | n/a |
| Overall Low Platelet Response (%) | 36% | n/a |
Data reported as median with inter-quartile ranges. Platelet aggregation was induced by agonist stimulation with adenosine diphosphate (ADP, via P2 receptors), collagen (via GpIa/IIa and GpVI receptors), thrombin receptor activating peptide-6 (TRAP, via PAR receptors), arachidonic acid (AA, via the cyclooxygenase pathway), or ristocetin (via vWF complex). Platelet adhesion to the electrodes was detected as increasing electrical impedance, measured by duplicate sets of sensor wires in each test cell. Platelet aggregation responses to multiple electrode platelet aggregometry are reported as area under the aggregation curve in units (U) over a 6-minute measurement period. p-values bolded for <0.05
Isolated TBI (iTBI) vs. TBI+Polytrauma (pTBI) vs. Non-TBI Cohorts
Twenty-three percent (54 patients) of the overall cohort had TBI (Table 1). The majority of those had iTBI (n=36), and the remainder had pTBI (n=18, Table 3a). The TBI groups (iTBI and pTBI) were older, had higher rates of blunt trauma, and were overall more injured with worse outcomes (higher ISS, transfusion rates, ARDS rates, and mortality rates; all p<0.0167 for multiple comparisons, Table 3a). The TBI groups had significantly higher rates of transfusion of all products. Eleven percent of the iTBI and 33% the pTBI patients were transfused platelets in the first 24 hours (compared to 4% in the non-TBI group, p<0.0167 for multiple comparisons, Table 3a). Despite this, there were no significant differences in the platelet aggregation response to ADP, collagen, TRAP, AA, or ristocetin stimulation between iTBI, pTBI, and non-TBI groups (Table 3b). The highest rate of coagulopathy (INR ≥1.3) was in patients with pTBI (24% vs 3%, p<0.05, Table 3b).
TABLE 3a.
Demographics and Outcomes by Isolated TBI (iTBI) vs. TBI + Polytrauma (pTBI) vs. Non-TBI
| Isolated TBI (n=36) |
TBI + Polytrauma (n=18) |
Non-TBI (n=179) |
p- value |
|
|---|---|---|---|---|
| Age (years) | 41 (27–53) | 39 (27–69) | 22 (24–45) | 0.04 |
| Male | 86% | 78% | 89% | 0.28 |
| BMI (kg/m2) | 25 +/− 5 | 26 +/− 4 | 27 +/− 5 | 0.01 |
| Blunt mechanism | 86% | 100% | 34% | <0.01 |
| AIS head | 4 (4–5) | 4 (3–5) | 0 (0–0) | <0.01 |
| Injury severity score | 22 (17–30) | 35 (29–41) | 5 (1–10) | <0.01 |
| Admit GCS | 11 (5–14) | 11 (3–14) | 15 (14–15) | <0.01 |
| Pre-hospital crystalloid (mL) | 0 (0–100) | 50 (0–100) | 0 (0–200) | 0.40 |
| Admit temperature (°) | 36.2 +/− 0.7 | 36.5 +/− 1.2 | 36.7 +/− 0.7 | <0.01 |
| Admit pH | 7.3 +/− 0.1 | 7.3 +/− 0.2 | 7.3 +/− 0.1 | 0.10 |
| Admit base deficit | 0.9 +/− 4.2 | 4.2 +/− 5.2 | 2.3 +/− 5.8 | 0.17 |
| Transfused in 24h | 31% | 61% | 20% | <0.01 |
| Transfused platelets in 24h | 11% | 33% | 4% | <0.01 |
| Total hospital days | 11 (3–18) | 9 (7–29) | 3 (1–6) | <0.01 |
| Total ICU days (28d) | 5 (2–8) | 5 (3–15) | 0 (0–2) | <0.01 |
| Ventilator free days (28d) | 25 (6–28) | 20 (2–26) | 28 (27–28) | <0.01 |
| ARDS | 8% | 22% | 2% | <0.01 |
| Multi-organ failure | 11% | 11% | 4% | 0.05 |
| Mortality at 24h | 8% | 11% | 0% | <0.01 |
| Mortality at discharge | 25% | 28% | 2% | <0.01 |
Patient demographics for the 233 patients. Data are mean +/− SD, median (inter-quartile range), or percentage. Data for skewed variables reported as median with inter-quartile ranges. Ventilator free days are counted for the first 28 days of hospitalization. Patients who expired received 0 ventilator free days. p-values bolded for <0.0167 for multiple comparisons
TABLE 3b.
Admit Multiple Electrode Platelet Aggregometry and Coagulation Results Isolated TBI (iTBI) vs. TBI + Polytrauma (pTBI) vs. Non-TBI
| Isolated TBI (n=36) |
TBI + Polytrauma (n=18) |
Non-TBI (n=179) |
p-value | |
|---|---|---|---|---|
| ADP AUC (U) | 60 (49–70) | 61 (48–73) | 60 (46–71) | 0.88 |
| Collagen AUC (U) | 49 (38–63) | 53 (36–64) | 49 (40–61) | 0.89 |
| TRAP AUC (U) | 99 (77–113) | 98 (83–125) | 97 (82–111) | 0.79 |
| AA AUC (U) | 61 (41–69) | 59 (40–76) | 61 (49–71) | 0.58 |
| Ristocetin AUC (U) | 57 (35–75) | 57 (39–85) | 69 (46–88) | 0.08 |
| Overall low platelet response (%) | 47% | 39% | 34% | 0.28 |
| Admit INR>1.3 | 3% | 24% | 4% | 0.01 |
| Admit INR | 1.1 (1.1–1.2) | 1.2 (1.1–1.3) | 1.1 (1–1.1) | <0.01 |
| Admit PTT (sec) | 27 (25–31) | 30 (28–33) | 28 (25–30) | 0.06 |
| Admit platelets (x 109/L) | 264 (202–286) | 288 (237–322) | 262 (224–306) | 0.47 |
Data reported as median with inter-quartile ranges. Platelet aggregation was induced by agonist stimulation with adenosine diphosphate (ADP, via P2 receptors), collagen (via GpIa/IIa and GpVI receptors), thrombin receptor activating peptide-6 (TRAP, via PAR receptors), arachidonic acid (AA, via the cyclooxygenase pathway), or ristocetin (via vWF complex). Platelet adhesion to the electrodes was detected as increasing electrical impedance, measured by duplicate sets of sensor wires in each test cell. Platelet aggregation responses to multiple electrode platelet aggregometry are reported as area under the aggregation curve in units (U) over a 6-minute measurement period. p-values bolded for <0.0167 for multiple comparisons
Any TBI (iTBI + pTBI) vs. no-TBI
Although there were no differences in the platelet aggregation responses to various stimulating agonists in iTBI vs. pTBI patients, the 54 patients with any TBI (iTBI + pTBI) had significantly impaired platelet aggregation response to ristocetin compared to those without TBI (median AUC 57 vs. 69, p=0.03, Table 3c). In multivariate analysis (controlling for extra-cranial injury severity [AIS in each category], gender, mechanism, and platelet count), there was a significant independent association of impaired platelet aggregation response to ristocetin stimulation with TBI. Patients with impaired platelet aggregation response to ristocetin stimulation were three times more likely to have TBI (OR 3.05, CI 1.01–9.15, p=0.04).
TABLE 3c.
Admission Multiple Electrode Platelet Aggregometry, Coagulation Results, and Factor Activity Any-TBI (iTBI + pTBI) vs. Non-TBI
| TBI (n=54) | Non-TBI (n=179) | p-value | |
|---|---|---|---|
| ADP AUC | 60 (48–70) | 60 (46–71) | 0.69 |
| Collagen AUC | 51 (38–63) | 49 (40–61) | 0.92 |
| TRAP AUC | 99 (82–118) | 97 (82–111) | 0.50 |
| AA AUC | 60 (40–69) | 61 (49–71) | 0.35 |
| Ristocetin AUC | 57 (38–77) | 69 (46–88) | 0.03 |
| Overall low platelet response (%) | 44% | 34% | 0.15 |
| Admit INR>1.3 | 10% | 4% | 0.14 |
| Admit INR | 1.1 (1.1–1.2) | 1.1 (1–1.1) | <0.01 |
| Admit PTT (sec) | 28 (26–32) | 28 (25–30) | 0.06 |
| Admit platelets (x 109/L) | 269 (217–310) | 262 (224–306) | 0.66 |
| Factor II (% activity) | 69 (59–80) | 72 (61–84) | 0.28 |
| Factor V (% activity) | 50 (36–68) | 61 (43–80) | 0.10 |
| Factor VII (% activity) | 70 (60–90) | 83 (62–100) | 0.05 |
| Factor VIII (% activity) | 215 (132–346) | 156 (97–221) | 0.01 |
| Factor IX (% activity) | 122 (105–151) | 137 (107–166) | 0.21 |
| Factor X (% activity) | 76 (60–83) | 79 (66–90) | 0.17 |
Data reported as median with inter-quartile ranges. Platelet aggregation was induced by agonist stimulation with adenosine diphosphate (ADP, via P2 receptors), collagen (via GpIa/IIa and GpVI receptors), thrombin receptor activating peptide-6 (TRAP, via PAR receptors), arachidonic acid (AA, via the cyclooxygenase pathway), or ristocetin (via vWF complex). Platelet adhesion to the electrodes was detected as increasing electrical impedance, measured by duplicate sets of sensor wires in each test cell. Platelet aggregation responses to multiple electrode platelet aggregometry are reported as area under the aggregation curve in units (U) over a 6-minute measurement period. p-values bolded for <0.05
The patients with TBI also had higher INR compared to those without TBI (Table 3c). Corresponding with this, the TBI patients trended toward lower procoagulant factor activity for all factors except factor VIII. Interestingly, TBI patients had significantly higher activity of factor VIII (215% vs. 156%, p=0.01, Table 3c).
DISCUSSION
This study examined platelet aggregation responses to various stimulating agonists measured by multiple electrode platelet aggregometry in injured patients, and found that patients with TBI (both isolated and polytrauma) have impaired platelet aggregation response to ristocetin stimulation compared to patients without TBI (even when controlling for extra-cranial injury severity, gender, mechanism, and platelet count). This finding is consistent with the overall hypothesis that disruption of the vWF rich blood-brain barrier may drive downstream alterations of the vWF-platelet axis. vWF is stored in secretory Weibel-Palade bodies of endothelial cells and is released upon injury into both plasma and basement membrane spaces. Because ristocetin is an antimicrobial substance which forms complexes with vWF causing a conformational change in vWF that induces binding to GP1b platelet receptors, the formation of ristocetin-vWF complexes leads to the aggregation of platelets (Figure 1, B1). Thus, it is known that a lack of platelet aggregation response to ristocetin stimulation can be caused by an absence of GPIb receptors on the platelet surface or by a decrease in the amount or function of vWF (Figure 1, B2/B3/B4).
Prior research suggests that certain injury patterns that are associated with significant endothelial damage may have a greater association with impaired platelet aggregation response to stimulating agonists in aggregometry assays. For example, Jacoby et al. identified specific associations of impaired platelet aggregation response to aggregometry in TBI patients (14). Furthermore, they identified that platelets were activated but had impaired aggregation which progressed at 24 hours after injury. Taken together, these results suggested that in TBI there is profound platelet activation leading to faster rates of adhesion and aggregation and a resultant prolonged period of impaired platelet responsiveness. This proposed mechanism may be induced by injury to the blood brain barrier causing a robust endothelial mediated activation of platelets. Additional groups have identified similar associations in TBI patients and patients with other injury patterns consistent with endothelial damage (high injury severity scores, shock states) (13, 27, 28, 30). Despite this, there have been no large prospective studies on the association of impaired post-injury platelet aggregation and biomarkers of endothelial injury (including vWF).
In this study, it is plausible that the impaired platelet aggregation response to ristocetin stimulation that we identified in TBI patients is due to a TBI-induced effect on vWF function (Figure 1, B4) or that the injury has caused the majority of the circulating vWF to be consumed in the process of binding to platelets in the clotting process (Figure 1, B3). This latter mechanism could potentially lead to a lack of circulating vWF left to complex with the ristocetin, and therefore a downstream impaired platelet aggregation response to ristocetin stimulation. Given the findings by De Oliveira et al. that plasma vWF antigen levels increase in TBI patients compared to controls (35), the latter mechanism seems most likely. Levels of measurable vWF antigen may be increased in TBI, but the ability for ristocetin to stimulate circulating vWF to bind to platelets may be diminished because the vWF-platelet binding has already endogenously occurred from the injury itself.
Additionally, we identified that patients with TBI had a trend toward lower factor activity for all procoagulant factors except for factor VIII, which was significantly higher. It is biologically plausible that the elevated factor VIII activity in the patients with TBI may be explained by the decreased amount or function of circulating vWF that we identified (Figure 1, B5). In studies of non-injured populations of patients, the ratio between circulating factor VIII and vWF antigen in several subtypes of von Willebrand Disease supports that the ratio of factor VIII:vWF is increased when vWF synthesis is reduced, thought to be secondary to decreased factor VIII carrying capacity (31). This is an important finding, as studies have demonstrated that elevated factor VIII levels are a common risk factor for venous thromboembolism (prevalence of 25% among thrombosis patients and a 5x increased risk) (36), which may partially contribute to the high rates of thromboembolic complications in TBI patients (37).
This study has multiple limitations. Due to the study design of a retrospective analysis of a prospectively followed cohort, we did not measure vWF antigen levels via enzyme-linked immunosorbent assays, which will be critical in the future to correlating vWF antigen levels to the mechanism of the impaired platelet aggregation response to ristocetin stimulation in this population. In addition, in order to control for the effects of thrombocytopenia on multiple electrode platelet aggregometry, the patients included in this study were only moderately injured (due to the exclusion of all patients with a platelet count <150 × 109/L on presentation), and the results cannot be generalized to a severely injured population with thrombocytopenia. Lastly, given that it is known that impaired platelet aggregation response to ristocetin stimulation can be caused by an absence of GPIb receptors on the platelet surface (consistent with Bernard-Soulier syndrome) or by an absence of vWF in circulation (von Willebrand’s Disease [vWD] type III), we recognize that there may be a small proportion of our patients who suffer from these diseases. However, these are rare conditions: Bernard-Soulier affects 1/1,000,000 patients and type III vWD is the affects 1/500,000 patients (38). Due to the rarity of these disease states, it is unlikely that they had significant effects on this study.
A better understanding of how injury modulates interactions between platelets and endothelial cells will advance progress towards targeted therapies for post-injury hemorrhage and trauma-induced coagulopathy. Our findings demonstrate that platelet aggregation response to ristocetin stimulation differs between injured patients with and without TBI. We identified an impaired platelet aggregation response to ristocetin stimulation with a corresponding increase in factor VIII activity in TBI patients. It is plausible that the impaired platelet aggregation response to ristocetin stimulation can be explained by a TBI-induced effect on vWF function or that the majority of vWF has already bound to platelets following disruption of the blood-brain barrier with resultant exposure of vWF. Either way, it may be that cerebral endothelial injury increases the circulating factor VIII activity in the setting of impaired vWF factor VIII carrier function. Future studies of platelet aggregation after TBI that measure both quantity and function of vWF and corresponding factor activity will advance our understanding of platelet and endothelial biology after injury.
Finally, of particular importance is that although decades of retrospective and prospective evidence have identified that resuscitation with balanced ratios of blood products (red cells: plasma: platelets) improves outcomes after injury for the treatment of hemorrhaging trauma patients (39), evidence suggests that the use of platelet transfusions following injury may not improve post-injury impairments in platelet aggregation (40). Furthermore, the appropriate timing and dosage of platelet transfusions after injury remains unclear. Transfusion of platelets first (prior to other blood products) in massive transfusion decreases death from hemorrhage by improving hemostasis following injury (33). However, early transfusion of platelets is strongly associated with the development of ARDS in patients with TBI (41). Additionally, with increasing evidence highlighting the importance of the role of repair of the blood-brain barrier endothelium for neuroprotection and long-term outcomes (15), improving resuscitation practices and targeting novel therapies for endothelial repair and maintenance of the platelet endothelial axis remains critical. Alam and colleagues recently identified that valproic acid, known to repair blood-brain barrier integrity in the setting of injury, may also have some effect on platelet aggregation (42).
Furthermore, there are multiple known therapies for vWF deficits outside the setting of injury including synthetic analogs of antidiuretic hormone like desmopressin (ddAVP), vWF replacement therapy with purified and recombinant vWF, and estrogens. Desmopressin reversal of anti-platelet medication induced impaired platelet aggregation has been described (43–46), although large studies have not been performed. In addition, studies of the use of ddAVP for intracranial hemorrhage (traumatic and atraumatic) in the setting of impaired platelet aggregation due to anti-platelet medication demonstrate improved platelet aggregation, increased levels of measureable vWF, and decreased hematoma volume (47–49). However, studies specific to the role of ddAVP in the reversal TBI-induced impaired platelet aggregation (and not anti-platelet induced impaired platelet aggregation) following injury are needed. In addition, the effects of direct replacement of vWF in this setting are unknown. Specific to this, future investigations of the thrombotic effects of these therapies in the setting of injury will also be pivotal. These lines of investigation are particularly compelling for vWF based therapeutic strategies in patients with TBI and hemorrhage.
ACKNOWLEDGEMENTS:
Diana Lim, digital scientific illustration for Figure 1.
Disclosure Information: Dr. Cohen is supported by DoD W911QY-15-C-0044 and NIH UM1HL120877, Dr. Callcut is supported by NIH K01ES026834 and DoD W911QY-15-C-0044, Dr. Calfee is supported by NIH HL110969, Dr. Hendrickson is supported by K23 HL133495.
Footnotes
AUTHOR CONTRIBUTIONS:
AJR, LZK, CMH, CSC, RAC, and MJC contributed to study design, data collection, data analysis, data interpretation, writing, and clinical revision.
ASC contributed to study design, data collection, and clinical revision.
ATF contributed to data interpretation, writing, and clinical revision
Disclosure outside the scope of this work: none.
Meeting presentation: Presented during the Earl G. Young Resident Paper Competition at the 48th annual meeting of the Western Trauma Association; February 25–March 2, 2018; in Whistler, British Columbia.
REFERENCES
- 1.National Center for Injury Prevention and Control (NCIPC). National Violent Death Reporting System. Available at: https://www.cdc.gov/injury/wisqars/LeadingCauses.html. Centers for Disease Control and Prevention; Atlanta, GA: Accessed June, 06, 2018. [Google Scholar]
- 2.Violence and Injury Prevention. Available at: http://www.who.int/violence_injury_prevention/key_facts/VIP_key_fact_4.pdf?ua=1. World Health Organization; Accessed June 06, 2018. [Google Scholar]
- 3.Norton R, Kobusingye O. Injuries. N Engl J Med. 2013;368(18):1723–30. [DOI] [PubMed] [Google Scholar]
- 4.Kauvar DS, Lefering R, Wade CE. Impact of hemorrhage on trauma outcome: an overview of epidemiology, clinical presentations, and therapeutic considerations. J Trauma Acute Care Surg. 2006;60(6 Suppl):S3–11. [DOI] [PubMed] [Google Scholar]
- 5.Murray CJ, Lopez AD. Mortality by cause for eight regions of the world: Global Burden of Disease Study. Lancet. 1997;349(9061):1269–76. [DOI] [PubMed] [Google Scholar]
- 6.Hess JR, Brohi K, Dutton RP, Hauser CJ, Holcomb JB, Kluger Y, Mackway-Jones K, Parr MJ, Rizoli SB, Yukioka T, et al. The coagulopathy of trauma: a review of mechanisms. J Trauma Acute Care Surg. 2008;65(4):748–54. [DOI] [PubMed] [Google Scholar]
- 7.White NJ, Ward KR, Pati S, Strandenes G, Cap AP. Hemorrhagic blood failure: Oxygen debt, coagulopathy, and endothelial damage. J Trauma Acute Care Surg. 2017;82(6S Suppl 1): S41–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pati S, Potter DR, Baimukanova G, Farrel DH, Holcomb JB, Schreiber MA. Modulating the endotheliopathy of trauma: Factor concentrate versus fresh frozen plasma. J Trauma Acute Care Surg. 2016;80(4):576–84; discussion 84–5. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez EG, Cardenas JC, Lopez E, Cotton BA, Tomasek JS, Ostrowski SR, Baer LA, Stensballe J, Holcomb JB, Johansson PI, et al. Early Identification of the Patient with Endotheliopathy of Trauma by Arrival Serum Albumin. Shock. 2017. [DOI] [PubMed] [Google Scholar]
- 10.Naumann DN, Hazeldine J, Davies DJ, Bishop J, Midwinter MJ, Belli A, Harrison P, Lord JM. Endotheliopathy of Trauma is an On-Scene Phenomenon, and is Associated with Multiple Organ Dysfunction Syndrome: A Prospective Observational Study. Shock. 2017. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez Rodriguez E, Ostrowski SR, Cardenas JC, Baer LA, Tomasek JS, Henriksen HH, Stensballe J, Cotton BA, Holcomb JB, Johansson PI, et al. Syndecan-1: A Quantitative Marker for the Endotheliopathy of Trauma. J Am Coll Surg. 2017;225(3):419–27. [DOI] [PubMed] [Google Scholar]
- 12.Johansson PI, Henriksen HH, Stensballe J, Gybel-Brask M, Cardenas JC, Baer LA, Cotton BA, Holcomb JB, Wade CE, Ostrowski SR. Traumatic Endotheliopathy: A Prospective Observational Study of 424 Severely Injured Patients. Ann Surg. 2017;265(3):597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kutcher ME, Redick BJ, McCreery RC, Crane IM, Greenberg MD, Cachola LM, Nelson MF, Cohen MJ. Characterization of platelet dysfunction after trauma. J Trauma Acute Care Surg. 2012;73(1):13–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacoby RC, Owings JT, Holmes J, Battistella FD, Gosselin RC, Paglieroni TG. Platelet activation and function after trauma. J Trauma Acute Care Surg. 2001;51(4):639–47. [DOI] [PubMed] [Google Scholar]
- 15.Nikolian VC, Dekker SE, Bambakidis T, Higgins GA, Dennahy IS, Georgoff PE, Williams AM, Andjelkovic AV, Alam HB. Improvement of Blood-Brain Barrier Integrity in Traumatic Brain Injury and Hemorrhagic Shock Following Treatment With Valproic Acid and Fresh Frozen Plasma. Crit Care Med. 2018;46(1):e59–e66. [DOI] [PubMed] [Google Scholar]
- 16.Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N Engl J Med. 2008;359(12):1261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baimukanova G, Miyazawa B, Potter DR, Gibb SL, Keating S, Danesh A, Beyer A, Dayter Y, Bruhn R, Muench MO, et al. The effects of 22°C and 4°C storage of platelets on vascular endothelial integrity and function. Transfusion. 2016;56:S52–S64. [DOI] [PubMed] [Google Scholar]
- 18.Baimukanova G, Miyazawa B, Potter DR, Muench MO, Bruhn R, Gibb SL, Spinella PC, Cap AP, Cohen MJ, Pati S. Platelets regulate vascular endothelial stability: assessing the storage lesion and donor variability of apheresis platelets. Transfusion. 2016;56:S65–S75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davenport RA, Brohi K. Coagulopathy in trauma patients: importance of thrombocyte function? Curr Opin Anaesthesiol. 2009;22(2):261–6. [DOI] [PubMed] [Google Scholar]
- 20.Brown LM, Call MS, Margaret Knudson M, Cohen MJ, Trauma Outcomes G, Holcomb JB, Wade CE, Brasel KJ, Vercruysse G, MacLeod J, et al. A normal platelet count may not be enough: the impact of admission platelet count on mortality and transfusion in severely injured trauma patients. J Trauma Acute Care Surg. 2011;71(2 Suppl 3):S337–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kornblith LZ, Kutcher ME, Redick BJ, Calfee CS, Vilardi RF, Cohen MJ. Fibrinogen and platelet contributions to clot formation: implications for trauma resuscitation and thromboprophylaxis. J Trauma Acute Care Surg. 2014;76(2):255–6; discussion 62–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li R, Elmongy H, Sims C, Diamond SL. Ex vivo recapitulation of trauma-induced coagulopathy and preliminary assessment of trauma patient platelet function under flow using microfluidic technology. J Trauma Acute Care Surg. 2016;80(3):440–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stansbury LG, Hess AS, Thompson K, Kramer B, Scalea TM, Hess JR. The clinical significance of platelet counts in the first 24 hours after severe injury. Transfusion. 2013;53(4):783–9. [DOI] [PubMed] [Google Scholar]
- 24.Wohlauer MV, Moore EE, Thomas S, Sauaia A, Evans E, Harr J, Silliman CC, Ploplis V, Castellino FJ, Walsh M. Early platelet dysfunction: an unrecognized role in the acute coagulopathy of trauma. J Am Coll Surg. 2012;214(5):739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solomon C, Traintinger S, Ziegler B, Hanke A, Rahe-Meyer N, Voelckel W, Schochl H. Platelet function following trauma. A multiple electrode aggregometry study. Thromb Haemost. 2011;106(2):322–30. [DOI] [PubMed] [Google Scholar]
- 26.Ding N, Chen G, Hoffman R, Loughran PA, Sodhi CP, Hackam DJ, Billiar TR, Neal MD. Toll-like receptor 4 regulates platelet function and contributes to coagulation abnormality and organ injury in hemorrhagic shock and resuscitation. Circ Cardiovasc Genet. 2014;7(5):615–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis PK, Musunuru H, Walsh M, Cassady R, Yount R, Losiniecki A, Moore EE, Wohlauer MV, Howard J, Ploplis VA, et al. Platelet dysfunction is an early marker for traumatic brain injury-induced coagulopathy. Neurocrit Care. 2013;18(2):201–8. [DOI] [PubMed] [Google Scholar]
- 28.Donahue DL, Beck J, Fritz B, Davis P, Sandoval-Cooper MJ, Thomas SG, Yount RA, Walsh M, Ploplis VA, Castellino FJ. Early platelet dysfunction in a rodent model of blunt traumatic brain injury reflects the acute traumatic coagulopathy found in humans. J Neurotrauma. 2014;31(4):404–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto K, de Waard V, Fearns C, Loskutoff DJ. Tissue distribution and regulation of murine von Willebrand factor gene expression in vivo. Blood. 1998;92(8):2791–801. [PubMed] [Google Scholar]
- 30.Briggs A, Gates JD, Kaufman RM, Calahan C, Gormley WB, Havens JM. Platelet dysfunction and platelet transfusion in traumatic brain injury. J Surg Res. 2015;193(2):802–6. [DOI] [PubMed] [Google Scholar]
- 31.Eikenboom JC, Castaman G, Kamphuisen PW, Rosendaal FR, Bertina RM. The factor VIII/von Willebrand factor ratio discriminates between reduced synthesis and increased clearance of von Willebrand factor. Thromb Haemost. 2002;87(2):252–7. [PubMed] [Google Scholar]
- 32.Sauaia A, Moore FA, Moore EE, Haenel JB, Read RA, Lezotte DC. Early predictors of postinjury multiple organ failure. Arch Surg (Chicago, Ill : 1960). 1994;129(1):39–45. [DOI] [PubMed] [Google Scholar]
- 33.Hendrickson CM, Howard BM, Kornblith LZ, Conroy AS, Nelson MF, Zhuo H, Liu KD, Manley GT, Matthay MA, Calfee CS, et al. The acute respiratory distress syndrome following isolated severe traumatic brain injury. J Trauma Acute Care Surg. 2016;80(6):989–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kutcher ME, Kornblith LZ, Vilardi RF, Redick BJ, Nelson MF, Cohen MJ. The natural history and effect of resuscitation ratio on coagulation after trauma: a prospective cohort study. Ann Surg. 2014;260(6):1103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Oliveira CO, Reimer AG, Da Rocha AB, Grivicich I, Schneider RF, Roisenberg I, Regner A, Simon D. Plasma von Willebrand factor levels correlate with clinical outcome of severe traumatic brain injury. J Neurotrauma. 2007;24(8):1331–8. [DOI] [PubMed] [Google Scholar]
- 36.Eikenboom JC. Determinants of increased levels of von Willebrand factor and coagulation factor VIII in patients with venous thrombosis. Neth Heart J. 2006;14(7-8):277–8. [PMC free article] [PubMed] [Google Scholar]
- 37.Skrifvars MB, Bailey M, Presneill J, French C, Nichol A, Little L, Duranteau J, Huet O, Haddad S, Arabi Y, et al. Venous thromboembolic events in critically ill traumatic brain injury patients. Intensive Care Med. 2017;43(3):419–28. [DOI] [PubMed] [Google Scholar]
- 38.R D-K. Inherited platelet disorders including GLanzmann thrombasthenia and Bernard-Soulier syndrome. Hematology. 2013:268–75. [DOI] [PubMed] [Google Scholar]
- 39.Spinella PC, Holcomb JB. Resuscitation and transfusion principles for traumatic hemorrhagic shock. Blood Rev. 2009;23(6):231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henriksen HH, Grand AG, Viggers S, Baer LA, Solbeck S, Cotton BA, Matijevic N, Ostrowski SR, Stensballe J, Fox EE, et al. Impact of blood products on platelet function in patients with traumatic injuries: a translational study. J Surg Res. 2017;214:154–61. [DOI] [PubMed] [Google Scholar]
- 41.Holcomb JB, Tilley BC, Baraniuk S, Fox EE, Wade CE, Podbielski JM, del Junco DJ, Brasel KJ, Bulger EM, Callcut RA, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA. 2015;313(5):471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bambakidis T, Dekker SE, Halaweish I, Liu B, Nikolian VC, Georgoff PE, Piascik P, Li Y, Sillesen M, Alam HB. Valproic acid modulates platelet and coagulation function ex vivo. Blood Coagul Fibrinolysis. 2017;28(6):479–84. [DOI] [PubMed] [Google Scholar]
- 43.Ranucci M, Nano G, Pazzaglia A, Bianchi P, Casana R, Tealdi DG. Platelet mapping and desmopressin reversal of platelet inhibition during emergency carotid endarterectomy. J Cardiothorac Vasc Anesth. 2007;21(6):851–4. [DOI] [PubMed] [Google Scholar]
- 44.McMillian WD, Rogers FB. Management of prehospital antiplatelet and anticoagulant therapy in traumatic head injury: a review. J Trauma Acute Care Surg. 2009;66(3):942–50. [DOI] [PubMed] [Google Scholar]
- 45.Flordal PA, Sahlin S. Use of desmopressin to prevent bleeding complications in patients treated with aspirin. Br J Surg. 1993;80(6):723–4. [DOI] [PubMed] [Google Scholar]
- 46.Gratz I, Koehler J, Olsen D, Afshar M, DeCastro N, Spagna PM, Ablaza SG, Larijani GE. The effect of desmopressin acetate on postoperative hemorrhage in patients receiving aspirin therapy before coronary artery bypass operations. J Thorac Cardiovasc Surg. 1992;104(5):1417–22. [PubMed] [Google Scholar]
- 47.Naidech AM, Maas MB, Levasseur-Franklin KE, Liotta EM, Guth JC, Berman M, Rosenow JM, Lindholm PF, Bendok BR, Prabhakaran S, et al. Desmopressin improves platelet activity in acute intracerebral hemorrhage. Stroke. 2014;45(8):2451–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kapapa T, Rohrer S, Struve S, Petscher M, Konig R, Wirtz CR, Woischneck D. Desmopressin acetate in intracranial haemorrhage. Neurol Res Int. 2014;2014:298767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim DY, O’Leary M, Nguyen A, Kaji A, Bricker S, Neville A, Bongard F, Putnam B, Plurad D. The Effect of Platelet and Desmopressin Administration on Early Radiographic Progression of Traumatic Intracranial Hemorrhage. J Neurotrauma 2015;32(22):1815–21. [DOI] [PubMed] [Google Scholar]