

Abstract
B cells are activated by two temporally distinct signals, the first provided by antigen binding to the B cell antigen receptor (BCR) and the second by T helper cells. Here we show that B cells responded to antigen by rapidly increasing metabolic activity including both oxidative phosphorylation and glycolysis. In the absence of a second signal B cells progressively lost mitochondrial function and glycolytic capacity leading to apoptosis. Mitochondrial dysfunction was a result of the gradual accumulation of intracellular calcium through calcium response activated calcium channels that was preventable for approximately nine hours after B cell antigen binding by either T helper cells or Toll-like receptor 9 signaling. Thus, BCR signaling appears to activate a metabolic program that imposes a limited time window in which B cells either receive a second signal and survive or are eliminated.
INTRODUCTION
Antigen-specific antibody responses are initiated by the binding of antigens to B cell antigen receptors (BCRs). Antigen binding alone initiates a cascade of signaling events that for many antigens is necessary but not sufficient to drive full B cell activation including proliferation and differentiation into antibody-secreting cells. For these antigens, full activation requires that B cells acquire a temporally distinct, second signal. Second signals are provided by antigen-specific T helper cells (TH cells) following processing and presentation of antigen by B cells to antigen-specific TH cells resulting in the formation of an immune synapse1–4. Ultimately, the engaged TH cell provides a critical second signal for the B cells through CD40 expressed by B cells binding to CD40L on the TH cells5. Second signals can also be delivered through pattern recognition receptors (PRRs) in the absence of T cells6–8. Toll-like receptor 9 (TLR9) that responds to unmethylated CpG oligonucleotides present in microbial genomes9 provides particularly potent survival and differentiation signals for antigen-activated B cells.
The requirement for acquisition of a second signal is a fundamental immune control mechanism to ensure that in the absence of antigen-specific TH cells or pathogen products, antigen binding alone will not promote B cell proliferation and differentiation to antibody-secreting cells. Despite the central role of the requirement for two signals in the generation of antibody responses, we have an incomplete understanding of the molecular nature of the consequences of each signal on B cells and the impact of the failure to acquire a second signal.
The requirements for the activation of lymphocytes are being increasingly viewed in the context of the transition of cells from a resting state to a highly active one. We now appreciate that the switch from a quiescent cell to a rapidly growing one requires metabolic reprogramming in order to both fuel the energy requirements of highly active cells and provide intermediates for biosynthesis10–12. Recent studies provided evidence that although B cells are able to consume glucose and fatty acids as sources of energy and for resting state biosynthesis, B cells stimulated through the BCR increase glycolysis and expression of the glucose transporter, GLUT1, through c-Myc- and phosphatidylinositol-3-OH kinase (PI3K)-dependent mechanisms10,11,13 but in addition continue to use oxidative phosphorylation11. The BCR-mediated boost in utilization of glucose is blunted by the inhibitory receptor, FcγRIIB,13 or by induction of hypo-responsive B cell states such as anergy10. The utility of understanding the metabolic demands on B cells during activation was highlighted by recent studies showing that B cell specific diversion of glucose carbons from glycolysis to the pentose phosphate pathway provided a target for treatment of B cell malignancies14.
Here, we provide the results of an extensive study of the metabolic reprogramming of activated B cells in which we discovered that antigen binding to the BCR activates a ‘metabolic clock’ that limits the time during which B cells must receive a second signal to survive.
RESULTS
Rapid metabolic changes accompany B cell activation
To assess metabolic changes in B cells following stimulation through the BCR using antibodies specific for IgM (anti-IgM) or through TLR9 using the TLR9 agonist, CpG, metabolic-stress tests were carried out15. Purified mouse splenic B cells were plated into the wells of a Seahorse extracellular flux analyzer to measure in real-time changes in B cells’ oxygen consumption rate (OCR), an indicator of oxidative phosphorylation (Fig. 1a) and the extracellular acidification rates (ECAR) an indication of the production of lactate during glycolysis (Fig. 1b). Stimulation of B cells with either CpG or anti-IgM or both induced a rapid increase in both the OCR and ECAR levels (Fig. 1a,b). The addition of oligomycin resulted in a drop in OCR levels (Fig. 1a) and the subsequent addition of 2,4-dinitrophenol (2,4-DNP), depolarized the mitochondrial membrane, resulting in an increase in the OCR levels. The difference between the baseline OCR and the OCR after 2,4-DNP addition represents the spare oxidative phosphorylation capacity of the cells that were similar for the different stimulations (Fig. 1a). The addition of oligomycin to wells also resulted and an increase in the ECAR levels (Fig. 1b) and the difference between the pre-oligomycin and post-oligomycin ECAR levels represents the augmented glycolytic reserves of the B cells that were similar in all cases.
Figure 1.
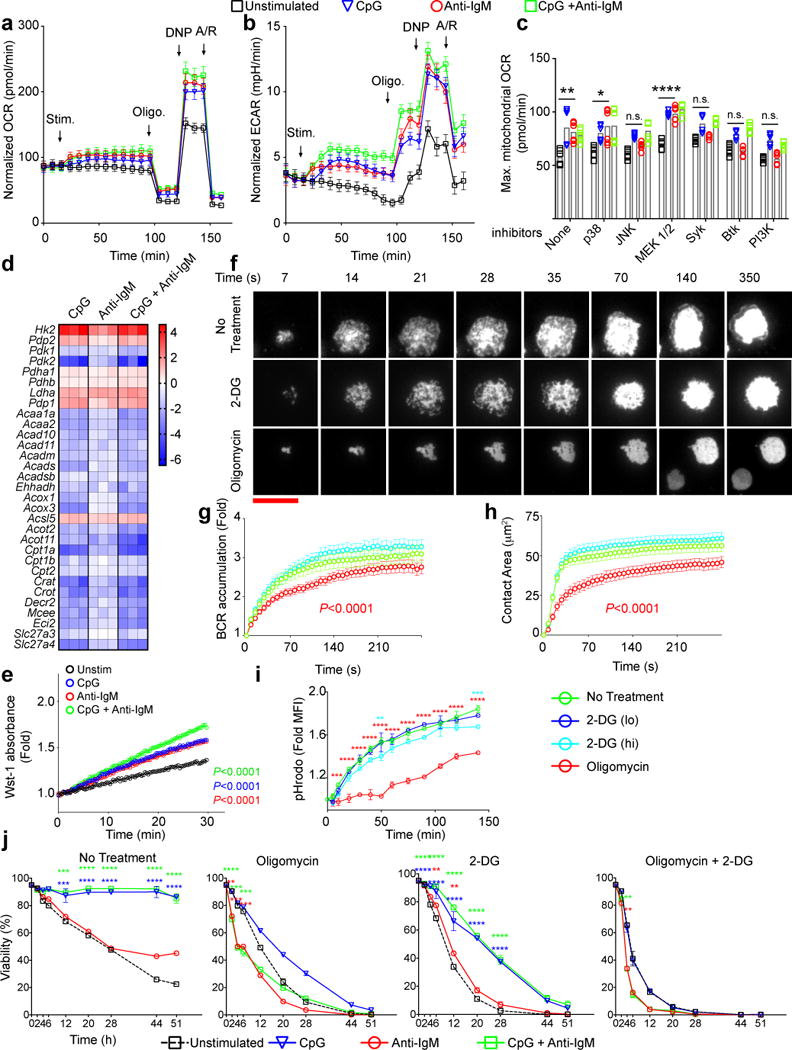
Similar metabolic changes occur immediately following B cell stimulation through either BCR or TLR9. a,b) Changes in Oxygen Consumption Rates (OCR) (a) and Extra Cellular Acidification Rates (ECAR) (b) were measured in real time for B cells stimulated with CpG (1 μM) and/or anti-IgM (5 μg/ml) or unstimulated. Arrows indicate the time points stimulants, oligomycin, DNP and antimycin/rotenone (A/R) were added to the wells. The data are pooled from 10 individual samples acquired in three independent experiments. Symbols and error bars represent the mean and standard error of the mean respectively. c) Maximum OCR levels following addition of 2,4-DNP were measured in B cell cultures in which the indicated inhibitors were added immediately after the stimulants. Bars indicate mean of ten individual samples, shown by symbols, pooled from two experiments. d) Heat maps show the log2 scale fold change in expression levels of a set of genes involved in glucose or fatty acid metabolism from RNA seq data16 of purified mouse splenic B cells that were cultured in media alone or media containing CpG (1 μM) and/or anti-IgM (5 μg/ml) in triplicates for 4h. e) Quantification of PDH activity in B cells following 6h in culture in media alone (unstimulated) or in media containing CpG alone, anti-IgM alone or both using colorimetric assay through Wst-1 reporter dye absorbance. f) HEL-specific MD4 B cells labeled with DyLight549-Fab anti-IgM to allow imaging of the BCRs were placed on HEL-containing PLBs without metabolic inhibitors (No Treatment) or with either 2-DG (55 mM) or Oligomycin (6 μM) and live cell time-lapse TIRF images were acquired for 30 min at 37°C. Representative TIRF images are shown. (scale bar = 10 μm) g) BCR accumulation in the contact area of the B cell with the HEL-PLB presented as the fold increase in BCR MFI above background at each time point. h) The contact area of the B cell with the HEL-PLB. Statistically significant decrease from unstimulated B cells are shown with a P value in the color of the symbol. i) HEL-specific B cells from MD4 mice were incubated with pHrodo-conjugated-HEL on ice, warmed to 37 °C in the presence or absence of 2-DG (lo=11mM, hi=55 mM) or Oligomycin (6 μM). Fold changes in pHrodo MFI compared to the initial time points are shown. Symbols and error bars refer to the mean of three replicates and standard deviation respectively. Data are representative of three independent experiments. j) Purified mouse splenic B cells were incubated in media alone or in media containing CpG (1 μM) and/or anti-IgM (5 μg/ml) in the presence or absence of 2-DG (11 nM) and/or Oligomycin (6 μM). Cell viability determined by staining with Live/Dead marker by flow cytometry is given as a function of time. Symbols and error bars refer to the mean of six replicates and standard error respectively. Data are pooled from two independent experiments. For all panels data points that are significantly different from the unstimulated (Panels a,b,g) or no-inhibitor conditions (Panels c,g,h,i,j) are shown with color-coded asterisks or as adjusted p values. (Panels i,j: two way ANOVA with Dunnet’s adjustment; Panel c: one way ANOVA with Tukey’s adjustment, Panel e: linear regression, Panels g,h: cubic regression with Bonferroni’s adjustment) (P>0.05 = n.s.; 0.01 <P ≤0.05 = *;0.001 <P ≤0.01 = **; 0.0001 <P ≤0.001 = ***; P ≤0.0001= ****)
BCR-induced increases in maximal OCR were blocked in the presence of the Syk inhibitor Piceatannol, the Btk inhibitor Ibrutinib, the PI3K inhibitor, Wortmannin, and the JNK inhibitor, SP600125 (Fig. 1c) directly linking BCR signaling to metabolic changes. TLR9-induced increases in OCR were unaffected suggesting TLR9-signaling was too robust or too redundant to be affected by the inhibitors used.
To determine if the immediate increases in OCR and ECAR required cellular remodeling, we measured changes in mitochondrial membrane potential, expression of the glucose transporters GLUT1 and GLUT3, the uptake of glucose and mitochondrial mass during the first 4h post stimulation. None of these parameters showed significant differences between unstimulated B cells and stimulated B cells (Fig. S1).
To gain insight into the metabolic reprogramming that occurs in activated B cells, we examined gene expression changes by RNAseq that occur within the first 4 h following stimulation by CpG, anti-IgM, both CpG and anti-IgM or medium alone16 focusing on changes in expression of genes encoding enzymes associated with cellular metabolism (Fig. 1d, Fig. S2). Overall, the patterns of differential gene expression relative to cultured unstimulated B cells were similar for treatment with CpG, anti-IgM or both. Two observations were notable. First, the transcription of the genes encoding key enzymes in glycolysis, including Hexokinase (Hk2) and Lactate Dehydrogenase A (Ldha) (Fig. S2) were upregulated consistent with the observed increase in glycolysis. Second, the transcription of Pdha1 and Pdhb, that encode subunits of the enzyme pyruvate dehydrogenase (PDHA1 and PDHB) that catalyzes pyruvate to acetyl CoA (Fig. S2) were not affected by any stimulation, suggesting that anaerobic glycolysis may be favored in activated B cells over glucose oxidation. However, the transcription of Pdp1 and Pdp2 that dephosphorylate and activate PDH were upregulated and the transcription of Pdk1 and Pdk2 that phosphorylate and inactivate PDH were downregulated (Fig. 1d, Fig. S2). We quantified PDH activity in the lysates of B cells cultured in vitro for 6h post-stimulation and found that each stimulant increased the PDH activity as compared to unstimulated B cells similarly (Fig. 1e) indicating an increase in glucose oxidation. Thus, we conclude that although B cells respond to BCR and TLR9 stimulation by increasing both glycolysis and oxidative phosphorylation relying on existing stores, they begin a process of cellular remodeling to increase mitochondrial and glycolytic capacities as early as 4 h post activation and that the shift towards anaerobic glycolysis is not complete and B cells continue to use oxidative phosphorylation to obtain energy from glucose.
Metabolic requirements for early antigen-induced BCR functions
The observed rapid increases in both oxidative phosphorylation and glycolysis following B cell stimulation led us to ask whether both metabolic pathways were equally required for early BCR signaling events and BCR internalization. To evaluate signaling, hen egg lysozyme (HEL)-specific B cells from MD4 mice were placed on planar lipid bilayers (PLBs) containing HEL in the presence or absence of inhibitors of glycolysis, 2-deoxyglucose (2-DG) and oxidative phosphorylation, oligomycin17 and imaged by total internal reflection fluorescence (TIRF) microscopy. In the absence of inhibitors, B cells rapidly spread over the HEL-containing PLB and accumulated BCRs in the contact area (Fig. 1f-h). Neither spreading nor BCR accumulation were diminished by inhibition of glycolysis, however, both processes required oxidative phosphorylation and were inhibited by oligomycin (Fig. 1f-h).
To quantify antigen internalization, HEL-specific B cells were incubated with HEL conjugated to pHrodo, a pH sensitive fluorescent dye that allows for the monitoring of HEL internalization and progression through intracellular acidic compartments (Fig. 1i). Neither standard (lo) (11 mM) nor high (hi) (55 mM) concentrations of 2-DG affected the increase in pHrodo MFI whereas oligomycin blocked pHrodo MFI increases indicating a block in HEL internalization and trafficking to acidic compartments (Fig. 1i, Fig. S3a). Thus, energy for the early events following B cell contact with antigen is provided primarily by oxidative phosphorylation, not glycolysis.
Only TLR9 signaling promotes B cell survival
We determined the metabolic requirements for anti-IgM- and CpG-induced proliferation and long term (up to 51 h) B cell survival. We observed no cell division at 24 h for any stimulation condition, however, by 48 h the majority of B cells stimulated with CpG alone or CpG plus anti-IgM had divided at least twice in contrast to B cells stimulated with anti-IgM alone of which very few had undergone more than a single round of division (Fig. S3b,c). B cells treated with anti-IgM alone maintained low viability through the 51 h time course measured by Live/Dead dye similar to unstimulated B cells (Fig. 1j). Cell death in both cases was nearly exclusively from apoptosis and not necrosis as determined by 7-AAD and FAM FLICA staining (Fig. S4a,b). In contrast, B cells treated with CpG alone or CpG plus anti-IgM maintained over 70% viability. For B cells stimulated with CpG alone, BCR alone or both, inhibitors of glycolysis and oxidative phosphorylation decreased both survival (Fig. 1j) and activation as measured by CD69 expression (Fig. S3d).
To gain insight into the metabolic state of B cells 24 h after stimulation, we measured the OCRs by a mitochondrial stress test and ECARs by a glycolytic stress test of live B cells FACS sorted from 24 h cultures. The baseline OCRs of B cells stimulated with CpG alone or CpG plus anti-IgM were considerably higher and showed greater maximum mitochondrial respiration capacity as compared to anti-IgM stimulated or unstimulated B cells (Fig. 2a). In the glycolytic stress test15, cells treated with CpG or CpG plus anti-IgM rapidly responded to the addition of glucose with an increase in ECAR and following the addition of Antimycin A and Rotenone showed further increases in ECAR, suggesting similar glycolytic reserves. In contrast, untreated or anti-IgM treated B cells showed little or no increase in ECAR levels in response to glucose addition (Fig. 2b).
Figure 2.
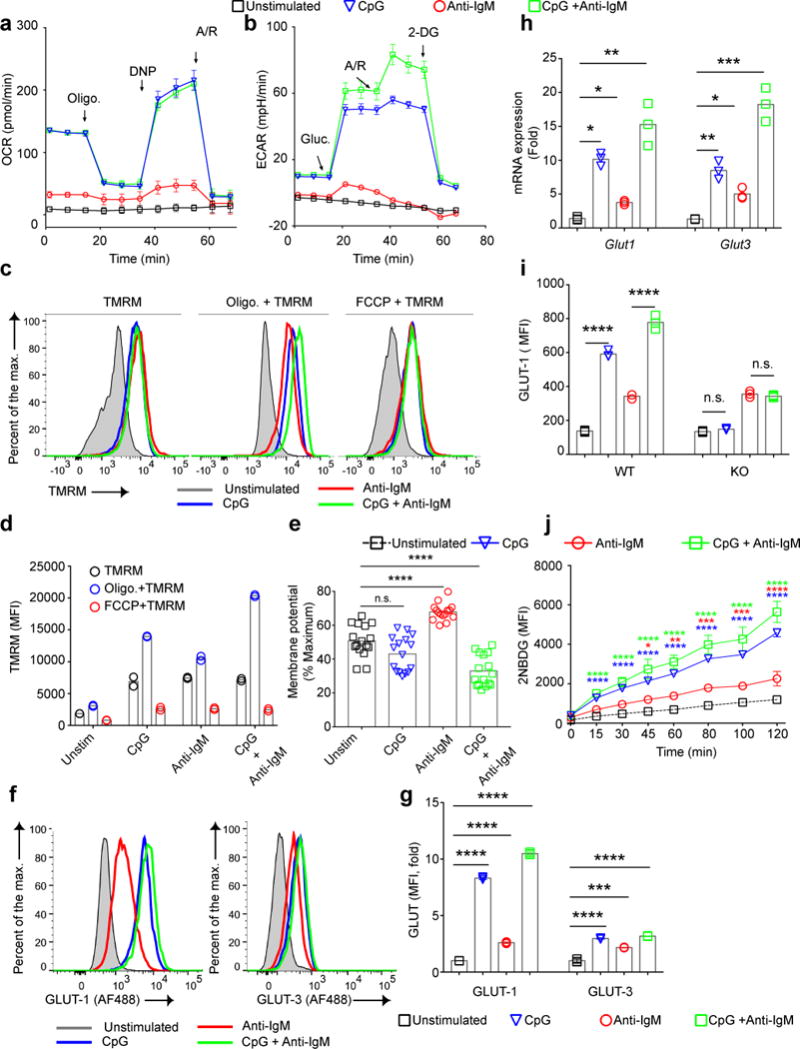
Glycolytic capacity and maximal mitochondrial respiration are correlated with B cell survival. a, b) Purified B cells were cultured in vitro in media alone or media containing CpG (1 μM) and/or anti-IgM (5 μg/ml) for 24 h. Cells were stained with a viability marker and FACS sorted for live cells. Changes in OCR by a mitochondrial stress test (a) and ECAR by a glycolytic stress test (b) are shown. Arrows indicate the time points glucose, 2-DG, Oligomycin, 2,4-DNP and Antimycin/Rotenone (A/R) were added. Data represent three independent experiments each carried out with triplicates. Symbols and error bars represent mean and standard error of the mean respectively. c-h) Purified B cells were stimulated with CpG (1 μM) and/or anti-IgM (5 μg/ml) or left unstimulated for 24h and mitochondrial membrane potentials were measured by staining with TMRM alone or in the presence of oligomycin or FCCP. Representative flow cytometry plots are shown (c) as are the average TMRM values (d) and the percent of the maximum membrane potential used by B cells under each stimulation condition, calculated from a pool of 5 independent experiments with each individual sample represented as a symbol (e). MFI of surface levels of GLUT-1 and GLUT-3 detected by flow cytometry are shown as representative FACS plots (f) and as average fold changes in MFI (g). The fold changes in GLUT-1 and GLUT-3 RNA levels detected by qPCR are given (h). i) MFI levels of GLUT-1 24h post stimulation are shown for B cells isolated from WT and TLR-9 KO mice stimulated with CpG and/or anti-IgM in vitro. j) B cells were cultured in media containing CpG and/or anti-IgM for 24h. During the last 2h of culture 2NBDG was added to the cultures and the MFI of 2NBDG taken up by the cells was measured by flow cytometry at intervals. Data represent three independent experiments each in triplicates. Bars indicate the mean of the triplicates. Data points that are significantly different from the unstimulated conditions are shown with asterisks. (Panels j: two way ANOVA with Dunnet’s adjustment; Panels d,e,g,h,i: one way ANOVA with Tukey’s adjustment) (P>0.05 = n.s.; 0.01 <P ≤0.05 = *;0.001 <P ≤0.01 = **; 0.0001 <P ≤0.001 = ***; P ≤0.0001= ****).
To further characterize the metabolic state of live B cells 24 h after stimulation we measured the mitochondrial membrane potential using TMRM staining in the presence or absence of oligomycin or FCCP in cells stained with a viability dye. Flow cytometry showed that the mitochondrial membrane potential for B cells under all stimulation conditions increased over 24 h as compared to unstimulated B cells (Fig. 2c,d). However, the percentage of the maximum mitochondrial potential used differed significantly between stimulation conditions (Fig. 2e) with anti-IgM treated B cells using the highest portion of their membrane potential (70%) to respond, suggesting mitochondrial stress.
We also determined if B cells had undergone cellular metabolic remodeling by 24 h after stimulation. CpG treatment alone or with anti-IgM resulted in increases in the expression of GLUT 1 and 3 both at the protein level (Fig. 2f,g) and mRNA level (Fig. 2h). The CpG-mediated increases in GLUT 1 and 3 expression were dependent on TLR9 signaling and were lost in B cells from TLR9 KO mice (Fig. 2i, Fig. S4c). Glucose uptake measured by 2NBDG between 22 and 24 h post-stimulation in vitro showed a hierarchy in glucose levels that reflected GLUT 1 and 3 expression (Fig. 2j, Fig. S4d). In contrast, B cells treated with anti-IgM alone showed only modest increases in GLUT 1 or 3 or glucose uptake as compared to untreated cells.
Both mitochondrial mass and volume also correlate with metabolic activity. We quantified mitochondrial mass by flow cytometry after staining with a Live/Dead dye and antibodies specific for the mitochondrial proteins TOM20, the voltage dependent anion channel 1 (VDAC1),18 and cytochrome C oxidase subunit IV (COXIV)19. The greatest increases in mitochondrial mass were evident in B cells stimulated with CpG alone or CpG plus anti-IgM (Fig. 3a, Fig. S4e). Immunoblot analysis of the mitochondrial proteins TOM20, Sirtuin 3 (Sirt3) and heat shock protein 60 (Hsp60) in the lysates of live B cells harvested from 24 h cultures confirmed these increases in mitochondrial mass (Fig. 3b,c).
Figure 3.
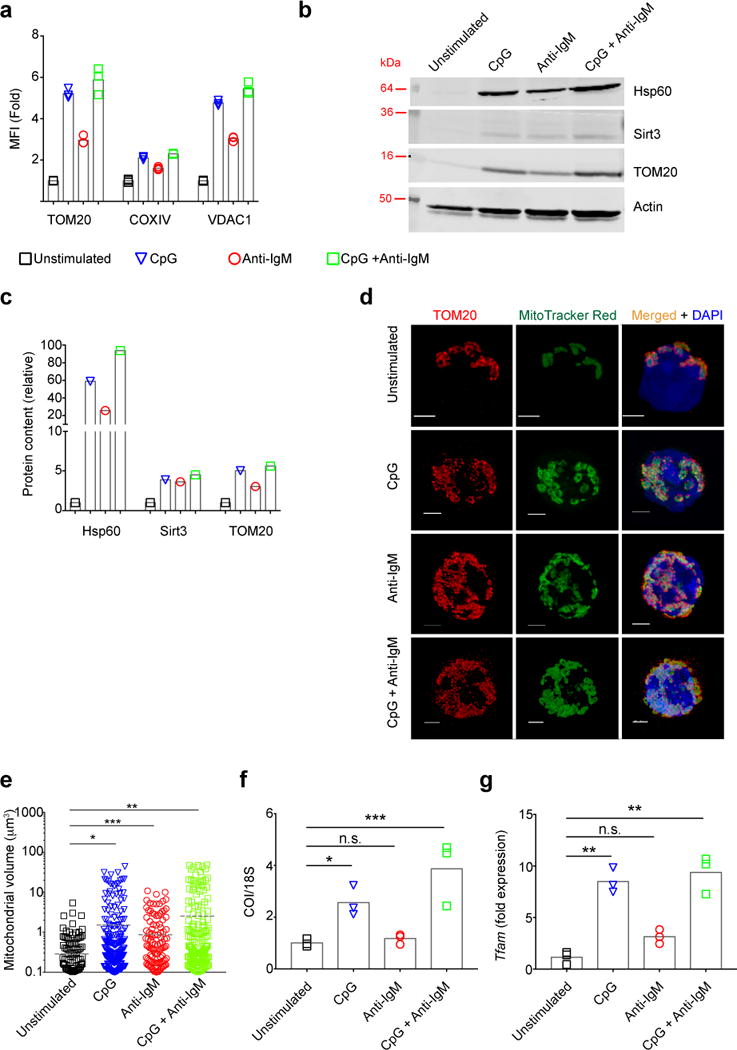
Increases in mitochondrial mass in response to activation through the BCR and/or TLR9. a) Purified B cells were stimulated with CpG (1 μM) and/or anti-IgM (5 μg/ml) or left unstimulated for 24h and stained with Live/Dead marker, fixed, permeabilized and stained with antibodies specific for the mitochondrial markers: TOM20; COXIV; and VDAC1. MFI levels quantified by flow cytometry are shown. Data represent three independent experiments each carried out with three replicates. Bars represent mean. b,c) B cells cultured in vitro for 24 h as in (a) were stained with Live/Dead marker and viable B cells were FACS sorted and lysates from equal number of cells were analyzed by immunoblot for: Hsp60; Sirt3, TOM20 and actin. The immunoblot was cropped guided by the appropriate molecular weight markers. Representative immunoblot (b) and quantification of the blot relative to actin (c) are given. Data represent three independent experiments. d,e) Purified mouse splenic B cells were cultured in vitro for 24 h in media containing CpG (1 μM) and/or anti-IgM (5 μg/ml), stained with Live/Dead marker and MitoTracker Red, washed and plated on poly-L-lysine coated coverslips. Cells were fixed, permeabilized and stained with TOM20-specific antibodies and DAPI and imaged by STED microscopy. Representative microscope images (d) and quantification of mitochondrial volume for each stimulation condition (e) are shown. Scale bar = 2 μm. Data represent two independent experiments. Images of at least 30 viable cells per experiment were analyzed for each condition. Dashed lines represent the mean values. f,g) Purified B cells cultured in vitro for 24 h as in (a) were harvested and total DNA and RNA were isolated. The relative levels of COI DNA to 18S DNA (f) and TFAM gene expressions compared to unstimulated condition were quantified by qPCR (g). Data represent three independent experiments each of which were carried out in triplicates. Bars represent mean. (P>0.05 = n.s.; 0.01 <P ≤0.05 = *;0.001 <P ≤0.01 = **; 0.0001 <P ≤0.001 = ***; P ≤0.0001= ****) (Panels a,f,g: One Way ANOVA with Tukey’s adjustment; Panel e: Mann-Withney test:).
Mitochondrial volumes were determined from 3D reconstructed high resolution stimulated emission depleted (STED) images of B cells cultured for 24 h and then labeled with TOM20-specific antibodies and the mitochondrial dye, MitoTracker CMXROS Red (Fig. 3d). The average volume of mitochondria increased for each stimulation condition with the largest increases observed for B cells stimulated with CpG alone or CpG plus anti-IgM (Fig. 3e).
To assess mitochondrial biogenesis, we determined by qPCR the ratio of mitochondria-encoded gene, cytochrome c oxidase subunit I (COI), and the nuclear gene encoding the 18S ribosomal subunit. We also measured the expression of the mitochondrial transcriptional factor A (TFAM)20, one of the master regulators of mitochondrial proliferation. We observed both an increased COI/18S ratio (Fig. 3f) and an upregulation of TFAM expression (Fig. 3g) for B cells stimulated in culture for 24 h with CpG alone or CpG plus anti-IgM indicating increases in mitochondrial biogenesis. In contrast, anti-IgM stimulation alone did not induce increases in mitochondrial biogenesis.
BCR signaling results in mitochondrial dysfunction in vitro
To further explore mitochondrial function in anti-IgM stimulated B cells we used a second mitochondrial dye, MitoTracker Green. MitoTracker Green is used as a measure of mitochondrial mass10,21–25 but staining can also indicate mitochondrial injury and swelling26,27. Only B cells activated with anti-IgM showed an increase in MitoTracker Green staining over 72 h of culture (Fig. 4a,b) despite the fact that we observed significant increases in mitochondrial mass in cells treated with CpG or CpG plus anti-IgM (Fig. 3a-c) Additionally, MitoTracker Green, as expected specifically localized to mitochondria only (Fig. S5a) and intensity differences between groups did not result from cell proliferation (Fig. S3b,c) rulling out non-mitochondrial causes that might lead to changes in staining and suggesting that the dye indicates mitochondrial injury or swelling not mass in B cells. In B cells from TLR9 KO mice, both anti-IgM and anti-IgM plus CpG treatment resulted in MitoTracker Green staining (Fig. 4c) (Fig. S5b) suggesting that TLR9 signaling prevented anti-IgM-induced increases in staining. MitoTracker Green staining appeared to be a direct consequence of BCR signaling as treatment of B cells with the Syk inhibitor Piceatannol or Btk inhibitor Ibrutinib 8 h post stimulation reduced the MitoTracker Green staining of anti-IgM treated cells at 24 h (Fig. 4d) (Fig. S5c).
Figure 4.
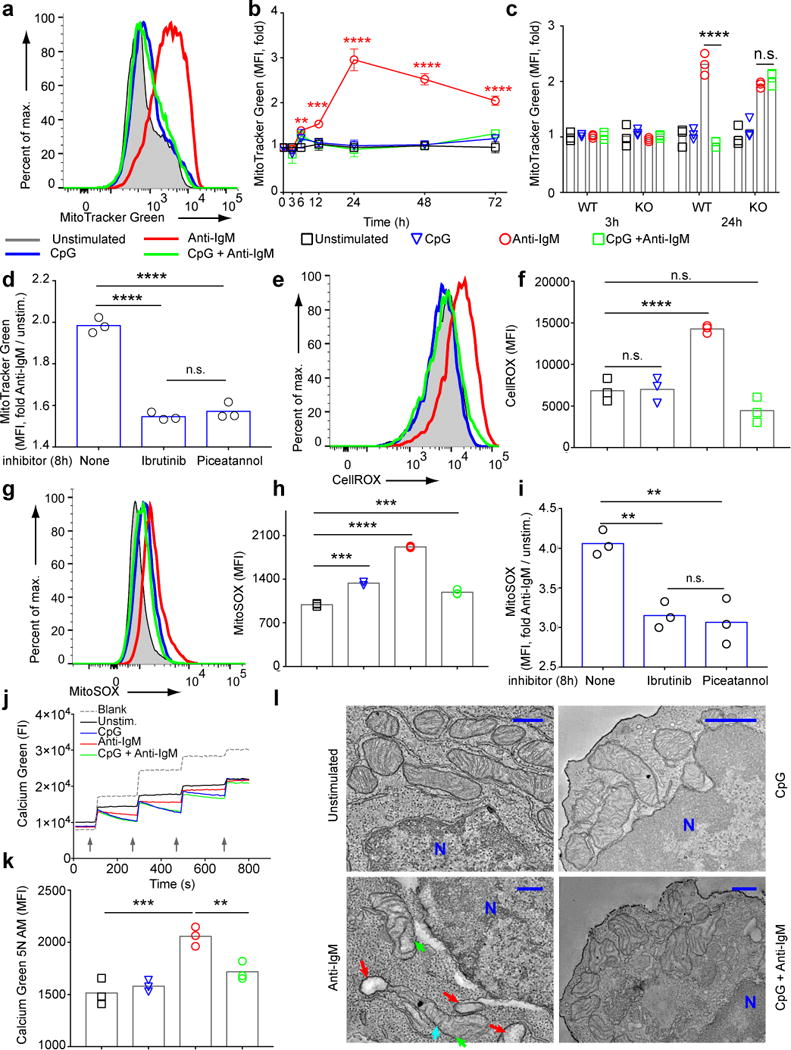
B cells stimulated only through their BCRs in vitro show mitochondrial dysfunction. a,b) Purified B cells were stimulated with CpG (1 μM) and/or anti-IgM (5 μg/ml) or left unstimulated. (a) Flow cytometry plots showing MitoTracker Green staining in the live B cell gate at 24 h post stimulation and (b) quantification of the fold change in MitoTracker Green MFI compared to unstained cells over the course of 72 h culture in vitro are shown. Symbols represent the means of each triplicate and error bars indicate SD. Data represent more than three independent experiments. c) B cells purified from WT and TLR9 KO mice and cultured as in (a) were stained with MitoTracker Green and Live/Dead at 3 h and 24 h post stimulation. Fold changes in MFI are shown. Bars indicate the mean values and error bars the SD of triplicate cultures. Data are representative of three independent experiments. d) B cells were cultured in media containing anti-IgM in the presence or absence of the Btk inhibitor Ibrutinib (1 μM) or the Syk inhibitor Piceatannol (5 μM) added to cultures 8 h post stimulation. Cells were incubated for an additional 16 h, harvested and analyzed for MitoTracker Green FI at 24 h. Fold MFI levels are shown. e-h) Purified B cells were cultured in vitro in the presence of CpG (1 μM) and/or anti-IgM (5 μg/ml) for 24 h. Levels of CellROX (e,f) and MitoSOX (g-i) staining at 24 h are shown as representative flow cytometry plots (e,g) and as the mean and SD of the MFI of triplicate samples (f,h). Each experiment was carried out at least three times with three samples per condition. i) B cells were cultured as in (d) and the fold MFI of MitoSOX staining determined by flow cytometry. j) Mitochondrial calcium uptake was measured in viable B cells purified following 24 h culture in media containing CpG and/or anti-IgM. Cells were pulsed four times with CaCl2 (20 μM) (indicated with arrow heads) and changes in Calcium Green 5N were measured in real time. Data represent three independent experiments. k) B cells were stimulated for 24 h in media containing CpG and/or anti-IgM, harvested and stained with Live/Dead dye and Calcium Green 5N AM to measure intracellular calcium accumulation in viable cells. Bars represent the mean. The experiment was repeated more than three times with at least three replicates per condition. l) B cells were cultured for 24 h in media containing CpG and/or anti-IgM, harvested, fixed and imaged by TEM. Two independent sets of samples were analyzed and at least 10 viable cells per condition were imaged. Swollen mitochondria (red arrow heads), vesicular swollen mitochondria (green arrow heads) and blunted cristae (cyan arrow heads) are shown. N indicates the nucleus. Scale bar = 100 μm. (P>0.05 = n.s.; 0.001 <P ≤0.01 = **; 0.0001 <P ≤0.001 = ***; P ≤0.0001= ****) (Panel b: Two way ANOVA with Dunnet’s adjustment; Panels d,f,i,k: One way ANOVA with Tukey’s adjustment).
It has been well-documented in multiple biological systems that dysregulation of reactive oxygen species (ROS) and/or calcium homeostasis lead to mitochondrial swelling and the resulting mitochondrial dysfunction further aggravates the ROS and calcium irregularities in a positive feed-back cycle leading to death of the cell28–32. Using CellROX, a fluorescent sensor of cellular oxidative stress33, we observed an increased MFI only in anti-IgM stimulated B cells 24 h post activation (Fig. 4e,f). Staining with MitoSOX, a fluorescent superoxide sensor for mitochondrial ROS production34, also increased under each stimulation condition as expected given the increased mitochondrial function but an excessive increase in MitoSOX staining was observed in B cells stimulated with anti-IgM alone (Fig. 4g,h). The anti-IgM-induced increase in MitoSOX staining was a direct consequence of BCR signaling and blocked by treatment with Piceatannol or Ibrutinib (Fig. 4i) (Fig. S5d). To assess the integrity of mitochondria, we measured the ability of the mitochondria to remove excess intracellular calcium using the Calcium Retention Capacity assay on permeabilized cells 35 measuring the fluorescent calcium sensor, Calcium Green 5N. B cells treated with CpG or CpG plus anti-IgM were able to remove excess pulsed calcium efficiently through the first three pulses (Fig. 4j). In contrast, B cells treated with anti-IgM had little capacity to remove excess calcium even after the first pulse. The inability of the mitochondria to remove excess calcium correlated with higher levels of intracellular calcium in B cells cultured for 24 h with anti-IgM as compared to untreated B cells (Fig. 4k) (Fig. S5e).
Electron micrographs of B cells cultured for 24 h in vitro with anti-IgM confirmed mitochondrial injury showing focal loss of cristae and swollen or vesicular swollen matrix areas, alternating with normal mitochondrial structures (Fig. 4l). In contrast, unstimulated B cells and B cells stimulated with CpG or CpG plus anti-IgM showed normal mitochondrial morphology.
Thus, BCR signaling induces mitochondria dysfunction, that correlated with MitoTracker Green staining, as shown by increased ROS production; increased percentage of maximum mitochondrial membrane potential; increased super oxide production; decreased capacity of the mitochondria to remove excess calcium; increased intracellular calcium levels and abnormal mitochondrial morphology.
Antigen induces B cell mitochondrial dysfunction in vivo
To determine if engagement of antigen alone by B cells induced mitochondrial dysfunction in vivo, we generated mice by adoptive transfer that contained e450 stained polyclonal B cells from CD45.1 WT mice or HEL-specific B cells from CD45.2 MD4 mice and immunized these mice 24 h post transfer with PBS, HEL or CpG alone or HEL plus CpG (Fig. S6a,b). Spleens were harvested 24 h later and the adoptively transferred B cells were analyzed by flow cytometry. Polyclonal WT B cells responded to CpG alone and CpG plus HEL but not to HEL alone as shown by an increase in expression of CD69 (Fig. 5a) but did not stain with MitoTracker Green (Fig. 5b,c). In contrast, HEL-specific B cells increased expression of CD69 in response to HEL alone and to CpG alone and showed an enhanced response to the combination of CpG and HEL (Fig. 5a). However, only HEL-specific B cells that responded to HEL alone stained with MitoTracker Green (Fig. 5b,c) and showed increases in both intracellular calcium as measured by Calcium Green (Fig. 5d,e) and ROS levels as measured using MitoSOX (Fig. 5f,g). Thus, the BCR-induced mitochondrial dysfunction observed in vitro can be recapitulated in vivo.
Figure 5.
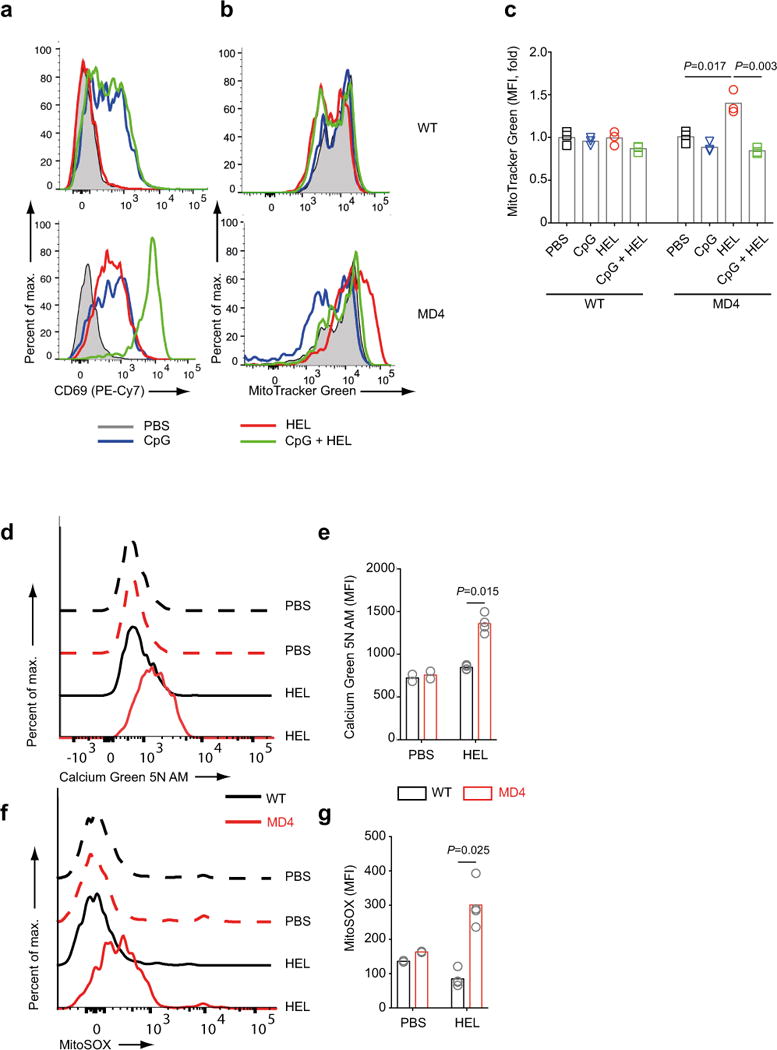
B cells stimulated with antigen alone in vivo show mitochondrial dysfunction. Fluorescently labelled B cells from CD45.1 WT and CD45.2 MD4 mice were mixed and adoptively transferred to WT mice as shown in Fig S6. Flow cytometry plots show the expression of CD69 (a) and MitoTracker Green (b) and quantification of the fold change in MitoTracker Green MFI for WT and MD4 B cells 24 h post i.v. injection with PBS alone, CpG alone, HEL alone or HEL plus CpG (c). Mice were injected with HEL in PBS (100 μg/mouse) or PBS alone and 24 h later splenocytes were stained for CD19, CD45.1, CD45.2 and Calcium Green 5N AM (d,e) and MitoSOX (f,g) and analyzed by flow cytometry. Shown are representative flow cytometry histograms (d,f) and the average MFI values as bar graphs (e,g). Each adoptive transfer experiment was repeated three times with three or more mice per stimulation condition. Bars represent the mean. Symbols represent individual mice. (Panel e: one way ANOVA with Tukey’s adjustment; Panels g,i: Welch’s t-test).
Quality of BCR stimulation dictates mitochondrial dysfunction
Mitochondrial dysfunction as measured by MitoTracker Green staining was significantly higher in B cells exposed to continuous 24 h anti-IgM stimulation as compared to B cells that received only a pulse of anti-IgM (Fig. 6a, Fig. S7a). We also compared the responses of HEL-specific B cells to HEL presented in three different contexts, namely on PLB or plasma membrane sheets (PMS)36 and to transfected cell lines expressing membrane-bound HEL on their surfaces37. HEL in each context induced similar mitochondrial dysfunction in HEL-specific B cells as measured by increases in MitoTracker Green MFI (Fig. 6b).
Figure 6.
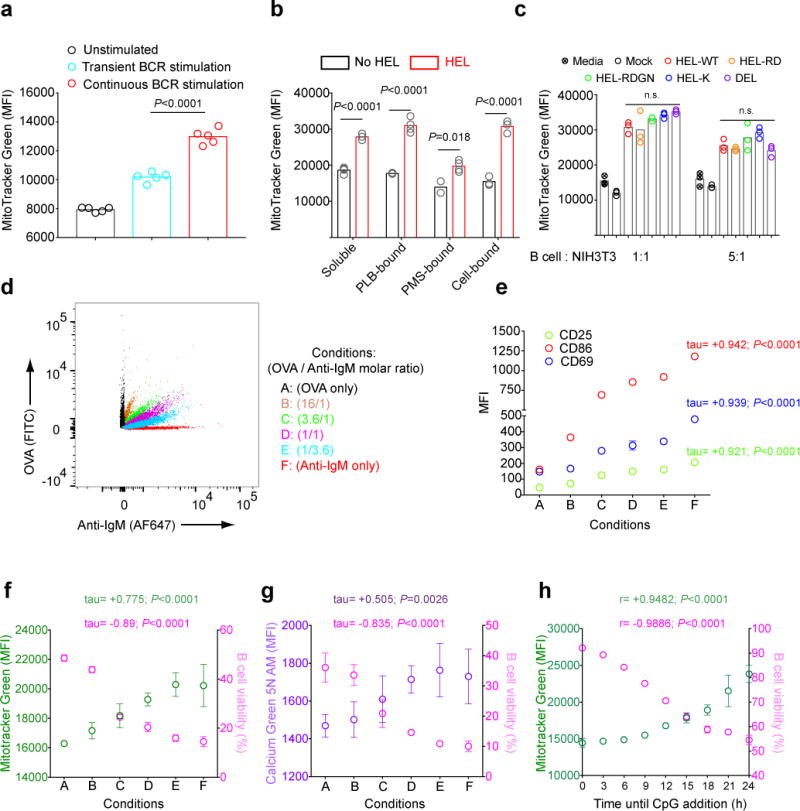
Antigen-induced mitochondrial dysfunction in B cells correlates with the strength and duration of the BCR stimulation a) Purified B cells were incubated on ice in media alone or in media containing anti-IgM (10 μg/ml), washed and resuspended in either media alone or in media containing anti-IgM (10 μg/ml). Cells were warmed to 37°C, cultured for 24 h in vitro. Live cells were obtained by FACS sorting and stained with MitoTracker Green. MFIs of MitoTracker Green in viable B cells are shown. Bars represent mean values and circles represent individual samples. b) HEL-specific B cells from MD4 mice were unstimulated or stimulated for 24 h with HEL in the following forms: soluble (1 μg/ml); PLB-bound; PMS-bound or expressed on the surface of NIH3T3 cells (cell-bound). The B cell MitoTracker Green MFI was measured by flow cytometry. Bars represent mean values. Data represent two independent experiments with three replicates. c) HEL-specific B cells purified from MD4 mice were co-cultured with untransfected NIH3T3 cells (mock) or NIH3T3 cells stably expressing the indicated proteins at cell ratios of 1:1 or 5:1 for 24 h and MitoTracker Green MFI of B cells measured by flow cytometry. Bars indicate mean values. Results are representative of two experiments. d-g) Biotinylated anti-IgM and OVA were mixed at different molar ratios and incubated with streptavidin coated beads (d). Beads were washed and the amounts of OVA versus anti-IgM on the bead surface was determined by flow cytometry using fluorescently labelled antibodies. B cells were incubated with the beads for 24 h in vitro and analyzed for: (e) the expression of the B cell activation markers, CD25, CD86 and CD69; (f) MitoTracker Green staining; (g) Calcium Green 5N AM staining and (f,g) cell viability using Live/Dead stain. Symbols and error bars indicate mean and SD values. Data are representative of three independent experiments each with three replicates per condition. h) B cells were incubated with anti-IgM (5μg/ml) in vitro and CpG was added to cultures at a final concentration of 1 μM at various times after the initiation of the cultures beginning at 3 h. Viability determined by Live/Dead stain and MitoTracker Green MFI were determined at 24 h. Data represent two independent experiments each with three replicates per condition. (P>0.05 = n.s.) (Panel a,c: one way ANOVA with Tukey’s adjustment; Panel b: Welch’s t-test; Panel e-g: Kendall’s tau correlation; Panel h: Pearson’s correlation)
To determine the effect of antigen affinity on the induction of mitochondrial dysfunction, B cells were incubated with NIH3T3 cells expressing equal amounts of HEL-CD4 chimeric proteins containing HEL, Duck Egg Lysozyme (DEL) or one of three different previously characterized mutant forms of HEL38–40 (Fig. S7b). The affinity of HEL-specific B cells for HEL, DEL and the three HEL mutants ranged over 3.5 × 103-fold. We observed no significant differences in the induction of mitochondrial dysfunction in B cells in response to antigens of different affinities (Fig. 6c), suggesting that this function of the BCR was relatively affinity-independent.
To test for the effect of antigen avidity and the strength of BCR cross-linking on BCR-induced mitochondrial dysfunction B cells were incubated with streptavidin coated beads that displayed increasing amounts of anti-IgM with OVA as a filler, as verified by flow cytometry (Fig. 6d). We observed a strong positive correlation between the density of anti-IgM displayed on the beads and the activation of B cells to express activation markers including CD69, CD86 and CD25, after a 24 h incubation (Fig. 6e). We observed a positive correlation between the density of anti-IgM displayed on the beads and mitochondrial dysfunction as measured by the MitoTracker Green (Fig. 6f) and Calcium Green 5N AM staining (Fig. 6g) and negative correlations with B cell viability (Fig. 6f,g).
It is of interest that the viability of B cells stimulated with anti-IgM beads was less than that of B cells incubated with OVA only beads at all concentrations of anti-IgM tested (Fig. 6f,g). Thus, in this culture system BCR-activation-induced cell death (AICD) of primary naïve B cells can be distinguished from death in unstimulated cells, providing the first evidence to our knowledge for AICD in primary B cells.
Both TLR9 signaling and T cell help block AICD
We asked how long CpG addition to anti-IgM treated B cells could be delayed and still maintain B cell viability and mitochondrial function at 24 h. In the absence of CpG (addition at 24 h) anti-IgM treated B cells showed high levels of MitoTracker Green FI and reduced viability (Fig. 6h). Cells to which CpG was added as late as 9 h after anti-IgM stimulation showed MitoTracker Green MFI close to background levels and nearly 80% viability at 24 h (Fig. 6h). After approximately 9 h, B cells showed increasing MitoTracker Green FI and decreasing viability even in the presence of CpG. Thus, anti-IgM induced mitochondrial dysfunction and cell death appear to be a cumulative process that can be blocked by early TLR9 signaling.
To determine whether TH cells could similarly block BCR-induced mitochondrial dysfunction, HEL-specific MD4 B cells were cultured either alone or together with HEL-specific TCR transgenic TH cells from 3A9 mice or 3A9 T cell hybridomas in the presence or absence of HEL for 24 h. The addition of HEL alone to HEL-specific B cells resulted in increased MitoTracker Green MFI (Fig. 7a,b), MitoSOX MFI (Fig. 7c,d) and intracellular calcium levels measured by Fluo-4FF AM MFI (Fig. 7e,f) indicating mitochondrial dysfunction that was not observed in the presence of HEL-specific TH cells. We also observed that CD40L alone was sufficient to block anti-IgM induced increases in MitoTracker Green staining (Fig. 7g,h) and cell death (Fig. 7i) to a similar extent as CpG.
Figure 7.
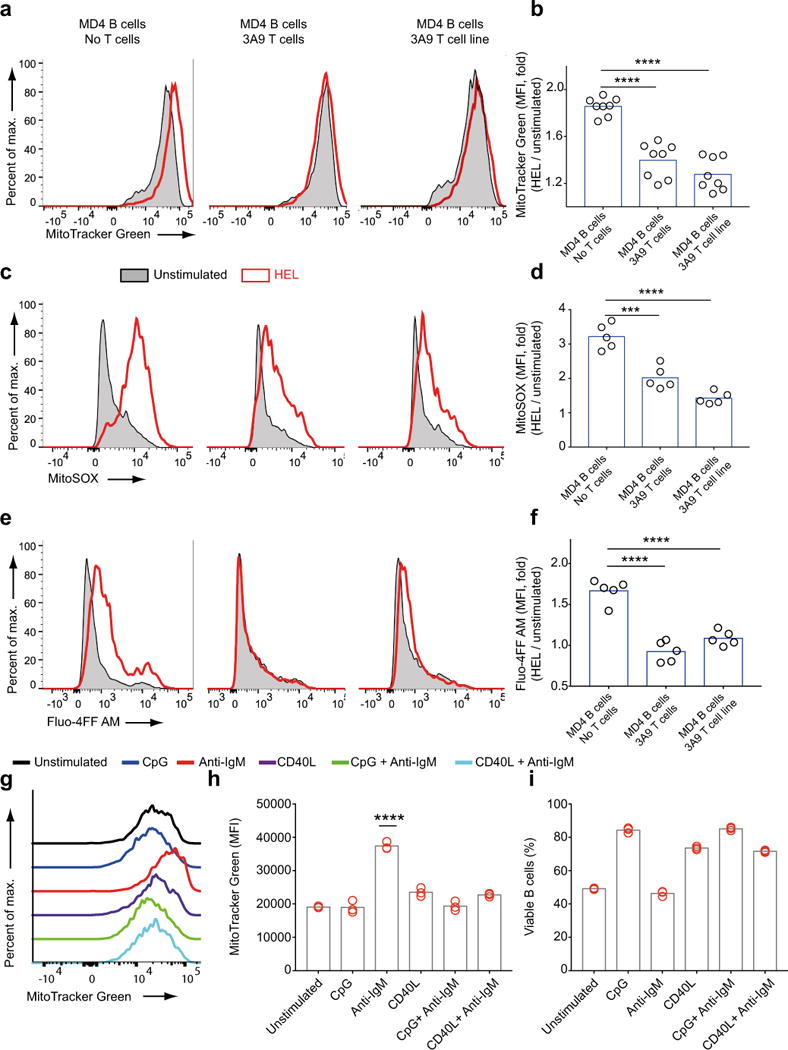
T cell help prevents antigen-induced mitochondrial dysfunction in B cells a-f) HEL-specific B cells from MD4 mice were cultured alone or with equal numbers of HEL-specific CD4+ T cells harvested from 3A9 mice or HEL-specific 3A9 T cell line in the presence or absence of HEL (1μg/ml) for 24h. Fluorescence of Mitotracker Green (a,b), MitoSOX (c,d) and Fluo-4FF AM (e,f) were measured by flow cytometry Representative flow cytometry plots (a,c,e) and fold change in MFI (b,d,f) are shown. Data was pooled from two independent experiments. Each circle represents a sample and bars indicate the means. g-i) Purified B cells were cultured for 24 h in vitro in media alone or media containing combinations of anti-IgM (5μg/ml), CpG (1 μM) and CD40L (1μg/ml), harvested and analyzed by flow cytometry for MitoTracker Green staining shown in representative plots (g). Bar graphs show changes in MitoTracker Green MFI (h) and B cell viability (i) for each condition. MFI values that are significantly different compared to the unstimulated controls are indicated by asterisks. Data are from more than three independent experiments each carried out with at least three replicates per condition. Bars indicate mean. (0.001 <P ≤0.01 = **; P ≤0.0001= ****) (One way ANOVA with Tukey’s adjustment).
Increases in intracellular calcium cause mitochondrial dysfunction
The results presented thus far demonstrate that prolonged BCR stimulation results in loss of mitochondria function. Well characterized causes of mitochondrial dysfunction include excessive increases in intracellular ROS and calcium both of which we demonstrated were induced by BCR signaling (Figures 4,5). To determine the contributions of ROS production to mitochondrial dysfunction, we analyzed by qPCR the transcriptional changes in the expression patterns of several cellular oxidases and antioxidants 4 h post stimulation. The most dramatic increases were in the expression of the genes encoding glutathione reductase (GSR) and both cytoplasmic and mitochondrial forms of superoxide dismutase (SOD-1 and SOD-2) (Fig. 8a) that occurred in B cells treated with CpG or CpG plus anti-IgM but not with anti-IgM alone. SOD-2 is of particular interest as it plays a central role in neutralizing superoxide produced by the activity of the ETC41. We measured SOD-2 activity in mitochondrial fractions of stimulated B cells and observed increases in B cells responding to CpG and CpG plus anti-IgM but not in response to anti-IgM alone (Fig. 8b). This observation suggested the possibility that mitochondrial dysfunction resulted from excess ROS accumulation in the absence of increases in SOD-2 in B cells stimulated through the BCR. If so, we predicted that treatment of anti-IgM activated B cells with antioxidants to reduce ROS levels should reduce mitochondrial dysfunction. We tested the antioxidants N-acetyl cysteine (NAC), ɑ Tocopherol, L-ascorbic acid, either alone or in combination and MitoTEMPO, over a range of concentrations and duration of exposure. None prevented mitochondrial dysfunction (Fig. 8c) suggesting that ROS production may be a consequence not a cause of mitochondrial dysfunction. In contrast, decreasing calcium levels in B cells activated by anti-IgM alone using the calcium chelator BAPTA AM 8h or 16h post-stimulation significantly reduced the MFI of both MitoSOX (Fig. 8d,e) and MitoTracker Green (Fig. 8f,g) staining. In addition, SKF96365, a broad range inhibitor of transient response potential channels42,43, and YM58483, a highly specific store operated calcium entry channel inhibitor that blocks calcium response activated calcium channels (CRACs)1,44 blocked anti-IgM induced increases in MFI of Calcium Green 5N AM (Fig. 8h) (Fig. S7c), MitoTracker Green (Fig. 8i) (Fig. S7d) and MitoSOX (Fig. 8j) (Fig. S7e) staining when added up to 8 h post stimulation for YM58483 (Fig. 8k). Taken together these results reveal a key role for increased intracellular calcium levels mediated by CRAC activity in BCR-induced mitochondrial dysfunction and suggest that increases in ROS production is a result not a cause of mitochondrial dysfunction.
Figure 8.
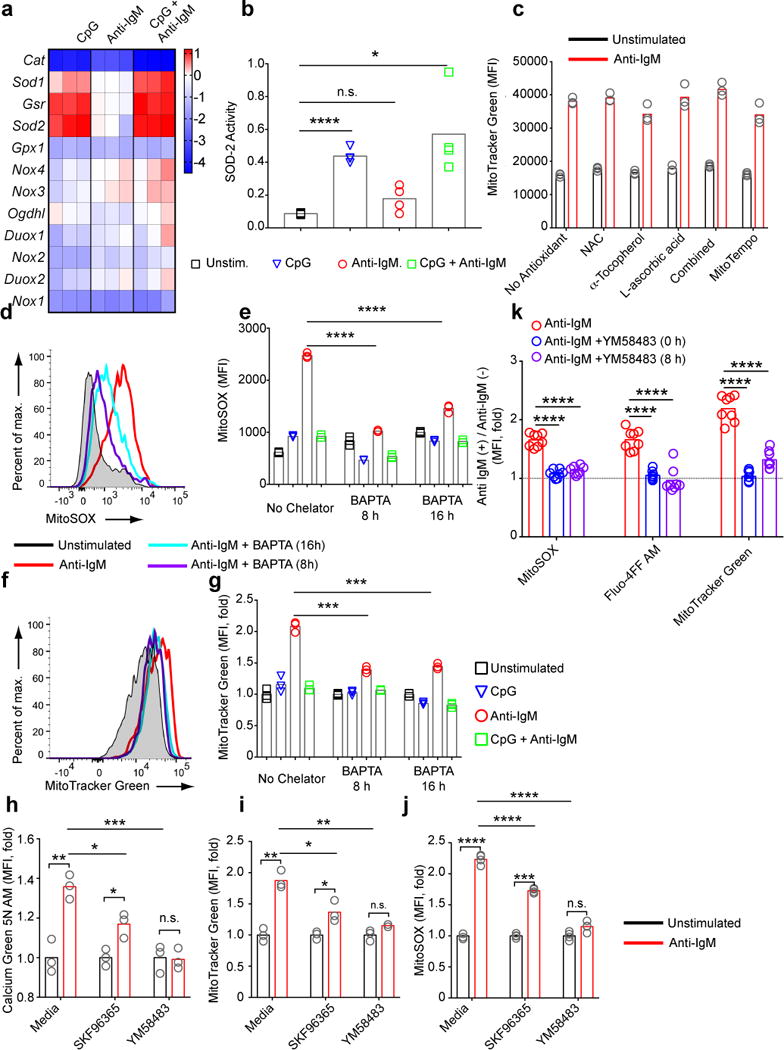
Antigen-induced mitochondrial dysfunction results from increases in intracellular calcium. a) B cells were cultured in media containing CpG (1 μM) and/or anti-IgM (5 μg/ml) for 4h, harvested and the expression levels of mRNAs encoding oxidants and anti-oxidants were measured using qPCR. The heatmap shows the log2 scale change in the expression of genes in stimulated B cells relative to unstimulated cells. Data represent two independent experiments. b) B cells were cultured in media containing CpG and/or anti-IgM for 24 h. Live cells were FACS sorted after Live/Dead staining and SOD-2 activity was measured in cell lysates using a colorimetric assay. Bars represent mean values. Data represent two independent experiments. c) B cells were cultured in media containing anti-IgM (5 μg/ml) in the presence of the compounds indicated for 24 h. Cells were harvested and analyzed in flow cytometry for MitoTracker Green staining. MFI values for each condition are shown. Bars represent mean. Representative of three independent experiments each of which contained three replicates shown with symbols per condition. d-g) B cells were incubated in vitro in media alone or in media containing CpG (1 μM) and/or anti-IgM (5 μg/ml). BAPTA AM (5 μM) was added to culture either 8 h or 16 h after the initiation of culture. Cells were harvested at 24 h and stained with MitoTracker Green (d,e) or MitoSOX (f,g) and for each, representative flow cytometry plots and bar graphs demonstrating the change in staining levels between conditions are shown. Data represent three independent experiments each carried out with three replicates. h-j) B cells were cultured in media or media containing anti-IgM (5 μg/ml) in the presence or absence of SKF96365 (20 μM) or YM58483 (20 μM) for 24h. Levels of Calcium Green 5N AM (h), MitoTracker Green (i), and MitoSOX (j) staining are shown. Bars represent means. Data are representative of more than three independent experiments each carried out with three replicates per condition. k) Purified mouse splenic B cells were cultured in the presence or absence of anti-IgM (5 μg/ml). YM58483 (20 μM) was added to samples at either 0 or 8 h post stimulation. Samples were stained for MitoSOX, Fluo-4FF AM and MitoTracker Green at 24 h post stimulation and bar graphs show the fold change in MFI of these stains. Each circle represents an individual sample and bars show the mean values. (P>0.05 = n.s.; 0.01 <P ≤0.05 = *;0.001 <P ≤0.01 = **; 0.0001 <P ≤0.001 = ***; P ≤0.0001= ****) (Panels b,e,g,k: one way Anova with Tukey’s adjustment; Panels h-j:Welch’s t-test for pair-wise comparisons and one way Anova with Tukey’s adjustment for multiple comparisons.).
DISCUSSION
Here, we presented the results of an extensive analysis of metabolic changes in B cells following BCR and TLR9 signaling that not only produced a detailed picture of the metabolic changes induced in B cells in response to activating signals but also provided new insights into the molecular mechanisms that underlie AICD and the two-signal requirement for full B cell activation. We demonstrated that upon engaging antigen, B cells rapidly increase their glycolytic and glucose oxidative metabolism from existing stores. At the same time B cells begin a process of cellular remodeling to increase their mitochondrial and glycolytic capacities as indicated by changes in transcriptional patterns of genes that mediate these metabolic processes. We speculate that these changes are in anticipation of a second signal provided either by antigen-specific TH cells or through TLR9 signaling that induce rapid proliferation and differentiation to antibody-secreting cells. However, in the absence of a costimulatory second signal, at 24h post-stimulation the mitochondria in BCR-activated B cells lose function and within 48h B cells undergo apoptosis. Mitochondrial dysfunction appeared to be the consequence of a gradual increase over hours in intracellular calcium through CRAC activity. We speculate that a positive feedback cycle between mitochondrial calcium levels and ROS production exacerbates the mitochondrial dysfunction leading to complete dysfunction of mitochondria and the eventual apoptotic death of the B cell. We showed that B cells provided with a second signal within approximately nine hours of the initiation of BCR signaling are rescued from mitochondrial dysfunction. We suggest that the gradual increase in intracellular calcium acts as a metabolic clock that limits the window of time during which BCR-activated B cells must acquire a second signal to survive.
Our description of the apoptosis of B cells following activation through the BCR in the absence of a costimulatory second signal is similar to the phenomenon AICD in lymphocytes. Understanding AICD in primary naïve B cells at a mechanistic level has been difficult in part, because both unstimulated B cells and B cells stimulated with anti-IgM undergo apoptosis at a similar pace in most conditions in vitro precluding the ability to correlate death with activation to soluble antigens. Consequently, our current understanding of AICD in B lymphocytes comes primarily from studies carried out using B cell lines45 that provided evidence that AICD was due to growth arrest. Here we asked whether the viability of BCR-activated B cells depends on the strength of BCR stimulation and if so, whether there is a point at which apoptosis of B cells activated through the BCR exceeds that of unstimulated B cells. We determined that B cell viability decreased with increases in the strength of BCR stimulation and that compared to unstimulated B cells, the survival rates of B cells stimulated with anti-IgM adsorbed to beads were significantly lower even for the lowest density of anti-IgM tested. To our knowledge this result provides the first evidence of AICD in primary naïve B lymphocytes. Furthermore, these data provide a mechanism for AICD showing that increased levels of intracellular calcium and mitochondrial dysfunction correlate with AICD.
Here we focused on the effect of BCR signaling on metabolic changes in naïve B cells and come to the conclusion that BCR signaling activates metabolic pathways in cells that prepare them for the prospect of receiving a second signal, from antigen-specific TH cells, for example, that will drive the cell into energy consuming proliferation and differentiation. In this sense the BCR is an activating receptor. However, if the B cell fails to acquire a second signal in a restricted time period, the very BCR signals that induced metabolic change, namely, calcium increases, begin to act as a ‘death clock’ that results in apoptosis if not blocked by a second signal. We see BCR-induced activation and apoptosis as two sides of the same coin. Another fascinating example of this duality is the ability of the BCR to rescue CD40-activated B cells from Fas-mediated apoptotic death46–48 . This mechanism is thought to play a role in antigen rescue of bystander B cells that have been inadvertently activated by CD4+ T cells. The findings presented here may also be pertinent to our recent observation that TLR9 signaling triggered by CpG antagonized the ability of naïve B cells to process and present antigen to TH cells both in vitro and in vivo and acquire a second signal that allows affinity maturation16. Taken together with the results presented here, we propose that TLR9 agonists block B cell presentation of antigen to T follicular helper cells and consequently block B cell acquisition of a second signal from T cells. At the same time TLR agonist independently provide a second signal and promote proliferation and differentiation to low affinity antibody secreting cells.
In summary, the results described here not only provide new information concerning the metabolic remodeling and its timeline that accompany B cell activation but also put these changes into the context of the two-signal model of B cell activation and AICD. We conclude that B cells undergo metabolic changes following antigen engagement in preparation for the energetic and metabolic requirements of proliferation and differentiation that result from provision of a second signal. However, as a safeguard, if BCR-activated B cells cannot acquire a second signal they undergo apoptosis triggered by the very events that prepare the cells for full activation.
ONLINE METHODS
Animals and Cells
C57BL/6 (Stock No: 00664), B6 CD45.1 (Pep Boy) (Stock No: 002014) and B10.BR (Stock No :00465) were purchased from Jackson Laboratory. Hen egg lysozyme (HEL)-specific BCR transgenic mice (MD4), were purchased from Taconic Farms. TLR9-deficient mice were provided by S. Bolland (NIAID, NIH). HEL-specific TCR transgenic mice (3A9) were provided by I. Gery (NEI, NIH). MD4 mice were either kept on MHC class II Ab haplotype –for in vitro B cell stimulation experiments and adoptive transfers– or in MHC class II Ab × Ak haplotype –for B cell- T cell co-culture experiments– by breeding with B6 or B10.BR, respectively. All mice were bred and maintained at NIAID animal facilities according to Animal Care and Use Committee (ACUC) standards. Experiments involving mice were approved by NIH ACUC.
The NIH/3T3 mouse fibroblast cell line (ATCC CRL-1658) and 3A9 mouse T cell hybridoma line (ATCC CRL-3293) were provided by O. Voss (NIAID, NIH) and P. Allen (Washington University), respectively. Phoenix Eco retroviral packaging cell line (ATCC CRL-3214) was purchased from American Type Culture Collection. B cells and naïve T cells were purified from mouse spleens using negative magnetic selection strategies explained in detail in49,50. Cells were maintained in vitro in complete culture media (RPMI 1640 media supplemented with 10% fetal calf serum, 10 mM HEPES pH7.3, 50 U/ml penicillin, 50 μM streptomycin, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate and 50 μM β-mercaptoethanol) in a humidified 5% CO2 tissue culture incubator at 37 °C unless otherwise specified.
Reagents and Antibodies
ODN 1826 (Invivogen) was used for TLR-9 stimulation. Fab’2 donkey Anti-mouse IgM (catolog no: 715-006-020) (Jackson ImmunoResearch) or Hen Egg Lysozyme (Sigma-Aldrich) were used for BCR stimulation. Recombinant multimeric mouse CD40L (AdipoGen Life Sciences) was used to simulate T cell help. Biotinylated anti mouse IgM (catalog no: 115-066-075) (Jackson ImmunoResearch), biotinylated OVA (Nanocs) and streptavidin-coated particles (SVM025-5H) (Spherotech) were used in bead-based stimulation assays. 2-deoxy-D-glucose (2-DG) (Sigma-Aldrich), oligomycin (Cell Signaling Technology) were used to inhibit metabolic pathways. Piceatannol, SB202190, SP600125, Wortmannin, PD98059, and Perifosine (InvivoGen) were used to inhibit signaling activities of Syk, p-38, JNK, PI3K, MEK1/2 and Akt, respectively. N-acetyl cysteine (NAC) (Sigma-Aldrich), alpha Tocopherol, L-ascorbic acid (Sigma-Aldrich) and MitoTEMPO (ThermoFisher) were used as anti-oxidants in certain experiments. Cell permeant calcium chelator BAPTA, AM (ThermoFisher) and ion channel blockers SKF96365, YM58483 (Sigma-Aldrich) were used to address the role of calcium in BCR-mediated mitochondrial changes. Flow cytometric assessment of the internalization/compartmentalization of BCR-bound antigen was carried out by coating MD4 B cells with pHrodo Red Avidin (ThermoFisher) conjugated biotinylated HEL (Sigma-Aldrich) on ice and then monitoring the change in red fluorescence due to internalization after bringing the cells back to 37 °C. 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose (2NBDG) (ThermoFisher) was used to assess the real time glucose uptake of cells by flow cytometry. Antibodies against CD19 (clone: 6D5), CD69 (clone: H1.2f3), CD4 (clone: RM4-5), CD45.1 (clone:A20), CD45.2 (clone:104), CD25 (clone: PC61), CD16/32 (clone 93) and B220 (clone:RA3-6B2), CD86 (clone: GL-1) (BioLegend); ovalbumin (catalog no: Ab85584),TOM20 (clone F10), COXIV (clone: 20E8C12), VDAC1 (clone: N152B/23) (Abcam); goat IgG light chain (catalog no:205-602-176) and mouse IgG2a (catalog no:115-607-186) (Jackson ImmunoResearch) were used in various flow cytometry experiments. Analysis of different stages of cell death was carried out using a necrosis vs apoptosis kit (Immunochemistry Technologies) according to manufacturer’s protocol.
Measurement of Mitochondrial Mass and Membrane Potential
Flow cytometric measurement of mitochondrial mass was carried out by staining the cells with a LIVE/DEAD fixable dead cell stain (ThermoFisher) and phenotyping antibodies, if required, followed by fixation and permeabilization using the BD Cytofix/Cytoperm™ kit (BD Biosciences). Permeabilized cells were then stained with antibodies against mitochondrial markers. For markers that require permeabilization of mitochondrial membranes a 1/100 dilution of Triton X-100 was used. For detection of mitochondrial mass using immunoblot, cultured cells were stained with the live dead marker and live cells were sorted by flow cytometry. Equal numbers of live cells from different conditions were lysed using RIPA buffer, separated by SDS-PAGE, and transferred to nitrocellulose membranes. The following antibodies were used: β-actin (A1978, Sigma-Aldrich), HSP60 (4870S, Cell Signaling Technology), SIRT3 (5490S, Cell Signaling Technology), and TOM20 (sc-11415, Santa Cruz Biotechnology). SeeBlue Plus2 Pre-stained protein standard (ThermoFisher) was used for band size estimation, Images were captured using the Odyssey system (Li-Cor).
For mitochondrial membrane potential measurement, cells were cultured in 96-well plate in phenol red-free culture media at a density of 2.5 × 105 cells per well at 37 °C for the required amount of time. A final concentration of 6 μM oligomycin or 5 μM FCCP was added each to one of the three wells of containing identically cultured cells. An equal volume of media was added to the third well. After 10 min of incubation at 37 °C, tetramethylrhodamine, methyl ester, perchlorate (TMRM) (ThermoFisher) was added to all wells at a final concentration of 30 nM and Sytox Blue Dead Cell Stain (ThermoFisher) was added at 1 μM. Following an additional 30 min incubation, cells were rapidly analyzed in flow cytometry. The percentage of the maximum mitochondrial potential was calculated using the algorithm: [100 × (MFI (TMRM alone)-MFI (TMRM+FCCP) )/(MFI (TMRM + Oligomycin) − MFI (TMRM+FCCP) )].
qPCR
For analysis of gene expression using qPCR, RNA was isolated from in vitro cultured B cells using RNeasy Plus Micro Kit (Qiagen), cDNA was generated using iScript™ Reverse Transcription Supermix (Bio-Rad) and depending on the choice of primers either iQ™ SYBR® Green Supermix (Bio-Rad) or Platinum® Quantitative PCR SuperMix-UDG (ThermoFisher) kits were used to amplify the target genes. Analysis of the fold change (target over control) was carried out using the comparative CT method. The list of primers and probes are shown in Supplementary Table 1. For quantification of the ratio between mitochondrial and genomic DNA, DNA was isolated from cultured B cells using DNeasy Blood and Tissue Kit (Qiagen) and qPCR was carried out with different dilutions of DNA using iQ™ SYBR® Green Supermix.
Measurement of ROS production and MitoTracker Green Fluorescence
For MitoTracker Green staining, cultured cells were re-suspended in RPMI without phenol red supplemented with 2% FCS, 1% HEPES, pH7.3 and 1/250 dilution of LIVE/DEAD Fixable Near IR dye (ThermoFisher). Cells were incubated at 4 °C for 20 min. After the incubation, cells were washed and resuspended with pre-warmed freshly made MitoTracker stain buffer (RPMI without phenol red supplemented with 2% FCS, 1% HEPES, pH7.3 and 40 nM MitoTracker Green (ThermoFisher)). Following incubation at 37 °C for 30 min, cells were spun down and resuspended with RPMI without phenol red supplemented with fresh 2% FCS and 1% HEPES (pH7.3) and immediately analyzed in flow cytometry. MitoSOX staining was carried out in HBSS supplemented with 5 μM MitoSOX Red (ThermoFisher) at 37 °C for 30 min. Cells were then washed three times with plain HBSS and resuspended with HBSS supplemented with 1 μM SYTOX Blue Dead Cell Stain for an additional 10 min incubation at 37 °C. At the end of this incubation cells were rapidly analyzed in flow cytometry. CellROX staining was carried out after washing the cultured cells once and resuspending them in full growth media prepared with phenol red free RPMI and supplemented with 500 nM CellROX (ThermoFisher). Cells were incubated at 37 °C for 45 min. Sytox Blue was added at 1 μM final concentration in the last 10 min of the incubation. Following this, cells were rapidly analyzed on a BD LSR II flow cytometer.
Injections and Immunizations
For adoptive transfer experiments, B cells purified from spleens of CD45.1 WT mice and CD45.2 MD4 mice were mixed at a 1:1 ratio, stained with e450 cell proliferation dye (ThermoFisher) according to the manufacturer’s recommendations, and resuspended in PBS. 200 μl PBS containing 4.5 × 106 e450-stained B cells were transferred into a CD45.2 WT mouse via tail vein injection. 24 h post-transfer mice were injected with 200 μl PBS alone or PBS supplemented with 95-100 μg/mouse ODN1826 and/or 100 μg/mouse HEL. 24 h post-stimulation spleens were harvested and splenocytes were used in downstream assays.
Extracellular Flux assay
For extracellular flux assays, we used a standardized protocol to measure OCR and ECAR by a Seahorse XF 96 analyzer as described recently15. Briefly, 5 × 105 cells B cells per well were resuspended in Seahorse medium and plated in 96-well Cell-Tak (Corning)-coated Seahorse plates. Cells were maintained at 37 °C in a non-CO2 incubator for at least 1 h before the assay. For the mitochondrial stress test, cells were treated sequentially with 1 μM oligomycin (an inhibitor of the ATP synthase), 0.1 mM 2,4-dinitrophenol (DNP, an uncoupler) and 1 μM Rotenone plus 1 μM antimycin A (inhibitors of complex I and III of the respiratory chain respectively). For the glycolysis stress test, glucose-starved cells were treated sequentially with 10 mM glucose, 1 μM rotenone plus 1 μM antimycin A and 50 mM 2-DG, an inhibitor of glycolysis.
SOD-2 and PDH activity assay
SOD activity was tested in equal numbers of live B cells using the SOD assay kit (Cayman Chemical). To ensure the detection of only Mn-SOD (SOD-2) activity, 5 mM potassium cyanide was added to the assay to inhibit cytosolic Cu/Zn-SOD (SOD-1) and extracellular Cu/Zn-SOD (SOD3) activities.
PDH activity was measured using PDH Enzyme Activity Microplate Assay Kit (Abcam) according to manufacturer’s guidelines using the lysates from 2.5 × 107 B cells stimulated for 6 h in vitro per condition.
Measurement of cellular calcium levels and mitochondrial calcium uptake
To measure changes in cellular calcium concentrations B cells were stained with 2-7 μM cell permeant Calcium Green, 5N AM (ThermoFisher) or 2-20 μM Fluo-4FF AM (ThermoFisher) together with Live/Dead stain and phenotyping antibodies at 4 °C for 20 min. Cells were then washed and analyzed by flow cytometry.
Mitochondrial calcium uptake was measured using a strategy described elsewhere 51. Briefly, in vitro cultured B cells were stained with Live/Dead Near IR and FACS sorted for live cells. 2 × 106 viable cells per condition were resuspended in 100 μl intracellular buffer (containing 10 mM HEPES, pH7.2, 0.25 M sucrose, 2 mM K2HPO4, 1 mM DTT). Cell impermeant-Calcium Green 5N (ThermoFisher) was added to the suspension at 0.1 μM final concentration. Digitonin was then added at 100 μM final concentration to permeabilize the cells, and mitochondria were energized by addition of succinate (5 mM) + rotenone (1 μM). A FLUOstar Omega Microplate Reader (BMG LABTECH) provided with temperature control (30 °C) and continuous stirring was used to monitor the fluorescence of Calcium Green (excitation 506 nm, emission 532 nm) and thus the extramitochondrial calcium. Cells were pulsed with 20 μM/pulse calcium chloride and calcium uptake into mitochondria was monitored in real time (as a decrease in fluorescence) until the onset of permeability transition.
Preparation of antigen containing plasma membrane sheets (PMS) and planar lipid bilayers (PLBs)
PMS were prepared from 293A cells as a protocol previously described in52 was carried out. PMS plated on coverglass were sequentially incubated with 0.5 μg/ml biotinylated Annexin V (BioLegend), 5 μg/ml Streptavidin (ThermoFisher) and 50 nM Alexa Fluor 488-conjugated biotinylated HEL with washing between steps with 15 mL HBSS solution containing 0.2% BSA and 2.5 mM Ca2+. PLBs containing biotinylated lipids were generated as previously described in53. 10 nM Alexa Fluor 488-labeled biotinylated HEL was added to PLB and was incubated at RT for 30 min in order to facilitate incorporation. At the end of this period, unbound HEL was removed by washing.
Preparation of PLB-bound antigen, TIRF imaging and image analyses
Time-lapse live cell total internal reflection fluorescence (TIRF) imaging of mouse splenic B cells from MD4 mouse labeled for surface BCR with goat Fab anti-mouse IgM conjugated with DyLight549 and placed on HEL-containing PLB was performed as described54. Image acquisition began right after the cells touched the surface and recorded with 7 s interval for 30 min. Image J software was used for the determination of fold change of BCR accumulation on the contact site and size of contact area. Background subtraction and then threshold setting for the positive signals above background noise were performed for all images before the measurement of BCR MFI and contact area using ImageJ FIJI software. BCR accumulation was determined by fold change of BCR MFI at a given time relative to that of the clusters at the first contact time point. The effect of metabolic inhibitors was compared by the B cell response in the absence or the presence of either 55 mM 2-DG or 6 μM oligomycin in PLB-Ag chamber.
Generation of NIH3T3 cell lines expressing recombinant lysozyme proteins
DNA constructs containing full length codon optimized sequences coding for WT Hen Egg Lysozyme (HEL) (Accession no: AAA48943.1) or WT Duck Egg Lysozyme (DEL) (Accession no: 0802160A) followed by a serine-threonine linker and then by the third and fourth transmembrane and intracellular domains of rat CD4 (residues 206-457) (Accession no: P05540.1) were synthesized by Geneart Gene Synthesis (ThermoFisher). Constructs containing different affinity mutations of HEL were generated simply by making the (K97A for HEL-K), (R21A, D101A for HEL-RD) and (R21A, D101A, G102A, N103A for HEL-RDGN) modifications on the initial WT HEL design. The numbers refer to the positions of the relevant amino acids after removal of the leader sequence. The final constructs were subcloned into a Pfb-Hygro retroviral vector. Phoenix Eco packaging system was used to generate viruses for transducing NIH3T3 cells. Transduction and hygromycin selection of NIH3T3 cell line was carried out as outlined in55.
STED microscopy
In vitro cultured B cells were stained with Live/Dead Green (ThermoFisher) and MitoTracker Red CMXROS (at 150 nM final concentration) (ThermoFisher) as explained above for MitoTracker Green. Cells were then, resuspended in HBSS and 106 cells were added to coverslips previously coated with poly-L-lysine (Sigma-Aldrich and were incubated at RT for 10 min. Unbound cells were removed and cover slips were blocked by washing twice with staining buffer (HBSS supplemented with 1% HEPES, pH7.3, 2% heat-inactivated FCS, and 0.09% sodium azide). Cells were then fixed and permeabilized using BD CytoFix/Cytoperm Kit (BD Biosciences). Intracellular staining for TOM20 was carried out using Rabbit anti mouse TOM20 (catalog no: 56783)(Abcam) followed by ATTO-647N-conjugated anti rabbit IgG secondary (catalog no: 40839) (Sigma-Aldrich). DAPI staining was done next in PBS containing 300 nM DAPI for 5 min. Following a wash, mounting solution, prepared by mixing Mowiol 4-88 (Milipore Sigma) with p-phenylenediamine according to the manufacturer’s guidelines, was used to mount the coverslips onto the slides. Images were collected on a Leica TCS SP8 STED 3X system equipped with white light and UV excitation lasers, a pulsed 775 nm depletion laser, a HC PL APO 100×/1.40 oil STED white objective, and gated HyD detectors. Images were further deconvolved using Huygens de-convolution software. Volumes smaller than 0.01 μm3 were arbitrarily excluded from the analysis but the results of excluded and non-excluded volumetric analysis were similar (data not shown).
Imaging of MitoTracker Green staining using confocal microscopy
B cells purified from WT C57BL/6 mice were in vitro cultured in various activation conditions for 24h. Cells were then washed with plain RPMI 1640 media without phenol red and stained with LIVE/DEAD Fixable reagent (ThermoFisher) at 4 °C for 30 min followed by MitoTracker Green staining. After one wash cells were plated onto poly-L-lysine coated 8-chamber Lab-Tek II chambered #1.5 German coverglass system (Nunc). After a 5 min incubation at 37 °C, media was aspirated and replaced with 400 μl/chamber complete culture media prepared with RPMI without phenol red. The plate was then placed into a Zeiss LSM 780 confocal microscope enclosed inside a 37 °C, 5% CO2 humidified incubation chamber and z-stack images were acquired.
Transmission Electron Microscopy
In vitro cultured B cells were fixed in Karnovsky’s fixative (Electron Microscopy Sciences) and processed according to the previously published microwave irradiation strategy56 with the following modifications: Cells were centrifuged at 800g between steps, cells were infiltrated in Araldite resin (SPI, Inc.) and tomography sections were cut at 150 nm thickness and imaged with no post-section staining. An Ultrascan 4000 camera (Gatan, Inc.) with SerialEM acquisition and control software (University of Colorado, Boulder, CO)57 was used to collect the tomographic tilt series.
Data Analysis
Flow cytometry data was analyzed using Flowjo software (Tree Star, Inc.). Confocal and STED microscopy data were analyzed using Imaris (Bitplane). Dead cells were excluded from both microscopy and flow cytometry analyses based on their staining with the relevant live dead marker. Statistical significance analyses were carried out using The R Project 3.4.3, JMP 13.2.0 and GraphPad Prism 7.03 software. The statistical methods used for each experiment were indicated in relevant figure legend. Briefly, many data sets were tested with repeated measures mixed model two-way ANOVA using Tukey-adjusted post hoc tests or Dunnett-adjusted post hoc tests against the unstimulated or no treatment control group. Fixed effect one-way ANOVA with Tukey-adjustment was used in single factor experiments, where repeated sampling over time was not used. Sometimes multiple fixed-effect one-way ANOVAs were paneled over multiple independent experiments. In other examples, fixed effect one-way ANOVA was only used on anti-IgM samples, because unstimulated controls were constrained to Fold MFI = 1 and included on the graph only as reference groups. Linear regression, cubic polynomial regression, Kendall’s tau correlations and Pearson’s correlations were used in situations where it seemed more relevant to compare the shape or direction of a relationship. Multiple Bonferroni-adjusted one-sided t-tests were used when it was most important to identify groups with responses higher than the unstimulated or untreated control group. Nonparametric Mann-Whitney tests were used when the normal distribution of data was in doubt. Welsh-corrected Student’s t-tests were used in situations where single independent experiments were compared, In all figures, **** P < 0.0001, *** P < 0.001, ** P < 0.01, * P < 0.05 and “n.s.” P > 0.05 are used to denote statistical significance.
Data availability statement
Further details of the experimental procedures are provided in the Life Sciences Reporting Summary. The data that support the findings of the study are available from the corresponding author upon request. RNA-seq data SRA database under project number: PRJNA422889.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Intramural Research Program, National Institute of Allergy and Infectious Diseases and National Heart, Lung, Blood Institute . Authors thank R. Kissinger for making the illustration in Supplementary Fig. 2; P.W. Sheehan, T. Leto, J. Brzostowski and J. Manzella-Lapeira for assistance and advice in various experiments.
Footnotes
AUTHOR CONTRIBUTIONS
Conceived the project: MA, SKP.
Secured Funding: SKP.
Designed the experiments: MA, SKP, ASR, JT, PM, BPT
Carried out experiments: MA, ASR, JT, PM, BA, BPT, MP, HS, MS, DWD
Analyzed data: MA, ASR, JT, PM, BPT, ED, BA, JK, HS, JS
Wrote the manuscript: MA
Edited Manuscript: SKP, MNS
COMPETING INTERESTS STATEMENT.
Authors declare no competing financial interests.
References
- 1.Scharenberg AM, Humphries LA, Rawlings DJ. Calcium signalling and cell-fate choice in B cells. Nat Rev Immunol. 2007;7:778–789. doi: 10.1038/nri2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol. 2015;15:137–148. doi: 10.1038/nri3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuseff MI, Pierobon P, Reversat A, Lennon-Dumenil AM. How B cells capture, process and present antigens: a crucial role for cell polarity. Nat Rev Immunol. 2013;13:475–486. doi: 10.1038/nri3469. [DOI] [PubMed] [Google Scholar]
- 4.Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol. 2015;15:203–216. doi: 10.1038/nri3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elgueta R, et al. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–172. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruprecht CR, Lanzavecchia A. Toll-like receptor stimulation as a third signal required for activation of human naive B cells. Eur J Immunol. 2006;36:810–816. doi: 10.1002/eji.200535744. [DOI] [PubMed] [Google Scholar]
- 7.Chaturvedi A, Pierce SK. How location governs toll-like receptor signaling. Traffic. 2009;10:621–628. doi: 10.1111/j.1600-0854.2009.00899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rawlings DJ, Schwartz MA, Jackson SW, Meyer-Bahlburg A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat Rev Immunol. 2012;12:282–294. doi: 10.1038/nri3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akkaya M, et al. B Cells Produce Type 1 IFNs in Response to the TLR9 Agonist CpG-A Conjugated to Cationic Lipids. J Immunol. 2017;199:931–940. doi: 10.4049/jimmunol.1700348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caro-Maldonado A, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. 2014;192:3626–3636. doi: 10.4049/jimmunol.1302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caro-Maldonado A, Gerriets VA, Rathmell JC. Matched and mismatched metabolic fuels in lymphocyte function. Semin Immunol. 2012;24:405–413. doi: 10.1016/j.smim.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lam WY, et al. Mitochondrial Pyruvate Import Promotes Long-Term Survival of Antibody-Secreting Plasma Cells. Immunity. 2016;45:60–73. doi: 10.1016/j.immuni.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doughty CA, et al. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood. 2006;107:4458–4465. doi: 10.1182/blood-2005-12-4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao G, et al. B-Cell-Specific Diversion of Glucose Carbon Utilization Reveals a Unique Vulnerability in B Cell Malignancies. Cell. 2018;173:470–484 e418. doi: 10.1016/j.cell.2018.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Traba J, Miozzo P, Akkaya B, Pierce SK, Akkaya M. An Optimized Protocol to Analyze Glycolysis and Mitochondrial Respiration in Lymphocytes. J Vis Exp. 2016 doi: 10.3791/54918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akkaya M, et al. Toll-like receptor 9 antagonizes antibody affinity maturation. Nat Immunol. 2018;19:255–266. doi: 10.1038/s41590-018-0052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fleire SJ, et al. B cell ligand discrimination through a spreading and contraction response. Science. 2006;312:738–741. doi: 10.1126/science.1123940. [DOI] [PubMed] [Google Scholar]
- 18.Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator–thinking outside the box. Biochim Biophys Acta. 2006;1762:181–190. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Park JS, Deng JH, Bai Y. Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J Bioenerg Biomembr. 2006;38:283–291. doi: 10.1007/s10863-006-9052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larsson NG, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 21.Onesto E, et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol Commun. 2016;4:47. doi: 10.1186/s40478-016-0316-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graves JA, et al. Mitochondrial structure, function and dynamics are temporally controlled by c-Myc. PLoS One. 2012;7:e37699. doi: 10.1371/journal.pone.0037699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sgarbi G, et al. Mitochondria hyperfusion and elevated autophagic activity are key mechanisms for cellular bioenergetic preservation in centenarians. Aging (Albany NY) 2014;6:296–310. doi: 10.18632/aging.100654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agnello M, Morici G, Rinaldi AM. A method for measuring mitochondrial mass and activity. Cytotechnology. 2008;56:145–149. doi: 10.1007/s10616-008-9143-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu J, et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci U S A. 2014;111:15514–15519. doi: 10.1073/pnas.1414859111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keij JF, Bell-Prince C, Steinkamp JA. Staining of mitochondrial membranes with 10-nonyl acridine orange, MitoFluor Green, and MitoTracker Green is affected by mitochondrial membrane potential altering drugs. Cytometry. 2000;39:203–210. doi: 10.1002/(sici)1097-0320(20000301)39:3<203::aid-cyto5>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 28.Contreras L, Drago I, Zampese E, Pozzan T. Mitochondria: the calcium connection. Biochim Biophys Acta. 2010;1797:607–618. doi: 10.1016/j.bbabio.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Celsi F, et al. Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim Biophys Acta. 2009;1787:335–344. doi: 10.1016/j.bbabio.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giorgi C, et al. Mitochondrial Ca(2+) and apoptosis. Cell Calcium. 2012;52:36–43. doi: 10.1016/j.ceca.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: A mutual interplay. Redox Biol. 2015;6:260–271. doi: 10.1016/j.redox.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mbaya E, et al. Calcium signalling-dependent mitochondrial dysfunction and bioenergetics regulation in respiratory chain Complex II deficiency. Cell Death Differ. 2010;17:1855–1866. doi: 10.1038/cdd.2010.51. [DOI] [PubMed] [Google Scholar]
- 33.Choi H, Yang Z, Weisshaar JC. Single-cell, real-time detection of oxidative stress induced in Escherichia coli by the antimicrobial peptide CM15. Proc Natl Acad Sci U S A. 2015;112:E303–310. doi: 10.1073/pnas.1417703112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson KM, et al. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santo-Domingo J, Demaurex N. Calcium uptake mechanisms of mitochondria. Biochim Biophys Acta. 2010;1797:907–912. doi: 10.1016/j.bbabio.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 36.Natkanski E, et al. B cells use mechanical energy to discriminate antigen affinities. Science. 2013;340:1587–1590. doi: 10.1126/science.1237572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Batista FD, Iber D, Neuberger MS. B cells acquire antigen from target cells after synapse formation. Nature. 2001;411:489–494. doi: 10.1038/35078099. [DOI] [PubMed] [Google Scholar]
- 38.Tze LE, Baness EA, Hippen KL, Behrens TW. Ig light chain receptor editing in anergic B cells. J Immunol. 2000;165:6796–6802. doi: 10.4049/jimmunol.165.12.6796. [DOI] [PubMed] [Google Scholar]
- 39.Batista FD, Neuberger MS. B cells extract and present immobilized antigen: implications for affinity discrimination. EMBO J. 2000;19:513–520. doi: 10.1093/emboj/19.4.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aucher A, Magdeleine E, Joly E, Hudrisier D. Capture of plasma membrane fragments from target cells by trogocytosis requires signaling in T cells but not in B cells. Blood. 2008;111:5621–5628. doi: 10.1182/blood-2008-01-134155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Traba J, et al. Prolonged fasting suppresses mitochondrial NLRP3 inflammasome assembly and activation via SIRT3-mediated activation of superoxide dismutase 2. J Biol Chem. 2017;292:12153–12164. doi: 10.1074/jbc.M117.791715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gees M, Colsoul B, Nilius B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb Perspect Biol. 2010;2:a003962. doi: 10.1101/cshperspect.a003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rae MG, Hilton J, Sharkey J. Putative TRP channel antagonists, SKF 96365, flufenamic acid and 2-APB, are non-competitive antagonists at recombinant human alpha1beta2gamma2 GABA(A) receptors. Neurochem Int. 2012;60:543–554. doi: 10.1016/j.neuint.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 44.Ohga K, Takezawa R, Arakida Y, Shimizu Y, Ishikawa J. Characterization of YM-58483/BTP2, a novel store-operated Ca2+ entry blocker, on T cell-mediated immune responses in vivo. Int Immunopharmacol. 2008;8:1787–1792. doi: 10.1016/j.intimp.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 45.Donjerkovic D, Scott DW. Activation-induced cell death in B lymphocytes. Cell Res. 2000;10:179–192. doi: 10.1038/sj.cr.7290047. [DOI] [PubMed] [Google Scholar]
- 46.Rathmell JC, et al. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature. 1995;376:181–184. doi: 10.1038/376181a0. [DOI] [PubMed] [Google Scholar]
- 47.Rothstein TL, et al. Protection against Fas-dependent Th1-mediated apoptosis by antigen receptor engagement in B cells. Nature. 1995;374:163–165. doi: 10.1038/374163a0. [DOI] [PubMed] [Google Scholar]
- 48.Lagresle C, Mondiere P, Bella C, Krammer PH, Defrance T. Concurrent engagement of CD40 and the antigen receptor protects naive and memory human B cells from APO-1/Fas-mediated apoptosis. J Exp Med. 1996;183:1377–1388. doi: 10.1084/jem.183.4.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akkaya B, et al. Ex-vivo iTreg differentiation revisited: Convenient alternatives to existing strategies. J Immunol Methods. 2017;441:67–71. doi: 10.1016/j.jim.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akkaya B, et al. A Simple, Versatile Antibody-Based Barcoding Method for Flow Cytometry. J Immunol. 2016;197:2027–2038. doi: 10.4049/jimmunol.1600727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Traba J, Del Arco A, Duchen MR, Szabadkai G, Satrustegui J. SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca(2+) buffering. Cell Death Differ. 2012;19:650–660. doi: 10.1038/cdd.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Natkanski E, et al. B cells use mechanical energy to discriminate antigen affinities. Science. 2013;340:1587–1590. doi: 10.1126/science.1237572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sohn HW, Tolar P, Brzostowski J, Pierce SK. A method for analyzing protein-protein interactions in the plasma membrane of live B cells by fluorescence resonance energy transfer imaging as acquired by total internal reflection fluorescence microscopy. Methods Mol Biol. 2010;591:159–183. doi: 10.1007/978-1-60761-404-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu W, Meckel T, Tolar P, Sohn HW, Pierce SK. Antigen affinity discrimination is an intrinsic function of the B cell receptor. J Exp Med. 2010;207:1095–1111. doi: 10.1084/jem.20092123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akkaya M, Aknin ML, Akkaya B, Barclay AN. Dissection of agonistic and blocking effects of CD200 receptor antibodies. PLoS One. 2013;8:e63325. doi: 10.1371/journal.pone.0063325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Offerdahl DK, Dorward DW, Hansen BT, Bloom ME. A three-dimensional comparison of tick-borne flavivirus infection in mammalian and tick cell lines. PLoS One. 2012;7:e47912. doi: 10.1371/journal.pone.0047912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Further details of the experimental procedures are provided in the Life Sciences Reporting Summary. The data that support the findings of the study are available from the corresponding author upon request. RNA-seq data SRA database under project number: PRJNA422889.