

Abstract
Objective
Atherosclerosis studies in Ldlr knockout mice require breeding to homozygosity and congenic status on C57BL6/J background, a process that is both time and resource-intensive. We aimed to develop a new method for generating atherosclerosis through somatic deletion of Ldlr in livers of adult mice.
Approach and Results
Overexpression of PCSK9 is currently used to study atherosclerosis, which promotes degradation of low density lipoprotein receptor (LDLR) in the liver. We sought to determine if CRISPR/Cas9 could also be used to generate atherosclerosis through genetic disruption of Ldlr in adult mice. We engineered Adeno-Associated Viral (AAV) vectors expressing Staphylococcus aureus Cas9 and a guide RNA targeting the Ldlr gene (AAV-CRISPR). Both male and female mice received either: 1) saline, 2) AAV-CRISPR, or 3) AAV-hPCSK9-D374Y. A fourth group of germline Ldlr-KO mice was included for comparison. Mice were placed on a Western diet and followed for twenty weeks to assess plasma lipids, PCSK9 protein levels, atherosclerosis, and editing efficiency. Disruption of Ldlr with AAV-CRISPR was robust, resulting in severe hypercholesterolemia and atherosclerotic lesions in the aorta. AAV-hPCSK9 also produced hypercholesterolemia and atherosclerosis as expected. Notable sexual dimorphism was observed, wherein AAV-CRISPR was superior for Ldlr removal in male mice, while AAV-hPCSK9 was more effective in female mice.
Conclusions
This all-in-one AAV-CRISPR vector targeting Ldlr is an effective and versatile tool to model atherosclerosis with a single injection, and provides a useful alternative to the use of germline Ldlr KO mice.
Keywords: Gene transfer, Ldlr, AAV-CRISPR, atherosclerosis, somatic genome editing, low-density lipoprotein, PCSK9
Subject Codes: Animal Models of Human Disease, Gene Therapy, Atherosclerosis
Graphical Abstract
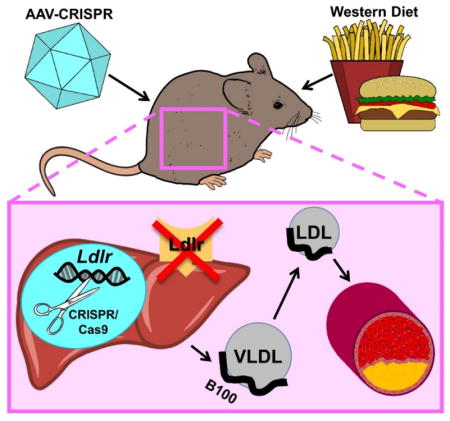
Introduction
The Apolipoprotein E (Apoe) and Low Density Lipoprotein Receptor (Ldlr) knockout (KO) mouse models have provided fundamental insights into the genetic, nutritional, and environmental factors contributing to atherosclerosis. Atherosclerosis is a highly polygenic disease, but testing the role of a new gene of interest requires extensive backcrossing to congenic status on C57BL/6J background, followed by breeding to homozygosity with Ldlr KO or Apoe KO mice1. This severely limits the rate at which candidate genes can be investigated and is further complicated when conditional alleles and reporter genes are required. Recent genetics studies have identified many loci implicated in atherosclerotic vascular disease in humans, yet our understanding of the underlying molecular mechanisms lags far behind2. The atherosclerosis field is in need of new, higher-throughput approaches that provide greater flexibility, lower costs, and faster completion times to advance our understanding of this complex disease.
Liver-directed methods have been particularly useful for studying lipid metabolism and metabolic disease. Antisense oligonucleotides have recently been used to knock down hepatic Ldlr expression as a reversible model of atherosclerosis and regression3. This new method is elegant in that the mechanism of reversal involves restoration of LDLR protein rather than compensatory inhibition of ApoB secretion, but requires weekly dosing of antisense oligos for the entirety of the study. Methods that provide single-dose, permanent knockdown of Ldlr would simplify atherosclerosis studies, reducing cost and stress on research animals. Adeno-associated viral (AAV) vectors can efficiently deliver transgenes to the liver, resulting in sustained expression for months to years4. AAV is currently being used to overexpress human or mouse Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) gain-of-function variants5 to generate atherosclerosis in adult mice without crossing to Ldlr KO background. This tool has gained popularity for its ease of use and rapid onset of hypercholesterolemia. In addition to showing the human D374Y variant can produce hypercholesterolemia6, the mouse D377Y variant has been used in modeling vascular calcification7, accelerated lesion formation using partial carotid ligation8, and for studying aortic aneurysm9. However, PCSK9 is overexpressed at supraphysiological levels by this method, which may not be ideal for all applications. It has been reported that at least a fraction of circulating PCSK9 resides on lipoprotein particles10–13, and this could be a confounding factor if the gene of interest is expected to alter lipoprotein metabolism, infiltration into the vessel wall, or uptake by monocytes and macrophages. Somatic methods that permanently delete Ldlr at the genetic level in adult mice would be preferred.
The Clustered Regularly-Interspaced Short Palindromic Repeats (CRISPR)/Cas9 system is an antiviral defense mechanism from bacteria that has been adapted for genome editing in mammalian cells14. A roughly 20 nucleotide small guide RNA (gRNA) directs the Cas9 nuclease to a specific DNA target site, where it generates a double-stranded break15. The double-stranded break is most often repaired by the error-prone non-homologous end joining repair system, causing small insertions and deletions (indels). When a coding exon is targeted, most indels result in a frameshift leading to a premature stop codon and nonsense-mediated decay of the message, effectively knocking out the gene. Previous experiments with Streptococcus pyogenes Cas9 (SpCas9) transgenic mice16 showed the potential for somatic genome editing for modeling atherosclerosis in vivo17. However, these experiments required the use of SpCas9-transgenic mice due to challenges in delivering this large nuclease to liver with AAV. An all-in-one AAV vector expressing both the gRNA against Ldlr and the Cas9 nuclease would enable atherosclerosis studies in any mouse model. To test this, we used a smaller Cas9 ortholog from Staphylococcus aureus (SaCas9) which we packaged into AAV with an Ldlr-targeting gRNA for in vivo editing (AAV-CRISPR). Here we demonstrate that liver-directed disruption of Ldlr with an all-in-one AAV-CRISPR vector is a robust and viable approach to study atherosclerosis in adult mice.
Material and methods
Cloning and virus production
We obtained pAAV-D374Y-hPCSK9 from Addgene (#58379)6,18. Oligos corresponding to the gRNA were annealed and cloned into the BbsI site of 1255_pAAV-U6-SA-BbsI-MluI-gRNA-CB-SACas9-HA-OLLAS-sPa to introduce the Ldlr gRNA to generate 1375_pAAV8-U6-SA-WTmLdlrEx14-gRNA2-N22-CB-SACas9-HA-OLLAS-spA. Plasmid 1375 encodes both S. aureus Cas9 as well as a guide RNA targeting exon 14 of the murine Ldlr gene. Complete vector sequences are included in the supplementary methods. Plasmids required for AAV packaging, Adenoviral helper plasmid pAdDeltaF6 (PL-F-PVADF6) and AAV8 packaging vector pAAV2/8 (PL-T-PV0007), were obtained from the University of Pennsylvania Vector Core. AAV-CRISPR and AAV-hPCSK9 were packaged using HEK 293T cells (ATCC, CRL-3216) using the triple transfection method19. Virus was purified by a single cesium chloride gradient. Virus-containing fractions were dialyzed against PBS using a 100 kDa MWCO Float-a-lyser (Spectra/Por G235059) to remove the cesium chloride. Dialyzed virus was concentrated using a 100 kDa Amicon (Millipore UFC210024) and stored at −80 degrees until titer determination or injection into mice. Viral titers were determined following DNase digestion by standard qPCR methods. All primers and oligos used in this study are included in Supplementary Table I.
Animals
Wild type C57BL/6J (stock number 000664) and Ldlr-KO (B6.129S7-Ldlrtm1Her/J, stock number 002207) mice were obtained from Jackson Laboratories and breeding colonies were maintained on a 14 hour light, 10 hour dark cycle. Until AAV administration, mice had free access to a standard mouse chow, irradiated PicoLab Select Rodent 50 IF/6F (LabDiet Product code: 5V5R). At six weeks of age, male and female C57BL6/J mice were injected intraperitoneally with saline or 5 × 1011 genome copies of AAV-CRISPR or hPCSK9 per mouse. Injection groups were randomly assigned within cages, except Ldlr-KO mice, which were maintained separately. On injection day, Ldlr-KO and AAV-injected mice were placed on Western Diet (Research Diets D12079B; containing 0.21% cholesterol w/w, 21% fat) for the duration of the 20-week study. To assess plasma lipids, blood from five-hour fasted mice was collected through the retro-orbital plexus using a heparinized Natelson tubes (Fisher, 0266810). Blood was centrifuged at 10,000 rpm for ten minutes to isolate plasma. Criteria for early euthanasia and exclusion from the study included health concerns like malocclusion, ulcerative dermatitis, or other unexplained illness not related to treatment. A small number of mice (6/92 total animals dissected, Supplemental Figure I), including saline-treated control animals, presented with damaged livers and enlarged spleens at time of final dissection and were excluded from all analyses due to an uncharacterized infection. These studies adhere to the guidelines for atherosclerosis studies described in the AHA statement20, as well as the ATVB council statement for considering sex as a biological variable21. All experiments were performed under protocol AN-7243 with the approval of the Baylor College of Medicine Institutional Animal Care and Use Committee.
Lipid analyses
Plasma was collected from five-hour fasted mice preceding AAV injection and at 4, 8, 12, 16, and 20 weeks post-injection. Total plasma cholesterol at all time points was measured using the Wako Cholesterol E kit (999-02601). Gel filtration chromatography was completed with 220 μL pooled plasma from mice with high plasma cholesterol. Pooled plasma was loaded into a 200 μL loop on an Amersham-Pharmacia ÄKTA chromatography system equipped with two Superose HR6 columns in tandem and eluted with TBS at a flow rate of 0.5 mL/min22. Lipoprotein cholesterol and triglyceride levels were determined using the Wako Cholesterol E (999-02601) and Infinity Triglycerides (TR22421) kits using 100 μL of each 1 mL fraction and expressed as micrograms per milliliter.
Atherosclerosis
At 20-week endpoints, aortae were cleared of fat and split from the aortic root to the illeal bifurcation. The split aortae were freed from the mouse and fixed in 10% formalin. Lesions were stained using Oil Red O (VWR AAA12989-14) as previously described23. Stained aortae were pinned using insect pins (Finescience.com 26002–20) on black paraffin under PBS and imaged using a Leica M80 dissection microscope and Leica IC80 HD camera. Group identifying information was removed from images and percent lesion area was quantified using ImageJ24.
Western Blotting
Approximately 100 mg of liver tissue were homogenized in ~10 volumes of radioimmunoprecipation buffer (50 mM Tris pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 150 mM sodium chloride, and protease inhibitors (Roche 11836153001)) using a Bead Blaster 24 (Benchmark D2400). The protein was quantified using bicinchoninic acid assay (Thermo-Pierce #23227). Liver protein (50 μg) was separated by SDS-PAGE using 4–12% gradient gels (Life Technologies NP0322BOX) and transferred to polyvinylidine fluoride membranes (Millipore IPFL00010). Membranes were blocked for one hour at room temperature in a 2:1 solution of Odyssey Blocking Buffer (Li-Cor 927-40000) and phosphate buffered saline plus 0.05% tween-20 (PBS-T). Primary antibodies to the c-terminus of LDLR (1:5000, rabbit, gift from Gene Ness25) and beta tubulin (1:500, mouse, University of Iowa Developmental Studies Hybridoma Bank E7) were incubated on membranes in PBS-T with 1% bovine serum albumin overnight at 4 degrees. For analysis of apolipoprotein content, FPLC fractions containing VLDL, LDL, HDL, and nonlipoprotein-containing fractions were pooled and separated by gel electrophoresis as above. Primary antibodies against Apolipoprotein B (1:5000, rabbit, Meridian, K23300R), Apolipoprotein E (1:5000, rabbit, Abcam, ab20874) or Apolipoprotein AI (1:5000, rabbit, Meridian, K23500R) were incubated overnight at 4 degrees. Appropriate goat anti-rabbit 680 nm and anti-mouse 800 nm antibodies (Rockland, 611-144-002-0.5 and 610-145-002-0.5) were incubated at room temperature for 2 hours and imaged using an Odyssey Classic (Li-Cor).
Quantitation of AAV genomes
Liver genomic DNA was isolated using the Qiagen DNeasy (Cat. 69506) kit. DNA (100 ng) was subjected to quantitative PCR (qPCR) using primers specific to the AAV-CRISPR and AAV-hPCSK9 vectors to assess the relative amount of virus present in the liver. A standard curve from plasmids used for virus preparation were used to quantify AAV genomes per microgram of DNA.
Analysis of on and off-target cutting by CRISPR/Cas9
Off-target sites for the Ldlr gRNA were determined using the online bioinformatics tool, COSMID at https://crispr.bme.gatech.edu/26. Searches were completed on the least stringent settings (up to three INDELS, two one-base deletions, two one-base insertions). In addition, we allowed for some leniency in the PAM motif by searching both NNGRRT and NNGRR PAM sequences. Four off-target sites were returned, so all sites were assessed for off-target INDEL formation. Primers specific to the Ldlr and off-target loci were used to amplify these genomic sites. Secondary PCR was completed using 2 μL of the primary PCR product to add barcode sequences and the Illumina P5 and P7 adapter sequences to each amplicon. The final barcoded amplicons were gel purified and pooled equally for deep sequencing analysis as previously described17.
Detection of AAV-CRISPR vector genome integrations
Forward and reverse integrations of AAV-CRISPR at the Ldlr target site were assessed using a PCR scheme where one primer is located within endogenous genomic sequence and the second primer is located within AAV-CRISPR. The primers used in this study are listed in Supplementary Table I. 50 μg of genomic liver DNA from Saline and AAV-CRISPR mice was subjected to 35 cycles of PCR (APEX TaqRed, 42-138) and bands were resolved by gel electrophoresis.
PCSK9 ELISA
Plasma hPCSK9 was measured using the Human Proprotein Convertase9/PCSK9 Quantikine ELISA from R&D Systems (DPC900) according to the manufacturer’s instructions. Plasma was diluted 1:20,000 in Calibrator Diluent RD5P for time point hPCSK9 assessment. FPLC fractions were diluted and assessed at 1:10 to 1:100 dilutions for lipoprotein hPCSK9 distribution. A best-fit line for hPCSK9 standards was determined by log-linear regression. The resulting equations were used to determine the concentration for each sample log OD and corrected for the dilution factor.
Statistics
Researchers were not blinded for animal work or data analysis, except for lesion quantification. For determining lesion area, all group information was removed from aorta images and a lab member who was uninvolved with aorta dissections quantified the percent lesion area. All data were assessed by either one-way ANOVA, or two-way ANOVA as appropriate, followed by Tukey’s posttest. Comparisons involving two groups were analyzed by a two-tailed student’s t-test. Data that was not normally distributed was analyzed by a Mann-Whitney test (Figure 1c). Individual data points are shown whenever possible, and the data is represented as the mean +/− standard deviation (S.D.). Graphs were generated using Graphpad Prism 6. In all cases significance is assigned at p<0.05.
Figure 1.

Efficiency and specificity of genome editing by AAV-CRISPR. A. Experimental design. Male and female mice were injected with saline or 5 × 1011 genome copies of AAV, placed on a Western diet, and followed or 20 weeks. Ldlr indel frequency in (B) male and (C) female mouse liver. D. Top ten representative indels from a single mouse. The WT sequence is shown at the top with the gRNA site underlined. The gRNA targets the bottom strand. E. Male and F. female on- and off-target indel frequency. Males: Saline n = 5, AAV-CRISPR n = 13. Females: Saline n = 5, AAV-CRISPR n = 9. *p<0.05 by standard student t-test (males) and Mann-Whitney test (females).
Results
Vector and Study Design
We sought to determine if somatic deletion of Ldlr in adult mice could be achieved with viral delivery of the CRISPR/Cas9 system as a means of studying atherosclerosis. To test this, we generated an all-in-one AAV vector expressing a small gRNA targeting exon 14 of Ldlr, as well as SaCas9 driven by the chicken beta actin promoter (AAV-CRISPR). We compared this approach to AAV-mediated overexpression of the human D374Y PCSK9 gain-of-function variant, as well as the “gold standard” Ldlr KO mice. AAV-CRISPR and AAV-hPCSK9 were packaged into AAV serotype 8 vectors, which have a high tropism for murine liver, and then delivered to six-week-old C57BL6/J mice at a dose of 5 × 1011 genome copies (GC) per animal. A group of control C57BL6/J mice were injected with saline alone and identically housed germline Ldlr KO mice were followed in parallel for comparison. Following AAV injection, mice in all groups were placed on a standard Western diet (21% fat, 0.21% cholesterol w/w), and plasma was collected at monthly intervals for lipid analysis. After 20 weeks, animals were sacrificed to assess Ldlr editing efficiency, hPCSK9 protein expression, plasma lipids, and atherosclerotic lesion burden. These studies were performed in both male and female mice to capture possible sex differences (Figure 1a).
Efficiency and specificity of genome editing by AAV-CRISPR
Targeted deep sequencing was performed to assess the efficiency of disruption of the Ldlr gene in mouse livers following the 20 week study. COSMID26 was used to predict the most likely genomic sites for off-target editing. SaCas9 has a more restrictive protospacer adjacent motif (PAM) than the commonly used SpCas9 system (NNGRRT vs. NGG; R = G or A), which occurs far less frequently in the mouse genome. Using the least stringent search terms, only four potential off-target sites were returned, so all were tested for off-target mutagenesis. The four off-targets and Ldlr on-target site were amplified and barcoded by PCR (Supplemental Table II). As expected, mice in the saline control group displayed no editing. Male AAV-CRISPR mice had 31.9 +/− 4.74%, and females had 33.1 +/− 15.1% indel formation on-target in Ldlr (Figure 1b,c). The most common indels observed in a representative animal with high editing efficiency are shown in Figure 1d. No off-target editing was observed at any of the four off target sites above the background levels detected in the control group, which includes PCR and sequencing error (Figure 1e,f). Interestingly, we also observed a small fraction of indels contained short fragments of the AAV vector inverted terminal repeats (ITRs) at the on-target cut site, similar to our previous findings with SpCas917 (Supplemental Figure II). This prompted us to ask whether larger AAV vector insertions (~5kb) also occurred which would not be captured by NGS. Indeed, evidence of whole AAV-CRISPR genome insertions was detected at the cut site in both forward and reverse orientations by PCR (Supplemental Figures III and IV).
Efficiency of overexpression by AAV-hPCSK9-D374Y
The levels of human PCSK9 protein were measured over time in the group receiving the AAV-hPCSK9 virus, as others have described previously6,18. There was considerable variability in plasma hPCSK9 protein levels within each group, which included a few animals with very low levels (Figure 2). Even though males and females received the same dose of AAV vectors, hPCSK9 levels were generally higher in the males, and relatively constant throughout the study (Figure 2a). In contrast, hPCSK9 plasma levels in the female mice tended to drop off over time (Figure 2b).
Figure 2.

Efficiency of overexpression by AAV-hPCSK9-D374Y. A. Male and B. female plasma hPCSK9 expression. Male AAV-PCSK9 n = 13. Female AAV-PCSK9 n = 8.
Knockdown of LDLR expression
To determine the effectiveness of AAV-CRISPR and AAV-hPCSK9 in knocking down LDLR, we measured liver LDLR protein levels by western blotting25 at the end of the 20 week study. LDLR was abundantly expressed in both male and female controls and did not vary by sex (Supplemental Figure V), but was absent in the Ldlr KO negative control samples. AAV-CRISPR was more effective at eliminating LDLR than PCSK9 overexpression in the male mice (Figure 3a, Supplemental Figure VI). Some mice in the hPCSK9 group showed little to no knockdown of LDLR, consistent with the highly variable plasma hPCSK9 levels. In female mice, AAV-CRISPR was less effective than AAV-hPCSK9 at knocking down Ldlr, with variable amounts of residual LDLR remaining in this group (Figure 3b, Supplemental Figure VI).
Figure 3.

Knockdown of LDLR protein expression. Liver lysates from representative male (A) and female (B) liver samples were blotted for LDLR and beta tubulin.
Plasma lipids
In the male control mice, plasma cholesterol increased from 123 +/− 13.9 mg/dL at the beginning of the study to 246 +/− 26.7 mg/dL after 20 weeks on Western diet. Mice treated with AAV-CRISPR and AAV-hPCSK9 began at similar levels and showed significant increases in plasma cholesterol as early as 4 weeks after AAV administration (Figure 4a). At the final time point, the AAV-CRISPR group (1408 +/− 473 mg/dL) had significantly higher total plasma cholesterol than the AAV-hPCSK9 group (993 +/− 481 mg/dL). Neither AAV-CRISPR nor AAV-hPCSK9 treated animals reached the plasma cholesterol levels seen in the Ldlr-KO mice (1966 +/− 412 mg/dL). Similarly, female control mice began the study at 94.1 +/− 9.0 mg/dL total plasma cholesterol and increased to 134 +/− 14 mg/dL after 20 weeks on Western diet. Female AAV-CRISPR and AAV-hPCSK9 mice had similar plasma cholesterol levels at the beginning of the study. After 20 weeks of Western diet, the AAV-hPCSK9 group reached 1142 +/− 155 mg/dL which was not significantly different from the Ldlr-KO group (1171 +/− 228 mg/dL). The AAV-CRISPR females had a slower onset of hypercholesterolemia, and lower final plasma cholesterol levels, reaching an average of 751 +/− 456 mg/dL (Figure 4b).
Figure 4.

Plasma lipids. A. Male and B. female plasma were collected following 5-hour fasts throughout the study and assessed for total cholesterol. Males: Saline n = 7, AAV-CRISPR n = 14, AAV-PCSK9 n = 13, Ldlr-KO n = 10. Females: Saline n = 7, AAV-CRISPR n = 9, AAV-PCSK9 n = 8, Ldlr-KO n = 8. *p<0.05 by two-way ANOVA. Cholesterol (C, D) and triglyceride (E,F) distribution amongst lipoprotein fractions in pooled plasma from a subset of male and female mice (n = 5 per group). Human PCSK9 distribution across separated lipoprotein fractions from male (G) and female mice (H) was determined by ELISA.
We used gel filtration chromatography to examine the lipoprotein cholesterol, triglyceride, and hPCSK9 distributions in both male and female pooled plasma samples. Both male and female saline controls showed cholesterol primarily in the HDL fractions, as expected for wild type C57BL6/J mice on Western diet. AAV-CRISPR males more closely tracked with the Ldlr-KO control mice than the AAV-hPCSK9 group, but all three groups showed the expected increases in VLDL, IDL, and LDL fractions (Figure 4c). Female AAV-CRISPR and AAV-hPCSK9 mice had a similar distribution with high amounts of VLDL, IDL, and LDL-associated cholesterol (Figure 4d). AAV-CRISPR, AAV-hPCSK9, and Ldlr deficient mice had elevated levels of triglycerides in the VLDL fractions relative to the control mice at the 20 week time point (Figure 4e,f). We also assessed the distribution of hPCSK9 across lipoprotein fractions by ELISA. PCSK9 circulates free in plasma, although a significant proportion has been reported to reside on lipoproteins, particularly on LDL10–12 and Lipoprotein(a) particles in humans27. Despite very high levels of VLDL, IDL and LDL, we were surprised to see a proportion of hPCSK9 eluted in the HDL-containing fractions. In male mice, similar amounts of hPCKS9 eluted in the HDL and non-lipoprotein fractions, but in females the hPCKS9 in the HDL fractions was considerably lower (Figure 4g,h). As expected, ApoB-48, ApoB-100, and ApoE were markedly elevated in the VLDL and LDL fractions from the AAV-CRISPR, AAV-hPCSK9 and Ldlr KO mice. In contrast, there were no major changes in ApoA-I content in the HDL fractions between any of the groups (Supplemental Figure VII).
Susceptibility to Atherosclerosis
Aortae were assessed for atherosclerotic plaque formation by en face Oil Red O staining (Figure 5a). None of the male or female control mice developed lesion, as expected for C57BL6/J mice fed this Western diet. Male AAV-CRISPR and hPCSK9 mice both had significant lesion burden, but these did not reach the level of the Ldlr KO mice (13.1 +/− 3.83%). Male mice treated with AAV-CRISPR had significantly more lesion than those in the AAV-hPCSK9 group (7.76 +/− 4.58% vs 3.89 +/− 3.91%, p<0.05). In female mice, AAV-hPCSK9 treated mice developed lesion levels similar to the Ldlr-KO (7.99 +/−1.48% vs 9.46 +/− 2.75%, n.s.). AAV-CRISPR females had significantly lower lesion area (2.84 +/− 3.36%) than both the AAV-PCSK9 and Ldlr-KO mice. Other groups have reported that AAV vectors are less effective at transducing liver in female mice relative to males28,29. Therefore, we examined transduction efficiency at the end of the study by qPCR using primers specific to the AAV-CRISPR and AAV-hPCSK9 vector genomes. Females receiving AAV-CRISPR had significantly lower vector genomes per microgram of DNA compared to males injected with the same dose of virus. This was not the case for AAV-hPCSK9, which was not significantly different between sexes (Supplemental Figure VIII).
Figure 5.

Susceptibility to atherosclerosis. A. Representative male aortas stained en face with Oil Red O. B. Quantification of male aorta lesion area. C. Representative female aortas stained en face with Oil Red O. D. Quantification of female aorta lesion area. Males: Saline n = 7, AAV-CRISPR n = 14, AAV-PCSK9 n = 13, Ldlr-KO n = 10. Females: Saline n = 7, AAV-CRISPR n = 9, AAV-PCSK9 n = 8, Ldlr-KO n = 8. *p<0.05 by one-way ANOVA.
Discussion
Here we report an all-in-one SaCas9-based AAV-CRISPR vector that is capable of highly efficient disruption of the Ldlr gene in adult mouse liver, producing severe hypercholesterolemia and atherosclerosis. We directly compared our AAV-CRISPR approach with the previously described AAV-PCSK9 method18 and the traditional Ldlr KO mouse model. AAV-CRISPR resulted in more complete removal of LDLR from liver and increased atherosclerotic lesion burden in male mice compared to overexpression of PCSK9. Interestingly, the opposite was true in female mice, where AAV-PCSK9 more closely mirrored the germline Ldlr KO model. Our data shows that the AAV-CRISPR system is a versatile, off-the-shelf tool that can be used to study atherosclerosis.
Adenoviral vectors have been used effectively to knock out Pcsk930,31 in the liver with CRISPR/Cas9, as well as to inactivate Pcsk932 and Angptl333 by base editing. Adenoviral vectors are known to elicit strong innate34 and adaptive immune responses35, making them unsuitable for human liver gene therapy36 or long-term experiments in mice. In contrast, AAV vectors have a favorable safety profile that has enabled numerous clinical trials, including liver-directed gene therapy of hemophilia37 and Familial Hypercholesterolemia38, as well as the first FDA approved gene therapy product in the U.S. for congenital blindness39. Due to the packaging constraints of this system, AAV delivery of the large SpCas9 (4.2 kb) to liver has been particularly challenging, and efficient editing has only recently been reported40,41. In our previous work, we showed that AAV delivery of gRNAs could efficiently edit the Ldlr and Apob genes in the livers of SpCas9 transgenic mice17. This provided proof-of-concept that AAV-CRISPR could be used to model atherosclerosis, but would require crossing to SpCas9 transgenic mice. Ran et al. developed a smaller Cas9 ortholog (3.3 kb) from S. aureus for use in mammalian systems and achieved highly efficient editing of Pcsk9 in mice with AAV delivery42. Here, we demonstrate that a single AAV-CRISPR vector encoding SaCas9 efficiently knocks out Ldlr in the liver.
Editing efficiency at the Ldlr target site averaged 31.9% in male mice and 33.1% in females by NGS. Despite the incomplete degree of editing, LDLR protein was virtually undetectable. Several factors likely account for this discrepancy. First, editing efficiencies of 100% are not possible because non-parenchymal cells constitute ~15% of the liver. Second, and most importantly, our deep sequencing strategy involves small PCR amplicons, so larger deletions and insertions are not captured. We explored the possibility that insertions of the AAV vector genome might also occur, in addition to the ITR fragments found by NGS. Conventional PCR identified whole AAV vector genome insertions at the cut site in both forward and reverse orientations. While it is not technically feasible to quantify these events, this suggests that the total rate of Ldlr mutagenesis is likely far greater than 32–33%. It is also worth noting that the frequency of Ldlr editing is lower than we obtained in the SpCas9 transgenic mice (54%)17. The indel spectrum generated by CRISPR/Cas9 is dependent on the sequence of the breakpoint. Since the gRNA, CRISPR/Cas9 system (SaCas9 vs. SpCas9), and the size of the AAV vectors (4.7 kb vs. 2.1 kb) differ, it is not possible to directly compare editing efficiency between these two studies. Aside from the unexpected on-target insertions, we did not detect off-target editing above background at the four predicted sites. The more restrictive PAM of the SaCas9 system (NNGRRT vs. NGG) may limit selection of on-target sites, but has the advantage of increased specificity since fewer possible off-target sites exist in the genome. Taken together, these data indicate that our AAV-CRISPR vector both efficiently and specifically disrupts the Ldlr gene.
In classic atherosclerosis studies in Ldlr KO mice, loss of Ldlr is guaranteed from birth and confirmed by genotyping. In the case of somatic removal of Ldlr with AAV-PCSK9 or AAV-CRISPR, further controls are needed. There can be significant differences in AAV titers, infectivity, and injections between laboratories, and insufficient dosing of CRISPR/Cas9 or PCSK9 is a principal concern. We examined multiple parameters in our mice to assess successful delivery and Ldlr removal. These included AAV vector genomes by Q-PCR, editing efficiency of the Ldlr locus, plasma PCSK9 levels, and LDLR protein in the liver. Based on these data, it is likely that some incomplete or failed deliveries occurred in our study (Supplemental Tables III and IV). We compared data from all mice to determine if guidelines could be established for exclusion of mice for others who may use this method. Plasma cholesterol generally tracked well with Ldlr editing efficiency as well as PCSK9 protein levels. Although plasma cholesterol can be measured from survival bleeds, this is often changed by genetic, dietary, or environmental interventions, and is therefore not a suitable criteria. PCSK9 levels can be measured in plasma, which could provide a useful short-term confirmation of delivery, although these were extremely variable between mice and time points. For both AAV-CRISPR and AAV-PCSK9, the abundance of vector genomes can be determined at the end of the study. We observed that mice with effective removal of Ldlr typically have 106 – 107 GC/μg genomic DNA when dosed at 5 × 1011 GC/mouse. Likewise, editing efficiency with AAV-CRISPR can be estimated using NGS or Sanger sequencing followed by TIDE43 or other web-based tools to calculate indel rates. While all of these are useful controls for AAV-CRISPR or AAV-PCSK9 delivery, the end goal is to remove LDLR from the liver. Therefore, there is no substitute for measurement of LDLR protein levels, which should be performed on all animals at completion of an atherosclerosis study.
This study shows that AAV-CRISPR is a novel method for generation of atherosclerosis in adult mice through liver-directed genetic removal of Ldlr. Both AAV-CRISPR and AAV-PCSK9 vectors require only a single AAV injection to remove LDLR in adult mice, eliminating the need to cross to Ldlr KO. These methods can also be performed in strains other than C57BL6/J or in mixed backgrounds. However, AAV transduction is known to vary by strain44, as well as by sex28,29. Therefore, the use of littermate controls is warranted, and comparisons across sex or strain should be avoided. AAV-CRISPR and AAV-PCSK9 could also be packaged into any AAV serotype provided they efficiently target the liver. Our studies used intraperitoneal injection of AAV8, which is effective in mice and can be accomplished with minimal technical training. There are likely differences in ability of other serotypes to transduce liver through this route of administration and these may require intravenous delivery. Age is also a consideration as AAV genomes are maintained episomally and lost through hepatocyte division45. Our study used six week old mice, a time when the liver is growing, but efficient Ldlr editing and PCSK9 overexpression can still be achieved. Studies should be initiated in age-matched adult mice to ensure comparable delivery within a given experiment.
AAV-CRISPR and AAV-PCSK9 are both capable of producing atherosclerosis on a Western diet, and either one may be preferable for particular applications. In our hands, AAV-CRISPR is more effective at generating atherosclerosis in male mice, while AAV-PCSK9 is more robust in females. AAV-PCSK9 has the benefit of a secreted protein that can be measured from survival bleeds to confirm successful delivery. In contrast, our AAV-CRISPR system more closely mimics the Ldlr KO mouse through genetic disruption of the gene. The AAV-CRISPR approach also avoids supraphysiological levels of PCSK9 in plasma. Given the high variability inherent in any atherosclerosis study, neither method will replace the time-tested Ldlr KO mouse model. However, we believe that both are valuable tools in the right setting. AAV-CRISPR may be particularly useful in 1) pilot studies to determine the feasibility of pursuing a new gene, 2) high throughput screening of candidate genes for atherosclerosis, 3) studies of genetically modified mice in strains other than C57BL6/J or mixed backgrounds, and 4) situations where multiple transgenes or targeted alleles are required, and crossing to Ldlr KO is impossible. We have made the AAV-CRISPR vector freely available, and believe that it will be an important addition to our arsenal of tools for atherosclerosis research.
Supplementary Material
Highlights.
Somatic disruption of Ldlr produces severe hypercholesterolemia and atherosclerosis.
An all-in-one AAV-CRISPR vector to knock out Ldlr in adult mice with a single injection, avoiding the need to cross to Ldlr KO mice.
Genetic disruption of Ldlr with AAV-CRISPR is efficient and specific.
AAV-CRISPR is a versatile tool for atherosclerosis studies which is most effective in male mice.
Acknowledgments
Sources of Funding: This work was supported by the American Heart Association 16BGIA26420102 (W.R.L.), the John S. Dunn Foundation Collaborative Research Award (G.B. and W.R.L.), the Cancer Prevention and Research Institute of Texas (CPRIT) grants RR140081 and RP170721 (G.B.), the National Institutes of Health T32 GM08231 (K.E.J.), T32 HL07676 (K.E.J.), HL129767 (H.J.P.), HL132840 (W.R.L.), and through the Texas Digestive Diseases morphology core (P30DK56338).
Abbreviations
- AAV
Adeno-Associated Virus
- Apoe
Apolipoprotein E
- CRISPR/Cas9
Clustered Regularly Interspaced Short Palindromic Repeats/Cas9
- gRNA
guide RNA
- Indels
insertions or deletions
- ITR
inverted terminal repeat
- LDLR
Low density lipoprotein receptor
- PCSK9
Proprotein Convertase Subtilisin/Kexin Type 9
- PAM
Protospacer Adjacent Motif
- SaCas9
Staphylococcus aureus Cas9
- SpCas9
Streptoccoccus pyogenes Cas9
Footnotes
Disclosures: The authors have no actual or perceived conflicts of interest to disclose.
References
- 1.Daugherty A, Tall AR, Daemen MJAP, Falk E, Fisher EA, García-Cardeña G, Lusis AJ, Owens AP, Rosenfeld ME, Virmani R. Recommendation on design, execution, and reporting of animal atherosclerosis studies: A scientific statement from the American Heart Association. Circ Res. 2017;121:e53–e79. doi: 10.1161/RES.0000000000000169. [DOI] [PubMed] [Google Scholar]
- 2.Nurnberg ST, Zhang H, Hand NJ, Bauer RC, Saleheen D, Reilly MP, Rader DJ. From Loci to Biology: Functional Genomics of Genome-Wide Association for Coronary Disease. Circ Res. 2016;118:586–606. doi: 10.1161/CIRCRESAHA.115.306464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basu D, Hu Y, Huggins L-A, Mullick AE, Graham MJ, Wietecha T, Barnhart S, Mogul A, Pfeiffer K, Zirlik A, Fisher EA, Bornfeldt KE, Willecke F, Goldberg IJ. Novel Reversible Model of Atherosclerosis and Regression Using Oligonucleotide Regulation of the LDL Receptor. Circ Res. 2018;122:560–567. doi: 10.1161/CIRCRESAHA.117.311361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lagor WR, Johnston JC, Lock M, Vandenberghe LH, Rader DJ. Adeno-associated viruses as liver-directed gene delivery vehicles: focus on lipoprotein metabolism. Methods Mol Biol. 2013;1027:273–307. doi: 10.1007/978-1-60327-369-5_13. [DOI] [PubMed] [Google Scholar]
- 5.Naoumova RP, Tosi I, Patel D, Neuwirth C, Horswell SD, Marais AD, Van Heyningen C, Soutar AK. Severe hypercholesterolemia in four British families with the D374Y mutation in the PCSK9 gene: Long-term follow-up and treatment response. Arterioscler Thromb Vasc Biol. 2005;25:2654–2660. doi: 10.1161/01.ATV.0000190668.94752.ab. [DOI] [PubMed] [Google Scholar]
- 6.Roche-Molina M, Sanz-Rosa D, Cruz FM, García-Prieto J, López S, Abia R, Muriana FJG, Fuster V, Ibáñez B, Bernal JA. Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler Thromb Vasc Biol. 2015;35:50–9. doi: 10.1161/ATVBAHA.114.303617. [DOI] [PubMed] [Google Scholar]
- 7.Goettsch C, Hutcheson JD, Hagita S, Rogers MA, Creager MD, Pham T, Choi J, Mlynarchik AK, Pieper B, Kjolby M, Aikawa M, Aikawa E. A single injection of gain-of-function mutant PCSK9 adeno-associated virus vector induces cardiovascular calcification in mice with no genetic modification. Atherosclerosis. 2016;251:109–118. doi: 10.1016/j.atherosclerosis.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar S, Kang D-W, Rezvan A, Jo H. Accelerated atherosclerosis development in C57Bl6 mice by overexpressing AAV-mediated PCSK9 and partial carotid ligation. Lab Invest. 2017;97:935–945. doi: 10.1038/labinvest.2017.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu H, Howatt DA, Balakrishnan A, Graham MJ, Mullick AE, Daugherty A. Hypercholesterolemia induced by a PCSK9 gain-of-function mutation augments angiotensin II-induced abdominal aortic aneurysms in C57BL/6 mice-brief report. Arterioscler Thromb Vasc Biol. 2016;36:1753–1757. doi: 10.1161/ATVBAHA.116.307613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kosenko T, Golder M, Leblond G, Weng W, Lagace TA. Low density lipoprotein binds to proprotein convertase subtilisin/kexin type-9 (PCSK9) in human plasma and inhibits PCSK9-mediated low density lipoprotein receptor degradation. J Biol Chem. 2013;288:8279–88. doi: 10.1074/jbc.M112.421370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun H, Samarghandi A, Zhang N, Yao Z, Xiong M, Teng B-B. Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low-density lipoprotein receptor. Arterioscler Thromb Vasc Biol. 2012;32:1585–95. doi: 10.1161/ATVBAHA.112.250043. [DOI] [PubMed] [Google Scholar]
- 12.Sun H, Krauss RM, Chang JT, Teng B-B. PCSK9 deficiency reduces atherosclerosis, apolipoprotein B secretion and endothelial dysfunction. J Lipid Res. 2017;59 doi: 10.1194/jlr.M078360. jlr.M078360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan D, Yancey PG, Qiu S, Ding L, Weeber EJ, Linton MF, Fazio S. Self-association of human PCSK9 correlates with its LDLR-degrading activity. Biochemistry. 2008;47:1631–9. doi: 10.1021/bi7016359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-Guide Human Genome Engineering via Cas9. Science (80- ) 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, Graham DB, Jhunjhunwala S, Heidenreich M, Xavier RJ, Langer R, Anderson DG, Hacohen N, Regev A, Feng G, Sharp PA, Zhang F. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jarrett KE, Lee CM, Yeh Y-H, Hsu RH, Gupta R, Zhang M, Rodriguez PJ, Lee CS, Gillard BK, Bissig K-D, Pownall HJ, Martin JF, Bao G, Lagor WR. Somatic genome editing with CRISPR/Cas9 generates and corrects a metabolic disease. Sci Rep. 2017;7:44624. doi: 10.1038/srep44624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bjørklund MM, Hollensen AK, Hagensen MK, Dagnæs-Hansen F, Christoffersen C, Mikkelsen JG, Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684–1689. doi: 10.1161/CIRCRESAHA.114.302937. [DOI] [PubMed] [Google Scholar]
- 19.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–32. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daugherty A, Tall AR, Daemen MJAP, Falk E, Fisher EA, García-Cardeña G, Lusis AJ, Owens AP, Rosenfeld ME, Virmani R American Heart Association Council on Arteriosclerosis Tand VB C on BCS. Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement From the American Heart Association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157. doi: 10.1161/ATV.0000000000000062. [DOI] [PubMed] [Google Scholar]
- 21.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of Sex Differences in Design and Reporting of Experimental Arterial Pathology Studies—Statement From ATVB CouncilHighlights. Arterioscler Thromb Vasc Biol. 2018;38:292–303. doi: 10.1161/ATVBAHA.117.309524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gillard BK, Courtney HS, Massey JB, Pownall HJ. Serum opacity factor unmasks human plasma high-density lipoprotein instability via selective delipidation and apolipoprotein A-I desorption. Biochemistry. 2007;46:12968–12978. doi: 10.1021/bi701525w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lagor WR, Fields DW, Bauer RC, Crawford A, Abt MC, Artis D, Wherry EJ, Rader DJ. Genetic manipulation of the ApoF/Stat2 locus supports an important role for type I interferon signaling in atherosclerosis. Atherosclerosis. 2014;233:234–241. doi: 10.1016/j.atherosclerosis.2013.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider Ca, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ness GC, Zhao Z. Thyroid hormone rapidly induces hepatic LDL receptor mRNA levels in hypophysectomized rats. Arch Biochem Biophys. 1994;315:199–202. doi: 10.1006/abbi.1994.1490. [DOI] [PubMed] [Google Scholar]
- 26.Cradick TJ, Qiu P, Lee CM, Fine EJ, Bao G. COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Mol Ther Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tavori H, Christian D, Minnier J, Plubell D, Shapiro MD, Yeang C, Giunzioni I, Croyal M, Duell PB, Lambert G, Tsimikas S, Fazio S. PCSK9 association with lipoprotein(a) Circ Res. 2016;119:29–35. doi: 10.1161/CIRCRESAHA.116.308811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davidoff AM, Ng CYC, Zhou J, Spence Y, Nathwani AC. Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood. 2003;102:480–488. doi: 10.1182/blood-2002-09-2889. [DOI] [PubMed] [Google Scholar]
- 29.Pañeda A, Vanrell L, Mauleon I, Crettaz JS, Berraondo P, Timmermans EJ, Beattie SG, Twisk J, van Deventer S, Prieto J, Fontanellas A, Rodriguez-Pena MS, Gonzalez-Aseguinolaza G. Effect of adeno-associated virus serotype and genomic structure on liver transduction and biodistribution in mice of both genders. Hum Gene Ther. 2009;20:908–17. doi: 10.1089/hum.2009.031. [DOI] [PubMed] [Google Scholar]
- 30.Ding Q, Strong A, Patel KM, Ng SL, Gosis BS, Regan SN, Cowan CA, Rader DJ, Musunuru K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. 2014;115:488–492. doi: 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Raghavan A, Chen T, Qiao L, Zhang Y, Ding Q, Musunuru K. CRISPR-Cas9 targeting of PCSK9 in human hepatocytes in vivo - Brief report. Arterioscler Thromb Vasc Biol. 2016;36:783–786. doi: 10.1161/ATVBAHA.116.307227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chadwick AC, Wang X, Musunuru K. In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler Thromb Vasc Biol. 2017;37:1741–1747. doi: 10.1161/ATVBAHA.117.309881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chadwick AC, Evitt NH, Lv W, Musunuru K. Reduced Blood Lipid Levels With In Vivo CRISPR-Cas9 Base Editing of ANGPTL3. Circulation. 2018;137:975–977. doi: 10.1161/CIRCULATIONAHA.117.031335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki M, Bertin TK, Rogers GL, Cela RG, Zolotukhin I, Palmer DJ, Ng P, Herzog RW, Lee B. Differential type i interferon-dependent transgene silencing of helper-dependent adenoviral vs. adeno-associated viral vectors in vivo. Mol Ther. 2013;21:796–805. doi: 10.1038/mt.2012.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu ZX, Govindarajan S, Okamoto S, Dennert G. Fas-mediated apoptosis causes elimination of virus-specific cytotoxic T cells in the virus-infected liver. J Immunol. 2001;166:3035–41. doi: 10.4049/jimmunol.166.5.3035. [DOI] [PubMed] [Google Scholar]
- 36.Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, Wilson JM, Batshaw ML. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80:148–158. doi: 10.1016/j.ymgme.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 37.George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J, Cuker A, Sullivan LM, Majumdar S, Teitel J, McGuinn CE, Ragni MV, Luk AY, Hui D, Wright JF, Chen Y, Liu Y, Wachtel K, Winters A, Tiefenbacher S, Arruda VR, van der Loo JCM, Zelenaia O, Takefman D, Carr ME, Couto LB, Anguela XM, High KA. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N Engl J Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ajufo E, Cuchel M. Recent Developments in Gene Therapy for Homozygous Familial Hypercholesterolemia. Curr Atheroscler Rep. 2016;18 doi: 10.1007/s11883-016-0579-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dias MF, Joo K, Kemp JA, Fialho SL, da Silva Cunha A, Woo SJ, Kwon YJ. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog Retin Eye Res. 2018;63:107–131. doi: 10.1016/j.preteyeres.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 40.Singh K, Evens H, Nair N, Rincón MY, Sarcar S, Samara-Kuko E, Chuah MK, VandenDriessche T. Efficient In Vivo Liver-Directed Gene Editing Using CRISPR/Cas9. Mol Ther. 2018;26 doi: 10.1016/j.ymthe.2018.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song C, Wang D, Jiang T, O’Connor K, Tang Q, Cai L, Li X, Weng Z, Yin H, Gao G, Mueller C, Flotte TR, Xue W. In vivo genome editing partially restores alpha1-antitrypsin in a murine model of AAT deficiency. Hum Gene Ther. 2018:1–10. doi: 10.1089/hum.2017.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ran FA, Cong L, Yan WX, Scott Da, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp Pa, Zhang F. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–190. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brinkman EK, Kousholt AN, Harmsen T, Leemans C, Chen T, Jonkers J, van Steensel B. Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res. 2018:218156. doi: 10.1093/nar/gky164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hordeaux J, Wang Q, Katz N, Buza EL, Bell P, Wilson JM. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol Ther. 2018;26:664–668. doi: 10.1016/j.ymthe.2018.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Y, Wang L, Bell P, McMenamin D, He Z, White J, Yu H, Xu C, Morizono H, Musunuru K, Batshaw ML, Wilson JM. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat Biotechnol. 2016;34:334–338. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.