

Abstract
Background and Purpose –
Aging and hypertension, co-morbidities prevalent in the stroke population, are associated with poor collateral status and worsened stroke outcome. However, underlying mechanisms by which these conditions affect stroke outcome are not clear. We studied the role of plasminogen activator inhibitor-1 (PAI-1) that is increased in aging and hypertension on brain and vascular expression of inflammatory factors and perfusion that may contribute to worse stroke outcomes.
Methods –
Aged (~50 weeks) and young (~18 weeks) spontaneously hypertensive rats (SHR) were subjected to ischemia by middle cerebral artery occlusion (MCAO, 2hr) and reperfusion (2hr) with or without treatment with the PAI-1 inhibitor TM5441. Changes in MCA and collateral perfusion territories were measured by multisite laser Doppler. Reactivity to TM5441 was studied using isolated and pressurized leptomeningeal anastomotic arterioles (LMAs). Brain injury was determined by 2,3,5-triphenyltetrazolium staining and quantitative immunohistochemistry of amyloid-β−42, PAI-1, and hemoglobin. Circulating inflammatory factors were measured by ELISA.
Results –
Changes in cerebral blood flow during MCAO were similar between groups, with both having poor collateral perfusion and incomplete reperfusion. However, aged SHR had greater brain injury vs. young (41±2 vs 23±2%, p<0.05) as well as increased brain deposition of amyloid-β−42 and circulating oxidized low-density lipoprotein. Erythrocyte aggregation and hemorrhage within the injured brain was observed in 50% of aged but no young SHR, with increased circulating PAI-1 in this subgroup of aged SHR (16±3 vs 6±2ng/ml, p<0.05). PAI-1 inhibition with TM5441 improved brain injury but did not affect hemorrhage. TM5441 increased collateral perfusion by 38±7% and dilated LMAs by 44±10%, which was abolished by nitric oxide synthase inhibition.
Conclusion –
Increased injury in aged SHR appeared to be related to poor collateral perfusion, hemorrhagic transformation, increased amyloid-β−42 and oxidative stress. PAI-1 inhibition reduced infarction in both groups of SHR that possibly due, in part, to increased collateral perfusion.
Keywords: acute stroke, aging, cerebral blood flow, focal ischemia, hypertension
INTRODUCTION
Many acute stroke patients are aged and with a history of chronic hypertension. For example, The International Stroke Trial of more than 17,000 patients revealed that 81.6% of patients were hypertensive and 84.1% were 60 years or older.1 Although slightly different among clinical studies, the majority of reported average age of first-ever stroke was approximately 70 years old or higher, further highlighting the large population of stroke patients that are aged.2, 3 Not only are aging and hypertension major risk factors and co-morbidities of stroke, patients who are aged and hypertensive have more severe stroke and worse stroke outcome, including larger infarct, and increased incidence of disability and death.1, 4 Importantly, larger infarct is associated with greater incidence of hemorrhagic transformation, although there is no direct evidence suggesting that patients who are aged and hypertensive are more likely to have this secondary injury after acute ischemic stroke.5, 6 Despite these epidemiological data, the effects of aging and hypertension on stroke outcome are under-studied as the majority of animal stroke research uses young male animals. In 2009, the Stroke Treatment Academic Industry Roundtable (STAIR) recommended the use of female, hypertensive, aged, and diabetic animals in pre-clinical stroke research to better mimic the human stroke population.7 The use of aged and hypertensive animals has been increasing for use in pre-clinical stroke studies, but the combined effect of aging and hypertension on stroke outcome is still largely unknown.
Both aging and hypertension are associated with increased tissue and circulating PAI-1, an endogenous serine protease inhibitor that regulates fibrinolysis, vascular fibrosis and endothelial cell senescence that could be an important contributor to stroke and stroke outcome. Increased PAI-1 reduces tissue (tPA)- and urokinase (uPA)-mediated plasmin production, leading to decreased fibrinolysis that is well-known in aging.8, 9 In addition, increased PAI-1 and decreased tPA has been found in patients with hypertension, suggesting a link between hypertension and hypofibrinolysis as well.10 PAI-1 also appears to have a role in stroke outcome. Overexpression of PAI-1 increased brain injury in a mouse model of thrombotic stroke but not permanent middle cerebral artery (MCA) ligation, suggesting increased ischemic brain injury may be related to PAI-1-mediated increase in coagulation and subsequent reduced reperfusion.11 In addition, inhibition of PAI-1 with an inhibitory antibody increased reperfusion, reduced infarct volume and fibrin deposition in a mouse model of acute ischemic stroke.12 These results demonstrate that PAI-1 inhibition may reduce ischemic brain injury through alleviating incomplete reperfusion by preventing coagulation, but it remains unknown in the setting of aging and hypertension when PAI-1 expression is high. Therefore, we studied the effect of pharmacological PAI-1 inhibition on cerebral collateral perfusion, incomplete reperfusion, and stroke outcome in aged compared to young hypertensive rats. We also investigated the role of elevated oxidative stress and chronic inflammation in aging and hypertension as they have major impacts on the cerebral circulation, neurons, and other brain cells that may be involved in worsening brain injury after MCAO in aged SHR.13, 14 Therefore, we measured mediators and markers of oxidative stress and inflammation that are known to be increased in aging and hypertension, including amyloid-β−42 protein (Aβ42), oxidized low-density lipoprotein (oxLDL), and tumor necrosis factor-α (TNFα).
MATERIALS AND METHODS
The data that support the findings of this study are available upon request. Please send request to the corresponding author.
Animals
Aged male SHR (50–52 weeks old, n=12) and control young male SHR (17–18 weeks old, n=18) were used in this study. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Vermont and complied with the National Institutes of Health guidelines for care and use of laboratory animals. Detailed animal descriptions are available in the Online Supplement (please see http://stroke.ahajournals.org).
Multi-site Laser Doppler Flowmetry
Dual laser Doppler probes (Perimed Inc., Ardmore, PA, USA) were used to measure changes in cerebral perfusion in the MCA and collateral perfusion territories, as described previously.15
Model of Transient Focal Ischemia and Treatment
A proximal MCAO filament model was used, as previously described.16 The MCA was occluded for 2 hours, followed by reperfusion for 2 hours. Some young (n=6) and aged (n=4) SHR were treated with a PAI-1 inhibitor TM5441 (Tocris, Minneapolis, MN, USA; 5 mg/kg, i.v. dissolved in DMSO diluted in saline) 10 minutes before reperfusion and compared to untreated groups. Detailed experimental procedures are available in the Online Supplement (please see http://stroke.ahajournals.org).
Stroke Outcome Determinations
Brain injury volume was determined by 2,3,5-triphenyltetrazolium chloride (TTC) staining, identified as white area of each section and measured with ImageJ software (NIH, Bethesda, MD, USA). Measurements were made by investigators who were blinded to experimental groups. Further microscopic observation for hemorrhagic transformation was performed using brain coronal sections stained with hematoxylin and eosin (H&E). Detailed experimental procedures are available in the Online Supplement (please see http://stroke.ahajournals.org).
Immunohistochemistry of Aβ42, PAI-1, Hemoglobin and Collagen IV
Formalin fixed, paraffin embedded brain coronal sections were cut into 5 μm sections for immunohistochemistry using standard techniques. Detailed experimental procedures and quantitative analyses are available in the Online Supplement (please see http://stroke.ahajournals.org).
Determination of Circulating Oxidative Stress and Inflammatory Markers
Commercially available ELISA kits were used for determining serum levels of Aβ42, PAI-1, oxLDL, and TNFα. Detailed experimental procedures are available in the Online Supplement (please see http://stroke.ahajournals.org).
Reactivity of Isolated Leptomeningeal Anastomoses
Leptomeningeal anastomotic arterioles (LMAs) identified as distal branches between anterior cerebral arteries (ACAs) and MCAs were isolated and pressurized in an arteriograph bath (Living System Instrumentation, St. Albans, VT, USA) containing circulating physiological saline solution (PSS) from young SHR (n=4), as previously described.17 LMAs were allowed to equilibrate for 1 hour at 20 mmHg to develop spontaneous tone. Myogenic reactivity of LMAs was determined by stepwise increase in pressure from 20–100 mmHg. One vessel was excluded because it did not develop sufficient spontaneous tone. Reactivity of LMAs to cumulative concentrations of TM5441 (10−8-10−5 mol/L) was tested first in the absence and then presence of Nω-nitro-L-arginine methyl ester (L-NAME, 10−4 mol/L; Sigma, St. Louis, MO, USA). This range of TM5441 concentration closely matched the dose in the in vivo treatment (maximum plasma concentration of 5 mg/kg TM5441 was ~1.8×10−5 mol/L).18 Zero calcium PSS was used to produce maximum dilation of LMAs and calculate tone and reactivity.
Data Calculations and Statistical Analysis
Calculation for percent tone and percent reactivity in isolated LMAs was described in detail previously.17 Results are presented as mean ± standard error of mean (SEM). Mann-Whitney or t-test was used to determine statistical differences between groups where appropriate using GraphPad Prism 7 (La Jolla, CA, USA). Repeated measures one-way analysis of variance (ANOVA) was used for changes of cerebral perfusion versus baseline. Two-way analysis of variance with a Tukey’s test for multiple comparisons was used for determining differences between groups. Differences were considered statistically significant at P<0.05.
Excluded Animals
A total of 5 animals (3 aged and 2 young SHR) were excluded due to insufficient drop in cerebral perfusion that was less than 70% during ischemia. One additional young SHR was excluded from isolated LMA studies due to insufficient tone development.
RESULTS
Physiological Parameters during Ischemia and Reperfusion in Young and Aged SHR
Mean arterial pressures in anesthetized animals were not different between young and aged SHR (Table II, Online Supplement). Arterial blood gases, measured at the beginning, middle, and end of the experiment, were in the physiological ranges throughout the experiment and were similar between the groups (Table II, Online Supplement).
Changes in Cerebral Perfusion during Ischemia and Reperfusion in Young and Aged SHR
Figure 1A shows changes in MCA flow during ischemia and reperfusion. Cerebral perfusion in the core MCA territory was decreased similarly during ischemia (0–120 minutes) between young and aged SHR, suggesting similar stroke severity. During reperfusion (120–240 minutes), cerebral perfusion was below baseline in both groups, indicating incomplete reperfusion that was similar between groups. Despite similar changes in CBF during ischemia and reperfusion, early brain injury volume was significantly increased in aged SHR (Figure 1B,C).
Figure 1. Cerebral blood flow (CBF) changes and acute injury during middle cerebral artery occlusion (MCAO) in young and aged SHR.
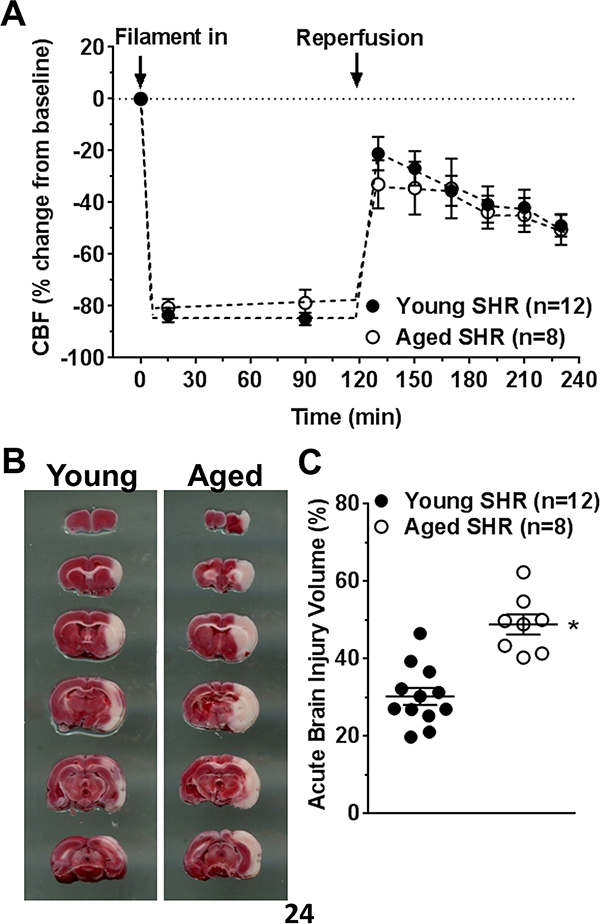
(A) Percent change in CBF during MCAO and reperfusion. Both young and aged SHR had similarly decreased CBF during filament occlusion and similar reperfusion that was incomplete in both groups. (B) Representative coronal sections stained with TTC and used for injury volume analysis. (C) Graph showing acute injury volume was increased in aged SHR. * p<0.05 vs. young SHR by Student’s t-test.
Brain capillary density was similar between young and aged SHR (Figure I, Online Supplement), suggesting increased brain injury volume in aged SHR was not due to capillary rarefaction in these animals.
Expression of Aβ42 and Circulating Markers of Oxidative Stress and Inflammation in Young and Aged SHR
Figure 2A and 2B show representative images of positive immunostaining of Aβ42 (blue) in brain tissue and capillaries, distinguished by positive collagen IV staining (green) in young and aged SHR, respectively. Micrographs of no primary antibody control for Aβ42 showed no non-specific staining (Figure IIA, Online Supplement). Percentage of capillaries that had positive Aβ42 staining was not different between groups (Figure 2C). However, when quantified over the whole image, the percentage of positive Aβ42 staining was significantly increased in aged when compared to young SHR (Figure 2D). Circulating Aβ42 was undetectable in both groups of rats (data not shown). However, circulating oxLDL levels were significantly increased in aged versus young SHR (3.93±0.44 vs. 1.85±0.32 ng/ml; p<0.05). In addition, circulating TNFα levels were insignificantly lower in aged versus young SHR (20.7±6.6 vs. 38.0±8.2 pg/ml).
Figure 2. Immunohistochemistry of Aβ42 in rat brain after ischemia and reperfusion.
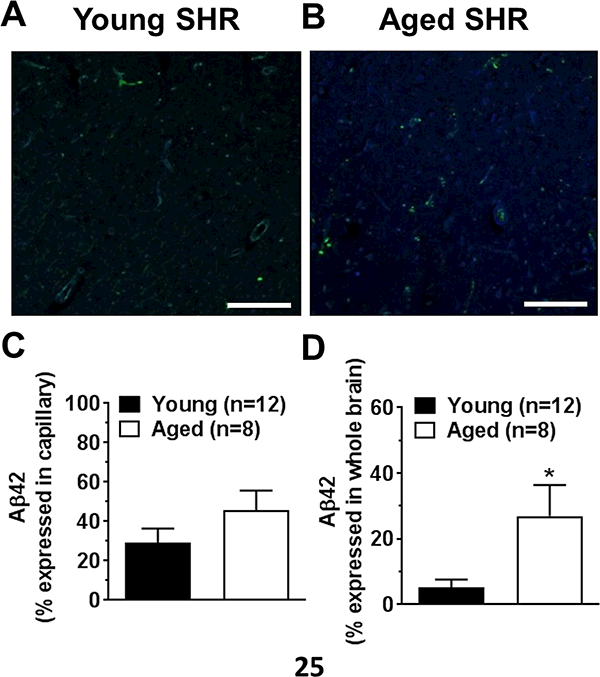
Representative images showing staining of collagen IV (green) and Aβ42 (blue) in young (A) and aged (B) SHR. Graphs showing average percentage of Aβ42 positive staining in capillaries (C) and brain tissue (D) in young and aged SHR. *P<0.05 versus young SHR by Student’s t-test. Scale bar: 100 μm.
Erythrocyte Aggregation, Hemorrhage and PAI-1 Expression in Young and Aged SHR
In addition to increased ischemic brain injury volume, 50% of aged SHR had pink coloration within the injury areas (Figure 3A, upper). Microscopic analysis with H&E staining showed the appearance of erythrocyte aggregation within blood vessels, suggesting red cell stasis and possibly early hemorrhagic transformation (Figure 3A, lower). To confirm that the pink coloration was erythrocytes, we performed immunohistochemistry for hemoglobin and quantified the fluorescence in the contralateral and ipsilateral brain regions (a representative image is shown in Figure IIF, Online Supplement). Figure 3B shows that hemoglobin fluorescence was detected in the contralateral hemispheres of both young and aged SHR. In addition, there was an increase in hemoglobin in the ipsilateral hemispheres that was not different overall with aging. However, when the aged brains were stratified by pink coloration vs. no coloration, there was a significant increase in hemoglobin in the brains with coloration, indicative of erythrocyte aggregation (Figure 3C). However, supplemental Figure III shows that hemoglobin was found outside the vasculature, suggesting at least some of the pink coloration was hemorrhage.
Figure 3. Erythrocyte aggregation and hemoglobin staining within the brain injury area and the role of PAI-1.
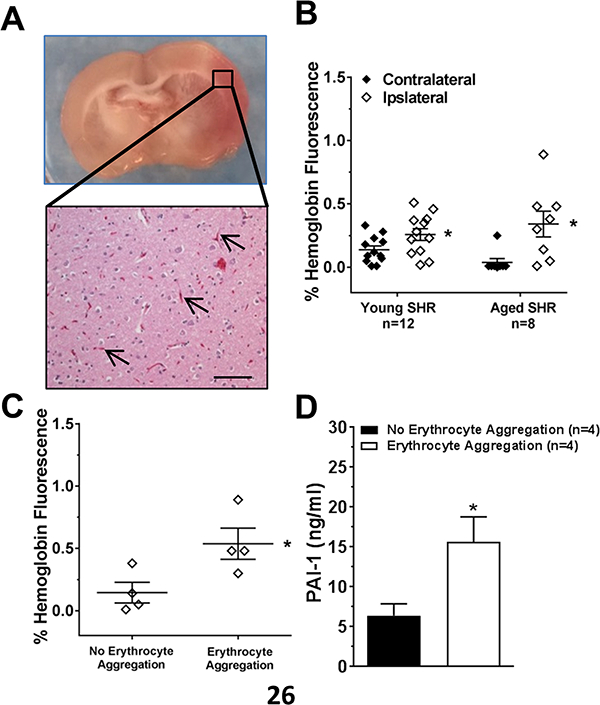
Representative images of a brain coronal section with pink coloration within the injury area (A, upper) and erythrocyte aggregation in brain capillaries with H&E staining (A, lower) in aged SHR. Scale bar: 100 μm (B) Graph showing quantification of hemoglobin staining by immunohistochemistry within the contralateral and ipsilateral side of occlusion for young and aged SHR. Hemoglobin was increased in both groups on the ipsilateral side. * p<0.05 vs. contralateral by Student’s t-test. (C) Graph showing comparison between aged SHR brains with and without pink coloration (erythrocyte aggregation). Brains with pink coloration had increased hemoglobin. * p<0.05 vs. no erythrocyte aggregation by Student’s t-test. (D) Comparison of PAI-1 in serum of aged SHR with and without pink coloration. * p<0.05 vs. no erythrocyte aggregation by Student’s t-test.
To determine if erythrocyte aggregation in capillaries in aged SHR were related to increased PAI-1, we measured circulating and tissue levels of PAI-1. Supplemental Figure IV shows there was no difference in cerebral microvessel expression of PAI-1 between aged SHR brains with and without pink coloration. However, serum PAI-1 levels were significantly increased in aged SHR with erythrocyte aggregation versus those without (Figure 3D). Thus, increased circulating PAI-1 levels, but not capillary expression of PAI-1, appeared related to erythrocyte aggregation in aged SHR after stroke.
Inhibition of PAI-1 during MCAO in SHR
The increase in circulating PAI-1 in aged SHR with hemorrhage led us to hypothesize that PAI-1 was involved in the increased brain injury in aged SHR. Therefore, the small molecule PAI-1 inhibitor TM5441 was given 10 minutes prior to reperfusion in both young and aged SHR. One aged SHR treated with TM5441 was excluded due to insufficient drop in CBF during filament occlusion that later was determined to have a secondary M1 branch of the MCA. The remaining animals all had CBF drops that were between 78 ± 4 and 84 ± 3% (p>0.05 for all). Figure 4A shows that acute treatment with TM5441 decreased acute brain injury volume in both young and aged SHR. Interestingly, PAI-1 inhibition did not affect the incidence of erythrocyte aggregation in aged SHR (Figure 4B). Moreover, PAI-1 inhibition did not significantly affect circulating levels of oxLDL (3.93±0.44 vs. 3.11±0.53 ng/ml; p>0.05) and TNFα (20.7±6.6 vs. 16.2±3.5 pg/ml) in aged SHR. In addition, treatment with TM5441 did not affect reperfusion CBF in the MCA territory that was below baseline and similar between groups (data not shown). However, TM5441 increased collateral flow in both young and aged SHR by 30–40 % relative to before treatment when the filament was still in place (Figure 5B). When compared to baseline (before filament insertion), TM5441 increased collateral CBF in young from −58±8 to −45±12% (p<0.05) and aged SHR from −43±12 to −28±17% (p=0.08).
Figure 4. The effect of PAI-1 inhibition on acute brain injury and erythrocyte aggregation in young and aged SHR during MCAO.
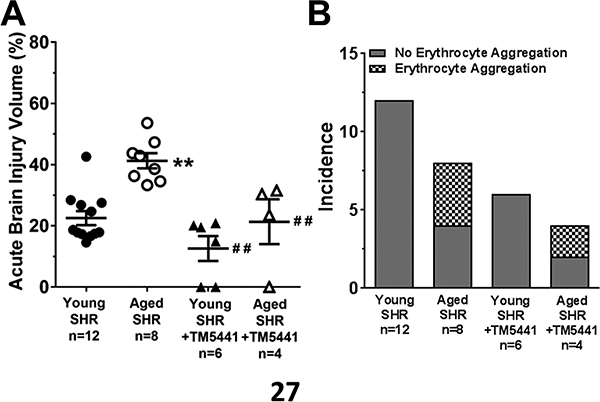
(A) TM5441 decreased the acute injury volume in both young and aged SHR. ** p<0.01 vs. untreated young SHR; ## p<0.01 vs. untreated aged SHR by two-way ANOVA with post hoc Tukey’s test. (B) Treatment with TM5441 did not decrease the incidence of erythrocyte aggregation in aged SHR.
Figure 5. Effect of PAI-1 inhibition on pial collaterals.
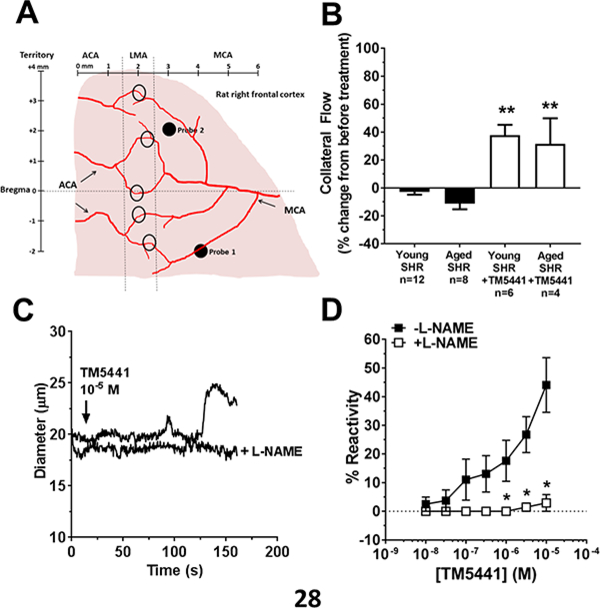
(A) Diagram showing placement of multi-site laser Doppler probes for measurement of core and collateral cerebral blood flow during MCAO and reperfusion. Shown also are leptomeningeal anastomoses (LMAs, black circles) between anterior (ACA) and middle cerebral arteries (MCA) that were dissected for reactivity studies. (B) Change in collateral flow (Probe 2) during MCAO and in response to treatment with TM5441. Both untreated young and aged SHR had little to no collateral flow that was increased to a similar extent with TM5441 treatment. ** p<0.01 vs. untreated by two-way ANOVA with Tukey’s multiple comparisons test. (C) Representative tracing of changes in diameter of isolated and pressurized LMAs. Addition of TM5441 caused concentration-dependent vasodilation that was prevented by nitric oxide inhibition with L-NAME. (D) Graph showing % reactivity of isolated pial collaterals to TM5441 in the absence and presence of L-NAME. * p<0.05 vs. without L-NAME by paired t-test; n=4.
To determine if the increase in collateral flow by TM5441 was due to vasodilation of LMAs (pial collaterals), an isolated vessel approach was used. As published previously, isolated and pressurized LMAs from SHR had considerable myogenic tone (43±4%).17 Figure 5C shows a representative tracing of LMA diameter changes to TM5441 in the absence or presence of L-NAME. TM5441 caused dose-dependent vasodilation of LMAs that was blocked in the presence of L-NAME (Figure 5D).
DISCUSSION
In this study, we used aged hypertensive rats to show that acute brain injury was significantly increased after ischemia and reperfusion when compared to their younger counterparts. Increased ischemic brain injury in aged SHR appeared to be multifactorial and was associated with increased oxidative stress, Aβ deposition, erythrocyte aggregation and hemorrhagic transformation. Pharmacological inhibition of PAI-1 during ischemia did not prevent aging-induced erythrocyte aggregation and hemorrhage, or incomplete reperfusion, but improved collateral perfusion that likely reduced ischemic brain injury (Figure 6).
Figure 6. Diagram summarizing potential mechanisms of increased brain injury in aged SHR after MCAO.
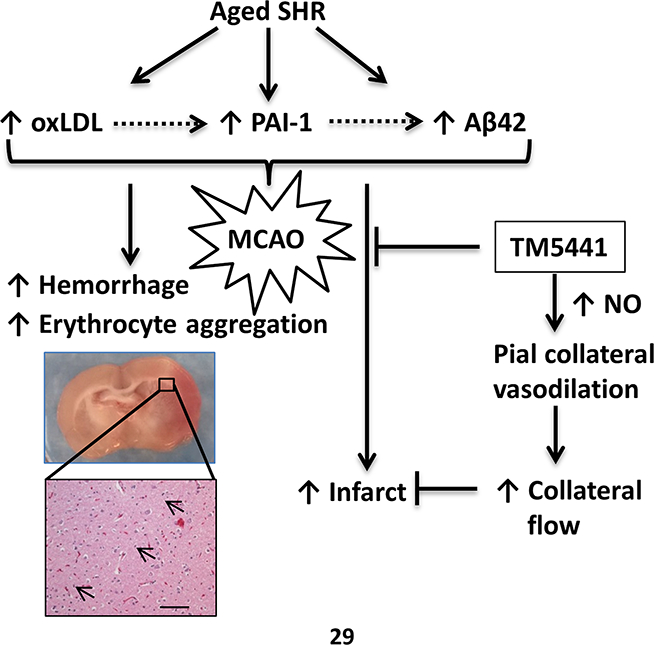
Aged SHR have increased oxidative stress as measured by high levels of oxLDL that can activate PAI-1 through NADPH oxidase. Increased PAI-1 could increase Aβ42 deposition through increased protease inhibition. During MCAO, impaired collateral perfusion in both young and aged SHR was increased by PAI-1 inhibition in a NO-dependent manner that likely contributed to the decrease in infarction. However, PAI-1 inhibition did not affect hemorrhage or erythrocyte aggregation in aged SHR.
PAI-1 is a serine protease inhibitor that regulates fibrinolysis by inhibiting plasminogen activators tPA and uPA.8 Circulating PAI-1 is considered a marker of aging and has been shown to be a mediator of cellular aging including vascular endothelial cells.9, 19 PAI-1 is also increased in hypertension through angiotensin type 1 receptor activation that can cause hemostatic and fibrinolytic dysregulation and worsened stroke outcome through impaired microvascular perfusion.10, 20 A previous study demonstrated that after acute embolic stroke in rats, PAI-1 was upregulated with increased fibrin obstruction of brain capillaries as early as 1 hour into ischemia.21 Another previous study showed that anti-PAI-1 antibody increased reperfusion cerebral blood flow, reduced fibrin deposit and ischemic brain injury measured sub-acutely between 6 to 24 hours of reperfusion.12 In the present study, we found that PAI-1 inhibition reduced acute brain injury, but not hemorrhage, suggesting the decreased infarction was not related to decreasing erythrocyte obstruction of brain capillaries. Further supporting this conclusion was that reperfusion cerebral blood flow was similarly low in both groups of SHR. However, we unexpectedly found that PAI-1 inhibitor TM5441 caused an increase in collateral perfusion acutely during ischemia, and dilated LMAs that was blocked by NOS inhibition.
In chronic hypertension, we have previously shown that LMAs from hypertensive rats were more constricted compared to their normotensive counterparts, suggesting collateral perfusion was limited during MCAO because of vasoconstricted LMAs and increased vascular resistance.17 Importantly, increased LMA constriction was not more severe in aged versus young SHR.17 The findings of the present study agree with the previous finding that changes in collateral perfusion during ischemia was close to zero or below in untreated young SHR and not worse in aged SHR. Interestingly, PAI-1 inhibition with TM5441 significantly increased collateral perfusion in young as well as aged SHR by 30–40%. To confirm TM5441 could directly increase collateral perfusion, we measured the vasodilatory response to TM5441 in isolated LMAs. We found TM5441 significantly dilated LMAs from young SHR that was abolished in the presence of the NOS inhibitor L-NAME. Therefore, it is possible that TM5441-mediated increased in collateral perfusion was due to vasodilation of LMAs through an NO-dependent mechanism.
The effect of TM5441 on NOS to cause vasodilation is not completely understood from this study. A previous study showed that in endothelial cell culture, PAI-1 inhibition with TM5441 reduced reactive oxygen species and prevented cellular senescence.19 Uncoupling of endothelial NOS (eNOS), which occurs due to decreased eNOS cofactor tetrahydrobiopterin (BH4) from substrate depletion, causes production of superoxide instead of NO. BH4 depletion and eNOS uncoupling occurs with post-ischemic reperfusion, as well as aging and hypertension.22, 23 Thus, it is possible that antioxidant properties of TM5441 reduced superoxide production that caused vasodilation of LMAs either directly or increasing bioavailability of NO. Regardless, the vasodilation of LMAs was consistent with this compound increasing collateral flow in the acute phase of stroke.
In addition to increasing collateral perfusion, TM5441 may reduce ischemic brain injury by exerting neuroprotective effects. Our previous study showed that Sanguinate™ (SG), a PEGylated carboxyhemoglobin gas transfer agent, increased collateral perfusion similar to TM5441, but only reduced brain injury when animals were treated after 30 minutes of ischemia, but not when given after 90 minutes of ischemia.15 In the current study, TM5441 reduced brain injury when administrated after 110 minutes of ischemia, a time point even later than SG. It is possible that TM5441 may be neuroprotective, possibly due to its ability to cross the blood-brain barrier and exert antioxidant and/or anti-inflammatory properties centrally.19, 24 Another possibility of PAI-1 inhibition was to reduce neutrophil Infiltration, which has been shown to cause brain injury during ischemia and reperfusion.25
Aβ is well-known to accumulate in brain tissue and blood vessels and is involved in Alzheimer’s Disease and vascular dementia, respectively.26 Importantly, serine proteases (including tPA and uPA) cleave Aβ42 and is believed to be one of the mechanisms for Aβ removal.27, 28 Thus, when tPA/uPA are inhibited by high PAI-1 in aging, removal of Aβ is decreased. Increased Aβ42 in the brain has been shown to activate neuronal cell apoptotic pathways by increasing oxidative damage.29, 30 In the current study, we found increased Aβ42 in brain tissue, but not microvasculature. Therefore, it is possible that increased brain injury in aged SHR was partly due to increased susceptibility of neurons to ischemic injury due to high oxidative stress. Although amyloid angiopathy appeared absent in these aged hypertensive animals (Aβ42 was not increased in the vasculature), we did not measure Aβ40, which is more soluble and likely to be involved in deposition in brain vasculature.26
Increased oxidative stress and chronic inflammation have also been shown to be important mechanisms of hypertension- and aging-related diseases.13, 14 A recent study demonstrated that elevated aldosterone in hypertensive animals may be the cause of increased oxidative stress and inflammation.31 oxLDL, one of the circulating markers of oxidative stress, was significantly increased in aged SHR compared to young. OxLDL has been shown to upregulate PAI-1 and therefore may possibly be a mechanism for increased circulating PAI-1 in aged SHR.32 PAI-1 inhibition by TM5441 did not reduce circulating oxLDL levels, agreeing with the idea that oxLDL is an upstream activator of PAI-1. On the other hand, the circulating pro-inflammatory cytokine TNFα was paradoxically decreased in aged SHR. Although a previous study showed an increase in circulating TNFα during aging, circulating soluble TNF receptors p55 and p75 are also elevated in aging may have reduced free circulating TNFα in aged SHR.33
There are notable limitations in this study. First, absolute cerebral blood flow was not measured and has been demonstrated to be lower in aged versus young SHR.34, 35 However, we chose to use multi-site laser Doppler flowmetry to allow us to continuously monitor changes in perfusion during ischemia, treatment, and reperfusion. The drawback of this approach was that we were unable to exclude the possibility that decreased baseline cerebral blood flow had an impact on worse stroke outcomes in aged SHR. Second, we studied stroke in these hypertensive animals in an anesthetized state that lowered blood pressure. Although chloral hydrate is known to have a larger effect on blood pressure than other anesthetics, we used it because pentobarbital and isoflurane are known to be neuroprotective and vasodilatory, respectively, that may profoundly impact our endpoint measurements. Although lower, blood pressures between groups were not significantly different and therefore the potential impact of blood pressure on collateral perfusion and stroke outcomes should have been minimal.
CONCLUSIONS
Acute ischemic stroke patients who are aged and hypertensive have worse stroke outcome. Despite the majority of stroke patients having these co-morbidities, the combined effect of aging and hypertension on stroke outcome is not clear because the majority of basic stroke studies have used young male animals that are usually normotensive. In the current study, the combined effect of aging and hypertension increased brain injury, including the incidence of erythrocyte aggregation and hemorrhagic transformation in a rat model of acute ischemic stroke. Increased brain injury was reduced by PAI-1 inhibition and vasodilation of LMAs and increased collateral perfusion during ischemia may be one of several mechanisms that contribute to a reduction in injury. Increased brain deposition of Aβ42 and circulating oxLDL may also have a role in worse ischemic brain injuries in aged SHR that deserves further investigation.
Supplementary Material
Acknowledgements
We would like to thank Dr. Abbie Johnson and Ms. Sarah Tremble (Department of Neurological Sciences, University of Vermont) for performing ELISA experiments.
Sources of Funding
This work was supported by NIH National Institute of Neurological Disorders and Stroke Grant R01 NS093289, the Totman Medical Research Trust, and Cardiovascular Research Institute of Vermont. We also acknowledge support from NIH National Center for Research Resources - award number 1S10RR019246 to the Microscopy Imaging Center, University of Vermont.
Footnotes
Disclosures
None.
REFERENCES
- 1.Leonardi-Bee J, Bath PM, Phillips SJ, Sandercock PA. Blood pressure and clinical outcomes in the international stroke trial. Stroke. 2002;33:1315–1320 [DOI] [PubMed] [Google Scholar]
- 2.Williams GR, Jiang JG, Matchar DB, Samsa GP. Incidence and occurrence of total (first-ever and recurrent) stroke. Stroke. 1999;30:2523–2528 [DOI] [PubMed] [Google Scholar]
- 3.Appelros P, Stegmayr B, Terent A. Sex differences in stroke epidemiology: A systematic review. Stroke. 2009;40:1082–1090 [DOI] [PubMed] [Google Scholar]
- 4.Bonita R Epidemiology of stroke. Lancet (London, England). 1992;339:342–344 [DOI] [PubMed] [Google Scholar]
- 5.Jaillard A, Cornu C, Durieux A, Moulin T, Boutitie F, Lees KR, et al. Hemorrhagic transformation in acute ischemic stroke. The mast-e study. Mast-e group. Stroke. 1999;30:1326–1332 [DOI] [PubMed] [Google Scholar]
- 6.Paciaroni M, Agnelli G, Corea F, Ageno W, Alberti A, Lanari A, et al. Early hemorrhagic transformation of brain infarction: Rate, predictive factors, and influence on clinical outcome: Results of a prospective multicenter study. Stroke. 2008;39:2249–2256 [DOI] [PubMed] [Google Scholar]
- 7.Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto K, Takeshita K, Saito H. Plasminogen activator inhibitor-1 in aging. Seminars in thrombosis and hemostasis. 2014;40:652–659 [DOI] [PubMed] [Google Scholar]
- 9.Vaughan DE, Rai R, Khan SS, Eren M, Ghosh AK. Plasminogen activator inhibitor-1 is a marker and a mediator of senescence. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:1446–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaughan DE, Lazos SA, Tong K. Angiotensin ii regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. The Journal of clinical investigation. 1995;95:995–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagai N, Suzuki Y, Van Hoef B, Lijnen HR, Collen D. Effects of plasminogen activator inhibitor-1 on ischemic brain injury in permanent and thrombotic middle cerebral artery occlusion models in mice. Journal of thrombosis and haemostasis : JTH. 2005;3:1379–1384 [DOI] [PubMed] [Google Scholar]
- 12.Denorme F, Wyseure T, Peeters M, Vandeputte N, Gils A, Deckmyn H, et al. Inhibition of thrombin-activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1 reduces ischemic brain damage in mice. Stroke. 2016;47:2419–2422 [DOI] [PubMed] [Google Scholar]
- 13.Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension (Dallas, Tex. : 1979). 2017;70:660–667 [DOI] [PubMed] [Google Scholar]
- 14.Buford TW. Hypertension and aging. Ageing research reviews. 2016;26:96–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cipolla MJ, Linfante I, Abuchowski A, Jubin R, Chan SL. Pharmacologically increasing collateral perfusion during acute stroke using a carboxyhemoglobin gas transfer agent (sanguinate) in spontaneously hypertensive rats. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2017:271678X17705567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cipolla MJ, Sweet JG, Chan SL. Effect of hypertension and peroxynitrite decomposition with fetmpyp on cbf and stroke outcome. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2017;37:1276–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan SL, Sweet JG, Bishop N, Cipolla MJ. Pial collateral reactivity during hypertension and aging: Understanding the function of collaterals for stroke therapy. Stroke. 2016;47:1618–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boe AE, Eren M, Murphy SB, Kamide CE, Ichimura A, Terry D, et al. Plasminogen activator inhibitor-1 antagonist tm5441 attenuates nomega-nitro-l-arginine methyl ester-induced hypertension and vascular senescence. Circulation. 2013;128:2318–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh AK, Rai R, Park KE, Eren M, Miyata T, Wilsbacher LD, et al. A small molecule inhibitor of pai-1 protects against doxorubicin-induced cellular senescence. Oncotarget. 2016;7:72443–72457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koh KK, Ahn JY, Han SH, Kim DS, Jin DK, Kim HS, et al. Pleiotropic effects of angiotensin ii receptor blocker in hypertensive patients. Journal of the American College of Cardiology. 2003;42:905–910 [DOI] [PubMed] [Google Scholar]
- 21.Zhang ZG, Chopp M, Goussev A, Lu D, Morris D, Tsang W, et al. Cerebral microvascular obstruction by fibrin is associated with upregulation of pai-1 acutely after onset of focal embolic ischemia in rats. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1999;19:10898–10907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forstermann U, Li H. Therapeutic effect of enhancing endothelial nitric oxide synthase (enos) expression and preventing enos uncoupling. British journal of pharmacology. 2011;164:213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang YM, Huang A, Kaley G, Sun D. Enos uncoupling and endothelial dysfunction in aged vessels. American journal of physiology. Heart and circulatory physiology. 2009;297:H1829–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pelisch N, Dan T, Ichimura A, Sekiguchi H, Vaughan DE, van Ypersele de Strihou C, et al. Plasminogen activator inhibitor-1 antagonist tm5484 attenuates demyelination and axonal degeneration in a mice model of multiple sclerosis. PloS one. 2015;10:e0124510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Praetner M, Zuchtriegel G, Holzer M, Uhl B, Schaubacher J, Mittmann L, et al. Plasminogen activator inhibitor-1 promotes neutrophil infiltration and tissue injury on ischemia-reperfusion. Arteriosclerosis, thrombosis, and vascular biology. 2018;38:829–842 [DOI] [PubMed] [Google Scholar]
- 26.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: Lessons from the alzheimer’s amyloid beta-peptide. Nature reviews. Molecular cell biology. 2007;8:101–112 [DOI] [PubMed] [Google Scholar]
- 27.Jacobsen JS, Comery TA, Martone RL, Elokdah H, Crandall DL, Oganesian A, et al. Enhanced clearance of abeta in brain by sustaining the plasmin proteolysis cascade. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:8754–8759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melchor JP, Pawlak R, Strickland S. The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-beta (abeta) degradation and inhibits abeta-induced neurodegeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:8867–8871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends in molecular medicine. 2001;7:548–554 [DOI] [PubMed] [Google Scholar]
- 30.Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, et al. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. Journal of neurochemistry. 2001;77:220–228 [DOI] [PubMed] [Google Scholar]
- 31.Harvey AP, Montezano AC, Hood KY, Lopes RA, Rios F, Ceravolo G, et al. Vascular dysfunction and fibrosis in stroke-prone spontaneously hypertensive rats: The aldosterone-mineralocorticoid receptor-nox1 axis. Life sciences. 2017;179:110–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sangle GV, Zhao R, Shen GX. Transmembrane signaling pathway mediates oxidized low-density lipoprotein-induced expression of plasminogen activator inhibitor-1 in vascular endothelial cells. American journal of physiology. Endocrinology and metabolism. 2008;295:E1243–1254 [DOI] [PubMed] [Google Scholar]
- 33.Himmerich H, Fulda S, Linseisen J, Seiler H, Wolfram G, Himmerich S, et al. Tnf-alpha, soluble tnf receptor and interleukin-6 plasma levels in the general population. European cytokine network. 2006;17:196–201 [PubMed] [Google Scholar]
- 34.Leoni RF, Paiva FF, Henning EC, Nascimento GC, Tannus A, de Araujo DB, et al. Magnetic resonance imaging quantification of regional cerebral blood flow and cerebrovascular reactivity to carbon dioxide in normotensive and hypertensive rats. NeuroImage. 2011;58:75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Shen Q, Huang S, Li W, Muir ER, Long JA, et al. Cerebral angiography, blood flow and vascular reactivity in progressive hypertension. NeuroImage. 2015;111:329–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.