

Abstract
Objective:
In osteoarthritis (OA), articular chondrocytes manifest mitochondrial damage, including mitochondrial DNA 4977-bp (mtDNA4977) deletion that impairs mitochondrial function. OA chondrocytes have decreased activity of AMPK, an energy biosensor that promotes mitochondrial biogenesis. Here, we tested if pharmacologic AMPK activation, via downstream activation of predominately mitochondrially localized sirtuin 3 (SIRT3), reverses existing decreases in mtDNA integrity and function in human OA chondrocytes and limits mouse knee OA development.
Design:
We assessed mitochondrial DNA (mtDNA) integrity and function including the common mtDNA4977 deletion and mtDNA content, mitochondrial reactive oxygen species (mtROS) generation, oxygen consumption and intracellular ATP levels. Phosphorylation of AMPKα, expression and activity of SIRT3, acetylation and expression of the mitochondrial antioxidant enzyme SOD2 and DNA repair enzyme 8-oxoguanine glycosylase (OGG1), and expression of subunits of mitochondrial respiratory complexes were examined. We assessed effect of pharmacologic activation of AMPK on age-related spontaneous mouse knee OA.
Results:
The mtDNA4977 deletion was detected in both OA chondrocytes and menadione-treated normal chondrocytes, associated with increased mtROS, decreased SIRT3, and increased acetylation of SOD2 and OGG1. AMPKα1 deficient chondrocytes exhibited significantly reduced SIRT3 activity. AMPK pharmacologic activation attenuated existing mtDNA4977 deletion and improved mitochondrial functions in OA chondrocytes via SIRT3 by reducing acetylation and increasing expression of SOD2 and OGG1, and limited aging-associated mouse knee OA development and progression.
Conclusions:
AMPK activation, via SIRT3, limits oxidative stress and improves mitochondrial DNA integrity and function in OA chondrocytes. These effects likely contribute to chondroprotective effects of AMPK activity.
Keywords: mitochondrial DNA, oxidative stress, AMPK, SIRT3, OGG1, SOD2
Introduction
In osteoarthritis (OA), the most common form of arthritis, there is convergence of aging, biomechanical injury, metabolic diseases, and other factors to promote changes in synovial joints that can culminate in progressive degeneration of articular cartilage (1). Chondrocytes, the sole cells in articular cartilage, are responsible for maintaining extracellular matrix homeostasis (1). However, changes in chondrocyte differentiation and function develop in OA cartilage, including several hallmarks of cell aging (2). These include accumulation of DNA damage (3,4), and impairment of mitochondrial function (5,6), decrease in the autophagic process that normally removes damaged intracellular proteins and organelles (7), and deregulated nutrient biosensing, exemplified by decreased activity of the metabolic biosensor AMPK and of several SIRT deactylase family members (8–11). Compromises in mitochondrial energy metabolism and other functions are particularly characteristic of aging-related diseases (2). OA chondrocytes demonstrate decreases in mitochondrial biogenesis, OXPHOS and cellular ATP levels, and increases in mitochondria-mediated oxidative stress and apoptosis, reduced antioxidant capacity, and enhanced catabolic responses to inflammatory cytokines (4–6,12–16).
Human mitochondrial DNA (mtDNA) is organized as 16.5 Kb double-stranded circular DNA encoding 13 polypeptides, 2 rRNAs and 22 tRNAs involved in OXPHOS and mitochondrial protein synthesis (17). Since mitochondria are a major source of reactive oxygen species (mtROS), mtDNA is particularly vulnerable to oxidative damage (18). Mitochondrial DNA damage causes mtDNA mutations, promoting mitochondrial respiratory chain dysfunction and augmenting ROS production (19). The most frequent mtDNA mutation in human tissues is a 4977-bp deletion (mtDNA4977), known as “the common deletion”, and occurring between two 13-bp direct repeats at nucleotide positions 13447–13459 and 8470–8482 (19). This mutation removes all or part of the genes encoding four complex I subunits, one complex IV subunit, two complex V subunits and five tRNA genes, which are indispensable for maintaining normal mitochondrial function (19). This deletion has been reported in multiple diseases and accumulates with age (19). Importantly, mtDNA damage accumulates in OA chondrocytes (4,15), and mtDNA4977 deletion is detected in human aged and OA knee cartilages (20).
Among sequelae of oxidative stress is oxidation of DNA guanine residues to 8-OHdG; addition of this adduct to DNA causes DNA damage (21). Amounts of 8-OHdG in mtDNA accumulate over the lifespan of individuals (21). The base excision DNA repair enzyme 8-oxoguanine glycosylase (OGG1), located in both the nuclear and mitochondrial compartments, is responsible for removing 8-OHdG (22). Deficiency of OGG1 can lead to elevated levels of 8-OHdG in mtDNA (23). The mtDNA isolated from OGG1 knockout mice have 20-fold more 8-OHdG than that from wild type mice (22,23). OGG1 is over 3-fold more active in mitochondria compared to nucleus (24). Mitochondrial OGG1 overexpression reduces mtDNA deletions via repair of 8-OHdG under oxidative stress conditions, and improves mitochondrial function in vitro (25–28). In addition, tissue-specific overexpression of OGG1 protects mitochondrial DNA following oxidative stress (28).
Protein acetylation in mitochondria typically promotes decreased mitochondrial integrity and function (29). SIRT3, a predominately mitochondria-localized nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylase, regulates the acetylation status of over 100 mitochondrial proteins (31), thereby regulating mitochondrial redox status and OXPHOS (31,32). For example, SIRT3 protects mitochondria from oxidative stress by deacetylating superoxide dismutase (SOD2), augmenting SOD2 anti-oxidative activity, and decreasing mtROS (31,32). SIRT3 also protects mitochondria from DNA damage by maintaining OGG1 levels, and deacetylating OGG1 to activate DNA repair (31,32). Moreover, SIRT3 mediates age-related changes in cartilage redox regulation and protects against earlystage OA (33).
Dysregulation of certain cell homeostasis signaling systems in aging-related diseases (2) is exemplified in OA chondrocytes by decreased AMPK and SIRT family member function (34). Such changes can impact mitochondrial function. In this context, pharmacologic AMPK activation reverses decreased mitochondrial biogenesis in cultured human OA chondrocytes, mediated by induction of the NAD+-dependent protein deacetylase SIRT1 and PPARgamma co-activator 1α (PGC-1α), a master regulator of mitochondrial biogenesis (6). Since activation of AMPK increases chondrocyte intracellular NAD+ levels, we tested and proved the hypothesis that AMPK activation exerts its chondroprotective role via NAD+-dependent SIRT3, which preserves mitochondrial DNA integrity and function through downstream targets SOD2 and OGG1.
Materials and Methods
Reagents
Chemical reagents including berberine chloride were obtained from Sigma-Aldrich, unless otherwise indicated. A-769662, a direct AMPK pharmacological activator was from LC laboratories. Antibodies to phospho-AMPKα (Thr172), AMPKα, SIRT3 and SOD2 were from Cell Signaling Technology, Inc. Antibodies to acetyl-SOD2 (K68), acetyl-OGG1 (K338+K341), OGG1 were from Abcam. Human SIRT3 siRNA and control siRNA were from Thermo Fisher Scientific.
Studies of human and mouse articular chondrocytes
Human chondrocytes were generously provided by Dr. Martin Lotz (Scripps Research Institute, La Jolla, CA). Studies were performed in compliance with institutional IRB reviewed and approved human subjects protocols at San Diego VA Medical Center. Human knee chondrocytes were isolated from autopsy donors that were graded macroscopically according to a modified Outerbridge scale (6) and cultured in Dulbecco’s modified Eagle’s (DMEM) high glucose medium with 10% fetal calf serum (FCS), 100 μg/ml streptomycin, and 100 IU/ml penicillin at 37°C. Human knee chondrocytes from both male and female normal (grade 0–1) and OA donors (grade 4) were used for the study. Mouse chondrocytes were isolated from 6–8 day-old mouse knees as described previously and cultured in the aforementioned media (35). Once cells reached confluence, they were either collected or re-plated in monolayer culture at 3 × 105 cells per well in 12 well plates or 7 × 105 cells per well in 6-well plates for all other experiments.
Studies of effect of AMPK activation on age-related spontaneous knee OA in mice
C57BL/6 male mice were used for the aging study in compliance with an institutionally reviewed and approved protocol by IACUC at the VA San Diego. Mice were housed in cages with access to food and water with a 12-h dark/12-h light cycle. Because the aging study was long-term (up to 24 months of age), we chose to use berberine, an orally bioavailable indirect AMPK activator. The mature 6 month-old of C57BL6 mice were randomly allocated to 3 groups (n-7–8 per group) and started receiving berberine chloride in the drinking water at 10–20 mg/kg/day, comparable to the one for humans. A common dosage recommendation for humans is 500 mg, 3 times a day. If average human body weight is 75 kg, the dose would be 500×3 mg/75 kg/day=20 mg/kg/day. The mice were sacrificed at 12, 18 and 24 months, after 6, 12 and 18 months of berberine treatment, respectively. Age and number matched C57BL6 male mice without berberine treatment were used as controls. Body weight of each mouse was measured at each time point. Knee joints from all groups of mice were resected, fixed in 10% zinc-buffered formalin (Z-Fix) for 2 days, decalcified in TBD-2 (Shandon) for 72 hours, paraffin embedded and coronally sectioned (5 μΜ). For each knee joint, 10–12 slides at ~75 micron intervals were stained with safranin-O and fast green for histologic scoring of the entire articular surface, using the OARSI grading system.
Detection of mtDNA4977 deletion
Total DNA, extracted using the DNeasy Blood & Tissue kit (Qiagen), was subjected to PCR for detection of mtDNA4977 deletion mutant as described in (20). The primers L8150 5′ CCGGGGGTATACTACGGTCA-3′ and H13650 5′-GGGGAAGCGAGGTTGACCTG-3′ were used to amplify 524-bp PCR product representing mtDNA4977 deletion mutant. The primers L3304 5′-AACATACCCATGGCCAACCT-3′ and H3836 5′-GGCAGGAGTAATCAGAGGTG-3′ were used to amplify a 533-bp DNA fragment representing total mtDNA. The PCR reaction consisted 35 cycles with denaturation at 94°C, for 15 seconds, annealing at 58°C for 15 seconds, and primer extension at 72°C for 40 seconds. The PCR products were separated by electrophoresis on a 1.5% agarose gel.
mtDNA content determination
Total DNA was isolated using the DNeasy Blood & Tissue kit (Qiagen). Quantitative PCR was performed using the efficiency-adjusted ΔΔCT method for mitochondrial DNA-encoded cytochrome c oxidase subunit I (COXI) (primers: forward 5’-GGCCTGACTGGCATTGTATT-3’, reverse 5’-TGGCGTAGGTTTGGTCTAGG-3’) and subunit II (COXII) (primers: forward 5’-GCCGACTAAATCAAGCAACA-3’, reverse 5’-CAATGGGCATAAAGCTATGG-3’) and nucleus-encoded 18S ribosomal DNA (18S rDNA) genes (primers: forward 5’-TAGAGGGACAAGTGGCGTTC-3’, reverse 5’-CGCTGAGCCAGTCAGTGT-3’). Ratio of COXI or COXII DNA copies to 18S rDNA represented relative mitochondrial copy number.
SDS-PAGE/Western Blot
Total cell lysates were prepared using RIPA buffer. Cytosolic and nuclear proteins were prepared by lysing the cytosolic and nuclear fractions of cells extracted using Cell Fractionation kit (Abcam). Proteins (10–15 μg) were separated by gradient 4–20% SDS-PAGE and transferred onto nitrocellulose membranes, probed with antibodies, exposed to chemiluminescent substrate, and visualized by autoradiography.
Immunohistochemistry (IHC)
Slides of mouse knee sections were treated with 3% H2O2 for 10 min, then blocked with 10% goat serum for 2 hours at room temperature. After washing with TBS, rabbit antibodies to phospho-AMPKα (Thr 172) (Thermofisher, 1:50), SIRT3 (Abgent, 1:50), SOD2 (Millipore, 1:50), OGG1 (Thermofisher, 1:100), 8-OHdG (Abbiotec, 1:50) and the negative control rabbit IgG (1:50) were applied to sections and incubated overnight at 4°C. Next, sections were washed with TBS, incubated with biotinylated goat antirabbit IgG secondary antibody for 30 min, and then incubated for 30 minutes using the Histostain Plus kit (Invitrogen, Carlsbad, CA). Last, sections were washed and incubated with 3,3’-diaminobenzidine (DAB) substrate for 2–5 min. Hematoxylin staining was included to verify cellularity in each section.
Knockdown and transfection of SIRT3, OGG1 and SOD2
Cultured primary human knee articular chondrocytes were transfected with either SIRT3 siRNA and nontarget control using the X-tremeGene siRNA transfection reagent (Roche), or the plasmids pCDNA4-Myc-HisB-Sirt3, pCMV/myc/mito-hOGG1, and pBI-EGFP-MnSOD, gifts from Toren Finkel, David Sidransky, and Bert Vogelstein (Addgene), respectively, and the vector controls (pcDNA4 and pCMV) using Lipofectamine 3000 transfection reagent (Life Technology).
Measurement of SIRT3 activity, oxygen consumption, cellular ATP, and nitric oxide (NO) release
SIRT3 activity assay kit (fluorometric, Abcam) was used for the quantitative measurement of SIRT3 activity in cell lysates (50 mg) in vitro. Oxygen consumption as a gauge for OXPHOS (presented as oxygen consumption rate calculated as a function of time (picomoles per minute) and intracellular ATP were quantified using the XF extracellular flux analyzer (Seahorse Bioscience) and Luminescent ATP Detection Assay Kit (Abcam), respectively. Conditioned media were tested for NO generation by Griess reaction assay to quantify nitrite.
Statistical analyses
For in vitro studies, due to limited human knee donors and limited amount of primary chondrocytes isolated from each donor, we were only able to use 3 donors with 3–4 replicates (sample size 9–12) per donor for each experiment. Based on the assumption of effect size=0.8 and significance level (alpha)=0.05, power calculation for comparing outcome of 2 groups between control and treatment or gene knockdown or overexpression was done by the paired student’s t-test (two tailed), which was 0.56–0.71 for sample size 9–12. Power calculation for determining outcome affected by two factors such as treatment and time was done by two-way ANOVA. Based on the assumption of effect size=0.8, confidence level=0.05, number of levels=2, and total number of levels of all other factors=2, power was 0.64–0.77 for sample size 9–12. All data from in vitro studies were expressed as mean±SD. Data from in vivo mouse study were expressed as mean±SEM. Statistical analyses were performed by either student’s t-test, one-way ANOVA or two-way ANOVA with either Tukey or Sidak’s post-hoc test using GraphPad PRISM 6. P values less than 0.05 were considered significant.
Results
mtDNA4977 deletion mutant in chondrocytes is associated with oxidative stress and SIRT3 deficiency
Detection of the common mtDNA4977 deletion mutant (20) in human knee OA chondrocytes (Figure 1A) was confirmed from greater than 60% of donors studied. In addition, generation of mtROS was greatly induced in normal human knee chondrocytes treated with menadione, a free radical inducer that forms superoxide anions during semiquinone reduction via redox cycling (Figure 1B). Menadione also induced generation of mtDNA4977 deletion mutant (Figure 1C, top panel). These effects were associated with reduced SIRT3 expression (Figure 1C, bottom panel), suggesting that oxidative stress is involved in downregulation of SIRT3 in normal chondrocytes.
Figure 1. Occurrence of mtDNA4977 deletion mutation in chondrocytes associated with excessive oxidative stress and SIRT3 deficiency.
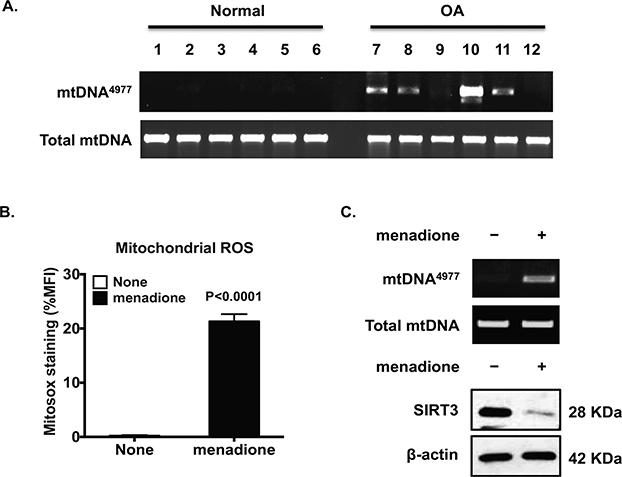
Total DNA was directly isolated from human knee cartilage from 6 normal and 6 OA donors (A), or cultured primary normal human chondrocytes treated with menadione (50 μM for 2 hours) (top panel, C). PCR analyzed mtDNA4977 deletion mutation and total mtDNA as described in the Methods. MitoSox staining, followed by flow cytometry, was performed to assess mitochondrial superoxide (ROS) generation (B). SIRT3 expression was examined by Western blot analysis (bottom panel, C). Data in A represent n= 24 donors studied with 12 normal donors (7 males and 5 females, the mean age was 46.4±7.2) and 12 OA donors (5 males and 7 females, the mean age was 66.5±2.9). Ages and genders in A were 29 male, 21 male, 22 female, 21 female, 28 male, and 22 male for normal donors (lane 1–6), and 76 female, 75 male, 65 female, 53 male, 82 male, and 64 female for OA donors (lane 7–12). Data in B represent n=12 normal donors studied. Student t-test was used for statistical data analysis in B comparing none to menadione-treated chondrocytes (the mean difference: 19.02±1.108, 95% confidence interval (CI): 16.72 to 21.32, p<0.0001). Data in C represent 3 individual experiments with 3 different normal donors (2 males and 1 female, age 33, 44 and 39, respectively).
SIRT3 siRNA knockdown induces mtDNA4977 deletion generation and mitochondrial dysfunction in normal chondrocytes
SIRT3 siRNA-transfected normal chondrocytes demonstrated hyperacetylation of SOD2 and OGG1, associated with slightly decreased expression of SOD2 and OGG1 (Figure 2A, top panel), suggesting reduced capacity in both anti-oxidative stress responsiveness and DNA repair. Notably, SIRT3 siRNA knockdown induced appearance of the mtDNA4977 deletion mutant (Figure 2A, bottom panel), with associated decrease in expression of mitochondrial respiratory complexes (Figure 2B), mtDNA content (Figure 2C), oxygen consumption (Figure 2D) and intracellular ATP levels (Figure 2E, consistent with mitochondrial dysfunction. Moreover, SIRT3 deficiency also augmented chondrocyte catabolic responses to IL-1β, with augmented release of NO and MMP3 induced by IL-1β (data not shown).
Figure 2. SIRT3 siRNA knockdown in normal chondrocytes promotes SOD2 and OGG1 hyperacetylation, generation of mtDNA4977 deletion mutation, and mitochondrial dysfunction.
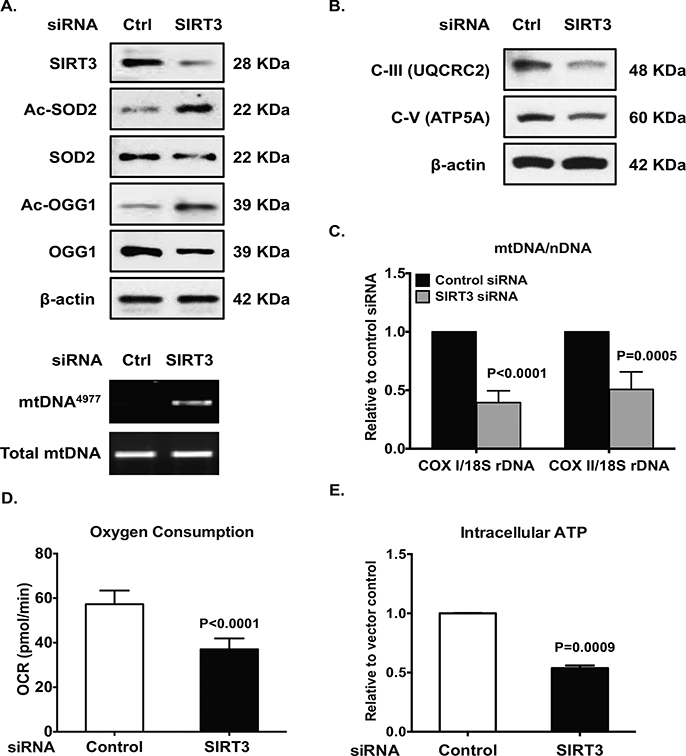
Cultured normal human chondrocytes (passage 1) was transfected with SIRT3 siRNA and control siRNA for 48 hours. Cell lysates were subjected to Western blot analysis for expression of SIRT3, acetylation status and expression of SOD2 and OGG1 (A, top panel), and subunits of mitochondrial respiratory complex III and V (B). PCR detected mtDNA4977 deletion mutation (A, bottom panel). Mitochondrial DNA content was determined as ratio of mtDNA (COXI or COXII) to nuclear DNA (18S rDNA) by PCR (C). Oxygen consumption (D) and intracellular ATP was quantified (E) as described in the Methods. Data in A and B represent 3 individual experiments with 3 different normal donors. Student t-test was used for statistical data analysis in C, D and E comparing control siRNA with SIRT3 siRNA chondrocytes (n=3 donors with 3–4 replicates for each donor, 2 males and 1 female, age 33, 44 and 39, respectively). The mean differences, 95% CIs and p values for COXI/18S dDNA, COXII/18S rDNA, oxygen consumption and ATP levels were 0.61 ± 0.04, 0.51 to 0.7, p<0.0001, 20.27 ± 1.52, 0.51 to 0.7, p<0.0001, and 0.46 ± 0.01, 0.43 to 0.5, p<0.0001, respectively.
Decreased AMPK-SIRT3 signaling in chondrocytes associated with aging and OA
IHC revealed that the numbers of chondrocytes stained positively for both phosphorylated AMPKα and SIRT3 in mouse knee tibial plateau articular cartilage were largely decreased at 18 and 24 months, compared to 12 months (Figure 3A). Notably, appearance of 8-OHdG was augmented particularly at 24 months, indicating increased DNA oxidative damage (Figure 3A). SIRT3 activity was much lower in AMPKα1KO compared to WT chondrocytes (Figure 3B), and the mean differences between AMPKα1KO and WT chondrocytes appeared to be significant as early as 4 minutes after addition of substrate (Figure 3B). A significant interaction between mouse genotype and time was observed (p=0.0004). Primary human knee OA chondrocytes also exhibited concurrent reduction of AMPKα phosphorylation and SIRT3 expression, correlated with increased acetylation of SOD2 and OGG1 (Figure 4A and 4B). These data suggest that AMPK plays a role in regulating SIRT3 expression and activity, and decreased AMPK and SIRT3 signaling may impair function of SOD2 and OGG1.
Figure 3. Aging-associated concurrent decrease in phosphorylation of AMPKα and SIRT3 expression in mouse knee cartilages in situ, and impaired SIRT3 activity in mouse chondrocytes deficient in AMPKα1.
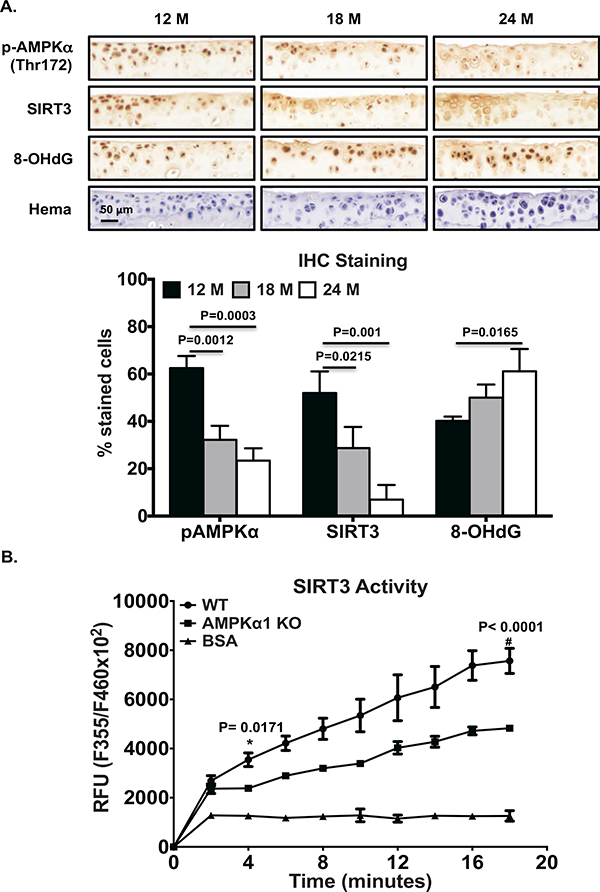
A. IHC analysis of AMPKα phosphorylation of and expression of SIRT3 and 8-OHdG in serial sections of knee tibial cartilages of 12, 18 and 24 month-old C57BL/6 mice. Hematoxylin staining assayed cellularity in cartilages (top panel). Positively stained cells in the non-calcified cartilage region were presented as percentage relative to total number of cells staining for hematoxylin (bottom panel). Data represent 4 donors from each age group. One-way ANOVA followed by Tukey’s post hoc test was used for statistical data analysis. The mean differences, 95% CI and p values for phosphorylated AMPKα between 12 and 18 months and between 18 and 24 months were 30.26, 16.71 to 43.8, p=0.0012, and 39.05, 25.5 to 52.6, p=0.0003, respectively. The mean differences, 95% CIs and p values for SIRT3 between 12 and 18 months and between 18 and 24 months were 23.28, 4.382 to 42.18, p=0.0215, and 43.03, 24.13 to 61.93, p=0.001, respectively. The mean difference for 8-OHdG between 12 and 24 months was 39.05, 95% CI was 25.5 to 52.6, p=0.0165. B. Cell lysates of mouse chondrocytes isolated from AMPKα1KO and WT control mice were subjected to SIRT3 activity assay. BSA was included as a negative control. Two-way ANOVA followed by Sidak’s post hoc test (recommended by GraphPad software) was used for statistical analysis of SIRT3 activity comparing WT with AMPKα 1 KO chondrocytes at all time points. The significant mean differences seen as early as 4 minutes after addition of the substrate (1162, 95% CI: 141.6 to 2183, p=0.0171).
Figure 4. Concurrent decrease in phosphorylation of AMPKα and SIRT3 expression in primary human knee OA chondrocytes.
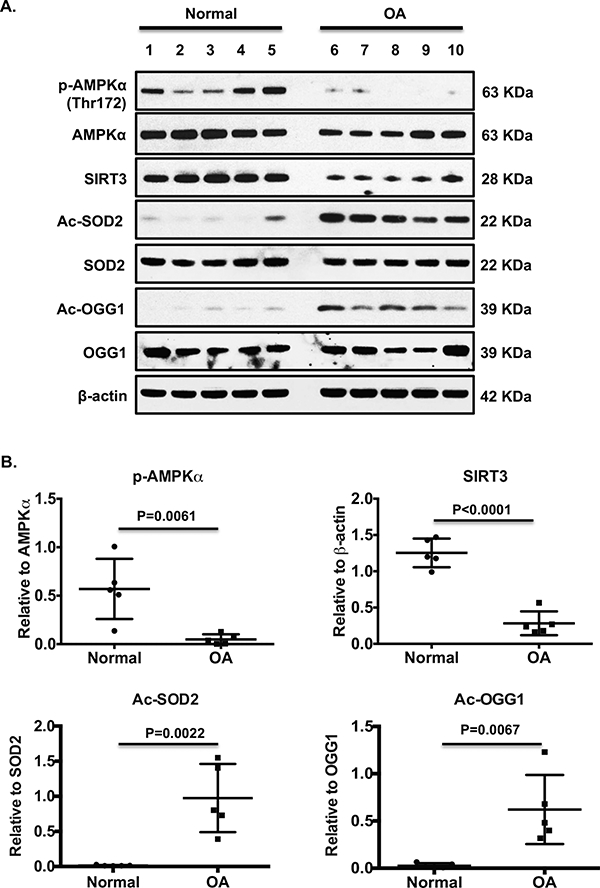
Primary human knee chondrocytes from both normal and OA donors were subjected to Western blot analysis for phosphorylation and expression of AMPKα, SIRT3 expression, and acetylation and expression of SOD2 and OGG1 (A). Ages and genders in A were 17 female, 44 male, 43 male, 49 male, and 61 female for normal donors (lane 1–5), and 60 female, 67 male, 56 female, 78 female, and 62 male for OA donors (lane 6–10). B. Densitometry analysis of levels of phosphorylation of AMPKα, SIRT3, acetylated SOD2 and OGG1 relative to AMPKα, β-actin, SOD2 and OGG1, respectively. Student t-test was used for statistical data analysis comparing normal with OA. The mean differences, 95% CIs, and p values for phosphorylation of AMPKα, SIRT3, acetylated SOD2 and OGG1 were 0.52±0.14, 0.2 to 0.85, p=0.0061, 0.97±0.11, 0.71 to 1.23, p<0.0001, 096±0.22, 0.46 to 1.46, p=0.0022, and 0.16±0.6, 022 to 0.97, p=0.0067.
Effect of pharmacologic activation of AMPK on mtDNA4977 deletion in chondrocytes
The selective pharmacological AMPK activator A-769662 greatly improved SIRT3 activity in OA chondrocytes, with earliest significant increase at 6 minutes after addition of the substrate (Figure 5A). There is a significant interaction between treatment and time (p<0.0001). In addition, A-769662 promoted increase in phosphorylation of AMPKα and SIRT3 expression (Figure 5B), decrease in acetylation of SOD2 and OGG1, and increase in expression of SOD2 and OGG1 (Figure 5B). Notably, A-769662 completely removed pre-existing mtDNA4977 deletion mutant in OA chondrocytes (Figure 5C). Furthermore, A-769662 attenuated menadione-induced generation of mtROS (Figure 5D) with a highly significant interaction between none and A-769662 treatment (p<0.0001) and the mtDNA4977 deletion mutant (Figure 5E).
Figure 5. Pharmacologic activation of AMPK eliminated pre-existing and oxidative stress-induced mtDNA4977 deletion mutation in OA and normal chondrocytes, respectively.
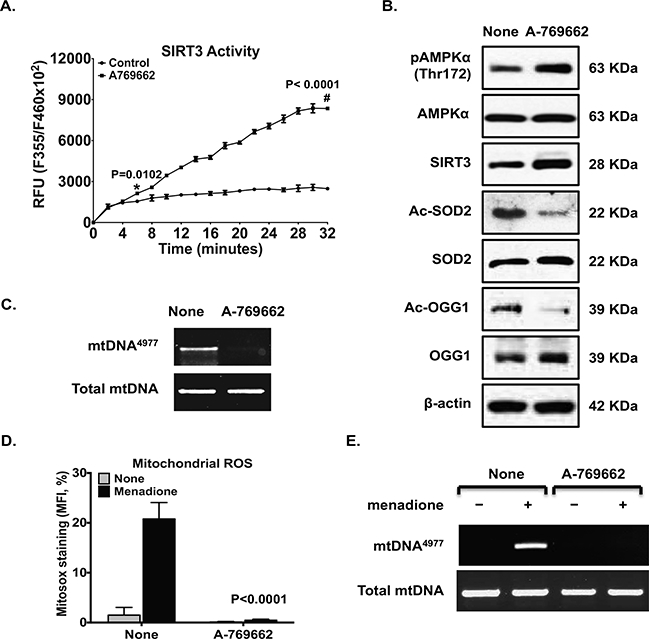
Cultured primary human knee OA chondrocytes were treated with AMPK pharmacological activator A-769662 (0.125 mM) for 2 hours (A and B). SIRT3 activity was measured (A), and phosphorylation and expression of AMPKα, SIRT3 expression, and acetylation and expression of SOD2 and OGG1 were examined by Western blot (B). The mtDNA4977 deletion mutation (C) was detected by PCR. Primary normal chondrocytes were pre-stimulated with A-769662 (0.125 mM) for 1 hour before treated with menadione (50 μM) for 2 hours; mtROS (D) and mtDNA4977 deletion mutation (E) were assessed. Data in A were the mean of 3 individual experiments with 3 different OA donors (2 females and 1 male, age 81, 58 and 64, respectively). Two-way ANOVA followed by Sidak’s post hoc test was used for statistical analysis of SIRT3 activity comparing control with A-769962 at all time points. The significant mean differences seen as early as 6 minutes after addition of the substrate (578.2, 95% CI: 91.22 to 1065, p=0.0102). Data in B and C represent 3 individual experiments with 3 different normal donors represent 3 individual experiments with 3 different OA donors (the same donors as in A). Data in D and E represent 3 individual experiments with 3 different normal donors (2 male and 1 female, age 34, 58 and 39, respectively). Two-way ANOVA followed by Tukey’s post hoc test was used in D comparing none with menadione alone (the mean differences: 19.29, 95% CI: 14.53 to 24.04, p<0.0001) and menadione vs menadione plus A-769662 (the mean differences: 20.32, 95% CI: 15.57 to 27.07, p<0.0001).
Transfection of SIRT3, mitochondria-targeted OGG1 (Mt-OGG1) or SOD2 in OA chondrocytes removed established mtDNA4977 deletion and improved mitochondrial function
Gain of function of SIRT3 via transfection in OA chondrocytes reduced acetylation of SOD2 and OGG1, increased expression of SOD2 and OGG1 (Figure 6A, top panel), and attenuated menadione-induced mtROS generation (data not shown). Importantly, overexpression of SIRT3 in OA chondrocytes removed detectable mtDNA4977 deletion mutant (Figure 6A, bottom panel). This was associated with significant improvement of mitochondrial function, supported by increased expression of subunits of mitochondrial respiratory complexes (Figure 6B,), mtDNA content (Figure 6C), oxygen consumption (Figure 6D) and intracellular ATP levels (Figure 6E). Gain of function of OGG1 in mitochondria via transfection of mt-OGG1 in OA chondrocytes showed comparable results (Figure 7A-C). Gain of function of SOD2 in OA chondrocytes also removed detectable mtDNA4977 deletion mutant and improved mitochondrial function (Figure 7D-E).
Figure 6. Gain of function of SIRT3 in OA chondrocytes eliminated pre-existing mtDNA4977 deletion mutation and improved mitochondrial function.
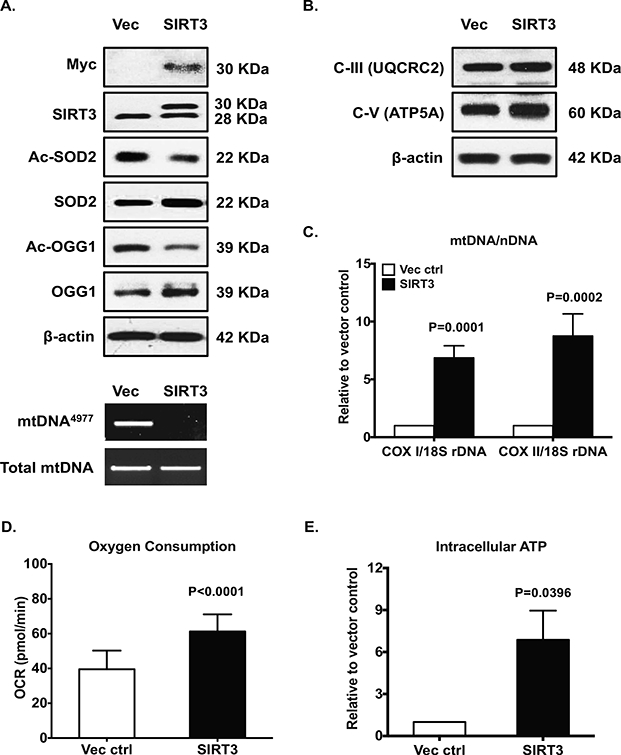
Cultured primary human knee OA chondrocytes were transfected with pCDNA4-Myc-HisB-Sirt3 and vector control for 48 hours. Expression of SIRT3 was confirmed by Western blot with antibodies to either Myc or SIRT3 (A, top panel). Acetylation and expression of SOD2 and OGG1 (A, top panel), and expression of subunits of mitochondrial respiratory complex III and V (B) were analyzed by Western blot. The mtDNA4977 deletion mutation (A, bottom panel), mtDNA content (C), oxygen consumption (D) and intracellular ATP (E) were determined as described in Figure 2 legend. Data in A and B represent 3 individual experiments with 3 different OA donors (2 females and 1 male, age 81, 58 and 64, respectively). Data in C, D and E were the mean of 4 individual experiments with 4 different OA donors (1 male and 3 females, age 67, 70, 56 and 68, respectively). Student t-test was used for statistical data analysis in C, D and E (n=3, 3–4 replicates for each donor) comparing vector control with SIRT3 overexpression. The mean differences, 95% CIs and p values for COXI/18S rDNA and COXII/18S rDNA: 6.106 ± 0.5714, 4.833 to 7.379, p<0.0001, and 7.747 ±0.7820, 6.004±9.489, p<0.0001, respectively. The mean differences, 95% CIs and p values for oxygen consumption and ATP levels: 55 ± 2.678, 11.16 to 21.95, p<0.0001, and 5.874 ± 1.205, 2.529 to 9.219, p=0.0082, respectively.
Figure 7. Transfection of mitochondria-targeted OGG1 (Mt-OGG1) or SOD2 eliminated preexisting mtDNA4977 deletion mutation and improved mitochondrial function.
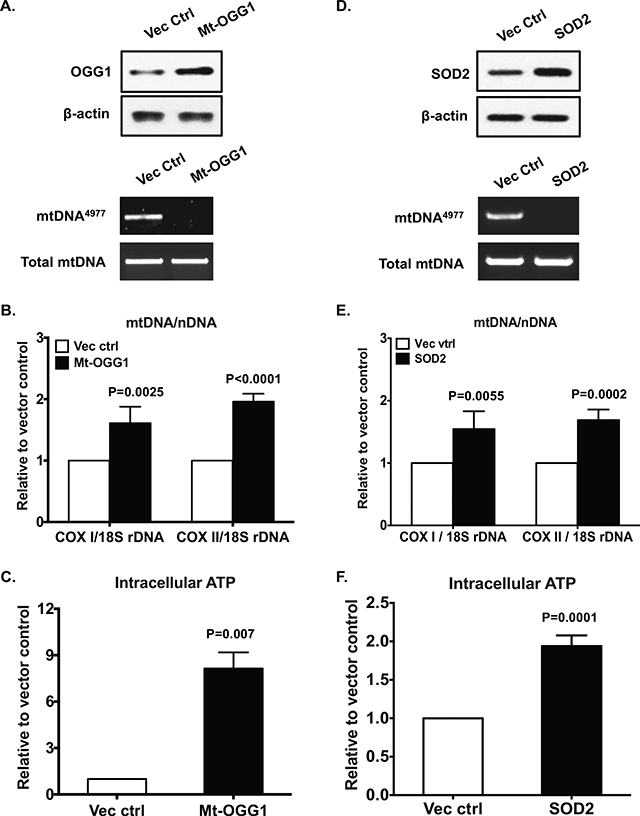
Primary human knee OA chondrocytes were transfected with pCMV/myc/mito-hOGGl or pBI-EGFP-MnSOD and the vector controls for 48 hours. Expression of OGG1 and SOD2 were examined by Western blot (A and D, top panel). The mtDNA4977 deletion mutation (A and D, bottom panel), mtDNA content (B and E) and intracellular ATP (C and F) were assayed as above. Data in A and D represent 3 individual experiments with 3 different OA donors (1 male and 2 females, age 68, 78 and 57, respectively). Data in B, C, E and F were the mean of 3 individual experiments with 3 different OA donors (the same donors as in A, n=3 with 3–4 replicates for each donor). Student t-test was used for statistical data analysis in B and C comparing the vector control with Mt-OGG1, and in E and F comparing the vector control with SOD2. The mean differences, 95% CIs and p values for COXI/18S rDNA and COXII/18S rDNA were 0.61±0.11, 0.37 to 0.85, p=0.0002 and 0.96±0.05, 0.84 to 1.08, p<0.0001 in B, and 0.54±0.12, 0.28 to 0.8, p=0.0009 and 0.69±0.07, 0.53 to 0.84, p<0.0001 in E. The mean differences, 95% CIs and p values for ATP levels were 7.14±0.6, 5.48 to 8.8, p=0.0003 in C and 0.94±0.06, 0.79 to 1.08, p<0.0001 in F.
Activation of AMPK-SIRT3 signaling pathway contributed to chondroprotection by berberine in age-related spontaneous murine knee OA
We performed an aging study to determine the effect of pharmacologic activation of AMPK by berberine, an indirect AMPK activator with oral bioavailability, on age-related spontaneous knee OA development in C57BL/6 male mice in vivo. We firstly confirmed that berberine, similar to A-769662, was capable of activating AMPK-SIRT3 signaling in human OA chondrocytes, demonstrated by increase in phosphorylation of AMPKα (Figure 8A), SIRT3 expression (Figure 8A) and activity with a significant interaction between treatment and time (p<0.0001) (Figure 8B), and expression of SOD2 and OGG1, and decrease in acetylation of SOD2 and OGG1 (Figure 8A). Next, we evaluated knee cartilage degradation in C57BL/6 male mice at 12, 18 and 24 months of age between the berberine treatment group and the age-matched non-treatment control group. No significant body weight differences were seen between these 2 groups at all time points. Mice in the control group exhibited cartilage degeneration (Figure 8C) progressing from little at 12 months to mild at 18 months and prominent at 24 months. The mean joint score was significantly higher at 24 months (n=7), compared to either 12 months (n=8, the mean difference: −2.463, 95% CI difference: −3.13 to −1.795, p<0.0001), or 18 months (n=8, the mean difference: −2.093, 95% CI difference: 1.812 to 3.104, p<0.0001). There is a significant interaction between treatment and mouse age (p=0.0153). Notably, the mean joint scores of mice in the berberine treatment group were significant lower, compared to the non-treatment control groups (Figure 8C) at 18 months (the mean difference: 0.6608, 95% CI difference: 0.01568 to 1.306, p=0.0408) and 24 months (mean difference: 1.236, 95% CI difference: 0.5462 to 1.925, p=<0.0001), suggesting chondroprotective effect. Since phosphorylation of AMPKα and expression of SIRT3 were significant lower in knee cartilage sections at both 18 and 24 months (Figure 3A), but cartilage degeneration was still mild at 18 months, compared to 24 months (Figure 8C), 18 month-old mouse knees appeared to be valuable for studying molecular changes in cartilage arisen from aging (before structural damage). IHC analysis showed that the numbers of cells stained positively for phosphorylated AMPKα, SIRT3, SOD2 and OGG1 were higher, and the numbers of cells stained positively for 8-OHdG were lower in mouse knee cartilages of berberine-treated group, compared to the control group at 18 months (Figure 8D), suggesting berberine restrained aging-associated reduced AMPK-SIRT3 signaling.
Figure 8. Activation of AMPK-SIRT3 pathway contributed to berberine chondroprotection in age-related spontaneous knee OA development in C57BL/6 mice.
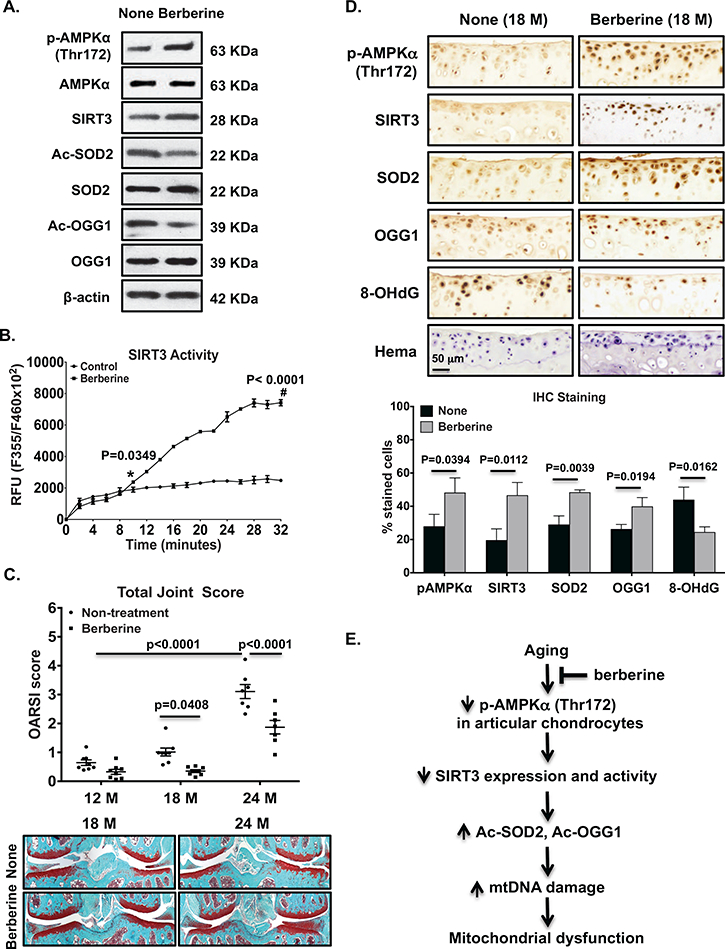
Cultured primary human knee OA chondrocytes were treated with berberine (25 μM) for 2 hours. Phosphorylation and expression of AMPKα, expression and activity of SIRT3, and acetylation and expression of SOD2 and OGG1 were examined (A and B). C57BL/6 male mice started receiving berberine treatment at 6 months of age, and were sacrificed after 6, 12 and 18 months of berberine treatment, which were equivalent to age of 12, 18 and 24 months, respectively (n=7–8 per group). The age-matched C57BL/6 male mice without berberine treatment were used as controls. Histological grading of cartilage degradation was performed using the OARSI scoring system (C). IHC analysis of AMPKα phosphorylation and expression of SIRT3, SOD2, OGG1 and 8-OHdG in 18 month-old mouse knee tibial cartilage (serial sections) with and without berberine treatment (D, top panel). Positively stained cells in the non-calcified cartilage region were presented as percentage relative to total number of cells staining for hematoxylin (D, bottom panel). Summary of results were depicted in E. Data in A and B represent 3 individual experiments with 3 different OA donors (1 male and 2 females, age 67, 68 and 57, respectively). Two-way ANOVA followed by Sidak’s post hoc test was used for statistical analysis of SIRT3 activity comparing control with berberine-treated chondrocytes for all time points. The mean differences became significant 10 minutes after addition of the substrate (480, 95% CI: 20.14 to 939.9, p=0.0349) and stayed significant with p<0.0001 for the rest of time points. Two-way ANOVA followed by Tukey post hoc test was used for analysis the joint scores among 12, 18 and 24 months in non-treatment control mice, or by Sidak’s post hoc test for analysis of the joint scores comparing nontreatment control with berberine-treated mice at each time point in C. Data in top panel of D were representatives of 4 different donors, and data in bottom panel of D were the densitometry data of IHC analysis from 4 different donors. Student t-test was used for statistical data analysis in D. The mean differences, 95% CIs and p values for pAMPKα, SIRT3, SOD2, OGG1 and 8-OhdG were 20.32±6.741, 1.6 to 39, p=0.0394, 27.1±6.1, 10.2 to 43.9, p=0.0112, 19.4±3.2, 10.4 to 28.5, p=0.0039, 13.6±3.6, 3.6 to 23.6, p=0.0194, −19.5±4.5, −33.03 to −5.95, p=0.0162, respectively.
Discussion
In this study, we found that the mitochondrial deletion mutant mtDNA4977 prevalent in OA chondrocytes was associated with oxidative stress and decreased expression of SIRT3 in vitro. These findings were in line with recent finding that human cells harboring mtDNA4977 deletion have reduced SIRT3 expression in response to oxidative stress (36). Here, in mechanistic analyses, we demonstrated that deficiency of SIRT3 greatly increased acetylation of SOD2 and OGG1 in chondrocytes, accompanied by generation of mtDNA4977 deletion mutant, leading to decreased expression of subunits of mitochondrial respiratory complexes, mtDNA content, oxygen consumption and intracellular ATP levels, therefore compromising mitochondrial function.
Decreased phosphorylation of AMPKα is a core event compromising OA chondrocyte mitochondrial biogenesis and function, mediated through down-regulation of PGC-1α (6), which also is decreased in OA chondrocytes (37). We demonstrated that AMPKα1 deficiency was associated with decreased SIRT3 activity in chondrocytes, and that primary human knee OA chondrocytes had decreased SIRT3, suggesting that AMPK regulates SIRT3 levels and activity. PGC-1a can promote Sirt3 gene expression by mediating estrogen related receptor-alpha (ERRα) binding to Sirt3 promoter (32). SIRT3 also can upregulate PGC-1α through a positive feedback mechanism by deacetylating liver kinase B1 (LKB1) (32), which in turn, phosphorylates and activates AMPK in chondrocytes and multiple other cell types (32). Consequently, activated AMPK increases PGC-1α expression (32). AMPK can also directly phosphorylate and activate PGC-1α (38). We did not test if SIRT3 deacetylates LKB1 in chondrocytes, but enhanced PGC-1a protein expression was observed in SIRT3-transfected chondrocytes (unpublished data). Thus, reduced SIRT3 could contribute to decreased mitochondrial biogenesis in OA chondrocytes through reduced LKB1-AMPK signaling, promoting mitochondrial dysfunction.
Reduced cartilage tissue SIRT3 expression is age-related, associated with elevated SOD2 acetylation and increased knee cartilage oxidative stress in rats (33). SIRT3 gain of function (by transfection) in OA chondrocytes increased SOD2 expression, inhibited SOD2 acetylation, and attenuated mtROS generation by menadione. SIRT3 also deacetylates isocitrate dehydrogenase 2 (IDH2) (39), an enzyme in the tricarboxylic acid (TCA) cycle for producing NADPH, a critical molecule for reducing oxidized glutathione (GSSG) to glutathione (GSH) (40). Increased acetylation of IDH2 has been shown in rat cartilage with age (33). SIRT3 has ability to promote the transcription of oxidative stress response genes as well. SIRT3 binds fork head box subgroup O 3a (FoxO3a) and promotes the transcription of catalase and SOD2 (32). In addition, SIRT3 deacetylates FoxO3a in the mitochondrial matrix, which allows FoxO3a binding to mtDNA and promoting up-regulation of all 13 mitochondrial-encoded genes (32), leading to increased mitochondrial respiration. We previously found that FoxO3a mediates the capacity of active AMPK to limit oxidative stress, by increasing protein expression of catalase and SOD2 in chondrocytes (37). Since we observed that pharmacologic activation of AMPK promoted increased SIRT3 levels and activity in this study, we speculate that SIRT3 cooperates with FoxO3a to mediate anti-oxidative stress effects of AMPK in chondrocytes.
SIRT3 can promote capacity of OGG1 to repair mtDNA by deacetylating OGG1 (32), which also stabilizes OGG1, as acetylated OGG1 is vulnerable to degradation by calpain, a calcium-dependent cysteine protease (41). We showed that pharmacologic activation of AMPK that increased SIRT3 level and activity or gain of function of SIRT3 in OA chondrocytes reduced acetylation of OGG1 and increased expression of OGG1. More importantly, both approaches, as well as overexpression of Mt-OGG1, eradicated the pre-existing mtDNA4977 deletion in OA chondrocytes, increased mitochondrial DNA content and intracellular ATP levels, therefore improving mitochondrial function. Notably, gain of function of SOD2 in OA chondrocytes also eliminated detectable mtDNA4977 deletion in OA chondrocytes. This may be partly owing to improved OGG1 function, as acetylation level of OGG1 was reduced under the same condition (unpublished data). Recent study revealed that mitochondrial aconitase (ACO-2), also an enzyme in the TCA cycle, plays an important role in stabilization of mtDNA in the oxidative stress setting (42). Interestingly, OGG1 can preserve ACO-2, whose activity is sensitive to the redox state of the cell, by chaperoning it from oxidative degradation (42). One question remains to be tested is if ACO-2 also participates in maintaining mtDNA integrity in chondrocytes.
One limitation of our in vivo study is that only male mice were used. We do not know if female mice would exhibit the similar age-related changes, which would be of interest of future study. Nevertheless, C57BL/6 male mice at 18 months, compared to 24 months, only exhibited mild cartilage degeneration but significant loss of phosphorylation of AMPKα and SIRT3 expression, indicating that loss of AMPK-SIRT3 signaling may be a key contributor to OA development related to aging. Since berberine was able to inhibit age-related loss of phosphorylation of AMPKα and expression of SIRT3, SOD2 and OGG1 in mouse knee cartilage, the approaches that activate AMPK and SIRT3 signaling may limit OA progression. This notion was supported by our observation that berberine, used at the same dose as in the aging study, was equally efficient to inhibit cartilage degeneration in the mouse destabilization of medial meniscus (DMM) model (unpublished data). Further dose-response study is needed to determine the necessary dose that exhibits the chondroprotective effect. Given that berberine is an isoquinoline alkaloid possessing anti-inflammatory, anti-diabetes and anti-□tumor properties (43), and has already been used in traditional medicine and as a dietary supplement, it has potential to be used to limit or delay OA development and progression.
SIRT3 can activate mitophagy, an autophagic process specifically removing damaged mitochondria and ineffective mitochondria with extensive mtDNA damage. Moreover, SIRT3 deficiency impairs mitophagy by increasing acetylation of Pink/Parkin and decreasing Parkin expression (44–46). Removal of damaged mitochondria by Parkin is necessary for mitochondrial quality control in chondrocytes (47). SIRT3 also regulates mitochondrial dynamics by directly binding and deacetylating OPA1, a mitochondrial protein (48). Because of mtDNA heteroplasmy, the exchange of mitochondrial contents by mitochondrial fusion enables the complementation of pathogenic (mutant) mtDNA in heteroplasmic cells, as introducing wild-type mtDNA dilutes the mutant mtDNA molecules and prevents them from reaching a crucial threshold in the cell, thereby diminishing mtDNA damage (49). Those mitochondria that cannot fuse would be removed through mitophagy (49). Future studies would be of interest to determine if mitophagy and mitochondrial fusion promoted by SIRT3 contribute to elimination of pre-existing mtDNA4977 deletion in OA chondrocytes.
In conclusion, concurrently reduced phosphorylation of AMPKα and expression of SIRT3 were prominent in human knee OA chondrocytes and in mouse knee cartilage with age. These changes could promote oxidative stress and reduce DNA repair capacity via increased acetylation of SOD2 and OGG1, with resultant mtDNA damage/mutation leading to mitochondrial dysfunction (Figure 8E). Activation of the AMPK-SIRT3 pathway eliminated pre-existing mtDNA mutation in human OA chondrocytes and improved mitochondrial function. Our results help further explain why pharmacologic approaches that act by promoting AMPK activity are chondroprotective.
Acknowledgments
Role of the funding source:
This study was supported by Department of Veterans Affairs Merit Review grant 1I01BX002234 (PI: Liu-Bryan), Arthritis Foundation (PI: Liu-Bryan), I01BX001660 (PI: Terkeltaub) and NIH grants AG007996 and T32 AR064194 (PI: Terkeltaub)
Studies were supported by the Department of Veterans Affairs grants 1I01BX002234, I01BX001660, Arthritis Foundation, and NIH grants AG007996 and T32 AR064194
Footnotes
Competing interests: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 2012;64:1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rose J, Söder S, Skhirtladze C, Schmitz N, Gebhard PM, Sesselmann S, Aigner T. DNA damage, discoordinated gene expression and cellular senescence in osteoarthritic chondrocytes. Osteoarthritis Cartilage. 2012;20:1020–8. [DOI] [PubMed] [Google Scholar]
- 4.Grishko VI, Ho R, Wilson GL, Pearsall AW 4th. Diminished mitochondrial DNA integrity and repair capacity in OA chondrocytes. Osteoarthritis Cartilage. 2009;17:107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maneiro E, Martín MA, de Andres MC, López-Armada MJ, Fernández-Sueiro JL, del Hoyo P, Galdo F, Arenas J, Blanco FJ. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheum. 2003;48:700–8. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Zhao X, Lotz M, Terkeltaub R, Liu-Bryan R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol. 2015;67:2141–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lotz MK, Carames B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat Rev Rheumatol. 2011;7:579–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terkeltaub R, Yang B, Lotz M, Liu-Bryan R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1β and tumor necrosis factor α. Arthritis Rheum. 2011;63:1928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petursson F, Husa M, June R, Lotz M, Terkeltaub R, Liu-Bryan R. Linked decreases in liver kinase B1 and AMP-activated protein kinase activity modulate matrix catabolic responses to biomechanical injury in chondrocytes. Arthritis Res Ther. 2013;15:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou S, Lu W, Chen L, Ge Q, Chen D, Xu Z, Shi D, Dai J, Li J, Ju H, Cao Y, Qin J, Chen S, Teng H, Jiang Q. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci Rep. 2017;7:43245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dvir-Ginzberg M, Mobasheri A, Kumar A. The Role of Sirtuins in Cartilage Homeostasis and Osteoarthritis. Curr Rheumatol Rep. 2016;18:43. [DOI] [PubMed] [Google Scholar]
- 12.Collins JA, Wood ST, Nelson KJ, Rowe MA, Carlson CS, Chubinskaya S, Poole LB, Furdui CM, Loeser RF. Oxidative Stress Promotes Peroxiredoxin Hyperoxidation and Attenuates Pro-survival Signaling in Aging Chondrocytes. J Biol Chem. 2016;291:6641–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang HS, Kim HA. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int J Mol Sci. 2015;16(11):26035–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gavriilidis C, Miwa S, von Zglinicki T, Taylor RW, Young DA. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis Rheum. 2013;65:378–87. [DOI] [PubMed] [Google Scholar]
- 15.Saita Y, Sasho T, Shirasawa T, Yokote K, Kaneko K, Shimizu T. Mechanical overloading causes mitochondrial superoxide and SOD2 imbalance in chondrocytes resulting in cartilage degeneration. Sci Rep. 2015;5:11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaamonde-García C, Riveiro-Naveira RR, Valcárcel-Ares MN, Hermida-Carballo L, Blanco FJ, Lopez-Armada MJ. Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheum. 2012;64:2927–36. [DOI] [PubMed] [Google Scholar]
- 17.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–65. [DOI] [PubMed] [Google Scholar]
- 18.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94:514–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park CB, Larsson NG. Mitochondrial DNA mutations in disease and aging. J Cell Biol. 2011;193:809–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang MC, Hung SC, Chen WY, Chen TL, Lee CF, Lee HC, Wang KL, Chiou CC, Wei YH. Accumulation of mitochondrial DNA with 4977-bp deletion in knee cartilage--an association with idiopathic osteoarthritis. Osteoarthritis Cartilage. 2005;13:1004–11. [DOI] [PubMed] [Google Scholar]
- 21.Bohr VA, Stevnsner T, de Souza-Pinto NC. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene. 2002;286:127–34. [DOI] [PubMed] [Google Scholar]
- 22.de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, Klungland A, Bohr VA. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001;61:5378–81. [PubMed] [Google Scholar]
- 23.Osterod M, Hollenbach S, Hengstler JG, Barnes DE, Lindahl T, Epe B. Age-related and tissue-specific accumulation of oxidative DNA base damage in 7,8-dihydro-8-oxoguanine-DNA glycosylase (Ogg1) deficient mice. Carcinogenesis. 2001;22:1459–63. [DOI] [PubMed] [Google Scholar]
- 24.Hollenbach S, Dhénaut A, Eckert I, Radicella JP, Epe B. Overexpression of Ogg1 in mammalian cells: effects on induced and spontaneous oxidative DNA damage and mutagenesis. Carcinogenesis. 1999;20:1863–8. [DOI] [PubMed] [Google Scholar]
- 25.Panduri V, Liu G, Surapureddi S, Kondapalli J, Soberanes S, de Souza-Pinto NC, Bohr VA, Budinger GR, Schumacker PT, Weitzman SA, Kamp DW. Role of mitochondrial hOGG1 and aconitase in oxidant-induced lung epithelial cell apoptosis. Free Radic Biol Med. 2009;47:750–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruchko MV, Gorodnya OM, Zuleta A, Pastukh VM, Gillespie MN. The DNA glycosylase Ogg1 defends against oxidant-induced mtDNA damage and apoptosis in pulmonary artery endothelial cells. Free Radic Biol Med. 2011;50:1107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torres-Gonzalez M, Gawlowski T, Kocalis H, Scott BT, Dillmann WH. Mitochondrial 8-oxoguanine glycosylase decreases mitochondrial fragmentation and improves mitochondrial function in H9C2 cells under oxidative stress conditions. Am J Physiol Cell Physiol. 2014;306:C221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Wang Q, Watson LJ, Jones SP, Epstein PN. Cardiac overexpression of 8-oxoguanine DNA glycosylase 1 protects mitochondrial DNA and reduces cardiac fibrosis following transaortic constriction. Am J Physiol Heart Circ Physiol. 2011;301:H2073–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baeza J, Smallegan MJ, Denu JM. Mechanisms and Dynamics of Protein Acetylation in Mitochondria. Trends Biochem Sci. 2016;41:231–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, Gibson BW. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci U S A. 2013;110:6601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kincaid B, Bossy-Wetzel E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front Aging Neurosci. 2013;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ansari A, Rahman MS, Saha SK, Saikot FK, Deep A, Kim KH. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell. 2017;16:4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu Y, Kinter M, Hudson J, Humphries KM, Lane RS, White JR, Hakim M, Pan Y,Verdin E, Griffin TM. Aging Promotes Sirtuin 3-Dependent Cartilage Superoxide Dismutase 2 Acetylation and Osteoarthritis. Arthritis Rheumatol. 2016;68:1887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu-Bryan R Inflammation and intracellular metabolism: new targets in OA. Osteoarthritis Cartilage. 2015;23:1835–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cecil DL, Terkeltaub R. Transamidation by transglutaminase 2 transforms S100A11 Calgranulin into a catabolic cytokine for chondrocytes. J. Immunol 2008;180:8378–8385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng Y, Ren X, Gowda AS, Shan Y, Zhang L, Yuan YS, Patel R, Wu H, Huber-Keener K, Yang JW, Liu D, Spratt TE, Yang JM. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013. July 18;4:e731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao X, Petursson F, Viollet B, Lotz M, Terkeltaub R, Liu-Bryan R. PGC-1□ and FOXO3a mediate chondroprotection by AMP-activated Protein Kinase. Arthritis Rheum. 2014;66:3073–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem. 2012;287:14078–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2007;292:H1227–36. [DOI] [PubMed] [Google Scholar]
- 41.Hill JW, Hu JJ, Evans MK. OGG1 is degraded by calpain following oxidative stress and cisplatin exposure. DNA Repair (Amst). 2008;7:648–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim SJ, Cheresh P, Jablonski RP, Williams DB, Kamp DW. The Role of Mitochondrial DNA in Mediating Alveolar Epithelial Cell Apoptosis and Pulmonary Fibrosis. Int J Mol Sci. 2015;16:21486–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang H, Zhu C, Ying Y, Luo L, Huang D, Luo Z. Metformin and berberine, two versatile drugs in treatment of common metabolic diseases. Oncotarget. 2017;9:10135–10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qiao A, Wang K, Yuan Y, Guan Y, Ren X, Li L, Chen X, Li F, Chen AF, Zhou J, Yang JM, Cheng Y. Sirt3-mediated mitophagy protects tumor cells against apoptosis under hypoxia. Oncotarget. 2016;7:43390–43400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu W, Gao B, Li N, Wang J, Qiu C, Zhang G, Liu M, Zhang R, Li C, Ji G, Zhang Y. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy. Biochim Biophys Acta. 2017; 1863:1973–1983. [DOI] [PubMed] [Google Scholar]
- 46.Wei T, Huang G, Gao J, Huang C, Sun M, Wu J, Bu J, Shen W. Sirtuin 3 Deficiency Accelerates Hypertensive Cardiac Remodeling by Impairing Angiogenesis.J Am Heart Assoc. 2017;6(8). pii: e006114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ansari MY, Khan NM, Ahmad I, Haqqi TM. Parkin clearance of dysfunctional mitochondria regulates ROS levels and increases survival of human chondrocytes. Osteoarthritis Cartilage. 2017. August 8 pii: S1063–4584(17)31125–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, Archer SL, Chan DC, Gupta MP. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol. 2014;34:807–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benigni A, Perico L, Macconi D. Mitochondrial Dynamics Is Linked to Longevity and Protects from End-Organ Injury: The Emerging Role of Sirtuin 3. Antioxid Redox Signal. 2016;25:185–99. [DOI] [PubMed] [Google Scholar]