

Abstract
Background:
Platelets have distinct roles in the vascular system as they are the major mediator of thrombosis, critical for restoration of tissue integrity and players in vascular inflammatory conditions. In close spatiotemporal proximity, the complement system acts as the first line of defense against invading microorganisms and is also a key mediator of inflammation. Whereas the fluid phase crosstalk between the complement and coagulation systems is well appreciated, the understanding of the pathophysiological implications of such interactions is still scant.
Methods:
We analyzed co-expression of the anaphylatoxin receptor C3aR with activated GPIIb/IIIa on platelets of 501 coronary artery disease patients using flow cytometry, detected C3aR expression in human or murine specimen by PCR, immunofluorescence, western blotting or flow cytometry and examined the importance of platelet C3aR by various in vitro platelet function tests, by in vivo bleeding time and intravital microscopy. The pathophysiological relevance of C3aR was scrutinized using disease models of myocardial infarction and stroke. To approach underlying molecular mechanisms, we identified the platelet small GTPase Rap1b using nanoscale liquid chromatography coupled to tandem mass spectrometry.
Results:
Interestingly, we found a strong positive correlation of platelet complement C3aR expression with activated GPIIb/IIIa in coronary artery disease patients and co-expression of C3aR with GPIIb/IIIa in thrombi obtained from patients with myocardial infarction. Our results demonstrate that the C3a/C3aR axis on platelets regulates distinct steps of thrombus formation such as platelet adhesion, spreading and Ca2+ influx. Using C3aR−/− mice or C3−/− mice with re-injection of C3a, we uncovered that the complement activation fragment C3a regulates bleeding time after tail injury and thrombosis. Notably, C3aR−/− mice were less prone to experimental stroke and myocardial infarction. Further, reconstitution of C3aR−/− mice with C3aR+/+ platelets and platelet depletion experiments demonstrated that the observed effects on thrombosis, myocardial infarction and stroke were specifically caused by platelet C3aR. Mechanistically, C3aR-mediated signaling regulates the activation of Rap1b and thereby bleeding arrest after injury and in vivo thrombus formation.
Conclusions:
Overall, our findings uncover a novel function of the anaphylatoxin C3a for platelet function and thrombus formation, highlighting a detrimental role of imbalanced complement activation in cardiovascular diseases.
Keywords: platelets, complement, thrombosis, inflammation, anaphylatoxin receptor, C3aR, innate immunity, myocardial infarction, stroke
Introduction
Upon any injury, defense and protection mechanisms are initiated, which aim to prevent further tissue damage and restore organ integrity. For instance, central systems of our body such as the complement cascade and platelet thrombus formation are initiated in close spatiotemporal and functional proximity.1 The complement system is an important part of innate immunity and consists of various tightly regulated soluble factors. It is activated by three distinct pathways converging in the formation of the C3 convertases C4bC2a and C3bBb, which mediate the cleavage of the central complement component C3 to C3b and C3a.2 Whereas C3b further propagates the downstream complement cascade, the anaphylatoxin C3a formed in parallel constitutes a highly active protein contributing to tissue remodeling.3, 4 Binding of C3a and its connate C5a fragment to their receptors C3aR or C5aR1 and C5aR2, respectively, on a variety of cell types mediates a broad range of effects during inflammation.5–7
Besides its function in innate immunity, complement regulates a wide range of processes during tissue-turnover, including angiogenesis, ischemia-reperfusion injury, sepsis or organ healing.3, 8, 9 While knowledge about the crosstalk between complement and platelet activation is scarce, several reports about intersection points between complement activation and the soluble coagulation system exist. For instance, complement activation results in generation of thrombin, which in turn mediates platelet activation.10 Very recently, it was shown that high concentrations of complement C3 are associated with an increased risk of venous thromboembolism11 and that complement factors C3 and C5 contribute to platelet activation and fibrin deposition.12 Furthermore, the human coagulation factor XIIa can activate the complement factor C1q and thereby initiate the classical pathway of complement activation.13 On the other hand, C1 esterase inhibition can suppress all three complement pathways and the intrinsic coagulation cascade via kallikrein and FXIIa.13, 14 It is known that thrombin can generate C5a in the absence of C3.15 Furthermore, C3aR-dependent formation of neutrophil extracellular traps (NETosis) on cellular and artificial surfaces triggers activation of the coagulation system, which links coagulation and complement activation to inflammation.16
Similar to the complement activation process, platelet thrombus formation is a structured and coordinated process, which involves adhesion to subendothelial matrix proteins, platelet spreading, and finally, formation of fibrinogen bridges between platelets recruited from the blood stream via activated glycoprotein (GP) IIb/IIIa (integrin αIIbβ3) receptors resulting in the development of a thrombotic clot.17
It has been shown that complement activation and the formation of C3b can occur on the platelet surface and that C3b can bind to the platelet receptor P-selectin, 18–20 which is expressed upon platelet activation. However, a role of C3a for platelet-complement crosstalk has not been addressed so far in vivo. A potential importance of this finding has been highlighted recently, as a whole-exome sequencing approach of a patient with severe and complex hemostatic abnormalities revealed a possible contributing frameshift mutation in C3aR1.21 A role of complement deposition on platelets has been suggested,20 although the exact mechanisms detailing how complement components bind to platelets, and a relevance of this binding for platelet functions remains elusive. As complement deposition on the platelet surface is of pathophysiological relevance in several immune mediated diseases with a prothrombotic state including paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic-uremic syndrome (aHUS) or atherosclerosis,22 it is important to characterize the underlying mechanisms connecting complement activation to thrombus formation. This is of particular importance in diseases featuring thrombo-inflammation such as stroke or myocardial infarction.22, 23 Already in the early phase of myocardial infarction, accumulation of the terminal complement complex was detected after reperfusion, and inhibition of the complement cascade was demonstrated to reduce myocardial damage after myocardial infarction.24, 25 In stroke, components of the complement system have been shown previously to be involved in tissue remodeling processes such as neural plasticity, and inhibition of complement components was proposed as a therapeutic approach after neural ischemia.26, 27 Interestingly, mice deficient for the central complement component C3 or mice treated with a C3aR antagonist are protected from transient focal cerebral ischemia,28 although the role of platelet derived anaphylatoxin receptors has not been addressed, so far.
Here, we sought to investigate whether expression of complement C3aR holds a pathophysiological relevance for platelet function, hemostasis and thrombosis in pathologies such as stroke or myocardial infarction.
Methods
Please see the online-only Data Supplement for expanded methods. Requests by researchers to access the data, analytic methods, and study materials for the purposes of reproducing the results or replicating procedures can be made to the corresponding author who manages the information.
Study approval
For experiments with human material, written informed consent was received from participants prior to inclusion in the study (approval number 270/2011B01).
All animal experiments were approved by governmental authorities and performed in accordance with the German law guidelines of animal care (approval number M10/12).
Immunofluorescence staining
Human thrombi harvested from aspirates of the coronary arteries of patients with myocardial infarction undergoing percutaneous coronary intervention (PCI) were analyzed by immunofluorescence microscopy. Samples were stained with primary antibodies: anti-human C3aR and anti-human or corresponding control IgG followed by a secondary antibody conjugated with ALEXA-fluorochromes.
Flow cytometry analysis
Human platelets in freshly-collected blood were evaluated for the surface expression of C3aR and P-selectin (CD62P) after gating for the platelet specific marker CD42b. Isolated murine platelets for flow cytometric analysis were stained with the anti-mouse antibodies FITC-anti-P-selectin, FITC-anti-GPVI, FITC-anti-GPIbα, FITC-anti-integrin αIIbβIII, FITC-anti-integrin α5 and FITC-anti-integrin β3. Activated GPIIb/IIIa was detected using fibrinogen-Alexa488 or PE-anti-integrin GPIIb/IIIa. Samples were analyzed with a FACSCalibur flow cytometer (Becton Dickinson, Heidelberg, Germany).
Mice
C57BL/6 mice and C3−/− mice were obtained from The Jackson Laboratory. C3aR−/− mice were a kind gift from Dr. Rick Wetsel (University of Houston, Texas, USA) and C5aR−/− mice were kindly provided by Dr. Craig Gerard (Harvard Medical School, Boston, USA). Spa.1−/− mice were provided by Dr. Yasutoshi Agata (Kyoto University, Kyoto, Japan) and bred with the C3aR−/− mice to obtain a Spa.1−/− x C3aR−/− double knockout strain. GPIbalpha−/− mice were kindly provided by Dr. Jerry Ware (University of Arkansas for Medical Sciences, USA). All strains were on a C57BL/6 background and genotypes were confirmed by PCR. Littermates were used in all experiments as control animals.
Isolation of human and murine platelets
Human platelets were isolated according to standard protocols. Murine platelet preparations were obtained using a modified protocol. For activation, isolated platelets were stimulated with 20 µM ADP or 0.01 U/mL thrombin and/or C3a (as indicated in figure legends) at room temperature.
Western blotting
Whole cell lysates were separated by SDS-polyacrylamide gel electrophoresis and subjected to western blot. Membranes were incubated with primary antibodies (anti-mouse C3aR, anti-human C3aR or anti-Spa-1, clone M-300 or anti-RAP1 or anti-GAPDH or anti-mouse phospho-PI3K or anti-mouse total PI3K or anti-Tubulin). Membranes were scanned with the Odyssey Infrared Imaging System (LI-COR, Bad Homburg, Germany) and analyzed.
Flow chamber assay
Isolated human platelets were stimulated with recombinant human C3a or scrambled C3a and perfused through a transparent flow chamber over a coated surface (collagen (20 µg/ml), vWF (20 µg/ml) or fibrinogen (50 µg/ml)) with high (1.700 s−1) shear rates for 10 min.29 Experiments were recorded with a video recorder linked to a microscope (optical objectives ×20 and ×40; Carl Zeiss) and adherent platelets were quantified.
Platelet Spreading
Isolated murine platelets were incubated on fibrinogen-coated and BSA-blocked coverslides for 30 minutes at room temperature. Platelets were fixed and permeabilized with PHEM buffer containing PFA and NP-40, stained with rhodamin-phalloidin and analyzed with an Axio Observer Z.1 microscope (Zeiss).
Scanning Ion Conductance Microscopy (SICM)
Isolated WT and C3aR−/− platelets were stimulated with thrombin and allowed to adhere for 30 min to fibrinogen-coated culture plates. Platelets were then fixed with 4% PFA and mounted into the SICM setup. Platelet morphology was analysed using a custom-written software. Briefly, the cell contour was determined automatically using a height threshold of 50 nm and processed to calculate the morphology parameters (area A, height, and circularity C = 4π A / P2 with perimeter P).
Immunoprecipitation
Resting and stimulated platelets were lysed by adding an equal amount of lysis buffer. Samples were incubated with anti-C3aR antibody. Protein G-Sepharose was added and samples were incubated over night at 4 °C with rotation. Immunoblotting was performed as indicated.
SDS PAGE and in-gel digestion
Complete protein eluates of the pull-down experiments were submitted to a short run (1cm in length) on 1D SDS-PAGE. The proteins were visualized by staining and the corresponding gel sectors were excised and subjected to tryptic in-gel digestion. The resulting peptide mixtures were desalted before Liquid chromatography/Mass spectrometry (LC/MS) measurement.
Nanoscale liquid chromatography coupled to tandem mass spectrometry (NanoLC-MS/MS) analysis
LC-MS analysis was carried out on a nanoLC (Easy-nLC , Thermo Fisher Scientific, formerly Proxeon Biosystems) coupled to a LTQ-Orbitrap-XL (Thermo Fisher Scientific). MS data were processed using the MaxQuant software suite, peptide sequences were retrieved by using the Andromeda search engine, data were searched against the Uniprot mouse database.
Rap1 activation and pulldown assay
For detection and pulldown of activated Rap1 (GTP-Rap1), a commercially available kit (affinity precipitation assay, Merck Millipore) was used.
Measurement of cytosolic Ca2+ concentration
Washed murine platelets were loaded with 5 μM fura-2 acetoxymethylester in the presence of 0.2 μg/ml pluronic F-127. Loaded platelets were activated with thrombin (0.01U) ± C3a (200 nM), ± C3aR Inhibitor SB290157 (10 µM). Calcium responses were measured under stirring with a spectrofluorimeter (LS 55; PerkinElmer) at alternate excitation wavelengths of 340 and 380 nm (37°C). The 340/380 nm ratio values were converted into concentrations of [Ca2+].
Platelet aggregometry
Aggregation of isolated human or murine platelets was estimated from light transmission determined with a luminoaggregometer (model 700; ChronoLog). Platelets were activated with 20 µM ADP, C3a in different doses or control protein (scrambled C3a) at the indicated concentrations. Analysis was performed with the Aggrolink8 software (ChronoLog).
Bleeding time
Mice were anesthetized and a 3-mm segment of the tail tip was removed with a scalpel. Tail bleeding was monitored by gentle absorption of the blood with filter paper at 20-s intervals without making contact with the wound site.
Intravital microscopy
Intravital microscopy and induction of platelet thrombus formation in vivo were carried out as described before.29
Stroke model
All stroke experiments were performed in accordance with the recently published ARRIVE guidelines (nc3rs) and performed as described before.30
Myocardial infarction model
All myocardial infarction experiments were performed as described before.31
Statistics
Data are provided as means±SD; n represents the number of experiments. All data were tested for significance using unpaired Student t-test and one-way or two-way ANOVA with Bonferroni’s post hoc test. Results with P < 0.05 were considered statistically significant.
Results
Various components of the complement cascade have previously been detected on platelets isolated from human subjects, particularly after platelet activation.20 Strikingly, in our large collective of patients with coronary artery disease (CAD), expression of C3aR was strongly correlated with the expression of the activated fibrinogen receptor GPIIb/IIIa (using an activation-specific antibody, r = 0.59, p < 0.001, Figure 1A, B). We furthermore observed a significant correlation in the subgroup of patients receiving no P2Y12 inhibitor (Spearman`s rank coefficient rs=0.60, p<0.001, n=410 patients) as well as in the subgroup of patients receiving P2Y12 inhibitors (Spearman`s rank coefficient rs=0.53, p<0.001, n=91 patients) as indicated in Supplemental Figure 1. The distribution of different P2Y12 inhibitors is depicted in Supplemental Table 1. In line with this finding, C3aR was detected in platelets isolated from both murine and human blood (Figure 1C – E). While a marginal expression of C3aR was observed on the surface of unstimulated platelets, ADP-induced platelet activation resulted in a significant increase in the expression levels of C3aR (Figure 1F, G). Overall expression of C3aR in lysed platelets showed no significant differences between resting, ADP- or C3a stimulated platelets (Supplemental Figure 2).
Figure 1. Platelets express the anaphylatoxin receptor C3aR.
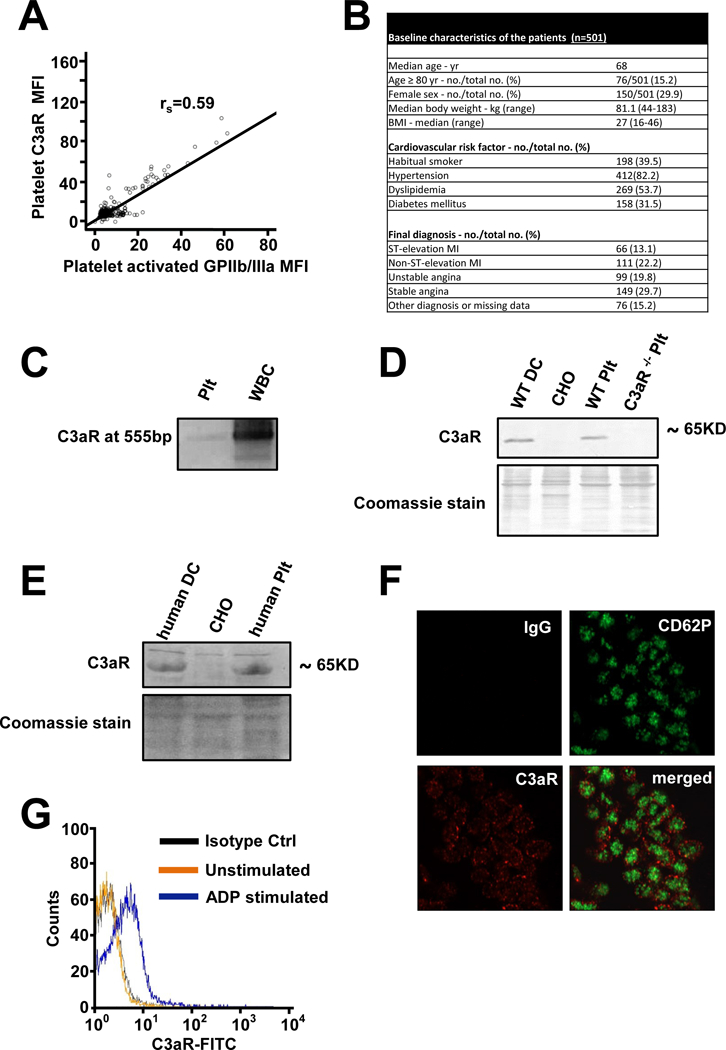
(A) Analyzing patients with coronary artery disease, expression of C3aR and activated GPIIb/IIIa (activation specific Ab PAC-1) was measured by flow cytometry. The correlation of C3aR with activated GPIIb/IIIa is depicted. Spearman`s rank coefficient rs=0.59, p<0.001, n=501 patients. (B) Baseline characteristics of the analyzed patients (C) Expression of C3aR on resting human platelets and WBC as control was detected by isolation of mRNA and RT-PCR of transcribed cDNA with specific primers. A C3aR specific band could be detected at 555bp on a 2% agarose gel. (D+E) Protein expression of C3aR on thrombin- stimulated (0.01 U/ml) murine (D) and human (E) platelets was analyzed by western blot. CHO cells served as negative control, murine bone marrow derived dendritic cells (DC) or human monocyte derived dendritic cells served as positive control. One of 3 representative blots is depicted. Coomassie Blue staining of the PVDF membranes after blotting served as loading control. (F) ADP-stimulated (20 µM) human platelets in PRP were fixed with 1% PFA and spun on poly-L-lysine coverslips. Permeabilized platelets were analyzed for expression of CD62-P (green) and C3aR (red) with a Zeiss LSM 800 microscope (63x objective. Area of interest cropped 4.7x) (G) Human platelets were isolated, stimulated with ADP (20 µM) and analyzed by flow cytometry for surface expression of C3aR. n=8, p<0.05 vs. resting platelets, one representative diagram is shown.
In light of the significant expression of C3aR on activated platelets, we assessed whether C3aR-mediated signaling impacts the function of platelets during bleeding. Initially, tail bleeding time was assessed in C3-deficient mice, as C3 is the central complement component and key for the activation of all three complement pathways. Prolonged bleeding time (3-times longer when compared with control mice) was observed in the absence of C3 (Figure 2A), which is in line with previous reports.32 Similar bleeding time was observed in GPIbalpha−/− mice, resembling the phenotype of the Bernard-Soulier Syndrome where increased bleeding time is seen as a result of thrombocytopenia and platelet dysfunction33 (Figure 2A). Given that C3 is activated on the surface of activated platelets with consequent generation of C3a 18 and the increased expression of C3aR (Figure 1G), we next analyzed bleeding time after tail injury in C3aR−/− mice. In line with the findings described above, mice deficient for C3aR showed a significantly prolonged time to bleeding arrest when compared with wildtype C57BL/6 (WT) littermate controls (Figure 2B). This effect could not be attributed to altered platelet numbers in these mice, as both WT and C3aR−/− mice showed comparable peripheral platelet counts (Supplemental Figure 3). In contrast to C3aR−/− animals, no significant increase in bleeding time was observed in mice deficient for the C5aR (Supplemental Figure 4), also a G protein-coupled receptor that binds the complement activation product C5a.3 Supporting a role for C3aR in the control of bleeding, pharmacological inhibition of C3aR with a small molecule antagonist (SB290157, 750 µg/mouse i.p.) resulted in prolonged bleeding time resembling the phenotype of C3−/− and C3aR-/- mice (Figure 2C). Interestingly, prolonged tail bleeding time in C3−/− mice was reversed upon i.v. administration of C3a peptide (15 µg/mouse), a peptide with scrambled amino acid sequence was used as negative control (Figure 2D).
Figure 2. Prolonged bleeding time in C3 deficient mice is mediated by C3a.
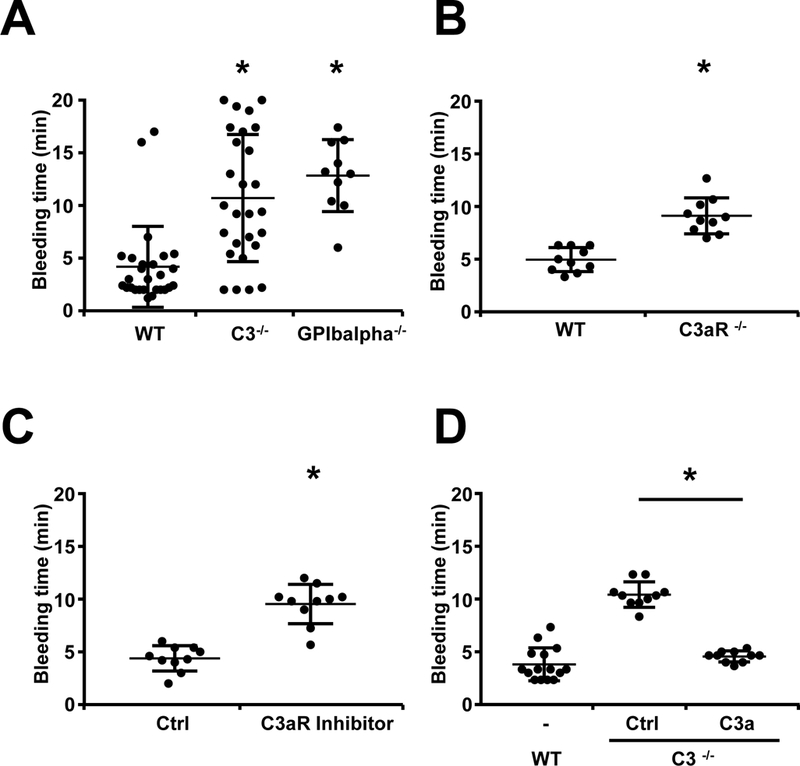
To assess the role of the C3aR in hemostasis, tail bleeding time was assessed in anesthesized C3−/− mice, C3aR−/− mice and WT mice after amputation of the tail tip. Data represent mean ± SD. (A) A prolonged bleeding time was observed in C3−/− mice compared to WT mice, GPIbalpha−/− mice served as positive control. n=27 (WT and C3−/−) and n=10 (GPIbalpha−/−) animals per group. *p<0.05 in comparison to WT mice. (B) Similarly, the bleeding time of C3aR−/− mice was significantly prolonged in comparison to WT mice. n=10 animals per group. *p<0.05 compared to WT mice. (C) WT mice were treated with a specific inhibitor of C3aR (SB290157) by i.p. injection (750 µg/mouse). Compared to control (DMSO) treated animals, bleeding time was significantly prolonged. n=10 animals per group. *p<0.05 compared to control peptide treated mice. (D) C3−/− mice were injected i.p. with C3a (15 µg/mouse) or control (scrambled C3a, 15µg/mouse). In mice injected with C3a, bleeding time was significantly reduced. n=15 (WT) and n= 20 (C3−/−; 10 with C3a and 10 with scrambled C3a) animals per group. *p<0.05 compared to control peptide.
As platelets have to firmly adhere to the extracellular matrix and spread before additional platelets can be recruited to the evolving thrombus,17 we tested whether C3aR-mediated signaling assists in the process of platelet adhesion. While C3a alone was not sufficient to mediate platelet adhesion (data not shown), a significant increase in platelet adhesion to fibrinogen-coated chambers under dynamic flow conditions was observed upon C3a stimulation (Figure 3A). Furthermore, co-immobilization of C3a with fibrinogen increased platelet adhesion (Supplemental Figure 5A), and adhesion of murine C3aR−/− platelets to fibrinogen was significantly reduced compared to WT platelets (Supplemental Figure 5B). The difference was not present any more, if C3a-des-arg7 was used (Supplemental Figure 6). Such differences in platelet adhesion were not a consequence of differential expression levels of adhesion molecules as platelets isolated from either WT or C3aR−/− mice showed similar expression of important platelet adhesion molecules (Figure 3B). In contrast, Ca2+ influx in response to thrombin, a key event during platelet adhesion to extracellular matrix proteins, was significantly reduced in platelets from C3aR−/− when compared to WT mice or in WT platelets treated with a C3aR inhibitor (Figure 3C, D). Furthermore, the deficiency of C3aR resulted in decreased spreading capacity of thrombin stimulated platelets (Figure 3E). A similar result was obtained using a C3aR inhibitor (Supplemental Figure 7). To investigate the influence of C3aR on platelet spreading on a single-cell level, WT and C3aR−/− platelets were analyzed using scanning ion conductance microscopy (SICM). SICM allows for a label-free, quantitative characterization of platelet 3D morphology with high resolution. After 30 minutes of adhesion on immobilized fibrinogen, thrombin-stimulated (0.01 U/ml) C3aR−/− platelets exhibited significantly reduced spreading area, increased height, and more irregular cell shape compared to WT platelets (Figure 3F).
Figure 3. C3aR is important for platelet function.
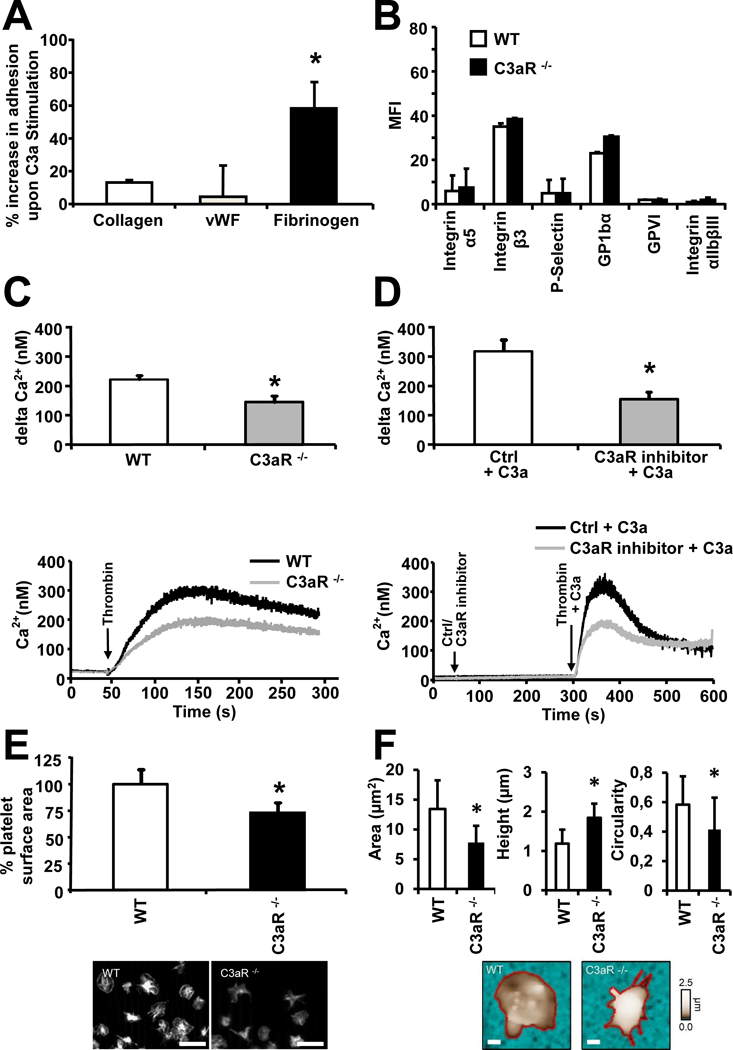
(A) Human isolated platelets were perfused over coverslides coated with different extracellular matrix proteins using a parallel-plate flow chamber. Under flow conditions (shear rates 1700s−1), the increase of adhesion upon stimulation with C3a (4 µM) in comparison to stimulation with control protein was measured. The increase in adhesion onto fibrinogen was significantly stronger in comparison to collagen and vWF. Data represent mean ± SD. n=5. *p<0.05 (B) Whole blood of WT and C3aR−/− mice was fixed with PFA and analyzed for platelet surface receptor expression by flow cytometry. Receptor expression is not altered between the two groups. Data represent mean±SD, n=5. (C, D) Ca2+-influx was analyzed in washed murine WT and C3aR−/− platelets activated with thrombin (0.01 U/ml) (C) or in washed WT platelets, treated with a C3aR Inhibitor (10 µM) or DMSO (Ctrl) and activated with thrombin (0.01 U/ml) +/− C3a (200 nM) (D). The measured 340/380 nm ratio values were converted into nanomolar concentrations of Ca2+. C3aR−/− platelets and C3aR-Inhibitor treated WT platelets show significantly reduced Ca2+ influx compared to WT (and control-treated WT) platelets. Data represent mean ± SD, n=5. *p<0.05 The corresponding curves show measurements representative for all experiments. (E) Isolated WT and C3aR−/− platelets were stimulated with 0.01 U/ml thrombin and allowed to adhere to immobilized fibrinogen, before the spreading area of rhodamin-phalloidin-stained platelets was measured by microscopy. Data represent mean ± SD, n=5. *p<0.05 for C3aR−/− platelets in comparison to platelets isolated from WT mice. scale bars: 10 µm (F) Isolated WT and C3aR−/− platelets were stimulated with 0.01 U/ml thrombin and adhesion to immobilized fibrinogen was analyzed after 30 minutes. Area, height, and circularity of PFA-fixed platelets were analyzed using SICM. Data represent mean ± SD, n = 26 platelets from n=4 independent experiments each. *p<0.05. Lower panel: shown are representative SICM topography images. The cell contour (red) was traced to quantify cell area, height and circularity. Scale bars: 1 µm.
Given our findings that C3aR-mediated signaling appears to modulate platelet adhesion and spreading consequently impacting thrombus formation, we next investigated the pathophysiological relevance of these observations using in vivo models of experimental myocardial infarction and stroke.30, 31 Focal cerebral ischemia was induced in WT and C3aR−/− mice. Notably, C3aR-/- mice showed reduced brain infarct volume and consecutively reduced functional impairment (Figure 4A–C) compared to WT mice, suggesting a deleterious role for C3aR during stroke. Interestingly, treatment of mice with platelet-depleting serum (10µg/mouse i.p., Supplemental Figure 8) before induction of brain infarction rescued the phenotype of reduced functional impairment associated with reduced infarct area in C3aR−/− mice compared to WT mice (Figure 4A–C). Addressing the role of C3aR in human heart disease, we could identify the presence of C3aR on platelets in thrombi aspirated from the coronary arteries of five patients with myocardial infarction (MI) during percutaneous coronary intervention (Figure 5A). Thus, we went on to study the role of the platelet C3aR in experimental MI. Similar to the results in experimental stroke, induction of myocardial infarction using an ischemia-reperfusion model resulted in significantly reduced myocardial necrosis and better LV-function in the C3aR−/− when compared with WT mice (Figure 5B − E, Supplemental Table 2, 3). If platelets were depleted, this difference was not observed any more, highlighting the cell-specific functional role of this receptor on platelets (Figure 5B, C). The difference between WT and C3aR−/− mice persisted, however, after neutrophil depletion (Figure 5 C). Reduced myocardial necrosis was associated with an increase in LV-function in C3aR−/− mice in comparison to WT mice, which was not observed in the absence of platelets (Figure 5D, E) suggesting an important role of the platelet C3aR in this setting.
Figure 4. Platelet derived C3aR contributes to experimental stroke.
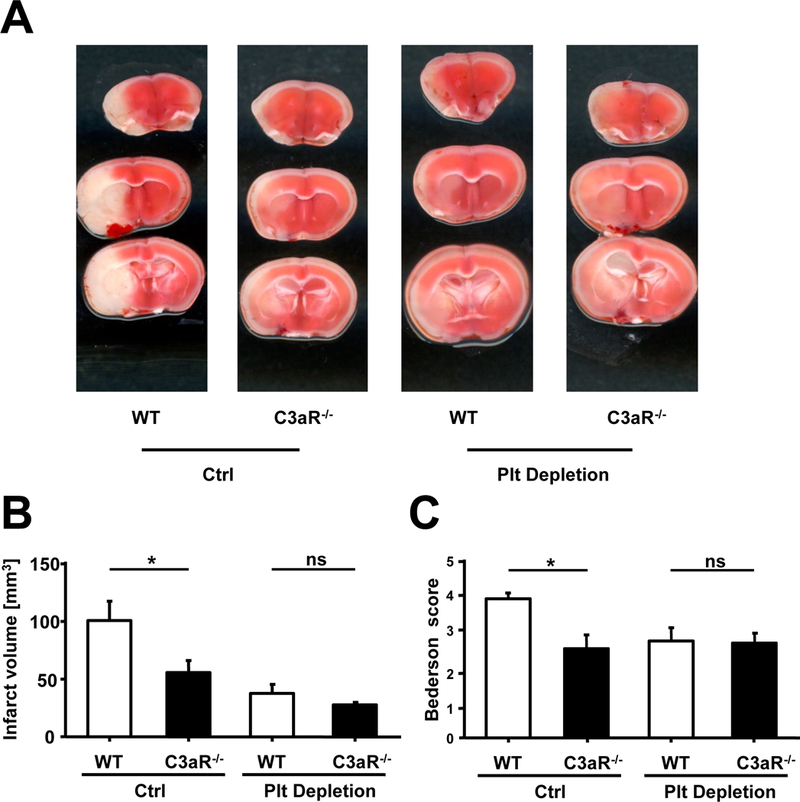
(A - C) Stroke was induced using the tMCAO model. (A) depicts sample images of TTC stainings for infarcted brain tissue 24h after stroke induction. (B) Brain infarct volumes in control and platelet-depleted mice 24 hours after tMCAO are given as mean ± SD. Compared to WT animals, C3aR−/− mice were protected from ischemic stroke. n=6 (WT) and n=8 (C3aR−/−). *p<0.05 compared to WT animals. No difference was observed between WT and C3aR−/− mice in the absence of platelets, ns=not significant. (C) Accordingly, the beneficial functional outcome observed in C3aR−/− compared to WT mice using the Bederson score disappeared after platelet depletion. n=6 (WT) and n=8 (C3aR−/−). *p<0.05 compared to WT animals, ns=not significant.
Figure 8. Role of Rap1b in C3aR mediated thrombus formation.

(A) Generation of Spa.1−/− x C3aR−/− double knockout (DKO) mice. Rap1 is activated (Rap1*GTP) by different ligands binding to G-Protein coupled receptors such as C3aR. Rap1 is inhibited by specific GTPase activating proteins (Rap1GAPs) such as Spa.1. In Spa.1−/− mice, lack of this Rap1GAP results in constantly active Rap1. In C3aR−/− mice, we observed a phenotype of reduced thrombosis and increased bleeding. To address any intersection point between these observations, we crossed Spa.1 knockout mice with C3aR−/− mice to generate double knockout (DKO) mice (C3aR−/− x Spa.1−/− mice). (B) Rap1 pull down assay for detection of activated Rap1 in murine platelet lysates of WT, C3aR−/− x Spa.1+/+, C3aR−/− x Spa.1−/− and Spa.1−/− mice stimulated with C3a. Activated Rap1 in murine platelet lysates was isolated with GST-tagged sepharose, subjected to SDS-PAGE, blotted and detected with an activation specific Rap1 antibody. Total Rap1 in platelet lysates was analyzed by western blot. One representative blot of three experiments is depicted. (C) Tail-bleeding time was measured in C3aR−/− x Spa.1−/− DKO mice, WT mice and C3aR−/− Spa.1+/+ mice. In C3aR−/− x Spa.1−/− DKO mice, bleeding time was significantly reduced compared to C3aR−/− x Spa.1+/+ mice. Data represent mean ± SD, n=15 (WT), n=9 (C3aR−/− x Spa.1+/+) and n=8 (C3aR−/− x Spa.1−/−). *p<0.05 (D) Similarly, FeCl3-induced thrombus formation was analyzed in these mice. After injury of mesenteric arterioles, time to vessel occlusion was significantly shorter in C3aR−/− x Spa.1−/− DKO mice compared to C3aR−/− x Spa.1+/+ littermate controls. Data represent mean ± SD, n=5 (WT) and n=7 (C3aR−/− x Spa.1+/+ and C3aR−/− x Spa.1-/-),*p<0.05.
Figure 5. Platelet derived C3aR contributes to experimental myocardial infarction.
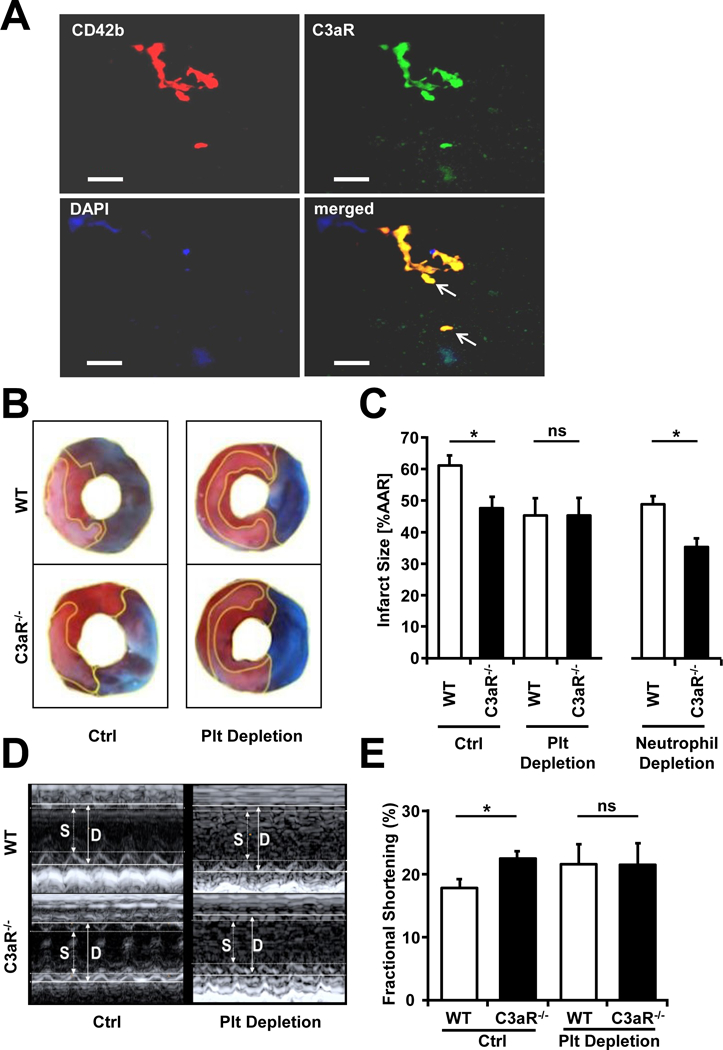
(A) Expression of C3aR on platelets in human thrombi from aspirates of the coronary arteries of patients with myocardial infarction undergoing PCI. Double immunofluorescence staining was carried out with anti-CD42b, a platelet marker, and anti-C3aR. Red fluorescence signal indicates platelets (CD42b positive), green fluorescence marks the C3aR. Nuclei were stained with DAPI. Arrows mark CD42b C3aR double positive platelets. Shown is one representative staining, 5 thrombi were analyzed. Scale bar 50 µm. (B - E) Myocardial infarction was induced in WT or littermate C3aR−/− mice by ligation of the left anterior descending artery (LAD). Additionally, mice were treated with platelet depleting serum, control serum (Ctrl), neutrophil granulocyte depleting antibody or a control antibody (intraperitoneally) as indicated. (B) Representative pictures of experiments in WT and C3aR−/− mice with injection of platelet depleting serum or control serum are depicted. The infarct area was quantified after Monolite blue/TTC staining. (C) The area of necrosis was quantified by a blinded investigator. We observed a significant reduction of necrosis in C3aR−/− animals compared to WT littermate controls treated with control serum. This difference was not present any more, when platelets were depleted. In neutrophil-depleted animals, we observed a significant reduction of necrosis in C3aR−/− animals compared to WT mice. Data represent mean ± SEM, n=7/8 (without depletion, one mouse died during the procedure), n=8 (platelet depletion), n=5/6 (neutrophil depletion, one mouse died during the procedure). *p<0.05. Values are presented as % of infarct size of the area at risk (AAR) compared to WT littermate controls. (D, E) Before mice were euthanized, echocardiography was carried out. Fractional shortening as a measure of LV-function was determined. We observed a significantly better LV-function in C3aR−/− animals compared to WT littermate controls only after injection of control serum. This difference was not present any more, when platelets were depleted. Data represent mean ± SEM, n=7/8 (without depletion, one mouse died during the procedure), n=8 (platelet depletion). *p<0.05. Values are presented as % increase in fractional shortening compared to WT littermate controls.
We next investigated whether the reduced infarct volume observed in C3aR−/− mice was associated with the expression of C3aR specifically on platelets. To this purpose, C3aR−/− mice were reconstituted with either C3aR−/− or C3aR+/+ platelets (1 × 108 platelets / mouse) prior to analyzing tail bleeding time as a measure of platelet thrombosis. The presence of the C3aR on platelets in C3aR−/− mice reconstituted with platelets from C3aR+/+ mice was verified by western blot 24 hours after adoptive transfer (data not shown). Interestingly, the prolonged bleeding time observed in C3aR−/− mice was reversed in the presence of WT platelets (Figure 6A). Consistent with our hypothesis, flow cytometric analysis of platelets showed lower expression of the activated GPIIb/IIIa fibrinogen receptor, critical for the triggering of the final events of platelet thrombus formation, in C3aR−/− mice reconstituted with WT platelets (1 × 108 platelets / mouse, Supplemental Figure 9). Further, in vitro isolated platelets showed reduced ADP-induced binding of Fibrinogen-Alexa 488 (20 µg/ml) and, thus, expression of activated GPIIb/IIIa in C3aR−/− when compared with C3aR+/+ platelets or after treatment with a C3aR inhibitor (SB290157, 10 µM, Figure 6B, C). To determine whether C3aR-mediated surface expression of activated GPIIb/IIIa is the mechanism underlying differential platelet function in the absence of C3aR, ADP-activated (20 µM) murine platelets of C3aR−/− and C3aR+/+ were stimulated with recombinant C3a. Stimulation of ADP-preactivated platelets with C3a resulted in increased platelet aggregation in a dose-dependent manner (Figure 6D). Similar results were obtained with human platelets (Supplemental Figure 10). The difference was not present any more, if C3a-des-arg34 (Supplemental Figure 11A) or other platelet agonists such as collagen and thrombin were used (Supplemental Figure 11B, C), suggesting specificity of our findings. To directly assess thrombus formation in vivo, an established intravital microscopy model of FeCl3-induced thrombus formation of the mesenteric vasculature was used.29 Supporting our previous findings, the time required to vessel occlusion was significantly prolonged in C3aR−/− when compared to WT or C5aR−/− mice (Figure 6E, F).
Figure 6. Platelet C3aR mediates thrombosis.
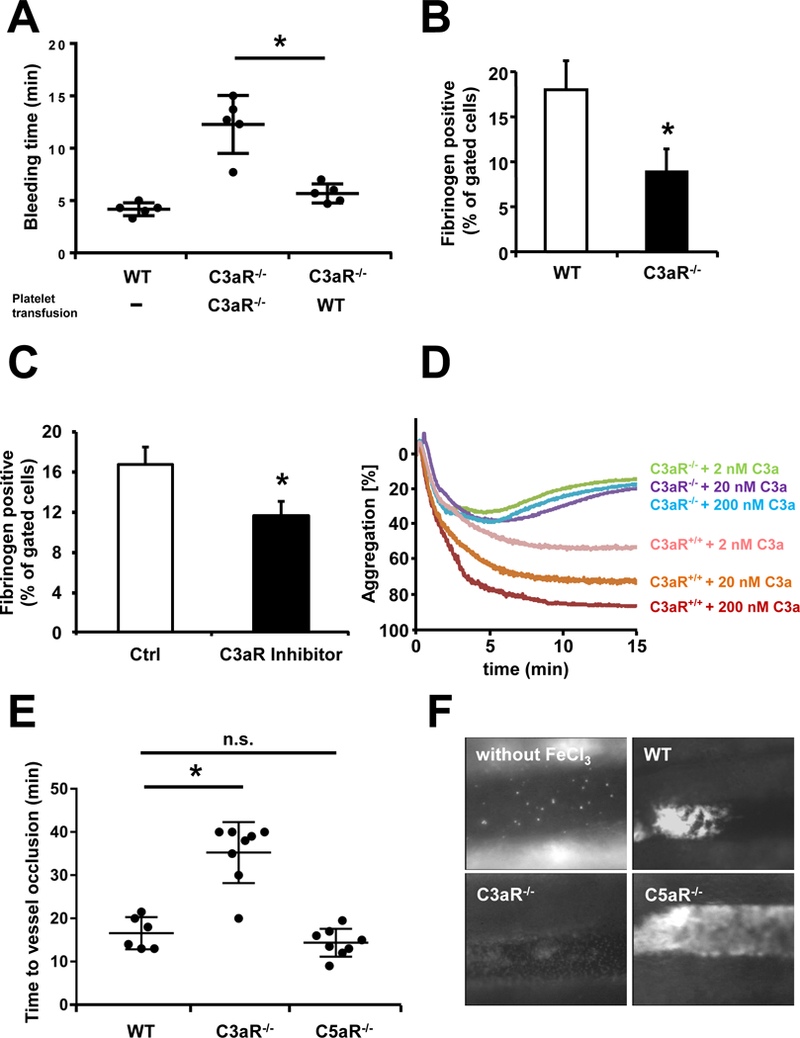
(A) Tail-bleeding time was measured in C3aR−/− mice after i.v. injection of isolated WT or C3aR−/− platelets (1 × 108 / mouse). In C3aR−/− mice transfused with WT platelets, bleeding time was significantly shorter than in those transfused with C3aR−/− platelets. Data represent mean ± SD, n=5. *p<0.05 (B) Detection of activated GPIIb/IIIa by flow cytometry. Isolated murine platelets were stimulated with 20 µM ADP and stained with Fibrinogen-Alexa488 (20 µg/ml). In C3aR−/− platelets, the amount of bound fibrinogen (and thus, activated GPIIb/IIIa) on platelets is lower than in WT mice. Data represent mean ± SD, n = 5. *p<0.05. (C) Detection of activated GPIIb/IIIa by flow cytometry. Isolated murine WT platelets were treated with a C3aR Inhibitor (10 µM) or DMSO (Ctrl), stimulated with 2 µM ADP and C3a (200 nM) and stained with Fibrinogen-Alexa488 (20 µg/ml). The binding of Fibrinogen-Alexa488 on activated platelets was inhibited by adding a C3aR inhibitor. Data represent mean ± SD, n=5. *p<0.05. (D) Platelet aggregation was analyzed in murine WT and C3aR−/− isolated platelets using conventional aggregometry. WT platelets pre-stimulated with 2 µM ADP show increased aggregation after addition of C3a (time lag: 5 seconds), which was dose-dependent. In C3aR−/− platelets, aggregation after stimulation with ADP and increasing doses of C3a was significantly reduced compared to WT platelets. (E, F) Mesenteric arterioles were injured by topical application of 20% FeCl3, and thrombus formation was monitored by intravital microscopy. In C3aR−/− mice, the time to vessel occlusion was significantly prolonged in comparison to WT mice. n.s. = no significant diference (WT in comparison to C5aR−/−mice). Data represent mean ± SD, n=6 (WT) n=8 (C3aR−/−) and n=8 (C5aR−/−). *p<0.05. (F) Representative images of the experiments are shown. Pictures were taken after 10 minutes.
Signaling via the GTPase Ras-related protein Rap-1b (Rap1b) is critical for thrombus formation in mice.35 Interestingly, Rap1 was detected after immuno-precipitation of platelet lysates using an anti-C3aR antibody (Figure 7A). Further proteins associated with C3aR are listed in Supplemental Table 4. A pull-down assay showed that stimulation of WT but not of C3aR−/− platelets with C3a (200 nM) reveals increased Rap1 activity (Figure 7B), and inhibition of C3aR using SB290157 (10 µM) reduces activated Rap1 in the presence of C3a and ADP (Figure 7C). To assess the influence of P2Y12 inhibition on C3aR expression, we analyzed platelets treated with inhibitors of P2Y1 and P2Y12 in vitro by flow cytometry. We observed a significantly reduced C3aR expression in platelets treated with the P2Y12 inhibitor, but not with the P2Y1 inhibitor (Supplemental Figure 12A). As inhibition of P2Y12 but not P2Y1 significantly reduces Rap1 activity in stimulated murine platelets, P2Y12 seems to be the relevant ADP-receptor in this setting (Supplemental Figure 12B). C3a is a cleavage product of C3,3 in the absence of C3 using platelets isolated from C3−/− mice, the effect of Rap1b-activation by C3a is still detectable (Supplemental Figure 13). To determine the role of C3aR for Rap1b signaling in platelets, Rap1b GTPase-activating protein Spa.1 (signal-induced proliferation-associated gene-1) knockout mice, which show constitutively active Rap1b,36 were backcrossed with C3aR−/− mice (Figure 8A). These C3aR−/− x Spa.1−/− double knock out mice, which also have constitutively activated Rap1b (Figure 8B), were tested in tail bleeding time experiments, in vitro adhesion and aggregation. Expression of Spa.1 was confirmed in isolated activated platelets (Supplemental Figure 14). In vitro analysis of C3aR−/− x Spa.1−/− double knock out platelets showed, that the phenotype of C3aR knockout (reduced spreading and reduced aggregometry) could be reversed by constitutively active Rap1 (Supplemental Figure 15 A, B). Strikingly, the phenotype of increased bleeding time previously observed in C3aR−/− mice was reversed in C3aR−/− x Spa.1−/− double knockout mice, but not in C3aR−/− x Spa.1+/+ littermate controls (Figure 8C), while Spa.1−/− mice show a regular bleeding time (Supplemental Figure 16). Additionally, using intravital microscopy, the time to vessel occlusion after induction of thrombus formation decreased in the presence of constitutively active Rap1b in C3aR−/− x Spa.1−/− mice (Figure 8D).
Figure 7. Treatment with C3a results in Rap1b activation.
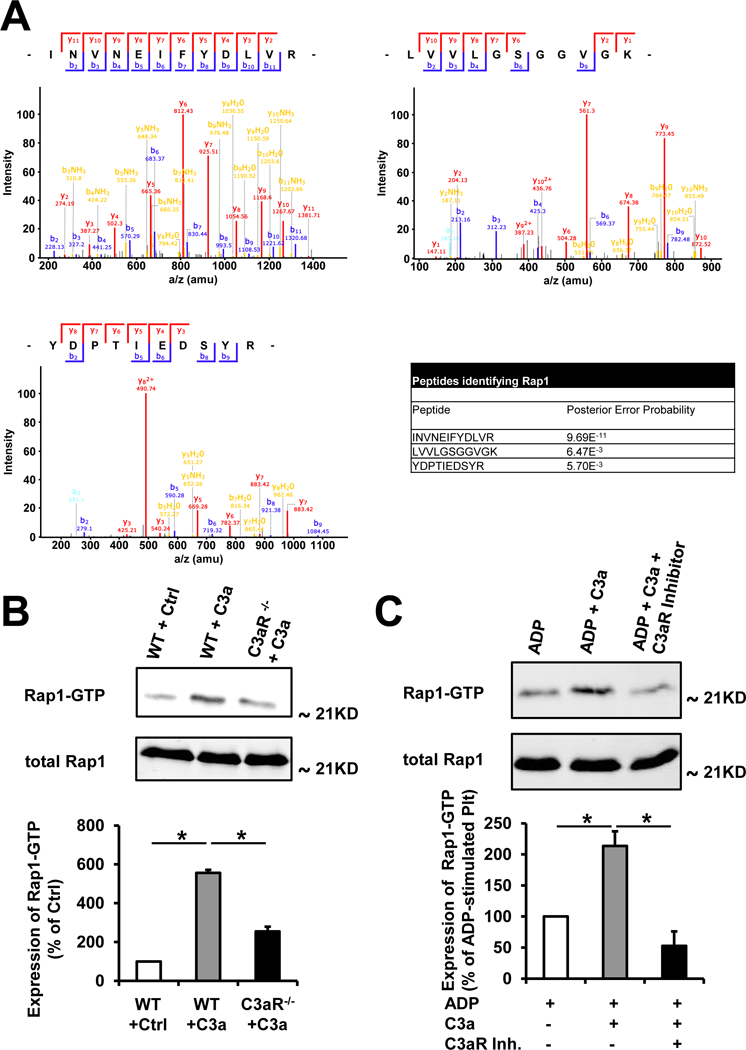
(A) Rap1-detection after immuno-precipitation of platelet lysates using an anti-C3aR antibody. Protein eluates of pull-down experiments using isolated murine platelets and an anti-C3aR antibody were subjected to Nano-LC MS/MS analysis. Processing of the retrieved MS data with Max Quant / Andromeda software suite revealed Rap1 as a C3aR receptor interaction partner. Shown are representative mass spectra of the 3 peptides INVNEIFYDLVR, LVVLGSGGVGK and YDPTIEDSYR identifying Rap1. The table shows the Posterior Error Probability (PEP) of the 3 peptides identifying Rap1. A PEP lower than 0.01 indicates that the peptide was identified with a probability of more than 99%. (B) Rap1 pull down assay for detection of activated Rap1 in murine platelet lysates of WT and C3aR−/− mice stimulated with C3a (200 nM) or control peptide (200 nM). Activated Rap1 in murine platelet lysates was isolated with Glutathione S-transferase (GST)-tagged sepharose, subjected to SDS-PAGE, blotted and detected with an activation specific Rap1 antibody. The amount of total Rap1 is equal in each sample. One representative blot of three experiments is depicted. The bar chart shows the densitometric analysis of Rap1-GTP expression. Data represent mean ± SD. n = 3. *p<0.01. Ctrl was set as the 100% value. (C) Rap1 pull down assay for detection of activated Rap1 in murine platelet lysates of WT mice stimulated with ADP (20 µM), or ADP and C3a (200 nM) or ADP and C3a and the C3aR inhibitor SB290157 (10 µM). Activated Rap1 in murine platelet lysates was isolated with GST-tagged sepharose, subjected to SDS-PAGE, blotted and detected with an activation specific Rap1 antibody. The amount of total Rap1 is equal in each sample. One representative blot of three experiments is depicted. The bar chart shows the densitometric analysis of Rap1-GTP expression. Data represent mean ± SD. n = 3. *p<0.01. ADP stimulated platelets were set as the 100% value.
From previous studies it is known that platelet stimuli such as ADP result in phosphorylation of Phosphoinositide 3-kinase (PI3K) and increased Rap1 activity.37 To further shed light on the signaling pathways involved in platelet C3aR signaling via Rap1b, we found that stimulation of platelets with C3a results in increased Rap1b activity on top of platelet stimulation by ADP. Accordingly, C3a and ADP-C3a co-stimulation resulted in increased phosphorylation of PI3K (Supplemental Figure 17).
Discussion
The complement cascade and platelet activation are important systems to preserve tissue integrity and organ function. Here, we show that the complement C3aR is expressed on mouse and human platelets and document a functional role of this receptor for platelet functions such as firm adhesion or platelet spreading, and importantly for thrombus formation in vivo. Furthermore, we show that the C3a-C3aR axis activates the small GTPase Rap1b, promotes GPIIb/IIIa activation and contributes to platelet aggregation and thrombus formation.
Early in vitro studies using guinea pig platelets suggested an expression of the anaphylatoxin receptor C3aR on platelets.38 Furthermore, the complement component C3 was found to be present in human plasma clots.39 A role of the platelet C3aR was demonstrated in the context of the prostaglandin metabolic pathway, before.40 Here, we demonstrate by western blot and flow cytometry that C3aR is expressed and upregulated on platelets upon activation.
Recently, it was observed that P2Y12 inhibitors resulted in modification of G-protein coupled receptors/GPIIbIIIa. Analyzing our patients, there was a significant correlation between C3aR expression and activated GPIIbIIIa, independent of the presence of a P2Y12 inhibitor. At this point, however, we cannot entirely rule out an influence of P2Y12-inhibition because of the small patient number, particularly in the group with no anti-platelet medication.
Expression of other platelet receptors relevant for platelet function was not different between WT and C3aR deficient animals. In vitro, C3aR on platelets regulates specific aspects of their function. For instance, we could measure a significant increase in platelet adhesion, spreading and aggregation after stimulation of platelets with a C3a peptide. Similarly, it was observed before that aggregation of human platelets after stimulation with C3a was more pronounced.41 One explanation is the reduced activation-dependent increase of the cytosolic Ca2+ concentrations in C3aR−/− platelets, which reflects an important step in platelet thrombus formation upon stimulation with different platelet agonists.42 A previous study had observed cleavage of C3 into C3b on the platelet surface,43 although the potential role of C3a or the C3aR for platelets was not addressed. Here, we demonstrated that C3aR deficient platelets show reduced spreading on fibrinogen, which is one of the later events during platelet thrombus formation before fibrinogen bridges are built by activated GPIIb/IIIa on aggregating platelets. Thrombosis is a determining factor and often decisive event in various (patho-) physiological settings such as provisional closure of an endothelial wound or vessel occlusion in stroke and myocardial infarction.44 Using aggregometry as gold standard for analyzing ex vivo thrombus formation, we observed a dose-dependent effect of C3a on pre-stimulated platelets on aggregation in vitro. In turn, C3aR−/− mice, but not C5aR−/− mice had a prolonged time to vessel occlusion after vascular injury. This was attributable to the presence of C3aR on platelets, as shown here by experiments with depletion of platelets in C3aR−/− mice and an additional approach with intravenous reconstitution of C3aR−/− mice with WT or C3aR−/− platelets as also carried out by others.45 Looking at tail bleeding time, C3aR−/− mice had a prolonged bleeding time, which could be reproduced with a pharmacological approach by the injection of a C3aR antagonist peptide. To identify potential mechanisms mediating the observed effects of the C3aR on platelet function, we analyzed proteins associated with the C3aR on platelets. Recently, generation of Rap1b knockout mice revealed a severely prolonged bleeding time phenotype.35 Rap proteins are small GTPases which belong to the Ras family 46 with Rap1b being the most abundant Ras protein in platelets.47 Very interestingly, we observed that Rap1b was associated with the C3aR on platelets and C3a treatment resulted in increased Rap1 activity in platelets. To follow up on these observations, we generated C3aR−/− x Spa.1−/− double knockout mice to verify these observations in vivo. Activated Rap1b (Rap1b-GTP) is inactivated (Rap1b-GDP) by Spa.1,48 and Spa.1 knockout mice have constitutively active Rap1.36 In line with our previous findings, C3aR−/− x Spa.1−/− mice showed a time to thrombosis significantly reduced compared to C3aR−/− x Spa.1+/+ littermate controls and similar to WT mice, indicating a role of this small GTPase for C3aR function in platelets. C3aR may activate Rap1b via PI3K signaling, as shown by our experiments detecting significantly increased amounts of phosphorylated PI3K in platelets costimulated with ADP and C3a.
The relevance of platelets in diseases with a prothrombotic state involving complement activation has been known for a long time, although the direct connection between platelets and the complement system remains largely uncharacterized.49 In experimental stroke, inhibition of complement activation and endothelial-platelet-leukocyte interactions using a sialyl Lewis x (sLex) -glycosylated complement inhibitory protein results in neuronal protection.26 A potential role for C3aR in stroke has also been proposed before, although the role of C3aR in this disease was so far mainly attributed to inflammation.50 C3−/− mice are protected from transient focal cerebral ischemia,28 but the relevance of platelet-derived complement has not been addressed, so far.
In cases in which clear complement targets can be identified (as in HAE or PNH), complement therapeutics have already been successfully introduced into the clinic and are applied in patients.51, 52 There are several human trials 53–56 using complement inhibitors in patients undergoing coronary artery interventions. A benefit, however, could only be shown in specific patient settings. As no drug targeting the C3a-C3aR axis has been applied in clinical studies with atherothrombosis patients so far, the available studies do not necessarily contradict our data. Many of the compounds tested for trials in coronary artery disease that have been discontinued, were designed to target underlying multifaceted and complex pathophysiologies such as vascular inflammation or ischemia/reperfusion injury.51 In these disorders, many parallel mechanisms are involved, and the influence of complement is still a matter of in depth investigation. Suddenly, the unsatisfactory results obtained for soluble complement receptor 1 (TP10)57 and pexelizumab56, which targets C5 downstream of C3a analyzed in our project, have left a bitter taste and may have had a negative impact on the field. For such complex indications, the key to success may be to use complement drugs not as single therapies but (in consonance with their name) as complementary therapies. Clearly, more basic research and additional tests are required in this context. Here, we provide an additional piece of knowledge to the puzzle, which may help to approach (specific) complement therapeutics in patients with cardiovascular diseases.
Here, we show that C3aR−/− mice have reduced stroke and myocardial infarction after ischemia-reperfusion injury, whereas there was no difference any more between WT and knockout mice in the absence of platelets indicating a functional role of the C3aR on platelets in this setting of thrombotic disease. Neutrophils and a crosstalk of platelets with neutrophils are known to contribute to I/R injury.58 Although the difference between WT and C3aR−/− mice persisted after neutrophil depletion in our experiments, at this point, we cannot entirely rule out any functional relevance of the C3aR on neutrophils for myocardial infarction, which will have to be addressed in future studies. Our findings support the hypothesis that the complement cascade, especially the C3-C3a-C3aR axis, is a “sweetspot” for therapeutic intervention and confirm the potential relevance of drugs modulating the complement cascade on the level of C3.59
In conclusion, our observations demonstrate - to the best of our knowledge for the first time - a functional role of the anaphylatoxin receptor C3aR expressed on platelets for thrombosis and hemostasis in vivo. Our findings suggest that targeting C3aR on platelets could be a potential point for therapeutic interventions and for the development of further complement inhibitors. This report, thus, identified a novel intersection point between two central systems of host defense and tissue repair - the complement cascade and platelet activation.
Supplementary Material
Clinical Perspective
What Is New?
Murine and human platelets express the anaphylatoxin receptor C3aR. It can be detected in aspirates of patients with myocardial infarction and its expression in patients with coronary artery disease correlates with the activated platelet fibrinogen receptor GPIIb/IIIa.
Using several in vitro, ex vivo and in vivo approaches and different genetic mouse models, we identified the anaphylatoxin receptor C3aR and its corresponding ligand C3a as platelet activators via intraplatelet signaling resulting in activated GPIIbIIIa, which mediates intravascular thrombosis, stroke and myocardial infarction.
We have identified a novel point of intersection between innate immunity and thrombosis with relevance for the thrombotic diseases stroke and myocardial infarction.
What Are the Clinical Implications?
Complement components involved in the development of atherothrombosis need to be further studied in translational and clinical settings of stroke and coronary heart disease.
The identified C3a-C3aR axis on platelets may be a promising diagnostic and therapeutic target to approach underlying thromboinflammatory mechanisms of stroke and myocardial infarction.
Acknowledgments
RJS and MS performed experiments, analyzed and compiled data and wrote parts of the manuscript. FNE, HN, SE, PK, PM, MM, JR and FE performed and analyzed experiments. TES, SGM, MG, TG, OB, CK and DD interpreted data and wrote parts of the manuscript. ESR and JDL provided reagents and scientific input and wrote parts of the manuscript. HFL conceived the project, analyzed data and wrote the manuscript.
We thank S. Gekeler for excellent technical support.
Sources of Funding
This work was supported by grants from the German Research Council (KFO 274 and CRC/TR 240) and the Volkswagen foundation (Lichtenberg program) and grants from the U.S. National Institutes of Health (AI068730) and the European Community’s Seventh Framework Programme under grant agreement number 602699 (DIREKT) to JDL. Furthermore, this work was supported by the Wilhelm Sander-Stiftung (2011.005.1).
Footnotes
Disclosures
J.D.L. is the inventor of patents and/or patent applications that describe complement inhibitors and/or their use for therapeutic purposes. J.D.L. is the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors for clinical applications. The remaining authors declare no competing financial interests.
References
- 1.Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, Lambris JD and Huber-Lang M. Interaction between the coagulation and complement system. Adv Exp Med Biol 2008;632:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol 2004;5:981–986. [DOI] [PubMed] [Google Scholar]
- 3.Ricklin D, Hajishengallis G, Yang K and Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol 2010;11:785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walport MJ. Complement. First of two parts. N Engl J Med 2001;344:1058–1066. [DOI] [PubMed] [Google Scholar]
- 5.Guo RF and Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol 2005;23:821–852. [DOI] [PubMed] [Google Scholar]
- 6.Kwan WH, van der Touw W, Paz-Artal E, Li MO and Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med 2013;210:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES and Kohl J. The role of the anaphylatoxins in health and disease. Mol Immunol 2009;46:2753–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langer HF, Chung KJ, Orlova VV, Choi EY, Kaul S, Kruhlak MJ, Alatsatianos M, DeAngelis RA, Roche PA, Magotti P, Li X, Economopoulou M, Rafail S, Lambris JD and Chavakis T. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood 2010;116:4395–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sjoberg AP, Trouw LA and Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol 2009;30:83–90. [DOI] [PubMed] [Google Scholar]
- 10.Ekdahl KN, Teramura Y, Hamad OA, Asif S, Duehrkop C, Fromell K, Gustafson E, Hong J, Kozarcanin H, Magnusson PU, Huber-Lang M, Garred P and Nilsson B. Dangerous liaisons: complement, coagulation, and kallikrein/kinin cross-talk act as a linchpin in the events leading to thromboinflammation. Immunol Rev 2016;274:245–269. [DOI] [PubMed] [Google Scholar]
- 11.Norgaard I, Nielsen SF and Nordestgaard BG. Complement C3 and High Risk of Venous Thromboembolism: 80517 Individuals from the Copenhagen General Population Study. Clin Chem 2016;62:525–534. [DOI] [PubMed] [Google Scholar]
- 12.Subramaniam S, Jurk K, Hobohm L, Jackel S, Saffarzadeh M, Schwierczek K, Wenzel P, Langer F, Reinhardt C and Ruf W. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 2017;129:2291–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, Brückner UB, Nilsson B, Gebhard F, Lambris JD and Huber-Lang M. Molecular Intercommunication between the Complement and Coagulation Systems. J Immunol 2010;185:5628–5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghebrehiwet B, Silverberg M and Kaplan AP. Activation of the classical pathway of complement by Hageman factor fragment. J Exp Med 1981;153:665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA and Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med 2006;12:682–687. [DOI] [PubMed] [Google Scholar]
- 16.Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, Bazolli B, Lu B, Penna G and Rescigno M. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun 2016;7:11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furie B and Furie BC. Mechanisms of thrombus formation. N Engl J Med 2008;359:938–949. [DOI] [PubMed] [Google Scholar]
- 18.Del Conde I, Cruz MA, Zhang H, Lopez JA and Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med 2005;201:871–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peerschke EI, Yin W, Grigg SE and Ghebrehiwet B. Blood platelets activate the classical pathway of human complement. J Thromb Haemost 2006;4:2035–2042. [DOI] [PubMed] [Google Scholar]
- 20.Hamad OA, Nilsson PH, Wouters D, Lambris JD, Ekdahl KN and Nilsson B. Complement component C3 binds to activated normal platelets without preceding proteolytic activation and promotes binding to complement receptor 1. J Immunol 2010;184:2686–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leinoe E, Nielsen OJ, Jonson L and Rossing M. Whole-exome sequencing of a patient with severe and complex hemostatic abnormalities reveals a possible contributing frameshift mutation in C3AR1. Cold Spring Harb Mol Case Stud 2016;2:a000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verschoor A and Langer HF. Crosstalk between platelets and the complement system in immune protection and disease. Thromb Haemost 2013;110:910–919. [DOI] [PubMed] [Google Scholar]
- 23.De Meyer SF, Denorme F, Langhauser F, Geuss E, Fluri F and Kleinschnitz C. Thromboinflammation in Stroke Brain Damage. Stroke 2016;47:1165–1172. [DOI] [PubMed] [Google Scholar]
- 24.Mathey D, Schofer J, Schafer HJ, Hamdoch T, Joachim HC, Ritgen A, Hugo F and Bhakdi S. Early accumulation of the terminal complement-complex in the ischaemic myocardium after reperfusion. Eur Heart J Suppl 1994;15:418–423. [DOI] [PubMed] [Google Scholar]
- 25.Vakeva A, Morgan BP, Tikkanen I, Helin K, Laurila P and Meri S. Time course of complement activation and inhibitor expression after ischemic injury of rat myocardium. Am J Pathol 1994;144:1357–1368. [PMC free article] [PubMed] [Google Scholar]
- 26.Huang J Neuronal Protection in Stroke by an sLex-Glycosylated Complement Inhibitory Protein. Science 1999;285:595–599. [DOI] [PubMed] [Google Scholar]
- 27.Stokowska A, Atkins AL, Moran J, Pekny T, Bulmer L, Pascoe MC, Barnum SR, Wetsel RA, Nilsson JA, Dragunow M and Pekna M. Complement peptide C3a stimulates neural plasticity after experimental brain ischaemia. Brain 2017;140:353–369. [DOI] [PubMed] [Google Scholar]
- 28.Mocco J, Mack WJ, Ducruet AF, Sosunov SA, Sughrue ME, Hassid BG, Nair MN, Laufer I, Komotar RJ, Claire M, Holland H, Pinsky DJ and Connolly ES Jr. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circ Res 2006;99:209–217. [DOI] [PubMed] [Google Scholar]
- 29.Lonsdorf AS, Kramer BF, Fahrleitner M, Schonberger T, Gnerlich S, Ring S, Gehring S, Schneider SW, Kruhlak MJ, Meuth SG, Nieswandt B, Gawaz M, Enk AH and Langer HF. Engagement of alphaIIbbeta3 (GPIIb/IIIa) with alphanubeta3 integrin mediates interaction of melanoma cells with platelets: a connection to hematogenous metastasis. J Biol Chem 2012;287:2168–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schleicher RI, Reichenbach F, Kraft P, Kumar A, Lescan M, Todt F, Gobel K, Hilgendorf I, Geisler T, Bauer A, Olbrich M, Schaller M, Wesselborg S, O’Reilly L, Meuth SG, Schulze-Osthoff K, Gawaz M, Li X, Kleinschnitz C, Edlich F and Langer HF. Platelets induce apoptosis via membrane-bound FasL. Blood 2015;126:1483–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von Elverfeldt D, Maier A, Duerschmied D, Braig M, Witsch T, Wang X, Mauler M, Neudorfer I, Menza M, Idzko M, Zirlik A, Heidt T, Bronsert P, Bode C, Peter K and von Zur Muhlen C. Dual-contrast molecular imaging allows noninvasive characterization of myocardial ischemia/reperfusion injury after coronary vessel occlusion in mice by magnetic resonance imaging. Circulation 2014;130:676–687. [DOI] [PubMed] [Google Scholar]
- 32.Gushiken FC, Han H, Li J, Rumbaut RE and Afshar-Kharghan V. Abnormal platelet function in C3-deficient mice. J Thromb Haemost 2009;7:865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kato K, Martinez C, Russell S, Nurden P, Nurden A, Fiering S and Ware J. Genetic deletion of mouse platelet glycoprotein Ibbeta produces a Bernard-Soulier phenotype with increased alpha-granule size. Blood 2004;104:2339–2344. [DOI] [PubMed] [Google Scholar]
- 34.Klos A, Wende E, Wareham KJ and Monk PN. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacol Rev 2013;65:500–543. [DOI] [PubMed] [Google Scholar]
- 35.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH and White GC 2nd. Rap1b is required for normal platelet function and hemostasis in mice. J Clin Invest 2005;115:680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishida D, Kometani K, Yang H, Kakugawa K, Masuda K, Iwai K, Suzuki M, Itohara S, Nakahata T, Hiai H, Kawamoto H, Hattori M and Minato N. Myeloproliferative stem cell disorders by deregulated Rap1 activation in SPA-1-deficient mice. Cancer cell 2003;4:55–65. [DOI] [PubMed] [Google Scholar]
- 37.Larson MK, Chen H, Kahn ML, Taylor AM, Fabre JE, Mortensen RM, Conley PB and Parise LV. Identification of P2Y12-dependent and -independent mechanisms of glycoprotein VI-mediated Rap1 activation in platelets. Blood 2003;101:1409–1415. [DOI] [PubMed] [Google Scholar]
- 38.Kohl J, Casaretto M, Gier M, Karwath G, Gietz C, Bautsch W, Saunders D and Bitter-Suermann D. Reevaluation of the C3a active site using short synthetic C3a analogues. Eur J Immunol 1990;20:1463–1468. [DOI] [PubMed] [Google Scholar]
- 39.Howes JM, Richardson VR, Smith KA, Schroeder V, Somani R, Shore A, Hess K, Ajjan R, Pease RJ, Keen JN, Standeven KF and Carter AM. Complement C3 is a novel plasma clot component with anti-fibrinolytic properties. Diab Vasc Dis Res 2012;9:216–225. [DOI] [PubMed] [Google Scholar]
- 40.Cosgrove LJ, d’Apice AJ, Haddad A, Pedersen J and McKenzie IF. CR3 receptor on platelets and its role in the prostaglandin metabolic pathway. Immunol Cell Biol 1987;65 ( Pt 6):453–460. [DOI] [PubMed] [Google Scholar]
- 41.Polley MJ and Nachman RL. Human platelet activation by C3a and C3a des-arg. The Journal of experimental medicine 1983;158:603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furie B and Furie BC. Thrombus formation in vivo. J Clin Invest 2005;115:3355–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saggu G, Cortes C, Emch HN, Ramirez G, Worth RG and Ferreira VP. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and C3(H2O). J Immunol 2013;190:6457–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stoll G, Kleinschnitz C and Nieswandt B. Molecular mechanisms of thrombus formation in ischemic stroke: novel insights and targets for treatment. Blood 2008;112:3555–3562. [DOI] [PubMed] [Google Scholar]
- 45.Elzey BD, Grant JF, Sinn HW, Nieswandt B, Waldschmidt TJ and Ratliff TL. Cooperation between platelet-derived CD154 and CD4+ T cells for enhanced germinal center formation. J Leukoc Biol 2005;78:80–84. [DOI] [PubMed] [Google Scholar]
- 46.Robbins SM, Suttorp VV, Weeks G and Spiegelman GB. A ras-related gene from the lower eukaryote Dictyostelium that is highly conserved relative to the human rap genes. Nucleic Acids Res 1990;18:5265–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klinz FJ, Seifert R, Schwaner I, Gausepohl H, Frank R and Schultz G. Generation of specific antibodies against the rap1A, rap1B and rap2 small GTP-binding proteins. Analysis of rap and ras proteins in membranes from mammalian cells. Eur J Biochem 1992;207:207–213. [DOI] [PubMed] [Google Scholar]
- 48.Kurachi H, Wada Y, Tsukamoto N, Maeda M, Kubota H, Hattori M, Iwai K and Minato N. Human SPA-1 gene product selectively expressed in lymphoid tissues is a specific GTPase-activating protein for Rap1 and Rap2. Segregate expression profiles from a rap1GAP gene product. J Biol Chem 1997;272:28081–28088. [DOI] [PubMed] [Google Scholar]
- 49.Licht C, Pluthero FG, Li L, Christensen H, Habbig S, Hoppe B, Geary DF, Zipfel PF and Kahr WH. Platelet-associated complement factor H in healthy persons and patients with atypical HUS. Blood 2009;114:4538–4545. [DOI] [PubMed] [Google Scholar]
- 50.Van Beek J, Bernaudin M, Petit E, Gasque P, Nouvelot A, MacKenzie ET and Fontaine M. Expression of receptors for complement anaphylatoxins C3a and C5a following permanent focal cerebral ischemia in the mouse. Exp Neurol 2000;161:373–382. [DOI] [PubMed] [Google Scholar]
- 51.Ricklin D and Lambris JD. Complement-targeted therapeutics. Nat Biotechnol 2007;25:1265–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ricklin D, Mastellos DC, Reis ES and Lambris JD. The renaissance of complement therapeutics. Nat Rev Nephrol 2018;14:26–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fitch JC, Rollins S, Matis L, Alford B, Aranki S, Collard CD, Dewar M, Elefteriades J, Hines R, Kopf G, Kraker P, Li L, O’Hara R, Rinder C, Rinder H, Shaw R, Smith B, Stahl G and Shernan SK. Pharmacology and biological efficacy of a recombinant, humanized, single-chain antibody C5 complement inhibitor in patients undergoing coronary artery bypass graft surgery with cardiopulmonary bypass. Circulation 1999;100:2499–2506. [DOI] [PubMed] [Google Scholar]
- 54.Granger CB, Mahaffey KW, Weaver WD, Theroux P, Hochman JS, Filloon TG, Rollins S, Todaro TG, Nicolau JC, Ruzyllo W, Armstrong PW and Investigators C. Pexelizumab, an anti-C5 complement antibody, as adjunctive therapy to primary percutaneous coronary intervention in acute myocardial infarction: the COMplement inhibition in Myocardial infarction treated with Angioplasty (COMMA) trial. Circulation 2003;108:1184–1190. [DOI] [PubMed] [Google Scholar]
- 55.Smith PK, Shernan SK, Chen JC, Carrier M, Verrier ED, Adams PX, Todaro TG, Muhlbaier LH, Levy JH and Investigators P- CI. Effects of C5 complement inhibitor pexelizumab on outcome in high-risk coronary artery bypass grafting: combined results from the PRIMO-CABG I and II trials. J Thorac Cardiovasc Surg 2011;142:89–98. [DOI] [PubMed] [Google Scholar]
- 56.Armstrong PW, Granger CB, Adams PX, Hamm C, Holmes D Jr., O’Neill WW, Todaro TG, Vahanian A and Van de Werf F. Pexelizumab for acute ST-elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. JAMA 2007;297:43–51. [DOI] [PubMed] [Google Scholar]
- 57.Li JS, Jaggers J and Anderson PA. The use of TP10, soluble complement receptor 1, in cardiopulmonary bypass. Expert Rev Cardiovasc Ther 2006;4:649–654. [DOI] [PubMed] [Google Scholar]
- 58.May AE, Langer H, Seizer P, Bigalke B, Lindemann S and Gawaz M. Platelet-leukocyte interactions in inflammation and atherothrombosis. Semin Thromb Hemost 2007;33:123–127. [DOI] [PubMed] [Google Scholar]
- 59.Mastellos DC, Yancopoulou D, Kokkinos P, Huber-Lang M, Hajishengallis G, Biglarnia AR, Lupu F, Nilsson B, Risitano AM, Ricklin D and Lambris JD. Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest 2015;45:423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.