

SUMMARY
Heterobifunctional small molecule degraders that induce protein degradation through ligase-mediated ubiquitination have shown considerable promise as a new pharmacological modality. However, we currently lack a detailed understanding of the molecular basis for target recruitment and selectivity, which is critically required to enable rational design of degraders. Here we utilize comprehensive characterization of the ligand dependent CRBN/BRD4 interaction to demonstrate that binding between proteins that have not evolved to interact is plastic. Multiple X-ray crystal structures show that plasticity results in several distinct low energy binding conformations, which are selectively bound by ligands. We demonstrate that computational protein-protein docking can reveal the underlying inter-protein contacts and inform the design of a BRD4 selective degrader that can discriminate between highly homologous BET bromodomains. Our findings that plastic inter-protein contacts confer selectivity for ligand-induced protein dimerization provide a conceptual framework for the development of heterobifunctional ligands.
INTRODUCTION
Rational design of synthetic chemical matter capable to induce selective protein dimerization is challenging. Significant progress has recently been made towards chemically induced targeted protein degradation using heterobifunctional small molecule ligands (often referred to as degraders or PROTACs for PROteolysis-TArgeting Chimeras)1–10. Targeted protein degradation refers to small molecule induced ubiquitination and degradation of disease targets, in which a small molecule simultaneously recruits both a ubiquitin E3 ligase and the target protein to be ubiquitinated (Supplementry Fig. 1a); therefore, representing a functional application of chemically induced protein dimerization8,11. Clinical proof of concept for targeted protein degradation is provided by the recent discovery that the potent anti-cancer drugs thalidomide, lenalidomide and pomalidomide (collectively known as IMiDs for immuno-modulatory drugs) exert their therapeutic effects through induced degradation of key efficacy targets, such as IKZF1, IKZF312–14, ZFP9115, or casein kinase 1 alpha (Ck1α)16,17. IMiDs bind CRBN, the substrate receptor of the CUL4-RBX1-DDB1-CRBN (CRL4CRBN) E3 ubiquitin ligase18–20, and act by redirecting the activity of the CRL4CRBN ligase to ubiquitinate these neo-substrates17.
PROTACs or heterobifunctional degrader molecules (hereafter referred to as degraders) typically comprise an E3 ligase binding scaffold (hereafter E3-moiety), often an analogue of thalidomide, or a ligand to the von Hippel-Lindau tumour suppressor (VHL) protein21, attached through a linker to another small molecule (hereafter target-moiety) that binds a target protein of interest (Fig. 1a and Supplementary Fig. 1b). Recruitment of this target protein to the E3 ubiquitin ligase facilitates ubiquitination and subsequent degradation of the target protein22. This principle has been successfully applied to several targets including the Bromodomain and Extra Terminal (BET) family (BRD2, BRD3, BRD4), RIPK2, BCR-ABL, FKBP12, BRD9, and ERRα2,4,6,23–26 and represents a promising new pharmacologic modality now widely explored in chemical biology and drug discovery.
Figure 1. Structure of the DDB1ΔB-CRBN-dBET23-BRD4BD1 complex.
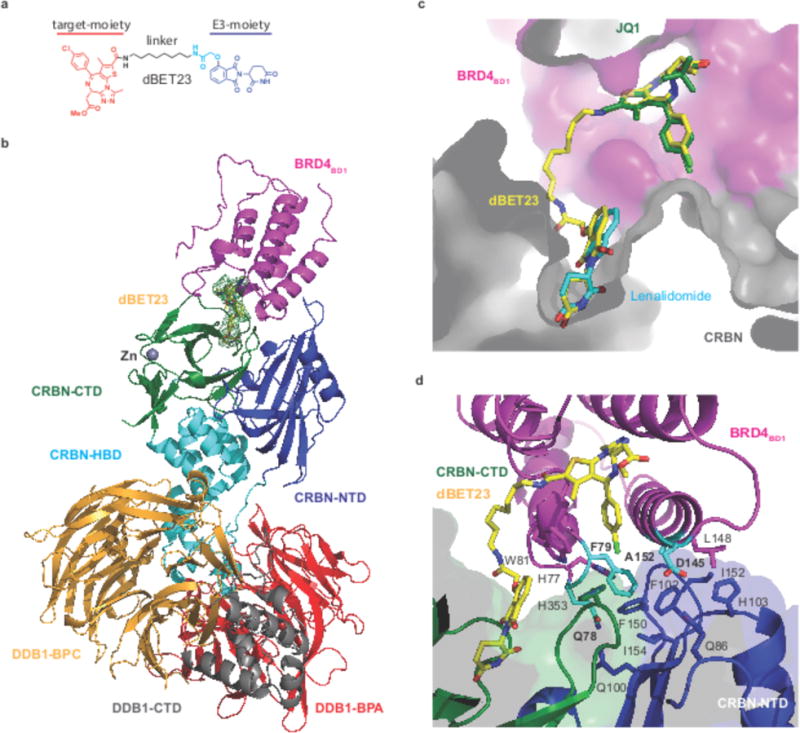
(a) The chemical structure of dBET23 is depicted with the target-moiety in red, the linker in black and cyan, and the E3-moiety in blue. (b) Cartoon representation of DDB1ΔB-CRBN-dBET23-BRD4BD1: DDB1 highlighting domains BPA (red), BPC (orange) and DDB1-CTD (grey); CRBN with domains NTD (blue), HBD (cyan) and CTD (green); BRD4BD1 (magenta). The Zn2+-ion is drawn as a grey sphere and dBET23 as sticks representation in yellow. The FO-FC map is shown as green mesh for dBET23 contoured at 3.0σ. (c) Superposition of DDB1ΔB-CRBN-dBET23-BRD4BD1 with CRBN bound to lenalidomide (pdb: 4tz4) and BRD4BD1 bound to JQ1-(S) (pdb: 3mxf). Surface representation for CRBN and BRD4BD1 are shown in gray and magenta, respectively. dBET23 is shown in yellow, JQ1 in green, and thalidomide in cyan. (d) Side-chain interactions between BRD4BD1, CRBN, and dBET23. Residues of BRD4BD1 mutated in this study are highlighted in cyan.
Small molecule induced protein degradation by degraders, requires ligand mediated binding of two proteins that have not evolved to interact. While this is evidently possible, the design of such molecules remains an empirical process in which molecules for new targets frequently fail27,28, likely due to insufficient understanding of the fundamental principles that govern these neo-interactions. Our structural understanding is limited to the recruitment of the second bromodomain of BRD4 (BRD4BD2) to the CUL2-RBX1-ElongB/C-VHL (CRL2VHL) ubiquitin ligase by the small molecule MZ1 (1)5, a degrader based on a VHL-ligand21 conjugated to the BRD4 ligand JQ1 (2)29. Positive cooperativity was observed for VHL-MZ1-BRD2/3/4BD2 complex formation, where additional contacts between VHL and BRD4BD2 as well as back-folding of the linker with additional linker-ligase/substrate contacts result in superior affinity (linkage cooperativity) over the individual affinities for the VHL and BRD4 binding moieties5. Whether such tight degrader mediated complexes are common and whether attractive inter-protein forces are required for effective degradation of target proteins that have not evolved to bind to the ligase remains an open question.
Other degraders targeting BRD4 utilize the CRL4CRBN targeting thalidomide moiety and it remains to be shown if these exhibit a similar ligase-substrate interface. In general, degraders have been found to exhibit different efficacy and selectivity profiles depending on the nature of the E3-moiety used, sometimes exhibiting improved selectivity over the parental target-moiety27,28,30. While positive cooperativity can explain certain cases – such as MZ1 –, it is unlikely to exist for a broad number of ligase-substrate pairs. We therefore set out to explore general principles that govern these interactions on a molecular level.
Through multiple X-ray crystal structures of degrader bound CRL4CRBN- BRD4 complexes, we demonstrate that plastic inter-protein contacts result in multiple distinct binding conformations depending on the bound degrader. We establish a model in which effective degradation does not require tight cooperative binding; however, distinct binding conformations are unique to ligase-substrate pairs and define selectivity. We further pioneer a computational approach to protein-protein docking using Rosetta and demonstrate the versatility of this approach through rational design of a degrader that can discriminate between the highly homologous BET bromodomains of BRD2/3/4, leading to synthesis of a highly effective and selective BRD4 degrader.
RESULTS
To study the molecular mechanism of substrate recruitment and since small changes to the degrader can result in dramatically altered cell permeability or solubility, we devised a synthetic system based on the recruitment of isolated BRD4 bromodomains to CRL4CRBN. Like other members of the BET family, BRD4 contains two bromodomains: bromodomain 1 (aa 75 –147 and referred to as BRD4BD1) and BRD4BD2 (aa 368 – 440), and sequence conservation between the two is limited (Supplementary Fig. 1c-e). These distinct domains bind the JQ1 based target-moiety with equal affinities29, hence establish a model system to understand how amino acid sequence and thereby protein surface properties influence protein dimerization. We further utilize a series of compounds, which were synthesized to bind CRBN and the bromodomains of BRD4 (see Supplementary Fig. 1b)6,10. These molecules comprise the E3-moiety thalidomide to bind to CRL4CRBN, a flexible linker of variable length and composition, and a target-moiety, JQ1, that binds to BRD4BD1 and BRD4BD2 with equal affinities29.
Crystal structure of a DDB1ΔB-CRBN-dBET23-BRD4BD1 complex
To determine the structural basis of BRD4 recruitment to CRBN, we reconstituted complexes comprising hsDDB1ΔB (harboring an internal deletion of the flexible BPB propeller17), hsCRBN, and hsBRD4BD1 bound to different degrader molecules. Initial crystals were obtained for the ~ 165 kDa DDB1ΔB-CRBN-dBET23-BRD4BD1 (dBET23 (3) comprises an 8-carbon linker attached to thalidomide via an oxy-acetamide linkage, and to the thiophene group of JQ1) complex and its structure was determined to 3.5 Å resolution by molecular replacement using a DDB1ΔB-CRBN search model (Fig. 1b and Supplementary Fig. 1f, pdb: 5fqd, see methods and Supplementary Table 1). The DDB1 β-propeller domains A and C (BPA and BPC) bind CRBN but do not contribute contacts to BRD4BD1. CRBN consists of three domains, the N-terminal domain (NTD), the helical-bundle domain (HBD) and the C-terminal domain (CTD), which harbors the thalidomide binding pocket20. The small molecule degrader dBET23 occupies the canonical binding sites on CRBN and BRD4BD1 for thalidomide and JQ1, respectively (Fig. 1c).
BRD4BD1 interacts with CRBN through contacts with the NTD domain of CRBN and with CRBN residues in direct proximity to the thalidomide-binding pocket (Fig. 1d). CRBN binds the BRD4BD1 αC helix (aa 145 – 161) and residues in the BRD4BD1 ZA loop (aa 76 – 104)31. The αC helix forms hydrophobic interactions with two loops in the CRBN-NTD (aa 101 – 104 and aa 147 – 154). Residues Leu148, Met149, Ala152, and Leu156 in the αC helix together with His77 and Phe79 in the ZA loop, form a hydrophobic patch that interacts with Phe102, His103, Phe150, Gly151, Ile152, and Ile154 in the CRBN-NTD. BRD4BD1 Gln78 forms an electrostatic interaction with Gln100 in the CRBN-NTD (Fig. 1d). Consequently, mutations of the BRD4BD1 residues Phe79Asp, Ala152Asp, and Gln78Ala all reduce tertiary complex formation as monitored by measuring the peak-height in a TR-FRET dimerization assay (Fig. 2a and Supplementary Fig. 2, see methods for details on quantification). We further noticed that Asp145 is buried in a hydrophobic environment, and accordingly, introducing an Asp145Ala mutation strengthens the binding of BRD4BD1 to CRBN (Fig. 2a). The interaction between CRBN and BRD4BD1 consists of a total buried surface area of ~ 550 Å2 32, comparable to that observed for CRBN-Ck1α (~ 600 Å2) and GSPT1 (~ 600 Å2)17,33.
Figure 2. Degrader mediated BRD4 recruitment is governed by negative cooperativity.
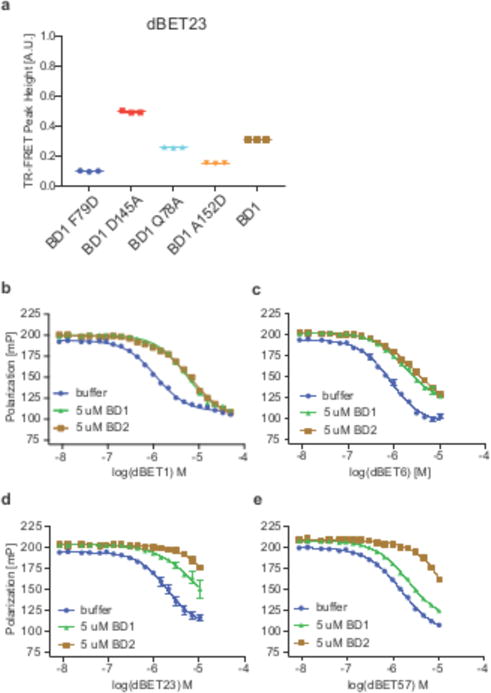
(a) TR-FRET. dBET23 titrated to DDB1ΔB-CRBNSPY-BODIPY, Terbium-Streptavidin and various BRD4BD1-biotin wild type and mutant proteins. The mean peak heights for dose response curves of three independent replicates are shown as dot-plot. Data in this figure is presented as means ± s.d. (n=3). (b) Competitive binding assay for dBET1 binding to DDB1ΔB-CRBN. Increasing concentrations of dBET1 titrated to preformed DDB1ΔB-CRBN-lenalidomideAtto565 complex in presence or absence of BRD4BD1 or BRD4BD2. (c) As in b but using dBET6, (d) dBET23, or (e) dBET57. All data in this figure are independent replicates presented as means ± s.d. (n=3).
In addition to dBET23, we also determined crystal structures with the related molecules dBET6 (4) (3.3 Å resolution), dBET70 (5) (4.4 Å resolution) – both have linkers of similar length – and significantly longer dBET55 (6) (4.0 Å resolution and crystallized with BRD4BD1(D145A)). The overall structures observed for these complexes are comparable to the structure obtained with dBET23 (Supplementary Fig. 2a). Since these structures were obtained from crystals with similar space group and unit cell dimensions, we cannot exclude that crystal packing may bias towards the observed binding conformation (see Supplementary Fig. 2 for discussion).
Inter-protein contacts are unique to BRD4BD1
The amino acid sequences of BRD4BD1 to BRD4BD2 are 49% similar (Supplementary Fig. 1d), yet none of the key residues in the αC helix or the ZA loop involved in contacts with CRBN are identical. We therefore asked whether this would manifest in reduced affinity of BRD4BD2 for CRBN in the presence of dBET6 or dBET23. While the determination of absolute binding affinities is difficult for a three body binding problem34, a qualitative measure of the relative affinities (or cooperativity of binding) can be indirectly obtained through CRBN-degrader binding assays in presence or absence of purified BRD4BD1 or BRD4BD2 protein. Using a lenalidomide-Atto565 fluorescent probe we measured binding of degraders to CRBN by competitive titration (Fig. 2b – e and Supplementary Fig. 3). Next, we carried out similar binding experiments in presence of increasing concentrations of either BRD4BD1 or BRD4BD2 to assess the cooperativity of ternary complex formation. We defined an apparent cooperativity factor alpha (αapp = IC50[binary]/IC50[ternary]), with positive cooperativity resulting in αapp > 1, and negative cooperativity in αapp < 1 (see methods and Fig. 2b – e and Supplementary Fig. 3). dBET6, exhibits an IC50 of ~ 0.8 μM in the absence of BRD4, which increases to an IC50 of ~ 1.8 μM (αapp = 0.6) in the presence of BRD4BD1, and an IC50 of ~ 4.1 μM (αapp = 0.2) in the presence of BRD4BD2 (Fig. 2c and Supplementary Fig. 3), indicative of negative cooperativity for both BRD4BD1 and BRD4BD2. For dBET23 and dBET57 (7) the difference between BRD4BD1 and BRD4BD2 is more pronounced, with αapp = 0.4 (dBET23) and αapp = 0.8 (dBET57) for BRD4BD1 and αapp < 0.1 for BRD4BD2 (the binding in presence of BRD4BD2 is too weak to quantify), indicating negative cooperativity and a preference for binding to BRD4BD1 (Fig. 2d, e and Supplementary Fig. 3).
To better understand the drivers of selectivity and to test whether the observed differences in cooperativity would result in differential degradation of isolated BRD4 bromodomains, we developed a system that allowed us to directly quantify cellular degradation of either BRD4BD1 or BRD4BD2. We treated reporter cells that stably express BRD4BD1-EGFP followed by a P2A splice site separated mCherry, with increasing concentrations of degrader molecules (Fig. 3a, b). This assay format enables quantitative readout of BRD4BD1 degradation with the GFP/mCherry ratio using flow cytometry (similar reporter cells were used for BRD4BD2, or a IKZF protein that has internal deletions Δ1-82, Δ197-239, and Δ256-519 hereafter referred to as IKZFΔ). We found that dBET6 (DC50/5h ~ 10 nM, with DC50/5h referring to half-maximal degradation after 5 hours of treatment), dBET23 (DC50/5h ~ 50 nM) and dBET70 (DC50/5h ~ 5 nM) exhibit the most potent effects on BRD4BD1 protein levels, followed by dBET1 (8) (DC50/5h ~ 500 nM) and dBET57 (DC50/5h ~ 500 nM) (Fig. 3a and Supplementary Fig. 4). For BRD4BD2, dBET70 (DC50/5h ~ 5 nM) has the most pronounced effects, followed by dBET6 (DC50/5h ~ 50 nM), dBET23 (DC50/5h > 1 μM) and dBET1 (DC50/5h ~ 1 μM). dBET57, which exhibits significant degradation of BRD4BD1, is inactive on BRD4BD2 (Fig. 3b and Supplementary Fig. 4). The cellular activity is thus largely proportional to the observed cooperativity factors (Supplementary Fig. 3), and dBET57 was found remarkably selective for BRD4BD1 in biochemical and cellular assays (Fig. 2e and Fig. 3a, b).
Figure 3. Quantitative assessment of cellular degradation for BRD4BD1 and BRD4BD2.
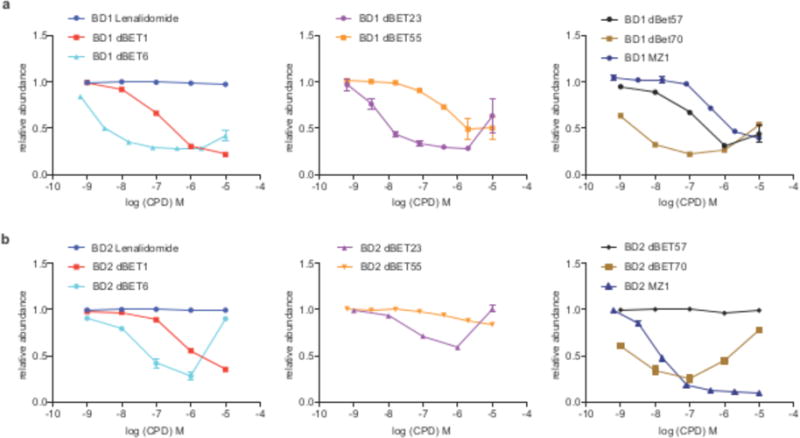
(a) Quantitative assessment of cellular degradation using a BRD4BD1-EGFP reporter assay. Cells stably expressing BRD4BD1-EGFP and mCherry were treated with increasing concentrations of dBET1, dBET6, dBET23, dBET55, dBET57, dBET70, MZ1 and lenalidomide and the EGFP and mCherry signals followed using flow cytometry analysis. (b) as in a but using cells that express a BRD4BD2-EGFP reporter along with mCherry. Data in a and b are EGFP and mCherry signals quantified using flow cytometry analysis. Data of independent cell culture replicates is presented as the means ± s.d. (n=3 for MZ1, dBET6, 23, 55; n=4 for dBET1, 57, 70; n=6 for lenalidomide).
Plastic binding confers selectivity to degraders
When comparing the CRBN-dBET23-BRD4BD1 structure to the previously determined structures of CRBN-Ck1a17, and CRBN-GSPT133, we find that these neo-substrates use different surfaces on CRBN to stabilize tertiary complex formation (Supplementary Fig. 5a). We also note that molecules with short linkers, such as dBET57, would not be able to dimerize CRBN and BRD4 in the conformation observed in the CRBN-dBET23-BRD4BD1 structure since a minimum of 8 carbons would be required to bridge the E3-moeity with the target-moiety and dBET57 comprises a 2-carbon linker (Supplementary Fig. 5c). We therefore asked whether degrader molecules incompatible with the observed binding mode, such as dBET57 or dBET1, would bind in a different overall conformation.
To explore potential differences in binding, we conducted mutational analysis. A set of single amino acid point mutations was introduced in CRBN and BRD4BD1 to obtain a mutational signature of binding. These CRBN mutations were previously shown to bind thalidomide with comparable affinities, except for the IMiD-binding deficient (IBD) control (CRBNP353G W386A)17. When comparing the mutational signatures of different degraders, we find that while dBET6 and 23 share similar profiles (Fig. 4a – c and Supplementary Fig. 2 and 5), the mutational signatures of dBET1 and dBET57 are distinct (Fig. 4d – f and Supplementary Fig. 5), consistent with distinct binding surfaces of dBET6/23 and dBET57 (Fig. 4b, e). This suggests that different degrader molecules – depending on linker length and linkage position – result in distinct binding conformations of CRBN-BRD4 complex formation.
Figure 4. Plasticity of CRBN-substrate interactions.
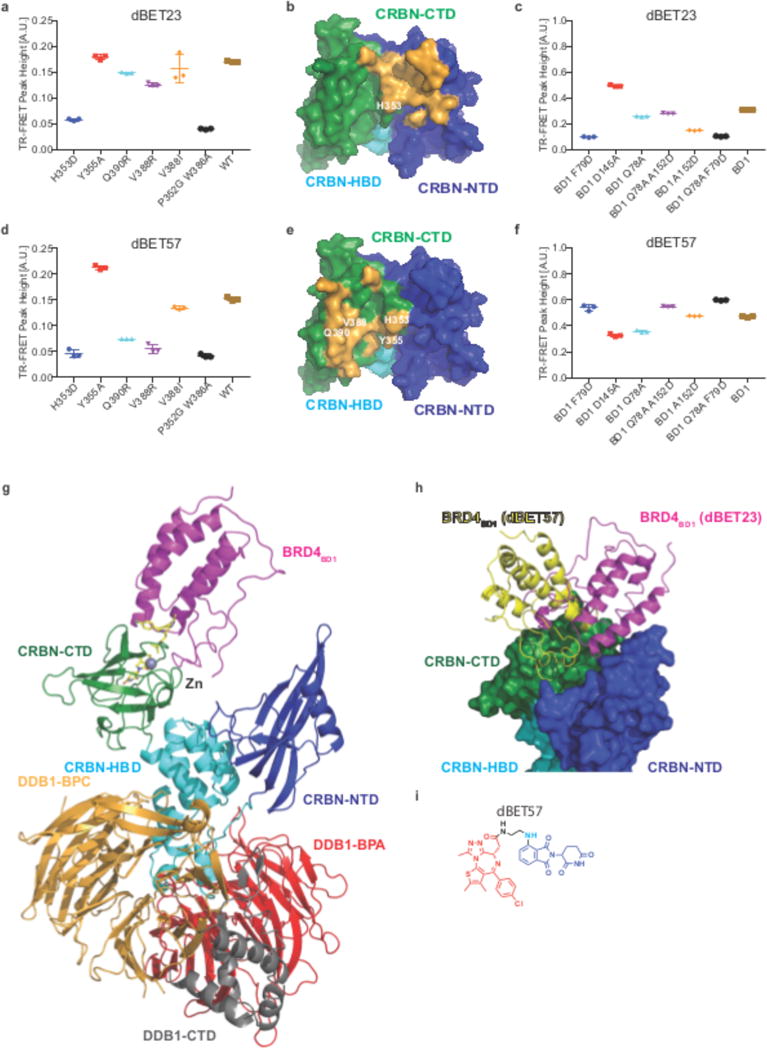
(a) TR-FRET. dBET23 titrated to BRD4BD1-SPYCATCHER-BODIPY, Terbium-antiHis antibody and various His6-DDB1ΔB-CRBN wild type and His6-DDB1-CRBN mutant proteins. The mean peak heights for dose response curves of three independent replicates are shown as dot-plot. TR-FRET data in this figure is presented as means ± s.d. (b) surface representation of CRBN highlighting the residues involved in dBET23 mediated BRD4BD1 binding in orange (residues Y59, L60, Q86, Q100, F102, H103, P104, D149, F150, G151, I152, I154, K156, P352, H353, E377, H378). CRBN interface residues mutated for biochemical assays are indicated. (c) TR-FRET. dBET23 titrated to DDB1ΔB-CRBNSPYCATCHER-BODIPY, Terbium-Streptavidin and various BRD4BD1-biotin wild type and mutant proteins. The mean peak heights for dose response curves of three independent replicates are shown as dot-plot. TR-FRET data in this figure is presented as means ± s.d. (d) as in a but titrating dBET57. (e) surface representation of CRBN highlighting the BRD4BD1 interacting residues for the dBET57 mediated recruitment in orange (residues: Q325, H353, Y355, H357, I371, G372, R373, E377, V388, Q390, C394, A395, S396, H397, T418, S420). CRBN interface residues mutated for biochemical assays are indicated. (f) as in b but titrating dBET57. (g) Cartoon representation of DDB1ΔB-CRBN-dBET57-BRD4BD1: DDB1 highlighting domains BPA (red), BPC (orange) and DDB1-CTD (grey); CRBN with domains NTD (blue), HBD (cyan) and CTD (green); BRD4BD1 (magenta). The Zn2+-ion is drawn as a grey sphere. dBET57 was not modelled in this structure but instead superpositions of lenalidomide (from pdb: 5fqd) and JQ1 (from pdb: 3mxf) are shown in yellow sticks. (h) Superposition of CRBN and BRD4BD1 for the dBET23 and dBET57 containing complexes. Superposition was carried out over the CRBN-CTD (residues 320 – 400). (i) The chemical structures of dBET57 is depicted with the target-moiety in red, the linker in black and cyan, and the E3-moiety in blue.
Figure 5. In silico docking to design degrader molecules.
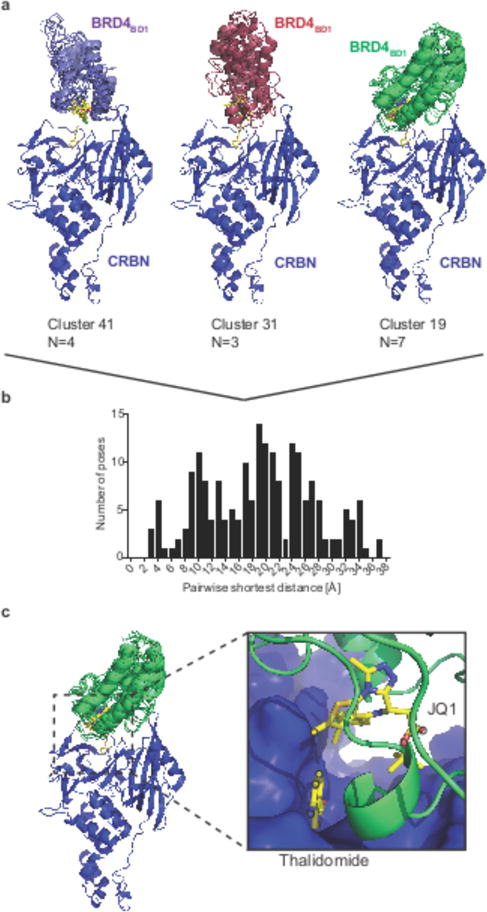
(a) Cartoon representations for representative clusters obtained by k-means clustering of the top 200 global docking poses between CRBN (pdb: 4tz4) and BRD4BD1 (pdb: 3mxf). (b) Histogram of the pairwise shortest distances for the top 200 docking poses. (c) Close-up view on the proximity of the JQ1 thiophene and lenalidomide that provided the rationale for synthesizing ZXH-2-147 and ZXH-3-26. Atoms used for calculation of the pairwise shortest distances between JQ1 and lenalidomide are highlighted in black circles.
To obtain insights into the molecular basis of this plastic CRBN/BRD4BD1 interactions, we set out to crystallize dBET57 (Fig. 4g – i), the molecule for which we observe the most pronounced selectivity for BRD4BD1 over BRD4BD2. We obtained crystals for a reconstituted DDB1ΔB-CRBN-dBET57-BRD4BD1 complex and determined the structure to 6.3 Å resolution (see Supplementary Fig. 6 for experimental validation of the structure). While the limited resolution prevents detailed interpretation of the molecular interactions that govern the CRBN-BRD4 interface, the overall binding mode is clearly resolved (Fig. 4g and Supplementary Fig. 6). In this complex, BRD4BD1 interacts with the CTD of CRBN, instead of the NTD as observed with dBET6/23 (Fig. 4g – h), which results in BRD4 now utilizing an entirely different set of residues for inter-protein contexts (compare Fig. 4b and Fig. 4e). Mutation of the BRD4 residues Lys91Asp, Lys91Trp, and Asp145Trp consequently reduce binding as measured by TR-FRET (Supplementary Fig. 6d). In the dBET57 bound structure, we observe that CRBN unfolds and the CRBN-NTD and CRBN-CTD domains no longer interact (Fig. 4g). This unexpected behavior could be due to the high salt crystallization condition (1.6 M Phosphate) or part of intrinsic CRBN plasticity. The binding mode observed with dBET57, however, is fully compatible with a closed CRBN conformation (Fig. 4h) and dBET57 mediated binding thus expected to occur with both CRBN conformations (see Supplementary Fig. 6 for additional discussion of CRBN conformation). The unexpected plasticity in degrader dependent binding of CRBN to the exact same protein, BRD4BD1, provides a rationale how degraders that share the same E3- and target-moieties can still exhibit different selectivity profiles. Depending on the linker chemistry and attachment location, different surface residues in the target protein may be involved in complex formation.
Protein docking reveals binding energy landscape
The mutational signatures obtained for different degrader molecules, the structural arrangements for dBET6/23/70 and dBET57 complexes, together with the absence of any co-evolution between CRBN and BRD4 let us hypothesize that BRD4 bromodomains can bind to CRBN in multiple different orientations depending on the ligand. Assessing such potential binding conformations to reduce chemical search space would be highly desirable. In silico protein-protein docking provides an attractive surrogate to in vitro experiments. We thus asked whether the Rosetta protein docking framework35 would allow modelling of such possible binding modes. One of the characteristics of Monte-Carlo docking algorithms is the stochastic sampling of low energy conformations, which frequently results in multiple solutions. While this often complicates the identification of evolved interactions between proteins, sampling of possible conformations provides an advantage in the study of degrader-induced binding modes since it enables exploration of the repertoire of low energy conformations.
We first confirmed that Rosetta can predict ligand mediated protein-protein interactions by docking Ck1α to the CRBN-lenalidomide complex (Supplementary Fig. 7). We next asked whether Rosetta docking would be able to provide models for possible degrader-induced binding modes by docking CRBN and the target BRD4BD1 in absence of a degrader molecule. Using the crystal structure of lenalidomide bound CRBN (pdb: 4tz4) and JQ1 bound BRD4BD1 (pdb: 3mxf), we performed a global docking experiment (20,000 models; see methods) using Rosetta docking (Supplementary Fig. 7). As predicted for a much weaker interaction between CRBN and BRD4BD1 in absence of a degrader, multiple low energy minima are found. Based on the hypothesis that the docking experiment will sample the repertoire of low energy binding conformations, clustering of the top 200 conformations provides a set of feasible binding modes (see Fig. 5 for three representative clusters). Within those top 200 lowest scoring conformations, we identified a conformation that closely resembles the conformation observed in the dBET23 crystal structure (Supplementary Fig. 7e, f), which confirms that some of the clusters represent conformations present in solution. While it remains to be shown whether docking can predict binding modes accurately, the overall conformational landscape provides a rationale for the design of required minimal linker lengths and suggest suitable linkage positions (Fig. 5). To test whether the docking information could be used to inform the design of degraders, we calculated the pairwise shortest distances between the selected solvent exposed atoms of JQ1 and lenalidomide for all top 200 poses and found a distance of 3-4Å, corresponding to linker of 2-3 atoms sufficient to bridge the two moieties (Fig. 5b, c). In theory, the shortest possible linker for a ligase-target pair should provide the most selective compound since it will restrict the degrees of freedom for BRD4BD1 binding to CRBN. This short linker length conformation was found to represent Cluster 19 (Fig. 5c). To test the prediction that such a short linker would result in a selective molecule, we next synthesized the according molecule (ZXH-2-147 (9)). Cellular degradation assays show that ZXH-2-147 is active on BRD4BD1, in accordance with the docking results (Supplementary Fig. 8a), however, with reduced activity compared to dBET6. To improve activity, we optimized the linker length by +2, +1, and -1 carbon and arrived at ZXH-3-26 (10) (Fig. 6a). Cellular degradation assays show that ZXH-3-26 exhibits a DC50/5h ~ 5 nM comparable to the best pan-BRD degrader dBET6 (Fig. 6b). To test whether these molecules exhibit isoform selectivity, we expanded our cellular reporter system to include the individual bromodomains of BRD2 and BRD3 and tested cellular degradation along with BRD4. We find that ZXH-3-26 shows activity exclusively on the first bromodomain of BRD4, and spares degradation of BRD2 or 3 at concentrations > 10μM (Fig. 6b), while dBET6 and MZ1 controls show activity on most bromodomains (Fig. 6c, d). Next, we assessed bromodomain degradation for dBET57 to test whether any short linker would result in selectivity for BRD4BD1. In contrast to ZXH-3-26, dBET57 is nearly equipotent on BRD3BD1 and BRD4BD1 (Fig. 6e). To test whether the selective ZXH-3-26 retains activity on endogenous full length BRD4, we treated HEK293T cells with increasing concentrations of ZXH-3-26. Immunoblot analysis confirms that ZXH-3-26 degrades endogenous BRD4 with comparable efficacy compared to the best pan-BET degrader dBET6 (Fig. 6f), while being inactive on BRD2 and BRD3 (Fig. 6g). Our docking results suggest that ZXH-3-26 would engage CRBN in a conformation distinct from what was observed with dBET6/23 or dBET57 (Supplementary Fig. 8d - h), which is confirmed by distinct mutational profiles (Supplementary Fig. 5). Interestingly, mutation of Q84 to R (as in BRD2 or K as in BRD3, see Supplementary Fig. 8d - j) decreases complex formation with CRBN mediated by ZXH-3-26, consistent with observed specificity for BRD2/3. Finally, whole proteome mass spectrometry confirmed that upon treatment with ZXH-3-26 BRD4 is the only significantly downregulated target (Fig. 6h, Supplementary Dataset 1), compared to dBET23 or dBET70 (Supplementary Fig. 9, Supplementary Dataset 1). ZXH-3-26 thus demonstrates that binding to a distinct conformation can yield a highly selective degrader molecule and that selectivity can be achieved across highly homologous domains such as the bromodomains of BET proteins.
Figure 6. Selective degradation of BRD4.
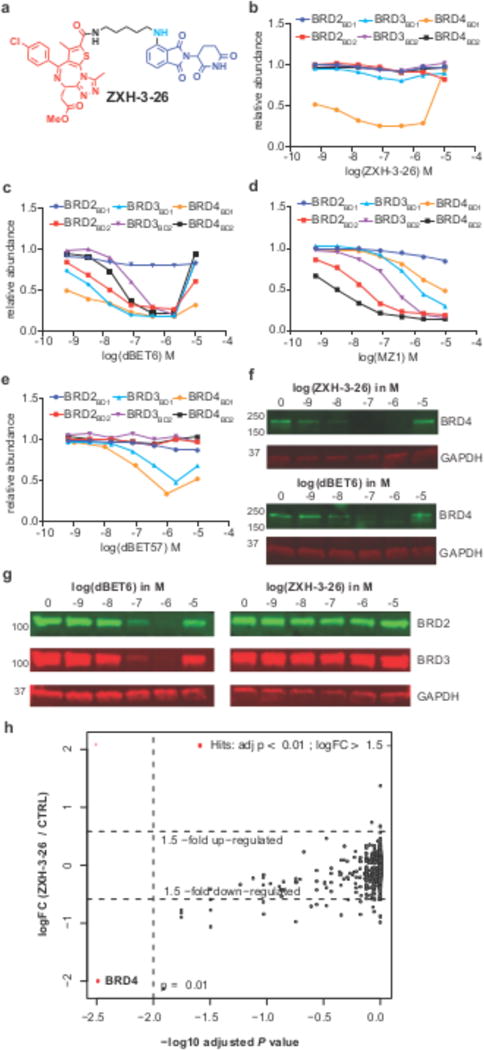
(a) Chemical structure of ZXH-3-26. (b) Quantitative assessment of cellular degradation using a EGFP/mCherry reporter assay. Cells stably expressing BRD4BD1-EGFP (or constructs harboring BRD2BD1, BRD2BD2, BRD3BD1, BRD3BD2, BRD4BD2) and mCherry were treated with increasing concentrations of ZXH-3-26 and the EGFP and mCherry signals followed using flow cytometry analysis (c) As in b but for dBET6, MZ1 (d), and dBET57 (e). Data in b-e are representative experiments out of at least three experiments. (f) Cellular degradation of endogenous BRD4. HEK293T cells were treated with increasing concentrations of ZXH-3-26 or dBET6 for 5 hours, and protein levels assessed by western blot. (g) As in f but assessing the degradation of BRD2 and BRD3 by western blot. Experiments in f and g are representative of two independent experiments. Full scans for all western blots are provided in Supplementary Fig. 10. (h) Scatter plot depicting the fold changes in relative abundance comparing 0.1 μM ZXH-3-26 to DMSO control treatment for 4 hours in MM.1s cells determined using quantitative proteomics. Negative log10 false discovery rate adjusted P Values are shown on the x-axis and log2 fold changes on the y-axis. BRD4 is significantly downregulated with a log2 fold change of -1.99, and a FDR adjusted P value of 0.0018. Data shown are of three cell culture replicates measured in a single 10-plex TMT experiment (showing 6311 proteins quantified by > 3 unique peptides). P Values were derived from a moderated t-statistic using the limma package and corrected for multiple hypothesis testing using a Benjamini-Hochberg approach (see methods for details)41.
DISCUSSION
Here, we use an integrated approach combining structural, biochemical, and cellular data to establish the molecular basis of degrader-mediated neo-substrate recruitment to the CRL4CRBN E3 ubiquitin ligase. We show that inter-protein contacts, while contributing relatively little binding affinity to the interaction, can be drivers of selectivity, and that highly effective degraders (e.g. the low nanomolar cellular activity of dBET6 or dBET70) can be achieved in absence of tight binding or positive cooperativity. Through multiple X-ray crystal structures together with comprehensive biochemical, cellular, and computational characterization, we show that binding between ligase and substrate is plastic and can adapt distinct conformations depending on linker length and position. We show that exploiting such ‘local’ energy/entropy minima underlies selectivity as seen for dBET57. We demonstrate that in silico protein docking can be used to reveal low energy binding modes and can guide development of the first heterobifunctional degraders that can discriminate between the highly homologous BET bromodomains, such as ZXH-2-147 and ZXH-3-26. Finally, we demonstrate that biochemical properties translate to cellular activity with respect to BRD4 on-target and IKZF1 off-target degradation, and that IKZF1 degradation can be tuned by IMiD linker composition (Supplementary Fig. 9).
Our data demonstrates that the same two proteins can bind in different overall conformations, which results in distinct surface patches on the ligase and target to interact. degraders therefore appear to exploit natural and widely occurring non-specific interactions by increasing the local concentration of the two protein binding partners. Non-specific interactions are widespread and thought to occur between any two proteins with affinities > 10 mM36. However, these interaction surfaces are not random as they require a certain degree of surface complementarity to avoid unfavorable contacts such as opposing charged surfaces. The constraints of relatively short linkers result in only few accessible inter-protein contact conformations. In theory, rationally designed linkers restricted to a specific binding mode unique to a ligase/substrate pair should be sufficient to drive selectivity since such a restricted conformation is unlikely to occur even in a close orthologue. We show that such can be achieved in practice with the tool compound ZXH-3-26.
The absence of positive cooperativity and the existence of multiple distinct binding conformations carries further important implications. The lack of a requirement for high affinity ligase-substrate interactions implies that a wide variety of E3 ligases can be explored to achieve desirable properties such as tissue specificity. With dBET57 and ZXH-3-26 we demonstrate that effective degraders can be designed to harbor relatively short linkers, which results in favorable and more ‘drug-like’ overall properties (Supplementary Fig. 1). We show that such short linker compounds exhibit high selectivity since the number of accessible binding conformations is reduced. Selectivity can also be further explored using different E3-moeities, as seen for CRBN- and VHL- targeting degraders (Fig. 3a). We demonstrate that computational modelling can provide an elegant surrogate, which depends only on a known structure for the individual components (ligase and target), and has the potential to enable initial predictions of possible linker length and trajectory to guide medicinal chemistry.
ZXH-3-26, to our knowledge represents the first small molecule to allow pharmacologic targeting of BRD4 without significant inhibition or degradation of BRD2/3. The notion that pharmacological selectivity can be achieved through induced protein-protein interactions distal from the immediate binding site of the small molecule affords new opportunities for the development of target-specific degrader molecules. The ability to selectively degrade BRD4 has potential implications for future drug development efforts since efficacy of BRD4 downregulation has been established for a variety of malignancies37,38. While pan BET inhibitors show toxicity, as observed in pre-clinical and clinical studies39, it is conceivable that selective degradation of BRD4 will retain efficacy, while significantly reducing pan-BET dependent toxicity for example in NUT midline carcinomas, which uniquely depend on the BRD4-NUT fusion protein. Such selective targeting of an oncogenic fusion protein has been shown as an effective treatment strategy in the case of BCR-ABL and Gleevec40.
ONLINE METHODS
Constructs and protein purification
Wild-type and mutant versions of human DDB1, human CRBN, and human IKZF1Δ were cloned in pAC-derived vectors42 and recombinant proteins were expressed as N-terminal His6 (DDB1ΔB, CRBN), StrepII-Avi (IKZF1Δ) or his6-3C-Spy43 (CRBN) fusions in Trichoplusia ni High-Five insect cells using the baculovirus expression system (Invitrogen). Wild-type and mutant BRD4BD1 and BRD4BD2 subcloned into E.Coli pET100/D-TOPO vector with N-terminal His6-Avi fusions were obtained from Invitrogen, BRD4BD1/2 were subcloned into N-terminal his6-MBP-TEV-Spy pETDuet vector and all expressed in BL21-DE3 or BL21-DE3 Rosetta cells using standard protocols. His6-TEV-BRD4BD1 construct used for selenomethionine derivative was a gift from Sirano Dhe-Paganon (originally AddGene: 38942) and was expressed in B834-DE3 cells in SeMet Medium (Molecular Dimensions, MD12-500) following the manufacturer’s instructions. For purification of His6 and GST tagged proteins, cells were resuspended in buffer containing 50 mM tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl) pH 8.0, 200 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1x protease inhibitor cocktail (Sigma) and lysed by sonication. Cells expressing StrepII-Avi-IKZF1Δ were lysed in the presence of 50 mM Tris-HCl pH 8.0, 500 mM NaCl, 1 mM TCEP, 1 mM PMSF and 1x protease inhibitor cocktail (Sigma). Following ultracentrifugation, the soluble fraction was passed over appropriate affinity resin Strep-Tactin Sepharose (IBA), Ni Sepharose 6 Fast Flow affinity resin (GE Healthcare), or Glutathione Sepharose 4B (GE Healthcare) and eluted with wash buffer (50 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM TCEP) supplemented with 2.5 mM D-Desthiobiotin (IBA), 100 mM imidazole (Fischer Chemical), or 10 mM glutathione (Fischer BioReagents), respectively. SeMet-BRD4BD1 affinity tag was TEV cleaved overnight. The affinity-purified protein was either further purified (CRBN-DDB1ΔB, IKZF1Δ, Spy-BRD4BD1) via ion exchange chromatography (Poros 50HQ) and subjected to size exclusion chromatography or concentrated and directly subjected to size exclusion chromatography in 50 mM HEPES pH 7.4, 200 mM NaCl and 1 mM TCEP. Biotinylation of IKZF1Δ and BRD4BD1, BRD4BD2 variants was performed as previously described44.
The protein-containing fractions were concentrated using ultrafiltration (Millipore) and flash frozen in liquid nitrogen (DDB1ΔB-CRBN constructs at 40-120 μM, biotinylated His6-Avi-BRD4 mutants and WT, and not biotinylated WT at ~25-100 μM, SeMetBRD4BD1 at 1mM, biotinylated StrepII-Avi-IKZF1 at ~20 μM concentration) and stored at -80°C or directly covalently labelled with BODIPY-FL-SpyCatcherS50C (His6-3C-Spy-CRBN-His6-DDB1ΔB, Spy-BRD4BD1) as described below.
SpyCatcher S50C mutant
Spycatcher43 containing a Ser50Cys mutation was obtained as synthetic dsDNA fragment from IDT (Integrated DNA technologies) and subcloned as GST-TEV fusion protein in a pET-Duet derived vector. Spycatcher S50C was expressed in BL21 DE3 and cells were lysed in the presence of 50 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM TCEP and 1 mM PMSF. Following ultracentrifugation, the soluble fraction was passed over Glutathione Sepharose 4B (GE Healthcare) and eluted with wash buffer (50 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM TCEP) supplemented with 10 mM glutathione (Fischer BioReagents). The affinity-purified protein was subjected to size exclusion chromatography, concentrated and flash frozen in liquid nitrogen.
Labelling of Spycatcher with BODIPY-FL-maleimide
Purified SpycatcherS50C protein was incubated with DTT (8 mM) at 4°C for 1 h. DTT was removed using a ENRich SEC650 10/300 (Bio-rad) size exclusion column in a buffer containing 50 mM Tris pH 7.5 and 150 mM NaCl, 0.1mM TCEP. BODIPY-FL-maleimide (Thermo Fisher) was dissolved in 100% DMSO and mixed with SpycatcherS50C to achieve 2.5 molar excess of BODIPY-FL-maleimide. SpyCatcherS50C labelling was carried out at room temperature (RT) for 3 h and stored overnight at 4°C. Labelled SpycatcherS50C was purified on a ENRich SEC650 10/300 (Bio-rad) size exclusion column in 50 mM Tris pH 7.5, 150 mM NaCl, 0.25 mM TCEP and 10% (v/v) glycerol, concentrated by ultrafiltration (Millipore), flash frozen (~40 μM) in liquid nitrogen and stored at -80°C.
BODIPY-FL-Spycatcher labelling of CRBN-DDB1ΔB and BRD4BD1
Purified His6-DDB1ΔB-His6-3C-Spy-CRBN or His6-Spy-BRD4BD1 was incubated overnight at 4°C with BODIPY-FL labelled SpyCatcherS50C protein at stoichiometric ratio. Protein was concentrated and loaded on the ENrich SEC 650 10/300 (Bio-rad) size exclusion column and the labelling monitored with absorption at 280 and 490 nm. The protein peak corresponding to the labeled protein was pooled, concentrated by ultrafiltration (Millipore), flash frozen (~9.6 μM for His6-DDB1ΔB-His6-3C-Spy-CRBNBODIPY SpyCatcher or ~22 uM for His6-Spy-BRD4BD1) in liquid nitrogen and stored at -80°C.
Crystallization and data collection
We have resorted to a previously developed DDB1 construct lacking WD40 propeller B (BPB, residues 396-705) domain (referred to as DDB1ΔB) successful in crystallization of a DDB1ΔB-CRBN-lenalidomide-CK1α complex17. For crystallization of His6-DDB1ΔB-His6-CRBN-dBET6/23/70-his6-BRD4BD1 and His6-DDB1ΔB-His6-CRBN-dBET55-His6-Avi-BRD4BD1(D145A) complexes 145 μM of dBET was mixed with 70 μM BRD4BD1 or BRD4BD1 D145A and 80 μM His6-DDB1ΔB-His6-CRBN and incubated for 15 min either on ice or at RT. Crystallization plates were set up in 3 sub-well plates (Intelli, Art Robbins) by vapor diffusion using NT8 (Formulatrix) at 20°C and images acquired using RockImager 1000 (Formulatrix). Crystals appeared in wells B9-F9 and H9 of Morpheus HT Screen (Molecular Dimensions) within few hours and were fully grown after 3 days. Single uniform crystals (length 80-100 μm) were present in condition C9 (10% (w/v) PEG20k, 20% (w/v) PEG550 MME, 0.1 M BICINE pH 8.5) in 2:1 or 1:1 protein to precipitant ratio in 150 or 225 nL drops. Further optimization of condition in Morpheus HT Screen C9 by SilverBullet (Hampton Research) additive screening in 1:10 additive to reservoir ratio resulted in optimal crystals for dBET6, dBET23, dBET55 and dBET70 in Silver Bullet wells D7, B5, G4 and F2 respectively, in 2:1 protein to precipitant ratio of 225 or 400 nL drops. Crystals were cryo-protected in reservoir solution supplemented with 25-30% PEG 400 containing 150-300 μM respective dBET and flash–cooled in liquid nitrogen. We have noticed that crystals harvested after 2-3 days resulted in optimal diffraction. Diffraction data were collected at the APS Chicago (beamline 24-ID-C) with a Pilatus 6M-F detector at a temperature of 100 K, or for dBET6 co-crystal structure at beamline 24-ID-E with a Eiger 16M detector at a temperature of 100 K. Data were indexed and integrated using XDS45 and scaled using AIMLESS supported by other programs of the CCP4 suite46 or RAPD pipeline (APS Chicago). Data processing statistics, refinement statistics and model quality parameters are provided in Supplementary Table 1.
dBET57 containing crystals were obtained by mixing His6-DDB1ΔB-His6-CRBN at 75 μM, with dBET57 at 140 μM and SeMet BRD4BD1 at 140 μM in condition B5 of the Hampton Index HT screen (1.26 M NaH2PO4, 0.14 M K2HPO4). Single crystals were harvested, stabilized by addition of 25% ethylene glycol containing dBET57 at 50 μM. Diffraction data were collected at the APS Chicago (beamline 24-ID-C) with a Pilatus 6M-F detector at a temperature of 100 K, at wavelength of 0.9724 Å. Data were indexed and integrated using RAPD pipeline (APS Chicago) and scaled using AIMLESS supported by other programs of the CCP4 suite46. Data processing statistics, refinement statistics and model quality parameters are provided in Supplementary Table 1.
Structure determination and model building
The DDB1ΔB-CRBN-dBET6/23/70-BRD4BD1 and DDB1ΔB-CRBN-dBET55-BRD4BD1/D145A quaternary complexes crystallized in space group P6522 with a single complex in the unit cell. PHASER47 was used to determine the structures by molecular replacement using a crystallographic model of DDB1ΔB-CRBN based on a crystal structure pdb: 5fqd. The initial model was iteratively improved with COOT and refined using PHENIX.REFINE48 and autoBUSTER49 with ligand restraints generated by Grade server (Global Phasing) or phenix.elbow50. Protein geometry analysis revealed 0.46%, 0.54%, 1.01%, 0.72%, 0.77% Ramachandran outliers, with 95.36%, 94.87%, 92.96%, 93.67%, 93.66% residues in favored regions and 4.17%, 4.58%, 6.04%, 5.60%, 5.56% residues in allowed regions for the complexes with dBET6, 23, 55, 57 and 70 respectively.
The DDB1ΔB-CRBN-dBET57-SeMetBRD4BD1 complex crystallized in space group I422 with a single complex in the unit cell. PHASER47 was used for molecular replacement using models of hsDDB1ΔB-hsCRBN-HBD derived from pdb: 5fqd, hsCRBN-NTD derived from pdb: 5fqd, and BRD4BD1 (pdb: 3mxf). The model was rigid body refined using PHENIX.REFINE48 and the hsCRBN-CTD was subsequently placed using Coot jiggle fit (part of Coot EM scripts from Alan Brown and Paul Emsley). The final model was rigid body refined using PHENIX.REFINE and autoBUSTER49. Anomalous maps were calculated with PHENIX.MAPS48.
Figures were generated with PyMOL (The PyMOL Molecular Graphics System, Version 1.8.6.0 Schrödinger, LLC) and model quality was assessed with MOLPROBITY51. Interaction surfaces were determined with PISA52. The IKZF1 homology model was taken from17.
Time-resolved fluorescence resonance energy transfer (TR-FRET)
Compounds in dimerization assays were dispensed in a 384-well microplate (Corning, 4514) using D300e Digital Dispenser (HP) normalized to 2% DMSO into 200 nM biotinylated His6-avi-bromodomain (WT or mutant, see Figure legends) or 80 nM biotinylated StrepII-avi-IKZF1Δ (See Figure legends), 100 nM His6-DDB1ΔB-His6-CRBNBODIPY-Spycatcher and 2nM terbium-coupled streptavidin (Invitrogen) in a buffer containing 50 mM Tris pH 7.5, 200 mM NaCl, 0.1% Pluronic F-68 solution (Sigma) and 2% DMSO (4% DMSO final). Compounds in CRBN mutants dimerization assay were dispensed as described above into 200 nM His6-DDB1-His6-CRBNmutants or 200 nM His6-DDB1ΔB-His6-CRBNWT, 100 nM BRD4BD1-BODIPY-SpyCatcher and 2 nM terbium-anti-HIS Ab (Invitrogen) in a buffer containing 50 mM Tris pH 7.5, 200 mM NaCl, 0.1% Pluronic F-68 solution (Sigma) and 2% DMSO (4% DMSO final). Before TR-FRET measurements were conducted, the reactions were incubated for 15 min at RT. After excitation of terbium fluorescence at 337 nm, emission at 490 nm (terbium) and 520 nm (BODIPY) were recorded with a 70 μs delay over 600 μs to reduce background fluorescence and the reaction was followed over 30 200 second cycles of each data point using a PHERAstar FS microplate reader (BMG Labtech). The TR-FRET signal of each data point was extracted by calculating the 520/490 nm ratio. The heterobifunctional nature of small molecule degraders results in a three-body binding equilibrium complicated by potential cooperativity or avidity effects arising from protein-protein interactions34, all of which precludes direct interpretation of the binding data. However, assuming constant concentrations of BRD4BD1, DDB1ΔB-CRBN, and fluorescent labels, as well as similar binding conformations, the peak height of the TR-FRET can be used as an indication for the amount of tertiary complex formation (containing BRD4BD1/BD2, dBET, and CRBN)34. The peak height of TR-FRET dBET dose response data was calculated in GraphPad Prism 7 using Area Under Curve analysis for three independent replicates (n=3) and the mean peak height and standard deviation calculated.
Counter titrations with unlabeled proteins were carried out by addition of solution of 200 nM His6-DDB1ΔB-His6-CRBNBODIPY-Spycatcher, 160 nM biotinylated His6-Avi-IKZF1Δ, 4 nM terbium-coupled streptavidin and 2 μM of dBET57, incubated for 15 min on ice, to equal volume of titrated unlabeled His6-Avi-BRD4BD1 or His6-Avi-BRD4BD2 to the final assay concentrations (see Figure legends).
The 520/490 nm ratios in IKZF1Δ TR-FRET assays were plotted to calculate the half maximal effective concentrations (EC50 – for unlabeled protein titrations) or IC50 (for compound titrations) assuming a single binding site using GraphPad Prism 7 variable slope equation. The standard deviation in IKZF1Δ TR-FRET compound titrations was calculated from three independent replicates (n=3), or as an average of 5 technical replicates of single experiment for unlabeled protein titrations.
Fluorescence polarization
Atto565-conjugated lenalidomide (10 nM) was mixed with increasing concentration of purified his6-DDB1ΔB-his6-CRBN (10 μM final top concentration, 2-fold, 23-point dilution and DMSO control) in 384-well microplates (Corning, 4514) and incubated for 15 min at RT. The change in fluorescence polarization was monitored using a PHERAstar FS microplate reader (BMG Labtech) for 20 min in 120 s cycles. The Atto565-lenalidomide bound fraction was calculated as described53 and the Kd was obtained from a fit in GraphPad Prism 7 from four independent replicates (n=4).
Compounds in Atto565-Lenalidomide displacement assay were dispensed in a 384-well microplate (Corning, 4514) using D300e Digital Dispenser (HP) normalized to 2% DMSO into 10 nM Atto565-Leanlidomide, 100 nM DDB1ΔB-CRBN, 50 mM Tris pH 7.5, 200 mM NaCl, 0.1% Pluronic F-68 solution (Sigma), 0.5 mg/ml BSA (Sigma) containing 2% DMSO (4% DMSO final). Compound titrations were performed in presence of 0, 1, 5, 20 μM of unbiotinylated his6-avi-BRD4BD1 or his6-avi-BRD4BD2 (see Figure legends) and incubated for 60 min at RT. The change in fluorescence polarization was monitored using a PHERAstar FS microplate reader (BMG Labtech) for 20 min in 200s cycles. Data from two independent replicates (n=2) was plotted and IC50 values estimated using variable slope equation in GraphPad Prism 7.
Cellular degradation assays
IKZF1Δ, BRD2BD1, BRD2BD2, BRD3BD1, BRD3BD2, BRD4BD1, and BRD4BD2 were subcloned into mammalian pcDNA5/FRT Vector (Ampicillin and Hygromycin B resistant) modified to contain MCS-eGFP-P2A-mCherry. Stable cell lines expressing eGFP-protein fusion and mCherry reporter were generated using Flip-In 293 system. Plasmid (0.3 μg) and pOG44 (4.7 μg) DNA were preincubated in 100 μL of Opti-MEM I (Gibco, Life Technologies) media containing 0.05 mg/ml Lipofectamine 2000 (Invitrogen) for 20 min and added to Flip-In 293 cells containing 1.9 ml of DMEM media (Gibco, Life Technologies) per well in a 6-well plate format (Falcon, 353046). Cells were propagated after 48 h and transferred into a 10 cm2 plate (Corning, 430165) in DMEM media containing 50 μg/ml of Hygromycin B (REF 10687010, Invitrogen) as a selection marker. Following 2-3 passage cycle FACS (FACSAria II, BD) was used to enrich for cells expressing eGFP and mCherry.
Cells were seeded at 30-50% confluency in either 24, 48 or 96 well plates (3524, 3548, 3596 respectively, Costar) a day before compound treatment. Titrated compounds (see Figure legends) were incubated with cells for 5h following trypsinization and resuspension in DMEM media, transferred into 96-well plates (353910, Falcon) and analyzed by flow cytometer (guava easyCyte HT, Millipore). Signal from minimal 3000 events per well was acquired and the eGFP and mCherry florescence monitored. Data was analyzed using FlowJo (FlowJo, LCC). Forward and side scatter outliers, frequently associated with cell debris, were removed leaving >90% of total cells, followed by removal of eGFP and mCherry signal outliers, leaving 88-90% of total cells creating the set used for quantification. The eGFP protein abundance relative to mCherry was then quantified as a ten-fold amplified ratio for each individual cell using the formula: 10 × eGFP/mCherry. The median of the ratio was then calculated per set, normalized to the median of the DMSO ratio.
Western Blot for cellular BRD2/3/4 degradation
HEK293T cells were seeded at 90% confluency in 12 well plates (353043, Falcon), left to attach for 1.5h, followed by the compound treatment for 5h. Primary and secondary antibodies used included anti-BRD4 at 1:1000 dilution (A301-985A-M, Bethyl Laboratories, Lot: 6), anti-BRD2 at 1:2,000 dilution (A302-582A, Bethyl Laboratories, Lot: 1), anti-BRD3 at 1:500 dilution (ab56342, Abcam, Lot: GR243549-8), anti-GAPDH at 1:10,000 dilution (G8795, Sigma, Lot: 065M4856V, Clone: GAPDH-71.1), IRDye680 Donkey anti-mouse at 1:10,000 dilution (926–68072, LiCor, Lot: C61116-05) and IRDye800 Goat anti-rabbit at 1:10,000 dilution (926–32211, LiCor, Lot: C70301-05).
Sample preparation TMT LC-MS3 mass spectrometry
MM.1s cell were treated with DMSO, 1 μM dBET23, or dBET70, or 0.1 μM ZXH-3-26 in cell culture triplicates for 5 hours and cells harvested by centrifugation. Lysis buffer (8 M Urea, 1% SDS, 50 mM Tris pH 8.5, Protease and Phosphatase inhibitors from Roche) was added to the cell pellets to achieve a cell lysate with a protein concentration between 2 – 8 mg mL−1. A micro-BCA assay (Pierce) was used to determine the final protein concentration in the cell lysate. 200 μg proteins for each sample were reduced and alkylated as previously described. Proteins were precipitated using methanol/chloroform. In brief, four volumes of methanol were added to the cell lysate, followed by one volume of chloroform, and finally three volumes of water. The mixture was vortexed and centrifuged to separate the chloroform phase from the aqueous phase. The precipitated protein was washed with one volume of ice-cold methanol. The washed precipitated protein was allowed to air dry. Precipitated protein was resuspended in 4 M Urea, 50 mM Tris pH 8.5. Proteins were first digested with LysC (1:50; enzyme:protein) for 12 hours at 25°C. The LysC digestion was diluted down in 1 M Urea, 50 mM Tris pH 8.5 and then digested with trypsin (1:100; enzyme:protein) for another 8 hours at 25°C. Peptides were desalted using a C18 solid phase extraction cartridges (Waters). Dried peptides were resuspended in 200 mM EPPS, pH 8.0. Peptide quantification was performed using the micro-BCA assay (Pierce). The same amount of peptide from each condition was labelled with tandem mass tag (TMT) reagent (1:4; peptide:TMT label) (Pierce). The 10-plex labelling reactions were performed for 2 hours at 25°C. Modification of tyrosine residue with TMT was reversed by the addition of 5% hydroxyl amine for 15 minutes at 25°C. The reaction was quenched with 0.5% TFA and samples were combined at a 1:1:1:1:1:1:1:1:1:1 ratio. Combined samples were desalted and offline fractionated into 96 fractions using an aeris peptide xb-c18 column (phenomenex) at pH 8.0. Fractions were recombined in a non-continuous manner into 24 fractions and every second fraction was used for subsequent mass spectrometry analysis.
Data were collected using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) coupled with a Proxeon EASY-nLC 1200 LC pump (Thermo Fisher Scientific). Peptides were separated on a 75 μm inner diameter microcapillary column packed with 35 cm of Accucore C18 resin (2.6 μm, 100 Å, ThermoFisher Scientific). Peptides were separated using a 3 hr gradient of 6–27% acetonitrile in 0.125% formic acid with a flow rate of 400 nL/min.
Each analysis used an MS3-based TMT method as described previously54. The data were acquired using a mass range of m/z 350 – 1350, resolution 120,000, AGC target 1 × 106, maximum injection time 100 ms, dynamic exclusion of 120 seconds for the peptide measurements in the Orbitrap. Data dependent MS2 spectra were acquired in the ion trap with a normalized collision energy (NCE) set at 35%, AGC target set to 1.8 × 104 and a maximum injection time of 120 ms. MS3 scans were acquired in the Orbitrap with a HCD collision energy set to 55%, AGC target set to 1.5 × 105, maximum injection time of 150 ms, resolution at 50,000 and with a maximum synchronous precursor selection (SPS) precursors set to 10.
LC-MS data analysis and statistics
Proteome Discoverer 2.1 (Thermo Fisher) was used to for .RAW file processing and controlling peptide and protein level false discovery rates, assembling proteins from peptides, and protein quantification from peptides. MS/MS spectra were searched against a Uniprot human database (September 2016) with both the forward and reverse sequences. Database search criteria are as follows: tryptic with two missed cleavages, a precursor mass tolerance of 50 ppm, fragment ion mass tolerance of 1.0 Da, static alkylation of cysteine (57.02146 Da), static TMT labelling of lysine residues and N-termini of peptides (229.16293 Da), and variable oxidation of methionine (15.99491 Da). TMT reporter ion intensities were measured using a 0.003 Da window around the theoretical m/z for each reporter ion in the MS3 scan. Peptide spectral matches with poor quality MS3 spectra were excluded from quantitation (< summed signal-to-noise across 10 channels and < 0.5 precursor isolation specificity).
Reporter ion intensities were normalized and scaled using in house scripts and the R framework55. Statistical analysis was carried out using the limma package within the R framework41. In short, a moderated t-test was applied across all proteins to assess statistical significance, where the standard errors are calculated using an empirical Bayes method utilizing information across all proteins to stabilize inference about individual proteins. This test assumes a normal distribution. The nominal p-values arising from the moderated t-statistic are corrected for multiple hypothesis testing by controlling the false discovery rate (FDR) as proposed by Benjamini and Hochberg41.
Protein docking
All protein docking was carried out using Rosetta 3.7 provided through SBGrid56. Input models were downloaded from the PDB (hsCRBN pdb: 4tz4; BRD4BD1 pdb: 3mxf, BRD4BD2 pdb: 2ouo, and hsCSNK1A1 pdb: 5fqd). Ligand conformers were generated using OpenEye Omega (OpenEye scientific) and parameter files generated using Rosetta ‘molfile_to_params.py’. Relevant pdb structure coordinates were combined into a single file and prepared for docking using the Rosetta ‘docking_prepack_protocol’ program. Initial global docking was performed using Rosetta ‘docking_protocol_mpi’ with the following command line options:
-
-
partners A_B - dock_pert 5 25 - randomize2 - ex1 ex2aro –nstruct 20000
providing the combined pdb and ligand specific parameter files as input.
For Ck1α, and the initial analysis of BRD4BD1, the two lowest scoring solutions were used for local perturbation docking with Rosetta ‘docking_protocol_mpi’ with the following command line options:
-
-
partners A_B - dock_pert 8 18 - ex1 ex2aro –nstruct 2000
To assess the landscape of possible binding modes for BRD4BD1 and BRD4BD2, the top 200 lowest scoring docking decoys were selected and hierarchical clustered according to the compound centroids and orientations. The shortest pairwise distance between selected set of atoms on JQ1 and set of atoms on lenalidomide (see highlighted atoms in Figure 6) was calculated in Pymol (The PyMOL Molecular Graphics System, Version 1.8.6.0 Schrödinger, LLC) as Euclidean distance for each of the top 200 poses. The histogram was obtained in GraphPad Prism 7 using Column Analysis - Frequency Distribution.
Data analysis and statistics for all steps were performed using the R framework55 or GraphPad Prism 7.
Supplementary Material
Acknowledgments
We thank Nicolas Thomä and Georg Petzold (Friedrich Miescher Institute for Biomedical Research) for providing constructs and purified protein for some of the CRBN mutants. We are grateful to Sirano Dhe-Paganon and Hyuk-Soo Seo (Dana-Farber Cancer Institute) for providing purified BRD4BD1 and BRD4BD2 protein and BRD4BD1 construct, and to Shiva Dastjerdi for help in the synthesis of dBET55. We thank Michael Eck for critical feedback on the manuscript. Financial support for this work was provided by NIH grant NCI R01CA214608 (grant to E.S.F.), The Harvard University William F. Milton Fund (grant to E.S.F.), the Friends of Dana Farber (grant to E.S.F.), the Claudia Adams Barr Program for Innovative Cancer Research and the Linde Family Foundation (both start-up funds to E.S.F.), and the Damon Runyon Cancer Research foundation (DRG-2196-14, fellowship to D.L.B). This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The authors would like to thank Diamond Light Source for beamtime, and the staff of beamlines I04-1 for assistance with crystal testing and data collection.
Footnotes
Author contributions
R.P.N, S.L.D. and E.S.F. initiated the project. R.P.N. and S.L.D. with help from C.M.P. conducted the protein purification and crystallization. R.P.N. collected, processed and refined X-ray data. R.P.N. conceived and performed biochemical assays. D.B. and M.I. synthesized the dBET series of compounds. Z.H. T.Z. synthesized other small molecules used in this study. K.A.D. and M.J. conducted the mass spectrometry experiments. J.A., N.S., C.M.P. and R.P.N. designed, constructed and performed the cellular reporter assays. R.P.N. and E.S.F. conceived and performed protein-docking experiments. J.D.M., N.S.G., J.E.B., and E.S.F. supervised all aspects of the project. R.P.N. and E.S.F. wrote the manuscript with input from all authors. All authors read, revised, and approved the manuscript.
Competing Financial Interest Statement:
E.S.F. is a member of the scientific advisory board of C4 Therapeutics and a consultant to Novartis Pharmaceuticals. N.S.G. is a founder and scientific advisory board member of C4 Therapeutics. J.E.B. is an executive and shareholder of Novartis Pharmaceuticals.
Data availability
Structural coordinates are deposited in the protein data bank and available under accession numbers DDB1ΔB-CRBN-dBET23-BRD4BD1 (pdb: 6bn7), DDB1ΔB-CRBN-dBET6-BRD4BD1 (pdb: 6boy), DDB1ΔB-CRBN-dBET70-BRD4BD1 (pdb: 6bn9), DDB1ΔB-CRBN-dBET55-BRD4BD1 (pdb: 6bn8), DDB1ΔB-CRBN-dBET57-BRD4BD1 (pdb: 6bnb). Mass spectrometry raw data files have been deposited in PRIDE Archive under the accession numbers PXD008674 for dBET70 and dBET23 and PXD008689 for ZXH-3-26.
Additional Information
Any supplementary information, chemical compound information and source data are available in the online version of the paper.
References
- 1.Gustafson JL, et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew Chem Int Ed Engl. 2015;54:9659–9662. doi: 10.1002/anie.201503720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bondeson DP, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckley DL, et al. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem Biol. 2015;10:1831–1837. doi: 10.1021/acschembio.5b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu J, et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gadd MS, et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017 doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winter GE, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakamoto KM, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kenten JH, Roberts SF. Controlling protein levels in eucaryotic organisms. 6306663. US patent. 2001;B1
- 9.Huang HT, et al. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chem Biol. 2017 doi: 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winter GE, et al. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol Cell. 2017;67:5–18. e19. doi: 10.1016/j.molcel.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer ES, Park E, Eck MJ, Thoma NH. SPLINTS: small-molecule protein ligand interface stabilizers. Curr Opin Struct Biol. 2016;37:115–122. doi: 10.1016/j.sbi.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu G, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343:305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kronke J, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gandhi AK, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN) British journal of haematology. 2014;164:811–821. doi: 10.1111/bjh.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.An J, et al. pSILAC mass spectrometry reveals ZFP91 as IMiD dependent substrate of the CRL4CRBN ubiquitin ligase. Nature communications. 2017 doi: 10.1038/NCOMMS15398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kronke J, et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature. 2015;523:183–188. doi: 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petzold G, Fischer ES, Thoma NH. Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4 ubiquitin ligase. Nature. 2016;532:127–130. doi: 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- 18.Chamberlain PP, et al. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat Struct Mol Biol. 2014;21:803–809. doi: 10.1038/nsmb.2874. [DOI] [PubMed] [Google Scholar]
- 19.Ito T, et al. Identification of a primary target of thalidomide teratogenicity. Science (New York, NY) 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 20.Fischer ES, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512:49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buckley DL, et al. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1alpha. Angew Chem Int Ed Engl. 2012;51:11463–11467. doi: 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raina K, Crews CM. Targeted protein knockdown using small molecule degraders. Curr Opin Chem Biol. 2017;39:46–53. doi: 10.1016/j.cbpa.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toure M, Crews CM. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew Chem Int Ed Engl. 2016;55:1966–1973. doi: 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- 24.Raina K, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2016;113:7124–7129. doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lai AC, et al. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed Engl. 2016;55:807–810. doi: 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Remillard D, et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew Chem Int Ed Engl. 2017;56:5738–5743. doi: 10.1002/anie.201611281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bondeson DP, et al. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chemical Biology. 2017 doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang HT, et al. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chemical Biology. 2017 doi: 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol. 2015;10:1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Filippakopoulos P, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 33.Matyskiela ME, et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature. 2016;535:252–257. doi: 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- 34.Douglass EF, Jr, Miller CJ, Sparer G, Shapiro H, Spiegel DA. A comprehensive mathematical model for three-body binding equilibria. J Am Chem Soc. 2013;135:6092–6099. doi: 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sircar A, Chaudhury S, Kilambi KP, Berrondo M, Gray JJ. A generalized approach to sampling backbone conformations with RosettaDock for CAPRI rounds 13-19. Proteins. 2010;78:3115–3123. doi: 10.1002/prot.22765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuriyan J, Eisenberg D. The origin of protein interactions and allostery in colocalization. Nature. 2007;450:983–990. doi: 10.1038/nature06524. [DOI] [PubMed] [Google Scholar]
- 37.Chau NG, et al. Intensive treatment and survival outcomes in NUT midline carcinoma of the head and neck. Cancer. 2016;122:3632–3640. doi: 10.1002/cncr.30242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuber J, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stathis A, et al. Clinical Response of Carcinomas Harboring the BRD4-NUT Oncoprotein to the Targeted Bromodomain Inhibitor OTX015/MK-8628. Cancer Discov. 2016;6:492–500. doi: 10.1158/2159-8290.CD-15-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchdunger E, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139–145. [PubMed] [Google Scholar]
- 41.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abdulrahman W, et al. A set of baculovirus transfer vectors for screening of affinity tags and parallel expression strategies. Analytical Biochemistry. 2009;385:383–385. doi: 10.1016/j.ab.2008.10.044. [DOI] [PubMed] [Google Scholar]
- 43.Zakeri B, et al. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc Natl Acad Sci U S A. 2012;109:E690–697. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cavadini S, et al. Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature. 2016;531:598–603. doi: 10.1038/nature17416. [DOI] [PubMed] [Google Scholar]
- 45.Kabsch W. XDS. Acta Crystallographica Section D. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallographica Section D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCoy AJ, et al. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallographica Section D. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.BUSTER version 2.10.2 v 2.10.2. Global Phasing Ltd; Cambridge, United Kingdom: 2011. [Google Scholar]
- 50.Moriarty NW, Grosse-Kunstleve RW, Adams PD. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallographica Section D. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krissinel E, Henrick K. Inference of Macromolecular Assemblies from Crystalline State. Journal of Molecular Biology. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 53.Marks BD, et al. Multiparameter Analysis of a Screen for Progesterone Receptor Ligands: Comparing Fluorescence Lifetime and Fluorescence Polarization Measurements. ASSAY and Drug Development Technologies. 2005;3:613–622. doi: 10.1089/adt.2005.3.613. [DOI] [PubMed] [Google Scholar]
- 54.McAlister GC, et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Team, R. C. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2013. [Google Scholar]
- 56.Morin A, et al. Collaboration gets the most out of software. Elife. 2013;2:e01456. doi: 10.7554/eLife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.