

Abstract
T cell antigen receptor (TCR) signaling requires the sequential activities of the kinases Lck and Zap70. Upon TCR stimulation, Lck phosphorylates the TCR, leading to the recruitment, phosphorylation, and activation of Zap70. Lck binds to and stabilizes phosho-Zap70 using its SH2 domain, and Zap70 phosphorylates the critical adaptors LAT and SLP76, which coordinate downstream signaling. It is unclear whether phosphorylation of these adaptors happens through passive diffusion or active recruitment. We report the discovery of a conserved proline-rich motif in LAT that mediated efficient LAT phosphorylation. Lck associated with this motif via its SH3 domain, and with phospho-Zap70 via its SH2 domain, thereby acting as molecular bridge to facilitate the co-localization of Zap70 and LAT. Elimination of this proline-rich motif compromised TCR signaling and T cell development. These results demonstrate the remarkable multi-functionality of Lck, where each of its domains has evolved to orchestrate a distinct step in TCR signaling.
INTRODUCTION
Signaling through the T cell antigen receptor (TCR) is the defining event for proper thymocyte development and mature T cell homeostasis, and TCR signaling is also critical for effective host responses to pathogens or tumors1–3. T cells interact with self-peptides bound to major histocompatibility complex proteins (self-pMHC) using their TCRs throughout their development and lifespan, acquiring survival signals and avoiding autoreactivity. At the same time, T cells must be capable of responding to pathogen- or tumor-derived antigenic peptides bound to MHC molecules (pMHC) to mount rapid and appropriate protective responses. Although the molecular discrimination of self-from non-self-pMHC by the TCR plays a critical role in dictating these responses, recent engineered T cell therapies for cancer, which rely on artificial antigen-recognition domains fused with native intracellular signaling molecules, further underscore the importance of downstream TCR-proximal signaling events in controlling the specificity and sensitivity of the T cell responses4.
Since the TCR has no intrinsic enzymatic activity, the tyrosine kinases Lck and Zap70 are tasked with initiating TCR signaling. A pool of Lck, a Src family kinase, is active in T cells prior to pMHC recognition5. The level of Lck activity upon TCR stimulation is controlled by multiple mechanisms1–3,6,7. For instance, the localization of Lck is regulated by non-covalent association with the cytoplasmic segments of the CD4 and CD8 coreceptors. Upon engagement of TCR with pMHC, the coreceptor co-engagement localizes active Lck to the engaged TCR8. There, Lck phosphorylates the paired tyrosines of the immunoreceptor tyrosine-based activation motifs (ITAMs) in the invariant CD3- and ζ-chains of the TCR complex9. If both tyrosines of an ITAM are phosphorylated, they form a highaffinity docking site for the tandem-SH2 domains of Zap7010,11. Binding to the ITAMs partially relieves Zap70 autoinhibition. Full activation of Zap70 also requires the phosphorylation by Lck of Zap70 to relieve its autoinhibition and to activate its catalytic activity since Zap70 cannot be activated by trans-autophosphorylation12–14.
Thus, recruitment and activation of Zap70 are absolutely reliant on Lck catalytic activity14. Moreover, the binding of the Lck SH2 domain to phospho-Y319 in interdomain B of Zap70 may serve to sustain Lck localization, its open active conformation and the catalytic activities of both kinases, thereby providing positive feedback6,15,16. However, despite their colocalization, the two kinases have mutually exclusive preferences for their substrates14,17. Lck cannot phosphorylate the substrates of Zap70, namely the adaptors LAT and SLP76. Zap70 phosphorylates LAT and SLP76 on multiple tyrosines, to form effective signaling complexes. LAT has four major tyrosine phosphorylation sites that serve as docking sites for the SH2-domains of downstream signaling effectors. The assembly of LAT-based signalosomes are essential to amplify TCR-induced signals that result in calcium mobilization, mitogen-activated protein kinase activation, and actin polymerization18.
While many mechanisms prevent premature and inappropriate LAT phosphorylation, T cells must ensure rapid and specific LAT phosphorylation following agonist pMHC stimulation of the TCR18. However, the prompt and specific phosphorylation of LAT following agonist pMHC stimulation of the TCR presents a hurdle, considering that LAT has not been known to directly associate with the TCR, where Zap70 is localized. It has been suggested that stimulated and activated Zap70 may be induced to dissociate and diffuse away from the engaged TCR before the activated kinase encounters LAT19. However, such a mechanism could potentially decouple Zap70 activity from the TCR recognition event and lead to inappropriate downstream signaling and amplification or premature termination of Zap70 activity via phosphatases or ubiquitin ligases20,21. This raises the question: how is Zap70 catalytic effector function appropriately coupled to TCR recognition?
Here we report that Lck uses each of its functional domains to ensure the agonist pMHC engaged TCR trigger efficient signal transduction leading to LAT phosphorylation. Our model suggests that Lck uses its SH2 domain to interact with TCR-bound Zap70 molecules which it has phosphorylated and activated, and subsequently via its SH3 domain serves to link Zap70 to its substrate LAT to facilitate efficient TCR signal transduction. We identified a proline-rich motif in LAT, which is highly conserved across mammalian species and specifically interacts with the SH3 domain of Lck to enhance the LAT phosphorylation by Zap70. Mutation of this proline-rich motif in LAT impeded TCR signal transduction and resulted in the significant reduction in thymocyte development. Our findings provide new insights into how efficient signal transduction by the TCR is coupled to the recognition of a bona-fide agonist pMHC.
RESULTS
LAT’s proline-rich region is important for TCR signaling
To understand how Zap70 catalytic effector function mediates efficient LAT phosphorylation in response to TCR recognition, we compared the amino acid sequences of LAT from 42 mammals, and identified an evolutionarily conserved amino acid sequence in the N-terminal membrane proximal cytoplasmic segments of mammalian LAT molecules (Fig. 1a, Supplementary Fig. 1a and Supplementary Table 1). These sequences revealed a strikingly high degree of conservation, especially in the distribution and positioning of proline residues. The disproportionate presence of proline residues in LAT was also evident after we quantified the frequency of each amino acid in human LAT relative to other proteins (Supplementary Fig. 1b–d). Notably, no structural or functional properties of this proline-rich region of LAT have previously been reported.
Fig. 1. A conserved proline-rich region in the membrane proximal region of LAT is important for TCR signaling.
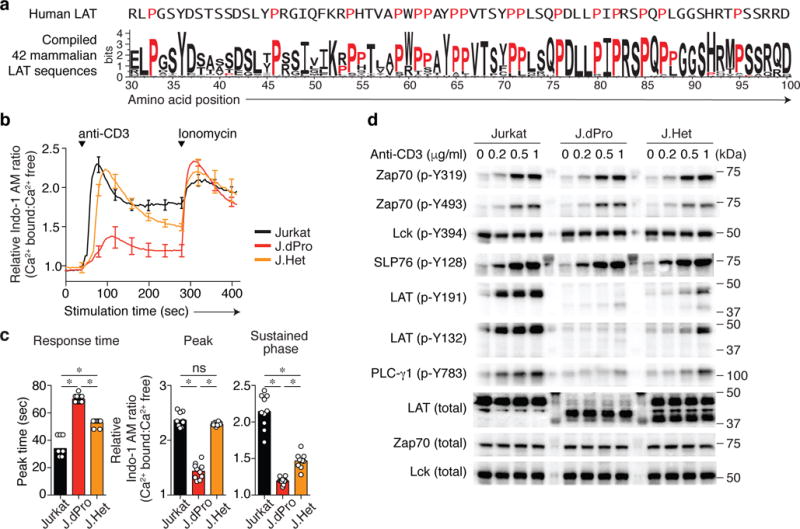
(a) Sequence conservation is depicted in the membrane proximal region of LAT in 42 mammalian species. The amino acid position is numbered based on human LAT isoform 2, the sequence of which is shown above the sequence logo. Proline residues are indicated in red. (b and c) Intracellular free calcium changes, expressed as the relative calcium-sensitive fluorescence ratio of Jurkat and CRISPR/Cas9-generated Jurkat variants (J.dPro and J.Het) lacking the proline-enriched region in response to anti-CD3 stimulation (0.5 μg/ml), as assessed with calcium indicator dye Indo-I AM. (b) Representative calcium-sensitive fluorescence ratios in the indicated cells over time. Data are normalized to the basal calcium-sensitive ratio in unstimulated Jurkat cells at t = 0 (mean ± s.d.). n = 3 technical replicates. Data are representative of at least two experiments. (c) Quantification of time (sec) required for Jurkat, J.dPro or J.Het cells from addition of anti-CD3 to reach the maximal Indo-1 AM ratio (left), or the magnitude change of Indo-I AM ratio at peak or sustained phase from 200 sec to 284 sec (right). Each symbol represents an individual technical replicate sample. Data are representative of duplicate samples in five independent experiments (n = 10, mean ± s.d.). *P < 0.0001 (exact); ns, not significant; two-tailed Mann-Whitney test. (d) Immunoblot analysis of phosphorylation of or total TCR-proximal signaling molecules in Jurkat, J.dPro or J.Het cells were left unstimulated or stimulated with anti-CD3 (concentration indicated above blots) for 1.5 min. Data are representative of at least four independent experiments. Lanes separating cell lysate samples contain mobility marker proteins. Lanes separating cell lysate samples contain mobility marker proteins.
To explore the potential functional significance of this conserved sequence in LAT, we employed CRISPR/Cas9 to generate mutants that lack the membrane-proximal segment in the LAT genomic locus using the Jurkat human T cell leukemic line, which is frequently used for TCR signaling studies. In one such mutant line, J.dPro, the resulting indels deleted the proline-region encoding sequences in both alleles (Supplementary Fig. 2a,b). The deletion of this proline-enriched segment did not interfere with the expression of the LAT protein (Supplementary Fig. 2a and Fig. 1). J.dPro exhibited a delayed and markedly diminished cytoplasmic calcium increase in response to anti-CD3 stimulation (Fig. 1b,c). The calcium response was also affected, albeit much more moderately, in a heterozygous Jurkat variant clone (called J.Het hereafter), which contained only one allele of a nearly identical internal deletion of LAT (Fig. 1b,c, and Supplementary Fig. 2b). Immunoblot analysis further revealed TCR signaling defects in J.dPro mutant cells (Fig. 1d). The phosphorylation of LAT, as assessed on either Y132 or Y191, was markedly impaired in J.dPro cells. Presumably, as a consequence of its recruitment to LAT phospho-Y132, phosphorylation of the phospholipase PLC-γ1 was likewise diminished and markedly delayed, consistent with the defective calcium responses observed. As expected, CD3-mediated signaling events upstream of LAT were intact, as evidenced by anti-CD3 stimulation of the mutant J.dPro cells induced tyrosine phosphorylation of the Zap70 activation loop (phospho-Y493) and phosphorylation of SLP76. These results suggest that this evolutionarily conserved proline-enriched membrane-proximal region of LAT is critical for productive TCR signaling through LAT.
PIPRSP motif in LAT facilitates LAT’s phosphorylation
The enrichment of prolines in the deleted segments of LAT suggested functional importance, but may be explained by two hypotheses. First, perhaps through yet unknown structural properties of LAT, the proline-enriched segment in LAT may provide a minimal distance away from the plasma membrane necessary for TCR induced phosphorylation of its more C-terminal tyrosines. Alternatively, proline-enriched peptides may have important roles in intracellular signaling through their ability to interact in a sequence specific context, for instance, with SH3 domains22. Thus, the proline-rich region in LAT may interact with an SH3-containing protein that facilitates the ability of LAT to interact with Zap70, to relocate to engaged TCR complexes, or to assemble LAT clusters which might promote LAT phosphorylation. To distinguish between these possibilities, we explored the Eukaryotic Linear Motif resource to identify potential functional SH3 domain-binding sites in the membrane-proximal segment of LAT23. There were three potential proline-rich motifs in LAT predicted to interact with SH3 domains (Fig. 2a). Using CRISPR/Cas9, we generated a LAT-deficient Jurkat clone (J.LAT, Supplementary Fig 2c,d), and reconstituted these cells with cDNAs encoding wild-type LAT or mutant LATs in which these three proline-enriched motifs had proline to alanine substitutions (Fig. 2a). Alanine mutations at the most evolutionarily conserved C-terminal PIPRSP motif markedly abrogated its ability to reconstitute a substantial calcium increase following anti-CD3 stimulation (Fig. 2b,c). Cells expressing the mutant AIARSA motif exhibited impaired TCR-induced phosphorylation of LAT at both Y132 or Y191 residues and diminished phosphorylation of PLC-γ1, whereas the other more N-terminal motif mutant motifs (AWAA or AAYAA) did not substantially impair tyrosine phosphorylation of these signaling proteins (Fig. 2d). The AIARSA mutation consistently impaired TCR induced phosphorylation of LAT and PLC-γ1 over a wide range of titrated CD3 stimuli (Supplementary Fig. 3a). Interestingly, in time course experiments, the responses of cells expressing the AIARSA mutant motif eventually caught up to the responses observed with wild-type LAT (Supplementary Fig. 3b). Since these site-directed mutagenesis experiments preserved the length of mutant LAT, the PIPRSP motif in LAT likely interacts with other signaling molecules to promote LAT phosphorylation, excluding the possibility that the proline rich region only functions as a structural spacer.
Fig. 2. A PIPRSP motif in LAT promotes the phosphorylation of LAT.
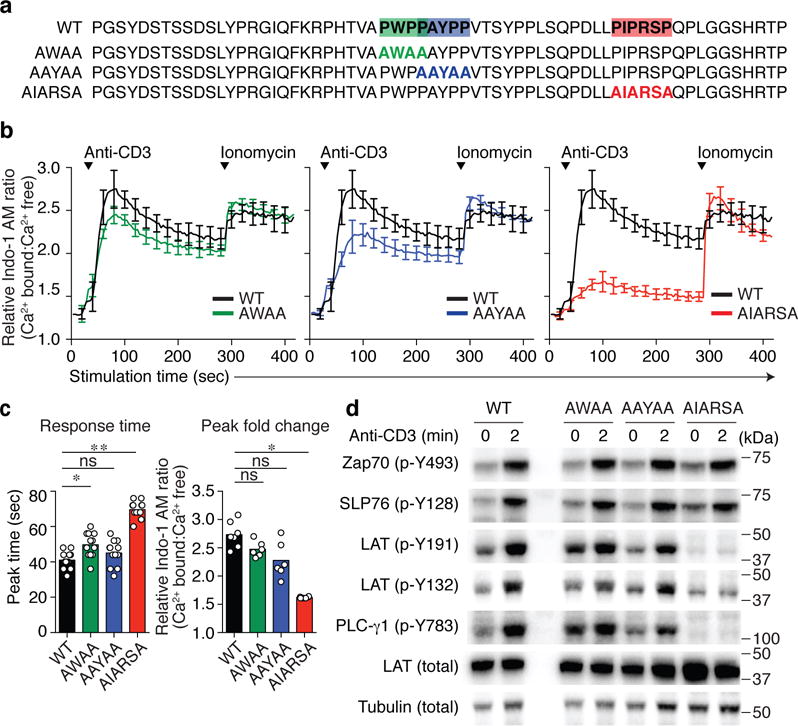
(a) Location of three proline-rich motifs, PWPP, PAYPP and PIPRSP, highlighted in green, blue and red respectively. Proline to alanine mutations in each of the motifs are shown below the wild type sequences (WT) of human LAT. (b) CRISPR/Cas9 generated LAT-deficient J.LAT cells were reconstituted with wild type LAT (WT), or each proline-rich motif mutant LAT (AWAA, AAYAA, or AIARSA). Cells were loaded with Indo-1 AM, stimulated with 0.5 μg/ml of anti-CD3 and the changes in relative calcium-sensitive fluorescence ratios over time for 400 sec are shown in b (mean ± s.d.; n = 6 technical replicates). Data are representative of five independent experiments with similar results. (c) Bar graph (left) shows the analysis of response time to reach the peak (mean ± s.d.; n = 12 technical replicates in five independent experiments). Experiments have been done at least five times with similar results. *P = 0.0029, **P < 0.0001 (exact value). ns, not significant; two-tailed Mann-Whitney test. Bar graph (right) shows the quantification of the Indo-I AM ratio fold change in peak (mean ± s.d.; n = 7 technical replicates in two experiments for WT, n = 6 technical replicates in two experiments for AWAA, AAYAA, AIARSA). Data are representative for at least five independent experiments. *P = 0.0012, ns, not significant; two-tailed Mann-Whitney test. (d). J.LAT cells reconstituted with WT, or mutant AWAA, AAYAA, AIARSA LAT were left unstimulated or treated with 0.5 μg/ml anti-CD3 for 2 min. Immunoblot analyses are shown. Data are representative of three experiments. Numbers to the right of cropped blots indicate mobilities of molecular mass marker proteins (kDa).
Thymic selection signals require the PIPRSP motif in LAT
To further examine the functional significance of the PIPRSP motif in LAT, we utilized a mouse model to explore whether primary mouse T lineage cells also rely on the PIPRSP motif-containing LAT for pre-TCR and TCR signal transduction during thymocyte development. During thymic development, immature thymocytes require pre-TCR and TCR signals of appropriate strength to pass β selection (mediated by the pre-TCR) and positive/negative selection (mediated by the mature TCR)24, 25. LAT-deficient mice exhibit a severe developmental block at the β-selection stage, resulting from the inability of immature CD4−CD8− double negative (DN) thymocytes that express the pre-TCR to transmit signals necessary for downstream translational and transcriptional programs required for proliferation and differentiation26, 27. Thus, LAT-deficient mice provided a useful model to evaluate the functional ability of a PIPRSP motif mutant LAT in vivo in restoring LAT function during development. We transduced CD45.2+Sca-1+c-Kit+ LAT-deficient hematopoietic stem cells with lentiviruses expressing either wild-type LAT-P2A-mCherry or the mutant AIARSA LAT-P2A-mCherry. The mCherry was produced through the self-cleaving P2A peptide downstream of LAT, allowing us to identify cells expressing wild-type or mutant LAT (Fig. 3a). The comparable mean fluorescence intensities of mCherry expression, together with our studies of this mutant in the Jurkat line (Supplementary Fig. 3) suggested the expression of the wild-type or AIARSA LAT were similar (Fig. 3a). After 6 – 8 weeks, expression of wild-type LAT successfully rescued LAT-deficient thymocytes from the LAT-deficient developmental block (Fig. 3b), whereas untransduced donor HSCs (mCherry negative cells) remained blocked at the DN stage (data not shown). Thymocytes expressing wild-type LAT completed β and positive selection, and matured into single-positive CD4+ or CD8+ (CD4SP or CD8SP) thymocytes (Fig. 3b–d and Supplementary Fig. 4a). In marked contrast, the expression of the mutant AIARSA LAT allowed only some DN immature thymocytes to pass β selection and become DP cells, albeit inefficiently, resulting in a relative accumulation of DN cells. Analysis of AIARSA-expressing DN subpopulations revealed a significant decrease in the DN4 stage and concomitant increase in the DN3 stage, suggesting a partial block in DN3 to DN4 transition where β selection takes place (Fig 3e,f and Supplementary Fig. 4b). The lack of the proline-rich motif in LAT also reduced positive selection as evidenced by a significant two-fold decrease in the CD4SP thymocyte population, and a similar loss in CD24−TCR+ mature CD8SP populations (Fig. 3b–d). During the positive-selection process, immature thymocytes upregulate expression of TCR and CD69, becoming TCRhi and CD69+. In cells expressing the AIARSA mutant LAT, an approximate three-fold reduction of TCRhiCD69+ DP cells was observed (Fig 3g,h and Supplementary Fig. 4c).
Fig. 3. Ectopic expression of LAT containing the PIPRSP mutant motif, AIARSA, impairs thymocyte β- and positive-selection in vivo.
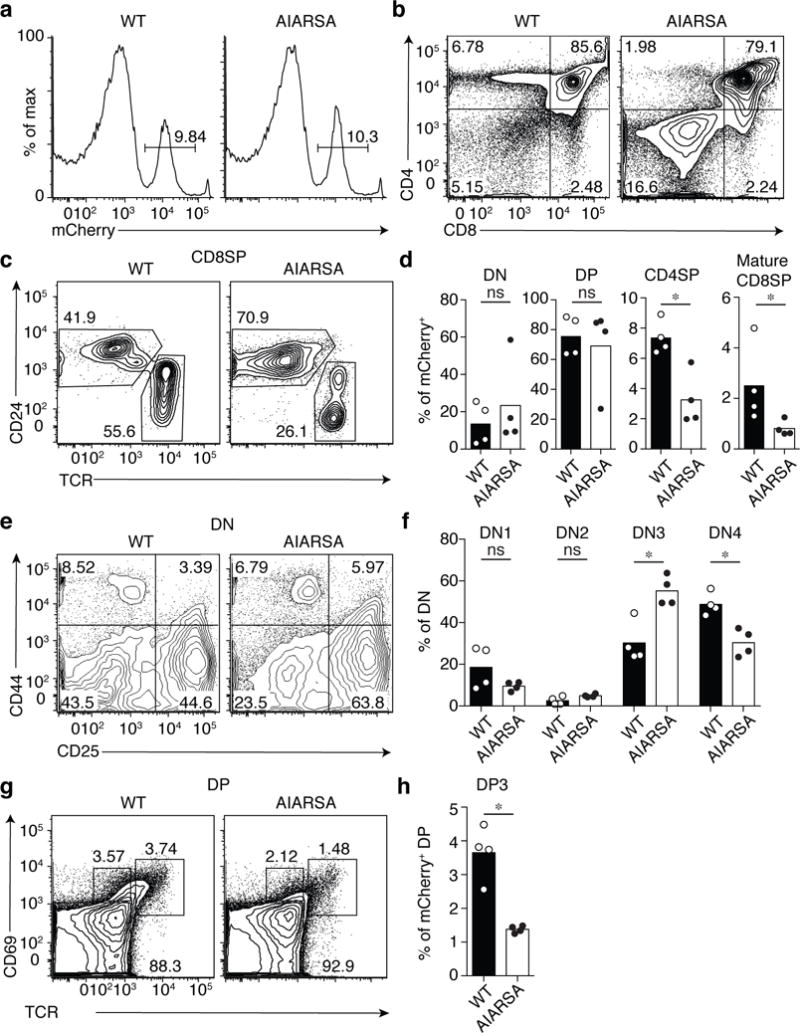
Flow cytometric analysis of thymocytes from lethally irradiated bone marrow BoyJ (CD45.1+) chimeras reconstituted with CD45.2+ LAT-deficient B6 hematopoietic stem cells transduced with lentiviruses expressing either WT LAT-P2A-mCherry (WT; n = 4 recipients) or mutant AIARSA LAT-P2A-mCherry (AIARSA; n = 4 recipients). Recipient mice were analyzed 6 – 8 weeks later. (a) Representative mCherry expression of thymocytes, gated on CD45.2+ cells. Numbers indicate percentages of mCherry+ cells in each gate. (b) Flow cytometric analysis of CD45.2+ mCherry+ thymocytes. Numbers in each quadrant indicate percent of cells in each. (c) Flow cytometry analysis of CD24 versus TCR expression of CD45.2+ mCherry+ CD8SP thymocytes. (d) Bar graphs representing the frequencies of DN, DP, CD4SP and CD24−TCR+ mature CD8SP thymocytes among CD45.2+ mCherry+ cells. Each symbol represents an individual mouse. Bars indicate the mean± s.d. n = 4 independent animals in two independent experiments. *P = 0.0286; ns, not significant; two-tailed Mann-Whitney test. (e) Flow cytometric analysis of DN1 to DN4 cell development based on CD44 and CD25 expression, gated on CD45.2+ mCherry+ DN thymocytes. (f) Frequency of DN1 (CD4+CD25+), DN2 (CD44+CD25+), DN3 (CD44−CD25+), DN4 (CD44−CD25−) of CD45.2+ mCherry+ DN T cells in each quadrant of (e). Bars indicate the mean± s.d. n = 4 independent animals in two independent experiments. *P = 0.0286; ns, not significant; two-tailed Mann-Whitney test. (g) Flow cytometric analysis of DP progression through thymic positive selection based on CD69 and TCR expression, gated on CD45.2+ mCherry+ DP thymocytes. (h) Frequency of post-selection DP3 (CD69+TCRhi) DP cells. Bars indicate the mean± s.d. n = 4 independent animals in two independent experiments. *P = 0.0286; two-tailed Mann-Whitney test. All data are representative of two independent experiments with similar results.
When DP thymocytes were stimulated by crosslinking anti-CD3 mAb, cells expressing the AIARSA LAT mutant exhibited a markedly reduced calcium response (Fig. 4a), and impaired induction of Erk phosphorylation (Fig. 4b). Expression of CD5 can also be used as a surrogate marker for the TCR signal strength during thymic development. Despite similar TCR surface expression, CD4SP and CD8SP cells expressing the AIARSA LAT mutant had substantially lower CD5 protein expression (Fig. 4c), supporting the notion that the AIARSA LAT mutant transduces weaker downstream signals. Thus, the absence of the PIPRSP motif in LAT led to partial developmental blocks at β and positive selection, presumably due to the inefficient LAT-dependent signal transduction. These data establish the importance of the PIPRSP proline-rich motif of LAT in T cell lineage development in vivo.
Fig. 4. Mutation of the PIPRSP motif in LAT impedes TCR signal transduction of DP thymocytes.
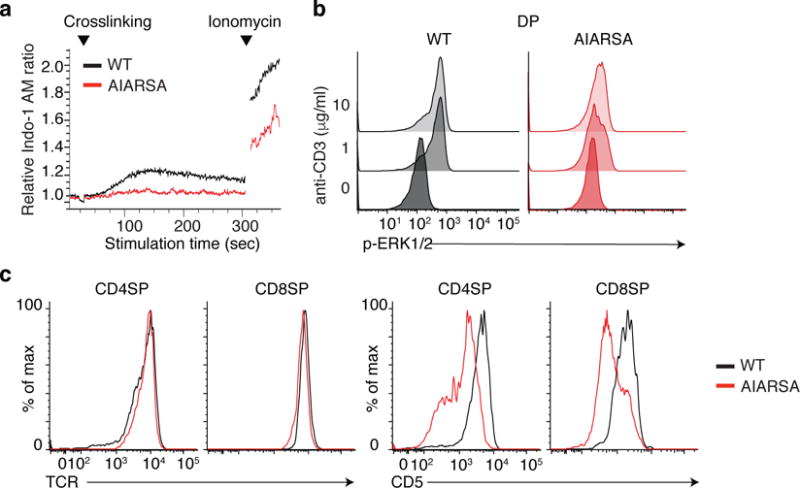
(a) Flow cytometric analysis of calcium-sensitive fluorescence changes of CD45.2+mCherry+ DP T cells, loaded with Indo-I AM and stimulated with crosslinked anti-CD3 over time. (b) Flow cytometric analysis of phosphorylated Erk in CD45.2+mCherry+ DP thymocytes, in response to crosslinked anti-CD3 stimulation. (c) Flow cytometric analysis of surface expression of the TCR (left) or CD5 (right) in CD45.2+ mCherry+ CD4SP or CD8SP thymocytes. Experiments were repeated independently twice with four independent animals and yielded similar results.
In peripheral lymphoid tissues, we did not observe significant differences in the frequencies of CD4+ or CD8+ T cell populations in AIARSA-expressing versus wild-type LAT-expressing bone marrow chimeras (Supplementary Fig. 5a). However, the expression of AIARSA consistently resulted in a lower CD5 expression in peripheral CD4+ and CD8+ T cells as was seen in thymocytes, although to a lesser extent (Supplementary Fig. 5b). In AIARSA LAT-expressing CD4+ and CD8+ T cells, the CD44hi CD62Llo memory-like population was considerably enlarged compared to the wild-type LAT-expressing T cells (Supplementary Fig. 5c,d). The enlarged memory-like population may be a consequence of increased peripheral homeostatic proliferation in the AIARSA LAT-expressing bone marrow chimeras. Thus, the mutations in PIPRSP motif in LAT impaired thymic selection, leading to fewer CD4SP and CD8SP cells developed in the thymus, and may have contributed to lymphopenia-induced T cell proliferation in the periphery.
Lck associates with LAT in a PIPRSP motif-dependent manner
Our data demonstrate the importance of the PIPRSP motif in LAT in TCR signal transduction. Absence of the PIPRSP motif impaired the tyrosine phosphorylation of LAT itself, and the activity of downstream signaling pathways important for T cell function and development. To explore the potential mechanism underlying this phenotype, we evaluated the role of the PIPRSP motif in mediating protein interactions with LAT. We stimulated the TCR on Jurkat cells expressing wild-type or AIARSA LAT proteins containing C-terminal Myc epitope tags, isolated LAT and its interacting proteins by immunoprecipitation, and utilized mass spectrometry to analyze the profiles of LAT-associated proteins. Mass spectrometry identified many proteins that were differentially represented in wild-type or AIARSA LAT-associated interactomes (Supplementary Table 2). Surprisingly, only 10 of these proteins contained SH3 domains (Supplementary Table 3). Among these, Lck was noteworthy because of the abundance of its peptides, and because it exhibited differential capability to interact with the proline-rich PIPRSP motif (Supplementary Fig. 6a). A previous study of the importance of the SH3 domain of Lck in downstream TCR signaling was of particular interest28. When a mutation at Trp97, which disrupts the function of the Lck SH3 domain, was introduced into mice, the mutation resulted in a significant decrease in β selection and positive selection in thymocytes as well as reduced tyrosine phosphorylation of LAT, PLC-γ1 and Erk28. In contrast, the phosphorylation of Zap70 and SLP76 was unperturbed. These results are consistent with our findings with the AIARSA LAT mutant. Moreover, immunoblot analysis of LAT immunoprecipitates from TCR-stimulated cells further demonstrated that Lck inducibly associated with wild-type LAT to a greater degree than the AIARISA mutant LAT (Supplementary Fig. 6b). Taken together, our results suggested Lck is an SH3-containing protein that interacts with LAT’s PIPRSP motif and may mediate a functionally important interaction.
Lck bridges LAT and Zap70 to enhance LAT phosphorylation
Given the concordance of our results with the AIARSA mutant LAT and previous studies on the inactivation of the Lck SH3 domain, we hypothesized a mechanism to explain the role of the proline-rich PIPRSP motif in LAT for TCR signal transduction (Supplementary Fig. 6c). In our model, upon TCR engagement, Lck may play multiple roles dictated by both its catalytic and non-catalytic biochemical activities. First, in response to coreceptor-pMHC-TCR engagement, Lck has its well-established function as the protein tyrosine kinase that phosphorylates ITAM motifs in CD3- and ζ-chains which lead to Zap70 recruitment. Second, it activates Zap70 by tyrosine phosphorylation. Third, Lck operates as a scaffold protein that stabilizes the active form of Zap70 through the previously described interaction between its SH2 domain with phospho-Y319 in Zap70, thereby also maintaining Lck in its open active conformation. Fourth, as described here, the SH3 domain of Lck binds the PIPRSP motif of LAT. Thus, through the latter two functions, Lck might act as a bridging molecule facilitating the interaction of Zap70 with LAT and promoting LAT phosphorylation.
To test this potential bridging function of Lck, we expressed Lck, LAT, and Zap70 in a heterologous cell system. Human embryonic kidney cells 293 (HEK293), which do not express Lck, Zap70 or LAT, were used to study the mechanism of the putative interactions and functional consequences, i.e., LAT phosphorylation. To separate the dual roles of Lck, we utilized a kinase-inactive form of Lck (a Lys273 to Arg mutation in the catalytic domain) that can only function as a scaffold protein (Supplementary Fig. 7a). We also took advantage of a recently identified mutation in Zap70, Lys362 to Glu, which disrupts autoinhibition and results in a weak activating effect of Zap70 catalytic activity (Supplementary Fig. 7a). To test our hypothesis, we expressed LckK273R, Zap70K362E, with either wild-type or mutant AIARSA LAT in HEK 293 cells, and examined the phosphorylation of two tyrosines on LAT, Y132 and Y191 (Supplementary Fig. 7b). We titrated the transfection ratio of Zap70 and LAT, and identified a dose range where the weakly-activated Zap70 displayed minimal phosphorylation of LAT. In the presence of the catalytically inactive LckK273R, the phosphorylation of LAT Y132 and Y191 residues by Zap70K362E were considerably increased (Fig. 5b). Notably, the increased phosphorylation of LAT highly depended on its PIPRSP motif, since the proline to alanine mutations impaired the phosphorylation of LAT (Fig. 5b). In addition to the PIPRSP motif in LAT, the binding function of both the SH3 and SH2 domains of catalytically inactive Lck were pivotal for its enhancing effect on the phosphorylation of LAT (Fig. 5c). Inactivating point mutations of either the SH3 or the SH2 domain of catalytically inactive Lck diminished phosphorylation of LAT. Thus, the SH3 domain of Lck, which binds to the PIPRSP proline-rich motif of LAT, and the SH2 domain of Lck, which binds to pY319 in interdomain B of Zap7015, together potentially facilitate the formation of a transient LAT-Lck-Zap70 complex. Interestingly, the catalytically inactive Src-family kinase FynK299R, also expressed in T cells, did not exhibit a similar enhancing effect on the phosphorylation of LAT (Fig. 5c). Although structurally similar to Lck, having SH3 and SH2 domains, the binding specificities of these domains of Fyn are different from those of Lck29. Thus, the interaction between the SH3 domain of Lck and PIPRSP motif of LAT exhibits a degree of specificity. These results support our hypothesis that upon T cell stimulation, Lck interaction with PIPRSP motif of LAT may facilitate the ability of Zap70 to phosphorylate LAT.
Fig. 5. The interaction of the Lck SH3 domain and the LAT PIPRSP motif enhances Zap70-dependent phosphorylation of LAT.
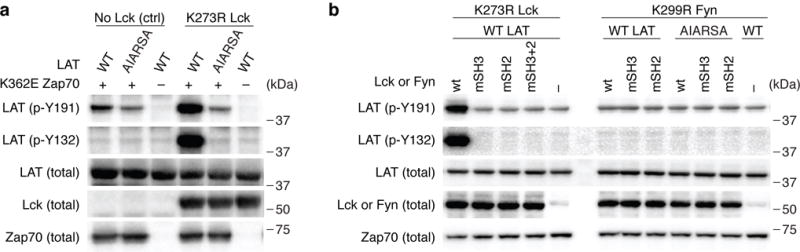
A reconstituted system in HEK 293 cells was used to examine the regulation of LAT phosphorylation (also in Supplementary Fig. 7). Transient transfection of WT or the AIARSA mutant LAT, Zap70K362E, and/or LckK273R or FynK299R were performed in HEK 293 cells as indicated. Data are representative of at least three independent experiments. (a) Immunoblot analysis of phospho-LAT on Y132 and Y191 in HEK293 cells reconstituted with WT or AIARSA mutant LAT, along with the presence of kinase inactive Lck (K273R mutant) or control protein GFP, and weakly-autoactivated Zap70 (K362E mutant). Data are representative of three experiments. (b) Immunoblot analysis of phosphorylation of LAT in HEK 293 cells reconstituted with WT or the AIARSA mutant LAT, Zap70K362E, as well as LckK273R or FynK299R with additional mutations of SH3 or SH2 domains as indicated above the blots. wt: kinase inactive Lck or Fyn with functional SH3 and SH2 domains (LckK273R or FynK299R); mSH3: mutant SH3 domain (LckW97A.K274R or FynW119A.K299R); mSH2: mutant SH2 domain (LckR154K.K273R or FynR176A.K299R); mSH3+2: mutant SH3 and SH2 domain (LckW97A.R154K.K274R). Data are representative of three experiments.
PIPRSP motif promotes localization of LAT to pMHC-TCR
To explore the possibility that TCR stimulation by agonist pMHC triggers the relocalization of LAT to engaged TCR complexes during antigen recognition, we took advantage of engineered Jurkat cells that express the murine OT-I TCR and human CD8 which either do or do not express human Lck (J.OT-I.hCD8.Lck KO or J.OT-I.hCD8.Lck-FLAG, respectively). The co-expression of human CD8 facilitates the interaction of mouse OT-I TCR with mouse MHCI H-2Kb (Supplementary Fig 8a,b), with comparable affinity as mouse CD830. We stimulated each of these cell lines with H-2Kb OVA MHC class I tetramers, and used anti-ζ chain mAb to immunoprecipitate proteins that associated with engaged coreceptor-TCR complexes. First, the use of the Lck-deficient line demonstrated a requirement for Lck in inducible phosphorylation of the ζ-chain and associated proteins. In cells expressing Lck, H-2Kb OVA tetramers induced signaling, as evidenced by ζ-chain phosphorylation and the induction of interacting phosphoproteins (Fig. 6a). We observed TCR stimulation induced-association of Zap70 and Lck with the ζ-chain immunoprecipitates (Fig. 6a). Moreover, we detected an increase in phospho-LAT in these ζ-chain immunoprecipitates. These data are consistent with LAT relocalization to the stimulated TCR complexes, where activated Zap70 and Lck both are localized. With mouse OT-II CD4+ T cells, we also observed similar results (Fig. 6b). Furthermore, we used CRISPR/Cas9 to generate LAT-deficient J.OT-I.hCD8.Lck-FLAG Jurkat variants, and then reconstituted cells with either wild-type or the AIARSA mutant LAT. With H-2Kb OVA tetramer stimulation followed by ζ-chain immunoprecipitation, wild-type LAT, but not AIARSA LAT, associated with ζ-chain (Fig. 6c). This is consistent with our working model in which the PIPRSP motif of LAT may contribute to relocalization of LAT to engaged TCR complexes.
Fig. 6. TCR engagement triggers the association of LAT with TCR signaling complexes.
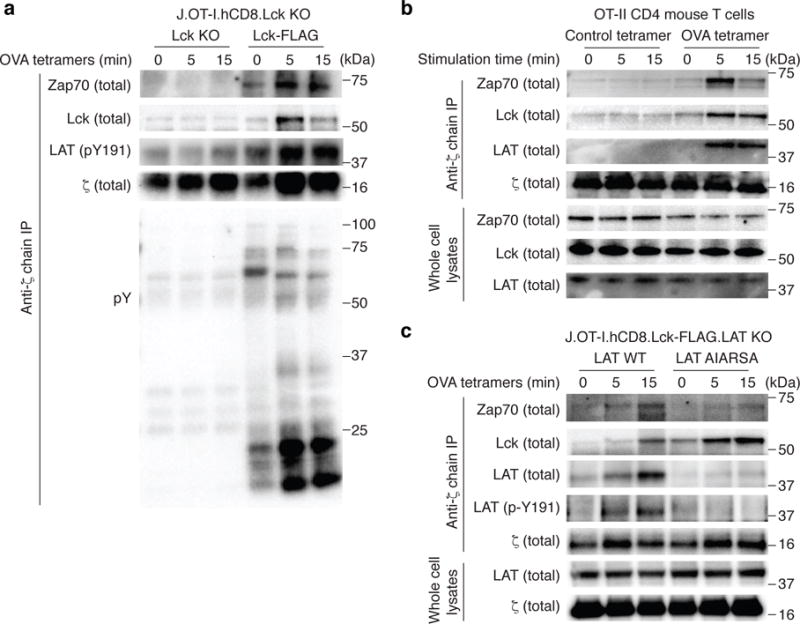
(a). Lck deficient J.Lck cells that were reconstituted with mouse OT-I TCR, human CD8, with or without the FLAG-tagged human Lck. Cells were stimulated with H-2Kb OVA tetramers (SIINFEKL) for the indicated times. Lysates were prepared and immunoprecipitated with anti-ζ chain mAb (6B10 clone). Immunoprecipitates were analyzed by immunoblot and probed for Zap70, FLAG (Lck), phospho-LAT, and phospho-tyrosine as indicated. Data are representative of three experiments. (b). Mouse OT-II CD4 T cells were stained with H-2Ab OVA tetramers or control tetramers on ice, and stimulated by warming at 37°C for the indicated times. Following lysis in NP-40, lysates were immuoprecipitated with anti-ζ chain mAb which were performed and analyzed with immunoblots as in (a). Data are representative of two experiments. (c). LAT-deficient J.OT-I.hCD8.Lck-FLAG cells were reconstituted with wild type or AIARSA LAT. Cells were stimulated, lysed and immunoprecipitated with anti-ζ chain mAb as in (a). Immunoprecipitates or whole cell lysates were analyzed by immunoblot and probed for Zap70, FLAG (Lck), LAT, and phospho-LAT as indicated. Data are representative of four experiments.
DISCUSSION
Our study revealed a mechanistic explanation for how Zap70 phosphorylates one of its key substrates, LAT. This process is mediated by an unanticipated scaffolding function of Lck, a kinase whose catalytic activity is critical to many events in the initiation of TCR signaling. In our model, every structural feature of Lck is specifically accommodated in TCR recognition of agonist pMHC. First, the acylation sites of Lck provide membrane anchorage. Second, the cysteine-rich motif within its unique domain provides its linkage to CD4 or CD8 coreceptors. Third, the kinase domain promotes the phosphorylation of ITAMs and Zap70, leading to the eventual catalytic activation of Zap70. The Lck SH2 domain, which can interact with Y505 to promote autoinhibition, can also bind to phospho-Y319 in Zap70 to maintain the open and active state of both Zap70 and Lck during TCR signaling. The Lck SH3 domain not only participates in its autoinhibition, but as shown here also provides a unique link to the proline-rich motif in LAT, thereby recruiting activated Zap70 to LAT. Accommodating every structural feature of an SFK in a single cellular process is rare and suggests that Lck has uniquely evolved to serve these functions in TCR recognition and signaling. Since Lck is associated with coreceptors, which are involved in the recognition of pMHC, our model envisions that Zap70 and LAT are linked to TCR recognition of pMHC by the ability of Lck to function both as a kinase and as a bridging scaffold that orchestrates efficient TCR signaling.
The segment of LAT which links its transmembrane segment to key tyrosines had no previously identified function. We found the evolutionarily conserved segment contained one key functionally important proline rich motif, PIPRSP. Mutation of this motif partially impeded TCR signaling and calcium responses. This PIPRSP motif supported efficient T cell development and function, as evidenced by bone marrow chimera studies. LAT is a major TCR signaling hub that coordinates many signaling effectors; thus, the role of PIPRSP motif in facilitating LAT phosphorylation contributes to a pivotal event in TCR signal transduction.
The T cell developmental and functional deficiency resulting from the expression of the proline motif mutant LAT was remarkably consistent with a previously published study. An inactivating SH3 domain mutant of Lck (LckW97A) impeded thymocyte development, resulting from partial blocks in β and positive selection28. Although similar numbers of peripheral T cells in LckW97A mice as in wild-type mice were reported, the mutant cells were functionally impaired, as evidenced by diminished LAT and PLCγ1 phosphorylation31. These results are consistent with ours, and support our proposed interaction between Lck and LAT during the initiation phase of TCR signal transduction. Notably, SLP76 was normally phosphorylated in both studies and may reflect the role of another adaptor proteins, such as CD632.
Thymocytes reconstituted with the AIARSA mutant LAT exhibited impairments in ability to transition across pre-TCR and TCR developmental checkpoints. However, T cells in the periphery exhibited some evidence of accumulation of effector memory cells. This could reflect the effects of an altered repertoire being inefficiently selected in the setting of impaired TCR signaling, which also mirror previous work on dysfunctional LAT mutant, such as LAT Y136F knock-in mice33,34. T cells can utilize LAT-independent signaling pathways to promote the activation of Erk35–42, and could explain why the AIARSA mutant in LAT had less impact on the phosphorylation of Erk.
Signaling by the TCR critically depends on the integrated functions of the Lck and Zap70 kinases. The Zap70 kinase uses its tandem SH2 domains to be recruited to the phosphorylated ITAMs of the stimulated TCR. Protein interaction domains of cytoplasmic tyrosine kinases were thought to be responsible for their substrate specificity by conferring their colocalization to substrates. Recent studies of Zap70 suggests the kinase domain alone is sufficient for substrate specificity14. However, it was not clear how activated Zap70, bound to the TCR, efficiently interacts with its substrate, LAT. Recent imaging studies suggested that activated Zap70 is inducibly released from ITAM motifs19. A mobile but membrane associated active pool of Zap70 may diffuse to and possibly phosphorylate distant LAT molecules, allowing multiple Zap70 molecules that transiently interacted with the stimulated TCR to amplify signaling. Whereas it is clear from our study that the LAT proline rich motif is important for phosphorylation of LAT by Zap70, we cannot exclude the possibility that a TCR independent complex of Zap70 and Lck, or Lck and LAT, might also exist distal from the pMHC-bound TCR. However, the proposed release of Zap70 uncouples its function from TCR recognition of agonist pMHC and does not offer it a means of protection from inactivation by cellular phosphatases or ubiquitin ligases when released from the TCR, raising concerns regarding how Zap70 would remain active. Moreover, our biochemical data with pMHC tetramer stimulation suggested LAT was recruited to the pMHC-stimulated TCR complex which also contained Lck and Zap70. This argues that at least some LAT interacted directly with the stimulated TCR complex. Further work will be needed to assess the site at which Zap-70/Lck/LAT form a functional signaling unit and the stability and the longevity of these interactions. A recent report also suggested the potential interaction between Lck and LAT may fine tune proximal TCR signaling43. Nonetheless, the evolutionarily conserved and functionally important proline-rich motif in LAT that we identified provides previously unappreciated insights into how components of the initial TCR signal machinery that is assembled transduces information downstream via newly identified direct interactions.
METHODS
Mice and cell lines
Mice used in these studies were housed in the specific pathogen-free facilities at the University of California, San Francisco, and were treated according to protocols that were approved by UCSF animal care ethics and veterinary committees, and are in accordance with NIH guidelines. The LAT-deficient mouse line (on a C57BL/6 background) was a gift from L. Samelson and C. Sommers (NIH). BoyJ (CD45.1) mice were obtained from Jackson (B6.SJL-Ptprca Pepcb/BoyJ). The human leukemic Jurkat T cell line, or Jurkat variants with LAT deficiency or knock-in mutations were maintained in RPMI culture medium supplemented with 5% fetal bovine serum and 2 mM glutamine. Cells lines that were reconstituted with wild-type LAT or various mutant LAT constructs were maintained in RPMI culture medium supplemented with 5% fetal bovine serum, 2 mM glutamine and 0.5 mg/ml of the aminoglycoside geneticin (G418). HEK293 cells were obtained from the ATCC and maintained in DMEM supplemented with 10% fetal bovine serum and 2 mM glutamine.
Compilation and analysis of orthologous LAT sequences
The mammalian LAT sequences used to generate Fig. 1 and Supplementary Fig. 1 were compiled through a series of protein-protein BLAST searches in the NCBI non-redundant protein database as previously described14. A total of 42 mammalian LAT sequences were identified. Sequence logos were generated using the online tool WebLogo44.
Generation of LAT deficient or knock-in mutant Jurkat variants
The Jurkat variants J.LAT (deficiency in LAT), J.dPro and J.Het (hemizygous or heterozygous mutant LAT that lacks membrane-proximal, proline-rich region in LAT) were generated by transiently expressing Cas9, a guide sgRNA against the LAT proline-rich region and ssODN repair template, as previously described44. In brief, guide sgRNAs were cloned into the pU6-(BbsI)_CBh-Cas9-T2A-BFP vector (Addgene Plasmid #64323), and electroporated into Jurkat T cells along with a ssODN repair template. Single cells were sorted into 96-well plates the next day. Successful knock-in mutant clones were selected by screening with immunoblots probed for anti-C terminal LAT antibody (Santa Cruz Biotechnology, clone # M19, catalog # sc-5320), as shown in Supplementary Fig 1. LAT deficient clones (J.LAT) were also selected from the screen. Genomic DNA of J.LAT, J. dPro and J.Het cells were purified and used as templates for genotyping. PCR products amplified from genomic DNA of J.Pro and J.Het were TOPO cloned into pCR2.1 vectors, and prepared for sequencing. Additionally, cDNA products of J.dPro and J.Het cells were prepared, and LAT cDNA from J.Pro or J.Het cells were amplified by PCR reactions, TOPO cloned, and sequenced. Oligonucleotides used in these studies are as followings: LAT sgRNA sense: 5′-CACCGCCATCTTCCCGGCGGGATTCTG A-3′; antisense: 5′-AAACTCAGAATCCCGCCGGGAAGATGGC-3′; LAT ssODN HDR template: 5′-TCCTGCCTCACCAGCCCTCTCTTTCCCAGGCTCCTACGACTACCCATACGATGTTCCAGATTACGCTAGCACATCCTCAGATAGGTGAGTCCGCCCCAGCATAGGCCTGGCCTGAGCTGACTTAGTCTCCCTCTCACCCTCTCTTTGAAGCCAACAGTGTGGCGAGCTACGAGAACGAGGGTGCGTCTGGGATCCGAGGT-3′; LAT gDNA screen sense: 5′AGGTGAGTGGGAAACTGGTG-3′; antisense: 5′-GCCTGGGTTGTGATAGTCGT-3′; LAT cDNA screen sense: 5′-GCTCCTGCTGCTGCCCATCC-3′; antisense: 5′-AGTCTTAGCCGCTCCAGGAT-3′
Reconstituted LAT-deficient J.LAT cells with LAT mutants
Reconstituted LAT-deficient (J.Lat) lines were generated by electroporating J.Lat cells with a pEF vector to express WT LAT, AWAA, AAYAA, AIARSA mutant LAT followed by selection with 2 mM G418. Individual clones were single cell sorted into 96-cell plates, and later assessed for comparable CD3 expression by flow cytometry analysis and comparable LAT expression assessed by immunoblot analysis.
Intracellular calcium measurements
Jurkat cells and LAT mutant variants were washed with PBS twice, and loaded with 1 μM Indo-1 AM calcium indicator dye (Thermo Fisher Scientific #I1223) at 37 °C for 30 min in RPMI medium. After loading with Indo-I, cells were washed with PBS twice, and transferred into 96 well plates. Changes in the fluorescence ratio (violet/blue) following the addition of anti-CD3 (OKT3 clone) or ionomycin were recorded using a Flex Station II (Molecular Probes) using SoftMax Pro software. Data were imported into GraphPad Prism software for analysis and production of graphs. Calcium responses in primary thymocytes from bone marrow chimera studies were analyzed by flow cytometry. Cells were loaded with 1.5 μM Indo-I AM at 37 °C in RPMI medium supplemented with 5% fetal bovine serum for 30 min. After loading with Indo-I, cells were stained with CD4, CD8, and CD45.2 antibodies. Cells were analyzed by flow cytometry and stimulated with 1 μg/ml anti-CD3 (clone 145-2C11), followed by crosslinking with 50 ng/ml goat anti-Armenian hamster IgG antibodies (Jackson ImmunoResearch #127-005-099). Change in intracellular calcium concentration were monitored as the ratio of Indo-I (blue/violet) and displayed as a function of time.
Immunoblot analysis
Jurkat and derivative cells were washed with PBS, and resuspended at 5 × 106 cells/ml and rested for 30 min at 37 °C. Cells were left unstimulated or stimulated with anti-CD3 (OKT3) over time as described in each experiment. Cells were lysed by directly adding 10 % NP-40 lysis buffer to the final concentration of 1% NP40 (containing inhibitors of 2 mM NaVO4, 10 mM NaF, 5 mM EDTA, 2 mM PMSF, 10 μg/ml Aprotinin, 1 μg/ml Pepstatin and 1 μg/ml Leupeptin). Lysates were placed on ice and centrifuged at 13,000 g to pellet cell debris. Supernatants were run on NuPAGE 4%–12%, 8% or10% Bis-Tris Protein Gels (ThermoFisher Scientific) and transferred to PVDF membranes using a Trans-Blot Turb Transfer System (Bio Rad). Membranes were blocked using TBS-T buffer containing 3% BSA, and probed with primary antibodies as described, overnight at 4 °C or 2 h at room temperature 25°C. The following day blots were rinsed and incubated with HRP-conjugated secondary antibodies. Blots were detected using a chemiluminescent substrate and a BioRad Chemi-Doc imaging system.
Production of lentivirus expressing WT or AIARSA LAT
Murine WT LAT was cloned into the pHR backbone under the expression of EF1A promoter. Murine AIARSA LAT mutant was generated using a QuickChange Lightening site-directed mutagenesis kit (Agilent #210518). A C-terminal P2A self-cleaving peptide followed by mCherry was incorporated to assess transduction efficiency and expression levels. Packaging vector pCMV dR8.91, envelope vector pMD 2.G, and pHR Lck constructs were transiently co-transfected into LX-293T cells using TransIT-LT1 reagent (Mirus Bio Mio2300). Supernatants containing virus particles were collected 48 h after transfection, filtered and concentrated by PEG 8000 precipitation. The virus particles were resuspended in PBS, and stored at −80°C.
Bone marrow chimeras
CD45.2+ B6 hematopoietic stem cells were enriched by EasySep Mouse Hematopoietic Progenitor Cell Enrichment Kit (StemCell Technology #19856), stained for c-Kit and Sca-1, and sorted to enrich an c-Kit+Sca-1+ population on an FACSAriaII. Hematopoietic stem cells were resuspended at a density of 1 × 104 cells per 200 μl DMEM-F12, containing 10% FCS, 1× nonessential amino acids, 2 mM GlutaMax, 1 mM sodium pyruvate, 0.05% gentamicin, 50 μM 2-mercaptoethanol, 50 ng/ml stem cell factor and 50 ng/ml thrombopoietin per well of a 96-well round-bottom plate. After 18 – 24 h of culture, 5 μl lentivirus stock was added to each well. Cells were adoptively transferred 24 h later into lethally irradiated CD45.1+ BoyJ mice by tail injection on day 0 (at least 1 × 104 hematopoietic stem cells per mouse). After 6 – 8 weeks of reconstitution, cells were recovered from the thymus and spleen of each recipient mouse and analyzed on a BD Fortessa flow cytometer and FlowJo software.
Immunoprecipitation and mass spectrometry analysis
3 × 104 J.LAT cells reconstituted with WT or AIARSA LAT with the myc tag fused to the C-termini were harvested and washed with PBS twice. Cells were resuspended in serum-free RPMI at the concentration of 5 × 107 cells/ml, and split into three samples. Cells were rested at 37 °C for 15 min, and were unstimulated or stimulated with anti-CD3 for 2 min, or 10 min. The stimulation was stopped with ice cold PBS on ice. Cells were washed with ice cold PBS, resuspended in 1 ml 1% NP-40 lysis buffer containing inhibitors of 2 mM NaVO4, 10 mM NaF, 5 mM EDTA, 2 mM PMSF, 10 μg/ml Aprotinin, 1 μg/ml Pepstatin and 1 μg/ml Leupeptin. Cells were centrifuged at 13,000 g, and supernatants were transferred to clean tubes. Anti-myc magnet beads were washed with 1% NP-40 lysis buffer, and added to each sample. Samples were rotated at 4 °C for 2 h. Beads were washed five times with Tris-NaCl buffer (20 mM Tris pH 7.4, 120 mM NaCl), and resuspended in 100 μl urea elution buffer (8 M urea, 20 mM Tris pH 7.5, and 100 mM NaCl) and rotated for 30 min at 25°C with frequent agitation before gentle centrifugation. Beads were collected and supernatant samples were transferred to clean tubes, and frozen down at –20 °C for mass spectrometry analysis.
Briefly, mass spectrometry analysis was performed by a fully automated proteomic technology platform45–47. The tryptic peptides were separated by an Agilent 1200 Series Quaternary HPLC system (Agilent Technologies) and analyzed by a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific). Peptide spectrum matching of MS/MS spectra were searched against a human-specific database (UniProt; downloaded 8/4/2016) using MASCOT v. 2.4.1 (Matrix Science, Ltd). A concatenated database containing 185,156 “target” and “decoy reversed” sequences was employed to estimate the false discovery rate (FDR)5. Peptide assignments from the database search were filtered down to 1% false discovery rate (FDR) by MOWSE score filtering, as previously described45, 48.
Relative quantification of peptide abundance was performed via calculation of selected ion chromatograms (SIC) peak areas. Retention time alignment of individual replicate analyses was performed as previously described47, 49. A minimum SIC peak area equivalent to the typical spectral noise level of 1000 was required of all data reported for label-free quantitation. Individual SIC peak areas were normalized to the peak area of the exogenously spiked peptide “DRVYIHPF” added prior to reversed-phase elution into the mass spectrometer. The proteomic datasets have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD008258. This data is available for review (Username: reviewer21222@ebi.ac.uk and password: zI1qcJ3B)
Samples immunoprecipitated with anti-myc tagged LAT were also analyzed by immunoblot analysis, and probed for total LAT, Lck, or Grb2 to confirm the mass spectrometry analysis.
Immunoprecipitation of proteins associated with TCR complexes
Anti-zeta chain antibody (6B10) was conjugated to protein G agarose and rinsed twice with 0.2 M sodium borate (pH 9.0). The resin was then resuspended in borate buffer and solid dimethylpimelimidate (DMP) added to a concentration of 20 mM. The resin was then incubated for 30 min at room temperature 25°C with mixing and then rinsed with 0.2 M ethanolamine (pH 8.0). The resin was then incubated with ethanolamine for 30 min and washed twice with TBS and stored at 4 °C as a 50% slurry. Prior to immunoprecipitation, the resin was rinsed with wash buffer (0.1% NP-40 TBS). Lck-deficient J.Lck cells were transduced to express murine OT-I TCR, human CD8, and with or without FLAG-tagged human Lck. 1 × 108 cells were washed with PBS twice, and resuspended in serum free RPMI medium and rested at 37 °C for 15 min. Cells were stained with H-2Kb OVA tetramers (SIINFEKL) on ice for 1 h, washed twice with PBS, and resuspended again in serum-free RPMI medium and split into three samples. TCR stimulation was initiated by incubating samples at 37 °C for 5 min or 15 min, or remained on ice. Cells were directly lysed with 10% NP-40 lysis buffer to the final concentration of 1% NP-40 (containing inhibitors of 2 mM NaVO4, 10 mM NaF, 5 mM EDTA, 2 mM PMSF, 10 μg/ml Aprotinin, 1 μg/ml Pepstatin and 1 μg/ml Leupeptin). Lysates were placed on ice and centrifuged at 13,000 g to pellet cell debris. Supernatants was transferred to clean tubes, and anti-zeta chain (clone 6B10) resin (50% slurry) was added. Samples were incubated for 2 h at 4 °C before pelleting the resin and rinsing 3× with ice cold wash buffer in a refrigerated microfuge. Captured proteins were eluted by adding SDS sample buffer to the resin and incubating 5 min at 95 °C.
HEK293 cell based reconstitution system
HEK293 cells were transfected at ~ 60% confluency with the following constructs: pEF LAT WT or AIARSA (2 μg), pEF Zap70K362E (0.2 μg), pEF LckK273R (0.2 μg), and empty pcDNA-EGFP vector to a 2.4 μg of total DNA per well (6-well plate). Plasmids were combined in Opti-mem medium before addition of Lipofectamine 2000 (ThermoFisher) (10 μL per 4μ of DNA) as per manufacturer’s instructions. Plasmids were then added to cells for 4 h before adding full medium and culturing overnight. After 24 h, cells were harvested and rinsed, and lysed on ice.
Preparation of J.OT-I.hCD8.Lck KO cells
Lck-deficient Jurkat cells7 were sequentially transduced with hCD8β-T2A-hCD8α in a lentiviral pHR vector32 (a gift from W. Paster, Centre for Pathophysiology, Infectiology and Immunology, Medical University of Vienna), OT-I TCRβ and OT-I TCRα (amplified from OT-I TCR transgenic mouse DNA) in retroviral vectors MSCV-IRES-Thy1.1 and MSCV-IRES-GFP, respectively. Human Lck-encoding sequence (a gift from T. Brdicka, IMG, Prague) was fused with C-terminal FLAG tag on its C–terminus via PCR and cloned into MSCV-IRES-LNGFR.
Lentiviral particles were produced by transfecting Hek293 cells with CD8β-T2A-CD8α-pHR along with pLP1, pLP2, and pLP-VSVG packaging vectors using Lipofectamin 2000 (Thermo Fisher Scientific). Retroviral particles were produced by transfecting Phoenix-AMPHO cell line using polyethylenimine transfection. Supernatants were collected and filtered 48 h post-transfection and the Jurkat cells were transduced by centrifugation (45 min, 1200 g) in the presence of 5-8 μg/ml polybrene.
Transduced Jurkat cells were labeled with PE-conjugated anti-Vα2, APC-conjugated anti-Vβ5.1/5.2 (clone MR9-4, both BD Pharmigen), PE-Cy7-conjugated anti-Thy1.1 (clone HIS51, eBioscience), and/or APC-conjugated anti-LNGFR (clone ME20.4-1.H4, Miltenyi Biotec) antibodies and sorted using BD Influx sorter (BD Biosciences).
Statistics and Reproducibility
Statistical analysis was applied to technical replicates, or biologically independent mice for each experiment. All experiments described in this study have been performed at least twice, and the exact numbers of independent experiments with similar results are indicated in the figure legends. All statistical analyses of experiments were performed using non parametric, two-tailed Mann-Whitney tests. GraphPad Prism 6 Software (GraphPad Software) was used for data analysis and representation. All bar graphs show mean with overlaid scatter dots, or error bars (indicating s.d.), to show the distribution of the data, as indicated in each figure legend. P values for comparisons are provided as exact values or as P < 0.0001 (exact values). 95% confident levels were used to determine a statistically significant P value.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Mass spectrometry data have been deposited in the ProteomeXchange Consortium repository via PRIDE (Username: reviewer21222@ebi.ac.uk and password: zI1qcJ3B). The primary data for analysis of all figures and supplementary figures are available upon request.
Supplementary Material
Acknowledgments
We thank L. Samelson and C. Sommers (NIH) for sharing the LAT-deficient mouse line; T. Kadlecek for generating the J.Lck mutant Jurkat cell clone; A. Roque for animal husbandry; W. Paster (Medical University Vienna) for sharing the CD8-expression vector; T. Brdicka (IMG, Prague) for sharing the Lck-encoding DNA sequence; UCSF Parnassus Flow Cytometry Core for maintaining FACSArias instruments and services; NIH Tetramer Core Facility for providing the H-2Kb OVA tetramers and H-2Ab OVA tetramers; DL. Donermeyer, BB. Au-Yeung, H. Wang, and C. Morley for critical reading of the manuscript and comments. This work was supported by the Jane Coffin Childs Fund 61-1560 (to W.-L.L.), the Damon Runyon Cancer Research Foundation 2198-14 (to N.S.), the Czech Science Foundation GJ16-09208Y (to O.S.), the Howard Hughes Medical Institute (A.W. and J.K.) and NIH, NIAID P01 AI091580-06 (to A.W., J.K. and A.S.), R01 AI083636 and P30 GM110759 (to A.S.), and DRC Center Grant P30 DK063720 (UCSF Parnassus Flow Cytometry Core). All data to understand and access the conclusions of this study are available in the main text, the supplementary materials, and the indicated repositories.
Footnotes
AUTHOR CONTRIBUTIONS
W.-L.L., N.H.S., and A.W. designed the experiments, W.-L.L., N.H.S., N.A., and V.H. conducted the experiments; W.-L.L., N.H.S., N.A., A.R.S., J.K., and A.W. analyzed data and provided intellectual input; V.H. and O.S. provided advice and reagents. W.-L.L., N.H.S., N.A., V.H., O.J., A.R.S., and A.W. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.van der Merwe PA, Dushek O. Mechanisms for T cell receptor triggering. Nat Rev Immunol. 2011;11:47–55. doi: 10.1038/nri2887. [DOI] [PubMed] [Google Scholar]
- 2.Chakraborty AK, Weiss A. Insights into the initiation of TCR signaling. Nat Immunol. 2014;15:798–807. doi: 10.1038/ni.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malissen B, Bongrand P. Early T cell activation: integrating biochemical, structural, and biophysical cues. Annu Rev Immunol. 2015;33:539–561. doi: 10.1146/annurev-immunol-032414-112158. [DOI] [PubMed] [Google Scholar]
- 4.Hartmann J, Schussler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. 2017;9:1183–1197. doi: 10.15252/emmm.201607485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nika K, et al. Constitutively active Lck kinase in T cells drives antigen receptor signal transduction. Immunity. 2010;32:766–777. doi: 10.1016/j.immuni.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thill PA, Weiss A, Chakraborty AK. Phosphorylation of a Tyrosine Residue on Zap70 by Lck and Its Subsequent Binding via an SH2 Domain May Be a Key Gatekeeper of T Cell Receptor Signaling In Vivo. Mol Cell Biol. 2016;36:2396–2402. doi: 10.1128/MCB.00165-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Courtney AH, et al. A Phosphosite within the SH2 Domain of Lck Regulates Its Activation by CD45. Mol Cell. 2017;67:498–511.e496. doi: 10.1016/j.molcel.2017.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stepanek O, et al. Coreceptor scanning by the T cell receptor provides a mechanism for T cell tolerance. Cell. 2014;159:333–345. doi: 10.1016/j.cell.2014.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Oers NS, Killeen N, Weiss A. Lck regulates the tyrosine phosphorylation of the T cell receptor subunits and ZAP-70 in murine thymocytes. J Exp Med. 1996;183:1053–1062. doi: 10.1084/jem.183.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatada MH, et al. Molecular basis for interaction of the protein tyrosine kinase ZAP-70 with the T-cell receptor. Nature. 1995;377:32–38. doi: 10.1038/377032a0. [DOI] [PubMed] [Google Scholar]
- 11.Love PE, Hayes SM. ITAM-mediated signaling by the T-cell antigen receptor. Cold Spring Harb Perspect Biol. 2010;2:a002485. doi: 10.1101/cshperspect.a002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deindl S, et al. Structural basis for the inhibition of tyrosine kinase activity of ZAP-70. Cell. 2007;129:735–746. doi: 10.1016/j.cell.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 13.Yan Q, et al. Structural basis for activation of ZAP-70 by phosphorylation of the SH2-kinase linker. Mol Cell Biol. 2013;33:2188–2201. doi: 10.1128/MCB.01637-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah NH, et al. An electrostatic selection mechanism controls sequential kinase signaling downstream of the T cell receptor. Elife. 2016;5 doi: 10.7554/eLife.20105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pelosi M, et al. Tyrosine 319 in the interdomain B of ZAP-70 is a binding site for the Src homology 2 domain of Lck. J Biol Chem. 1999;274:14229–14237. doi: 10.1074/jbc.274.20.14229. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, et al. ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb Perspect Biol. 2010;2:a002279. doi: 10.1101/cshperspect.a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukherjee S, et al. Monovalent and multivalent ligation of the B cell receptor exhibit differential dependence upon Syk and Src family kinases. Sci Signal. 2013;6:ra1. doi: 10.1126/scisignal.2003220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balagopalan L, Coussens NP, Sherman E, Samelson LE, Sommers CL. The LAT story: a tale of cooperativity, coordination, and choreography. Cold Spring Harb Perspect Biol. 2010;2:a005512. doi: 10.1101/cshperspect.a005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katz ZB, Novotna L, Blount A, Lillemeier BF. A cycle of Zap70 kinase activation and release from the TCR amplifies and disperses antigenic stimuli. Nat Immunol. 2017;18:86–95. doi: 10.1038/ni.3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luis BS, Carpino N. Insights into the suppressor of T-cell receptor (TCR) signaling-1 (Sts-1)-mediated regulation of TCR signaling through the use of novel substrate-trapping Sts-1 phosphatase variants. FEBS J. 2014;281:696–707. doi: 10.1111/febs.12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang M, et al. K33-linked polyubiquitination of Zap70 by Nrdp1 controls CD8(+) T cell activation. Nat Immunol. 2015;16:1253–1262. doi: 10.1038/ni.3258. [DOI] [PubMed] [Google Scholar]
- 22.Mayer BJ. The discovery of modular binding domains: building blocks of cell signalling. Nat Rev Mol Cell Biol. 2015;16:691–698. doi: 10.1038/nrm4068. [DOI] [PubMed] [Google Scholar]
- 23.Dinkel H, et al. ELM 2016–data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res. 2016;44:D294–300. doi: 10.1093/nar/gkv1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michie AM, Zuniga-Pflucker JC. Regulation of thymocyte differentiation: pre-TCR signals and beta-selection. Semin Immunol. 2002;14:311–323. doi: 10.1016/s1044-5323(02)00064-7. [DOI] [PubMed] [Google Scholar]
- 25.Moran AE, Hogquist KA. T-cell receptor affinity in thymic development. Immunology. 2012;135:261–267. doi: 10.1111/j.1365-2567.2011.03547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang K, Zhou B, Wang Y, Rao L, Zhang L. The TLR4 gene polymorphisms and susceptibility to cancer: a systematic review and meta-analysis. Eur J Cancer. 2013;49:946–954. doi: 10.1016/j.ejca.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 27.Shen S, Zhu M, Lau J, Chuck M, Zhang W. The essential role of LAT in thymocyte development during transition from the double-positive to single-positive stage. J Immunol. 2009;182:5596–5604. doi: 10.4049/jimmunol.0803170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rudd ML, Tua-Smith A, Straus DB. Lck SH3 domain function is required for T-cell receptor signals regulating thymocyte development. Mol Cell Biol. 2006;26:7892–7900. doi: 10.1128/MCB.00968-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23:7990–8000. doi: 10.1038/sj.onc.1208074. [DOI] [PubMed] [Google Scholar]
- 30.Purbhoo MA, et al. The human CD8 coreceptor effects cytotoxic T cell activation and antigen sensitivity primarily by mediating complete phosphorylation of the T cell receptor zeta chain. J Biol Chem. 2001;276:32786–32792. doi: 10.1074/jbc.M102498200. [DOI] [PubMed] [Google Scholar]
- 31.McCoy ME, Finkelman FD, Straus DB. Th2-specific immunity and function of peripheral T cells is regulated by the p56Lck Src homology 3 domain. J Immunol. 2010;185:3285–3294. doi: 10.4049/jimmunol.0900027. [DOI] [PubMed] [Google Scholar]
- 32.Paster W, et al. A THEMIS:SHP1 complex promotes T-cell survival. EMBO J. 2015;34:393–409. doi: 10.15252/embj.201387725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aguado E, et al. Induction of T helper type 2 immunity by a point mutation in the LAT adaptor. Science. 2002;296:2036–2040. doi: 10.1126/science.1069057. [DOI] [PubMed] [Google Scholar]
- 34.Sommers CL, et al. A LAT mutation that inhibits T cell development yet induces lymphoproliferation. Science. 2002;296:2040–2043. doi: 10.1126/science.1069066. [DOI] [PubMed] [Google Scholar]
- 35.Miyaji M, et al. Genetic evidence for the role of Erk activation in a lymphoproliferative disease of mice. Proc Natl Acad Sci U S A. 2009;106:14502–14507. doi: 10.1073/pnas.0903894106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kortum RL, et al. A phospholipase C-gamma1-independent, RasGRP1-ERK-dependent pathway drives lymphoproliferative disease in linker for activation of T cells-Y136F mutant mice. J Immunol. 2013;190:147–158. doi: 10.4049/jimmunol.1201458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ravichandran KS, et al. Interaction of Shc with the zeta chain of the T cell receptor upon T cell activation. Science. 1993;262:902–905. doi: 10.1126/science.8235613. [DOI] [PubMed] [Google Scholar]
- 38.Ravichandran KS, Lorenz U, Shoelson SE, Burakoff SJ. Interaction of Shc with Grb2 regulates association of Grb2 with mSOS. Mol Cell Biol. 1995;15:593–600. doi: 10.1128/mcb.15.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ravichandran KS, Lorenz U, Shoelson SE, Burakoff SJ. Interaction of Shc with Grb2 regulates the Grb2 association with mSOS. Ann N Y Acad Sci. 1995;766:202–203. doi: 10.1111/j.1749-6632.1995.tb26663.x. [DOI] [PubMed] [Google Scholar]
- 40.Zhou MM, et al. Structure and ligand recognition of the phosphotyrosine binding domain of Shc. Nature. 1995;378:584–592. doi: 10.1038/378584a0. [DOI] [PubMed] [Google Scholar]
- 41.Ravichandran KS, et al. Evidence for a requirement for both phospholipid and phosphotyrosine binding via the Shc phosphotyrosine-binding domain in vivo. Mol Cell Biol. 1997;17:5540–5549. doi: 10.1128/mcb.17.9.5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pratt JC, et al. Requirement for Shc in TCR-mediated activation of a T cell hybridoma. J Immunol. 1999;163:2586–2591. [PubMed] [Google Scholar]
- 43.Arbulo-Echevarria MM, et al. A Stretch of Negatively Charged Amino Acids of Linker for Activation of T-Cell Adaptor Has a Dual Role in T-Cell Antigen Receptor Intracellular Signaling. Front Immunol. 2018;9:115. doi: 10.3389/fimmu.2018.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu K, Salomon AR. PeptideDepot: flexible relational database for visual analysis of quantitative proteomic data and integration of existing protein information. Proteomics. 2009;9:5350–5358. doi: 10.1002/pmic.200900119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu K, Salomon AR. HTAPP: high-throughput autonomous proteomic pipeline. Proteomics. 2010;10:2113–2122. doi: 10.1002/pmic.200900159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahsan N, Belmont J, Chen Z, Clifton JG, Salomon AR. Highly reproducible improved label-free quantitative analysis of cellular phosphoproteome by optimization of LC-MS/MS gradient and analytical column construction. J Proteomics. 2017;165:69–74. doi: 10.1016/j.jprot.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 49.Demirkan G, Yu K, Boylan JM, Salomon AR, Gruppuso PA. Phosphoproteomic profiling of in vivo signaling in liver by the mammalian target of rapamycin complex 1 (mTORC1) PLoS One. 2011;6:e21729. doi: 10.1371/journal.pone.0021729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry data have been deposited in the ProteomeXchange Consortium repository via PRIDE (Username: reviewer21222@ebi.ac.uk and password: zI1qcJ3B). The primary data for analysis of all figures and supplementary figures are available upon request.