

Abstract
Sequence-specific transcription factors (TFs) bind short DNA sequences in the genome to regulate the expression of target genes. In the last decade, numerous technical advances have enabled the determination of the DNA binding specificities of many of these factors. Large-scale screens of many TFs enabled the creation of databases of TF DNA binding specificities, typically represented as position weight matrices (PWMs). Although great progress has been made in determining and predicting binding specificities systematically, there are still many surprises to be found when studying a particular TF’s interactions with DNA in detail. Paralogous TFs’ binding specificities can differ in subtle ways, in a manner that is not immediately apparent from looking at their PWMs. These differences affect gene regulatory outputs and enable TFs to rewire transcriptional networks over evolutionary time. This review discusses recent observations made in the study of TF-DNA interactions that highlight the importance of continued in-depth analysis of TF-DNA interactions and their inherent complexity.
Keywords: Transcription factors, gene regulatory networks, evolution, DNA binding sites, motifs, transcription factor-DNA interactions, specificity
Graphical Abstract
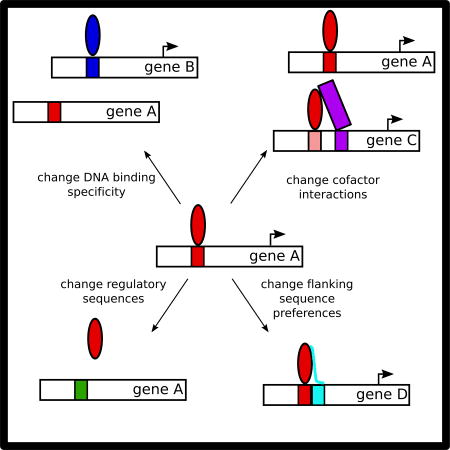
Transcription factor-DNA interactions can change by a variety of mechanisms in evolution, leading to changes in gene regulatory networks.
Introduction
Sequence-specific transcription factors (TFs) are proteins that regulate gene expression through binding to specific short DNA sequences in genomic regulatory elements(Vaquerizas, Kummerfeld, Teichmann, & Luscombe, 2009). The genes a TF can regulate depend on the DNA sequences to which it can bind, termed its DNA binding specificity, as well as where those DNA sequences reside within the genome. Given a TF’s binding specificity, one can identify potential binding sites in the genome, suggesting candidate target genes and providing a starting point to understand the connectivity of gene regulatory networks.
In recent years, techniques for characterizing the interactions between TFs and DNA binding sequences have flourished, enabling the determination of binding specificities for numerous TFs from diverse species (Berger et al., 2006; Jolma et al., 2013; Weirauch et al., 2014). Several databases of binding specificities currently exist (e.g. CIS-BP, HOCOMOCO, JASPAR, and UniPROBE), containing in vivo and in vitro learned specificity models for hundreds of TFs (Hume, Barrera, Gisselbrecht, & Bulyk, 2015; Khan et al., 2017; Kulakovskiy et al., 2016; Weirauch et al., 2014). These databases enable scientists to obtain position weight matrices (PWMs) and sometimes k-mer (DNA sequences of length k) data for their TFs of interest, allowing interpretation of in vivo binding and prediction of regulatory targets (reviewed in Inukai, Kock, & Bulyk, 2017). In vitro assays including PBMs, SELEX-seq, and MITOMI measure the inherent DNA binding preferences of a TF (Berger et al., 2006; Jolma et al., 2013; Maerkl & Quake, 2007). Chromatin immunoprecipitation followed by sequencing (ChIP-seq) experiments can be used to assess in vivo binding, which is complicated by the nuclear environment, including the presence of nucleosomes, chromatin modifications, and other TF co-factors (Johnson, Mortazavi, Myers, & Wold, 2007). Therefore, it is valuable to have precise in vitro understanding of binding specificity to help interpret and explain modes of in vivo binding.
Importantly, these techniques provide binding information for DNA sequences across a wide range of affinities, rather than just the highest affinity sites. This depth of systematic information is a major shift from the previous paradigm of looking solely at consensus sites. This depth enables a more complete understanding of the full spectrum of interactions between TFs and their cognate sites, which would be missed by simply looking at PWM representations of binding or considering only high affinity sites.
Given that these protein-DNA interactions are the specific connections that hold gene regulatory networks together, this detailed understanding of binding specificity leads to a more in-depth understanding of regulatory networks. Incorporating detailed, quantitative information on protein-DNA binding with gene expression data elucidates how networks integrate information to finely tune expression of target genes. Additionally, understanding how families of TFs have diversified their sequence preferences allows investigation of how these regulatory networks can evolve.
In this review, we present various features that contribute to specificity in protein-DNA interactions, highlighting how recent structural and mechanistic studies allow deeper insight. Additionally, we discuss ways that members of TF families have diversified their DNA binding specificities through evolutionary expansion, and contextualize the importance of biochemical and phylogenetic analyses for understanding gene regulatory network function, structure, and evolution.
TRANSCRIPTION FACTOR – DNA INTERACTIONS
Representations of Binding Specificity
Traditionally, a TF’s binding specificity was typically represented as a consensus site, indicating the best bound sites observed for that factor (Figure 1a) (Christy & Nathans, 1989). Now, given the ability to measure DNA binding in high throughput across a wide affinity range, more comprehensive ways of representing specificity have been developed. The binding specificity of a TF is typically represented as a motif, or position weight matrix (PWM), which indicates the likelihood of each nucleotide at each position in the binding site (Figure 1b) (reviewed in Stormo, 2013). Typical PWM models assume that the binding sites can be aligned with each other and that the positions in the binding sites are independent of each other – the presence of a nucleotide in one position does not affect the TF preference for particular nucleotides at another position. While this independence assumption provides a close approximation to the specificity of TFs, it is not always true (Benos, Bulyk, & Stormo, 2002). Therefore, protein-DNA interaction models that account for interdependencies between DNA positions in the binding site, instead of assuming this independence, can also be used to explain binding specificity (Riley, Lazarovici, Mann, & Bussemaker, 2015). Additionally, k-mer-based representations of binding specificity, including support vector regression models, have also been developed and can outperform PWM-based models in explaining binding specificity (Figure 1c) (Agius, Arvey, Chang, Noble, & Leslie, 2010). Recently, the importance of DNA shape in transcription factor-DNA recognition has been appreciated (Rohs et al., 2009). Including DNA shape features in models of TF binding specificity has been shown to improve the ability to explain and predict DNA binding (Ma, Yang, Rohs, & Noble, 2017; Zhou et al., 2015).
Figure 1. Representations of binding specificity.

(a) Groups of sequences bound by a TF can be used to create a consensus sequence, represented using IUPAC notation. The group of k-mers themselves can be used to denote sequences bound by the TF. (b) Here, bound sequences are aligned to create a motif, which indicates the probability of each nucleotide at every position within the binding site. Multiple algorithms exist for creating a PWM from high-throughput binding data (reviewed in Stormo, 2013). (c) Machine learning approaches can learn specificity models from binding data, incorporating short k-mer and DNA shape features of the DNA binding sites.
Additionally, surveys of the binding specificities of numerous TFs have shown that many TFs’ specificities are not represented well by just one binding model (Badis et al., 2009; Gordan et al., 2011). Some TFs that bind as dimers, recognizing a site composed of two half-sites, can bind sites with different spacer lengths between the half-sites. Some of these TFs have been shown to recognize these two different motifs in vivo as well (Johnson et al., 2007). Other TFs recognize distinct sites that cannot be aligned with each other or explained by differences in spacer lengths. For example, most forkhead factors (e.g. Foxa2 and Foxo3) can bind both a primary forkhead motif (RYAAAYA) and a secondary motif (AHAACA) (R = A or G, Y = C or T, H = A, C, or T) (Badis et al., 2009; Mariani, Weinand, Vedenko, Barrera, & Bulyk, 2017). While the overall prevalence of this phenomenon among TFs is debated, it is clear that for some TF classes, such diversity in the types of DNA sequences they can recognize is genuine (Morris, Bulyk, & Hughes, 2011; Siggers et al., 2012; Zhao & Stormo, 2011).
Transcription Factor Families
TFs typically contact DNA through a DNA binding domain (DBD). DBDs fall into evolutionarily related families, which adopt a similar structure to contact DNA, and TFs within the same TF family tend to bind similar DNA sequences (Badis et al., 2009; Harrison, 1991; Luscombe, Austin, Berman, & Thornton, 2000; Vaquerizas et al., 2009). A recent analysis of DNA binding specificities of 671 metazoan TFs found that when clustering DNA binding sequence preferences, most TFs cluster by family (Mariani et al., 2017). However, TF families were frequently subdivided into subfamilies with distinct specificities, largely consistent with previously observed specificity differences (Badis et al., 2009; Berger et al., 2008).
Some families of TFs, particularly C2H2 zinc finger (ZF) TFs have developed great diversity in binding specificity. Differences in specificity can be largely explained by changes in protein sequence at direct DNA-contacting positions (Figure 2a, 2b) (Wolfe, Nekludova, & Pabo, 1999). ZFs were thought to have a relatively straightforward DNA-recognition mechanism, whereby each finger contacts three to four DNA bases using canonical recognition positions in the ZF recognition helix; modular arrays of ZFs can recognize longer DNA sequences (M Elrod-Erickson, Rould, Nekludova, & Pabo, 1996; Monicia Elrod-Erickson & Pabo, 1999; Pavletich & Pabo, 1991). By combining different individual zinc fingers with different DNA-contacting amino acids, and therefore different binding preferences, members of this family can bind many diverse DNA sequences. However, this straightforward model of specificity determination does not always accurately capture the specificities observed in this family. Interactions between individual ZFs, the linker sequences that connect individual fingers, as well as protein sequences outside of the canonical recognition positions, can alter DNA binding preferences (Figure 2c) (Enuameh et al., 2013; Garton et al., 2015; Siggers, Reddy, Barron, & Bulyk, 2014). The mechanism of generating diversity in sequence specificity by using different amino acids at DNA-contacting positions in the recognition helix has also occurred in other TF families, such as homeodomains (HDs) (Hanes et al., 1989; Treisman, Gönczy, Vashishtha, Harris, & Desplan, 1989). For example, the K50 class of HDs, including the Drosophila TF Bicoid and human TF PITX2, have lysine at position 50 within the HD recognition helix, while most other HDs have glutamine at that position. This amino acid difference allows the K50 class to recognize TAATCC, while other HDs typically recognize TAATTA (Baird-Titus et al., 2006; Chaney, Clark-Baldwin, Dave, Ma, & Rance, 2005).
Figure 2. Structural differences between TFs enable divergence in DNA binding.

(a) Modular TF families, such as ZFs, contain members with different numbers and arrangements of individual DBDs. (b) Members of a family can contain different amino acids at DNA-contacting positions, as seen in both the fly and mouse HD specificity classes (Berger et al., 2008; Noyes et al., 2008). (c) Differences not at DNA-contacting positions can alter specificity through allosteric mechanisms, as observed in the human ETS factors SAP-1 and Elk-1 (Mo et al., 2000). (d) Protein loops can contact DNA flanking the core recognition motif, adding preferences for DNA shape features, as seen in the yeast S. cerevisiae bHLH proteins Cbf1 and Tye7 (Gordan et al., 2013). (e) DBDs that bind as dimers can recognize sites with different spacer lengths between half sites, as seen in yeast S. cerevisiae bZIP proteins (Gordan et al., 2011). (f) DNA binding along with a co-factor can change the specificity of a TF, as observed in the specificities of the fly Hox protein binding with the cofactors Exd and Hth (Slattery et al., 2011).
Additionally, more subtle specificity differences have been observed, where paralogs share some binding sites, but diverge in their preferences for other, potentially lower affinity, sites (Badis et al., 2009; Berger et al., 2008). For example, a survey of specificities of murine HDs showed that while many members of this family share binding to canonical HD sites (TAATTA), they also recognize sub-family specific moderate and lower affinity k-mers, such as TAATGA for Lhx2 and TAATCA for Lhx4. Characterizing the full range of binding preferences for TFs, especially how members of a class differ, is important, given that low affinity sites have been shown to be crucial for graded gene expression (Parker, White, Ramos, Cohen, & Barolo, 2011). For example, a pair of lower affinity binding sites for the HD TF Prep1 in a Pax6 transcriptional enhancer are evolutionarily conserved and tuned to Prep1 expression levels, ensuring Pax6 expression in the developing eye at the appropriate stage in mouse embryonic development (Rowan et al., 2010). This may be a generalizable principle: low affinity sites can limit expression to certain tissues where the TF is present at higher nuclear concentrations; furthermore, lower affinity sites may be unique to a TF, while higher affinity sites are often shared by more family members (Crocker et al., 2015; Farley et al., 2015). The HD TF CRX, involved in photoreceptor cell-fate specification, has been shown to act as an activator at high affinity sites, but as a repressor at clusters of lower affinity sites (M. A. White et al., 2016). Therefore, having models of TF specificity that fully capture this range of interactions across affinities is required to understand TF function.
Features that influence TF binding
In addition to this understanding of core motif recognition, it is important to consider other factors that may complicate protein-DNA binding specificity.
Co-factor influences on binding specificity
The regulatory regions in which TFs bind typically contain binding sites for multiple TFs, which together combinatorially regulate gene expression (reviewed in Spitz & Furlong, 2012). Several studies have shown that interactions between TFs can alter their binding specificities (Figure 2e, 2f). Co-binding with the HD proteins Exd and Hth was shown to reveal latent binding specificities of Drosophila Hox proteins, allowing for further diversity among the binding preferences within this TF family (Slattery et al., 2011). As monomers, eight Drosophila Hox proteins bound similar sequences. But, as trimeric complexes with Exd and Hth, the proteins preferred distinct minor groove widths in the binding site. The differences between individual TF binding sites and the binding motif for TF pairs has been documented for many TF pairs in in vitro large-scale studies (Jolma et al., 2015). These effects on specificity may be achieved through protein-protein interactions directly changing the protein conformation, or through DNA-induced allostery, whereby changes to the DNA shape induced by the binding of one TF affect the binding of other TFs (Kim et al., 2013).
Effects of flanking sequence on binding
Additionally, two TFs can have the same apparent specificity for their core binding motif, but differ in their preferences for flanking DNA, allowing them to recognize different genomic sites. For example, the two yeast basic helix-loop-helix (bHLH) factors Cbf1 and Tye7 both recognize the same core E-box sequence (CACGTG), but have distinct preferences for the DNA shape of the sequences flanking this motif, allowing them to occupy distinct genomic binding sites (Gordan et al., 2013). The domains of the protein responsible for these preferences are thought to be loops that do not make direct base contacts, but instead recognize DNA shape through an indirect readout mechanism (Figure 2d) (Otwinowski et al., 1988). This recognition mechanism is possible because DNA structure is also complex and varies with DNA sequence. The GC content of flanking DNA sequence can also distinguish between bound in vivo sites for a given TF, and non-functional genomic motif occurrences (Dror, Golan, Levy, Rohs, & Mandel-gutfreund, 2015; M. a White, Myers, Corbo, & Cohen, 2013). GC content is associated with DNA shape features and DNA flexibility, hinting that potential shape recognition mechanisms may explain these preferences.
Features that affect TF binding in vivo
In cells, a TF’s DNA binding can be affected by the presence of other DNA binding proteins. Nucleosomal occupancy of genomic DNA limits which DNA sites are available for the TF to recognize (Gross & Garrard, 1988). Some TFs, referred to as pioneer factors (e.g. the forkhead factor FoxA1), can recognize their binding sites even when the site occurs within a nucleosome, and are known to have important developmental roles by controlling chromatin accessibility (Cirillo et al., 2002; Ghisletti et al., 2010; Schulz et al., 2015). DNA modifications, particularly cytosine methylation, may also either decrease or increase TF binding depending on where they occur within the TF-DNA interface (Kribelbauer et al., 2017; Yin et al., 2017).
Insights into binding specificity from detailed studies of TF structure
Much effort has been expended trying to understand the molecular basis of sequence specificity within TF families. A thorough understanding of which positions in a DBD control binding specificity could allow for the prediction of the DNA binding specificities of TFs that have not been assayed experimentally (Badis et al., 2009). This could allow for prediction of gene regulatory network structure across species, as well as in networks involving less-studied proteins in the common model organisms. Given that TFs from the same families tend to recognize similar sequences, it has been suggested that a TF’s specificity can be inferred from the specificity of the family member with the closest protein sequence identity, allowing the prediction of DNA binding specificity for 34% of eukaryotic TFs (Weirauch et al., 2014). This approach has been used to create databases of inferred TF binding motifs, and has greatly expanded the coverage of specificities of TFs from certain species (Weirauch et al., 2014). However, given the intricacy and complexity of differences in binding specificity between family members as described above, this approach may overlook much of this subtlety in DNA recognition.
For some well-studied classes of TFs, structural knowledge of how the family contacts DNA can be used to identify which aspects of the protein sequence are most important for determining binding specificity. This information can be used to derive ‘recognition rules’, which allow one to predict the specificity of an uncharacterized TF from its amino acid sequence. For example, in the HD family, the amino acid positions in the protein that contact DNA have been identified, as have the binding specificities of many factors from organisms including fly and mouse (Berger et al., 2008; Noyes et al., 2008). This knowledge has been compiled to identify the specificity-determining amino acid positions in the domain, which can be used to predict the specificities of other HDs. Additionally, machine learning techniques can identify amino acid positions within DBDs that are predictive of binding specificity, from analyzing known DNA binding specificities and the corresponding DBD protein sequences (Christensen et al., 2012; Pelossof et al., 2015). These learned models can be used to predict specificities for uncharacterized DBDs, such as DBDs from other species or potentially DBDs with mutated protein sequence, allowing for better understanding of networks.
However, complexity in protein-DNA recognition has generally made defining these recognition rules difficult. Even for particularly well-studied classes of TFs with seemingly simple recognition rules, such as ZFs, there are exceptions and epistatic interactions within and between DBDs that make it very difficult to predict specificity de novo (Enuameh et al., 2013). The positions that lead to these specificity differences can occur far from the direct DNA-contacting positions, affecting the DBD conformation and consequently the DNA contacts, complicating the identification of which positions in the domain hold predictive power (Mo, Vaessen, & Johnston, 2000). This complexity suggests that a fuller understanding of recognition rules in TF families will not simply require the determination of the binding specificities of representative TFs from the family, but also in-depth investigations of the structure with which the DBD contacts DNA, the protein positions that correlate with differences in binding specificity, allostery within DBDs, and how mutations in DBDs affect DNA binding specificity.
In a survey of the effects on DNA binding of genetic variation within TF DBDs, variants at DNA-contacting positions were enriched among mutations associated with Mendelian disorders (Barrera et al., 2016). However, some protein positions that are not known DNA-contacting positions were associated with disease and affected DNA binding, showing the need for a clearer picture of which parts of DBDs are essential for proper DNA binding. In-depth studies of all possible variants of individual TFs, as has been performed for the nuclear hormone receptor TF PPARγ, are able to relate binding changes to disease phenotypes, but require knowledge of target genes and regulated binding sites in order to design a reporter assay (Majithia et al., 2016). Additionally, by assaying binding to only one binding site, coding variants that affect binding to only some sites will be missed. For example, a variant at a position that contacts DNA flanking the core binding site did not affect binding to a synthetic PPARγ binding site, but did disrupt PPARγ binding to endogenous enhancer sequences and was found in lipodystrophy patients (Majithia et al., 2016). These studies highlight the value of a comprehensive understanding of the binding mechanisms of TFs, including the full complexity of their DNA interactions.
EVOLUTION OF GENE REGULATORY NETWORKS
In addition to understanding how gene regulatory networks function in extant species, it is particularly interesting to consider how regulatory information evolves, given the complexity of the networks involved. It has been the dogma in the field that the potential for pleiotropic phenotypic effects of TF mutations should severely disfavor evolution of TF function in favor of cis-regulatory evolution (Figure 3a) (Hoekstra & Coyne, 2007). Cis-regulatory sequences in the genome, composed of a series of short TF binding sites, are thought to be more modular than trans-regulatory factors and therefore easier to change over evolutionary time(Wagner & Lynch, 2008). Hence, these cis-regulatory changes may be expected to occur over shorter evolutionary time-scales. Some large-scale studies support the idea that cis-regulatory changes are indeed the main source of regulatory differences between species (Wilson & Odom, 2009). For example, TF binding at genomic sites is largely not conserved between species, when comparing homologous tissues. Experiments in which a human chromosome was introduced into mouse demonstrated that the chromosome maintained the overall patterns of TF binding, histone modifications, and gene expression as in the native human context, rather than that of the orthologous chromosome in mouse, indicating that the trans-regulatory machinery is sufficiently conserved between these species to achieve the proper readout of the cis-regulatory sequences (Wilson et al., 2008). These findings have led to a general focus in the field on the mechanisms by which cis-regulatory sequences change between species.
Figure 3. Possible effects of cis- and trans- changes to gene regulatory networks.

(a) Cis-regulatory mutations to TF binding sites can add or remove genes from a TF’s regulon. (b) Changes to the specificity of trans-acting TFs can rewire the genes regulated by the TF. (c) Gain of co-factor interactions can recruit a TF to newly regulated genes, stabilizing interactions with low affinity binding sites. (d) Cis-regulatory sequences and TFs can co-evolve to maintain the same regulatory logic. (e) TFs can gain new regulatory domains, or interactions with co-factors with regulatory domains, changing the expression of the genes under their control. The arrows denote potentially multiple evolutionary steps.
However, changes to trans-acting regulators have also occurred through a variety of mechanisms (Figure 3b–3e). The spatiotemporal expression pattern of a TF can change, affecting the output of the genes under its control, as has been observed for the expression of BCL11A, the ZF TF that regulates the β-globin locus, in human and mouse (Sankaran et al., 2009). The cis-regulatory elements controlling expression of the HD Pitx1, an important developmental regulator, in stickleback fish have been shown to be frequently mutated and show signatures of positive selection, affecting the tissues where Pitx1 is expressed and leading to phenotypic differences (Chan et al., 2010).
TFs can also evolve the ability to interact with different co-factors, as has been observed in the evolution of mating gene regulation in yeast (Tsong, Tuch, Li, & Johnson, 2006). In-depth studies of the logic underlying the regulatory circuits governing expression of mating cell-type genes in different fungal species have revealed that evolution of co-factor interactions and changes to cis-regulatory sequences can both contribute to re-wiring of networks, leading to different network structures in different species (Baker, Booth, Sorrells, & Johnson, 2012). This highlights the value of considering the context of the entire regulatory circuit when studying TF evolution.
Changes in DNA binding specificity
Additionally, the DNA binding specificities themselves of the TFs can evolve, affecting which genes can be regulated by that factor. From surveys of TF binding specificities across different species, we know that TFs from the same families tend to have similar binding specificities, but can also diverge significantly (Badis et al., 2009; Berger et al., 2008; Jolma et al., 2013). It is thought that the expansion of TF families allowed for these changes in specificity, as the existence of paralogous factors could ameliorate the potential negative pleiotropic effects of changing specificity. In support of this, gene duplication and divergence has been shown to be a major contributor to the evolution of transcriptional networks in both E. coli and yeast (Teichmann & Babu, 2004). Paralogs may maintain conserved binding to a common set of high affinity DNA sites, while having divergent preferences for lower affinity sites, as was observed for Lhx family HD factors (Berger et al., 2008). In the Msn family of ZF TFs in the yeast S. cerevisiae, while all five family members share binding to the common AGGGG stress response element, different paralogs have gained comparably strong binding to their own preferred sites that are distinct from the common motif (Siggers et al., 2014). These examples demonstrate that modular protein activity can be a feature of TF evolution.
More drastic specificity differences, where binding specificity completely switches from one motif to another, have also occurred (Baker, Tuch, & Johnson, 2011; Sayou et al., 2014). In a particularly dramatic example, the forkhead family of TFs separately evolved the ability to recognize an alternate DNA motif in three subfamilies (Nakagawa, Gisselbrecht, Rogers, Hartl, & Bulyk, 2013; Schlake, Schorpp, Nehls, & Boehm, 1997; Zhu et al., 2009). This alternate motif (GACGC) cannot be aligned to the canonical forkhead motif (RYAAAYA) recognized by the majority of forkhead proteins. Interestingly, some individual forkheads can bind both the canonical and alternate motifs using the same DBD. Furthermore, there are no substitutions within the base-contacting recognition helix within this family that can explain how the alternate specificity arose. Substitutions elsewhere in the DBD may alter the protein conformation, thus allowing recognition of these different DNA sites (Figure 2c). Further structural studies promise to be insightful for elucidating the mechanism of DNA recognition in this TF family.
These highlighted examples show how binding specificity has evolved to give the current landscape of diverse TF DNA binding specificities. However, studies considering the effects of binding specificity changes on gene regulatory networks, and investigations into potential mechanisms that allow changes to these trans-regulators without wholesale disruptions to regulatory function, are needed. The complexity of these networks that makes it challenging to consider how any regulator could evolve without toppling the whole regulatory system may also provide redundancy and buffering necessary for evolutionary exploration of DNA-binding space. Genome sequencing studies have revealed that individual humans harbor many mutations within TF DBDs, some of which are predicted to alter DNA binding affinity or specificity(Barrera et al., 2016). The fact that this variation exists within populations suggests that regulatory networks may be robust to changes in TF function. Analogously, redundancy has been observed in cis-regulatory enhancers, which can provide developmental robustness to stressors (Hong, Hendrix, & Levine, 2008; Osterwalder et al., 2018). Multiple enhancers regulating expression of the same gene can allow evolutionary change within the individual enhancers while maintaining the conserved overall gene expression pattern, allowing for exploration of regulatory space (Wunderlich et al., 2015).
Some features of the way specificities have changed between paralogs may indicate how these changes are tolerated. The ability of factors to modularly gain new recognition sequences, while maintaining shared binding sites with other paralogs may allow them to explore new regulatory roles without disrupting existing networks (Siggers et al., 2014). The ability of TFs to bind DNA with co-factors could also let factors explore new specificities, as protein-protein interaction strength can reduce the necessity for optimal protein-DNA interactions (Baker et al., 2012). Also, cis-regulatory sequences have been observed to change in parallel with alterations to binding specificities, allowing homologs with different specificities to maintain the same regulatory roles across different species (Figure 3d) (Gasch et al., 2004).
Understanding evolution can help to understand mechanism
By carefully studying the mechanisms by which these changes in specificity have occurred during evolution, a better understanding of how specificity is encoded in a TF family can be achieved. Significant advances in this area have been achieved using an approach called ancestral reconstruction, in which the protein sequences of ancestral proteins are estimated using evolutionary models (reviewed in Thornton, 2004). The proteins are subsequently synthesized and examined using various functional assays to identify which set of mutations have led to functional differences between ancestral and extant states. For example, the evolution of both the ligand binding domain (LBD) and the DNA binding domain (DBD) of the steroid hormone receptor TF family has been studied extensively (Bridgham, Carroll, & Thornton, 2006; McKeown et al., 2014). This family experienced a gene duplication event and diversification of paralogs, after which the DBD in one subfamily switched its DNA binding specificity while the other paralog maintained the DNA binding preferences of the ancestor. By studying the differences between the ancestral state and the derived DBD, mutations that inhibited the interaction with the ancestral DNA binding site as well as others that enhanced binding to a new site were identified. Additionally, other mutations arose after the duplication that enhanced the DNA binding affinity of the TF non-specifically. These evolutionary studies therefore revealed important aspects of the steroid hormone protein-DNA recognition rules, including which positions are responsible for overall high affinity DNA binding as well as specific interactions with different DNA sequences. Further studies examining the evolution of the corresponding DNA recognition elements showed how epistatic interactions between the TF and its recognition element shape the possible evolutionary trajectories that this regulatory unit can pass through while still maintaining existing functions (Anderson, McKeown, & Thornton, 2015). Certain TF mutations act as permissive mutations by leading to increased degeneracy, preserving regulatory interactions despite changes to DNA sequence, while others restrict specificity. This interplay between the DNA binding sites and the TF is even more complex when considering the multiple genomic copies of these regulatory elements through which a TF coherently controls the expression of a group of genes. Overall, careful evolutionary studies such as these have shed light on how gene regulatory networks can evolve, while also revealing important information about the determinants of the TF’s DNA binding specificity.
Conclusion
In this review, we have discussed developments in the understanding of TF-DNA binding specificity, particularly the inherent complexity in the interactions between proteins and DNA binding sequences. Continued development of models that can incorporate the depth and intricacy of these interactions will be important for proper understanding of TFs’ interactions with the genome. A primary challenge that remains is predicting which of a TF’s many potential binding sites in the genome it will bind in each particular cell type in vivo. Further studies into TF-TF interactions, the role of pioneer TFs, DNA shape features, the influence of flanking DNA sequence, and DNA modification on protein binding will continue to reveal how TFs interact with the genome.
The work discussed here highlights the importance of studies into the mechanisms by which DNA binding proteins specifically interact with DNA, and which features of these proteins are involved in determining binding specificity. Specificity is not simply determined by the direct DNA base-contacting positions, but also can be modulated through allosteric effects of residues in other parts of the protein. Further structural and biochemical studies of DBDs will be required to parse out the protein sequence features of different TF families that determine DNA binding specificity. Importantly, these mechanisms may differ between TF subfamilies and individual TFs. Better mechanistic models will be learned from such detailed studies, which should allow us to better understand how these proteins interact with the genome. Additionally, these analyses will allow for better interpretation of the effects of genetic variation in TFs.
Lastly, we have discussed examples of TF evolution, particularly how DNA binding specificities have changed. Investigations of how changes to trans-acting factors affect the flow of information through regulatory networks promises to continue to be an interesting area of study. Combining a thorough understanding of the mechanisms of TF interactions with genomic sequence from an evolutionary perspective promises to yield novel discoveries into how the complex regulatory networks observed in extant species arose.
Acknowledgments
We would like to thank Stephen S. Gisselbrecht, Dr. Luca Mariani, Kian Hong Kock, Colin T. Waters, Dr. Sachi Inukai, Raehoon Jeong, and Kathryn Weinand for helpful feedback and discussion, and Professor Raluca Gordân for assistance in figure design. This work was supported by grant R01 HG003985 (M.L.B.)
Contributor Information
Julia M. Rogers, Email: julia.m.rogers@gmail.com, Division of Genetics, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, 02115, USA; Committee on Higher Degrees in Biophysics, Harvard University, Cambridge, MA, 02138, USA.
Martha L. Bulyk, Email: mlbulyk@genetics.med.harvard.edu, Division of Genetics, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, 02115, USA; Committee on Higher Degrees in Biophysics, Harvard University, Cambridge, MA, 02138, USA; Department of Pathology, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, 02115, USA.
References
- Agius P, Arvey A, Chang W, Noble WS, Leslie C. High resolution models of transcription factor-DNA affinities improve in vitro and in vivo binding predictions. PLoS Computational Biology. 2010;6(9) doi: 10.1371/journal.pcbi.1000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DW, McKeown AN, Thornton JW. Intermolecular epistasis shaped the function and evolution of an ancient transcription factor and its DNA binding sites. eLife. 2015;4(e07864) doi: 10.7554/eLife.07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Bulyk ML. Diversity and complexity in DNA recognition by transcription factors. Science (New York, N.Y.) 2009;324(5935):1720–3. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird-Titus JM, Clark-Baldwin K, Dave V, Caperelli CA, Ma J, Rance M. The solution structure of the native K50 Bicoid homeodomain bound to the consensus TAATCC DNA-binding site. Journal of Molecular Biology. 2006;356(5):1137–1151. doi: 10.1016/j.jmb.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Baker CR, Booth LN, Sorrells TR, Johnson AD. Protein modularity, cooperative binding, and hybrid regulatory states underlie transcriptional network diversification. Cell. 2012;151(1):80–95. doi: 10.1016/j.cell.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CR, Tuch BB, Johnson AD. Extensive DNA-binding specificity divergence of a conserved transcription regulator. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(18):7493–8. doi: 10.1073/pnas.1019177108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera LA, Vedenko A, Kurland JV, Rogers JM, Gisselbrecht SS, Rossin EJ, Bulyk ML. Survey of variation in human transcription factors reveals prevalent DNA binding changes. Science. 2016;351(6280):1450–1454. doi: 10.1126/science.aad2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benos PV, Bulyk ML, Stormo GD. Additivity in protein-DNA interactions: how good an approximation is it? Nucleic Acids Research. 2002;30(20):4442–51. doi: 10.1093/nar/gkf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Peña-Castillo L, Hughes TR. Variation in Homeodomain DNA Binding Revealed by High-Resolution Analysis of Sequence Preferences. Cell. 2008;133(7):1266–1276. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Philippakis AA, Qureshi AM, Fangxue HS, Estep PW, Bulyk ML. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nature Biotechnology. 2006;24(11):1429–1435. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgham JT, Carroll SM, Thornton JW. Evolution of hormone-receptor complexity by molecular exploitation. Science. 2006;312(5770):97–101. doi: 10.1126/science.1123348. [DOI] [PubMed] [Google Scholar]
- Chan YF, Marks ME, Jones FC, Villarreal G, Shapiro MD, Brady SD, Kingsley DM. Adaptive Evolution of Pelvic Reduction in Sticklebacks by Recurrent Deletion of a Pitx1 Enhancer. Science. 2010;327(5963):302–305. doi: 10.1126/science.1182213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaney BA, Clark-Baldwin K, Dave V, Ma J, Rance M. Solution structure of the K50 class homeodomain PITX2 bound to DNA and implications for mutations that cause Rieger syndrome. Biochemistry. 2005;44(20):7497–7511. doi: 10.1021/bi0473253. [DOI] [PubMed] [Google Scholar]
- Christensen RG, Enuameh MS, Noyes MB, Brodsky MH, Wolfe SA, Stormo GD. Recognition models to predict DNA-binding specificities of homeodomain proteins. Bioinformatics. 2012;28(12):84–89. doi: 10.1093/bioinformatics/bts202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christy B, Nathans D. DNA binding site of the growth factor-inducible protein Zif268. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(22):8737–8741. doi: 10.1073/pnas.86.22.8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Molecular Cell. 2002;9(2):279–89. doi: 10.1016/S1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- Crocker J, Abe N, Rinaldi L, McGregor AP, Frankel N, Wang S, Stern DL. Low affinity binding site clusters confer HOX specificity and regulatory robustness. Cell. 2015;160(1–2):191–203. doi: 10.1016/j.cell.2014.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror I, Golan T, Levy C, Rohs R, Mandel-gutfreund Y. A widespread role of the motif environment in transcription factor binding across diverse protein families. 2015. pp. 1268–1280. [DOI] [PMC free article] [PubMed]
- Elrod-Erickson M, Pabo CO. Binding Studies with Mutants of Zif268. The Journal of Biological Chemistry. 1999;274(27):19281–19285. doi: 10.1074/jbc.274.27.19281. [DOI] [PubMed] [Google Scholar]
- Elrod-Erickson M, Rould MA, Nekludova L, Pabo CO. Zif268 protein-DNA complex refined at 1.6 A: a model system for understanding zinc finger-DNA interactions. Structure. 1996;4(10):1171–1180. doi: 10.1016/S0969-2126(96)00125-6. [DOI] [PubMed] [Google Scholar]
- Enuameh MS, Asriyan Y, Richards A, Christensen RG, Hall VL, Kazemian M, Wolfe SA. Global analysis of Drosophila Cys 2 -His 2 zinc finger proteins reveals a multitude of novel recognition motifs and binding determinants. 2013. pp. 928–940. [DOI] [PMC free article] [PubMed]
- Farley EK, Olson KM, Zhang W, Brandt AJ, Rokhsar DS, Levine MS. Suboptimization of developmental enhancers. Science. 2015;350(6258):325–328. doi: 10.1126/science.aac6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garton M, Najafabadi HS, Schmitges FW, Radovani E, Hughes TR, Kim PM. A structural approach reveals how neighbouring C2H2 zinc fingers influence DNA binding specificity. Nucleic Acids Research. 2015;43(19):9147–9157. doi: 10.1093/nar/gkv919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Moses AM, Chiang DY, Fraser HB, Berardini M, Eisen MB. Conservation and evolution of cis-regulatory systems in ascomycete fungi. PLoS Biology. 2004;2(12) doi: 10.1371/journal.pbio.0020398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, Natoli G. Identification and Characterization of Enhancers Controlling the Inflammatory Gene Expression Program in Macrophages. Immunity. 2010;32(3):317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Gordan R, Murphy K, McCord R, Zhu C, Vedenko A, Bulyk M. Curated collection of yeast transcription factor DNA binding specificity data reveals novel structural and gene regulatory insights. Genome Biology. 2011;12(12):R125. doi: 10.1186/gb-2011-12-12-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordan R, Shen N, Dror I, Zhou T, Horton J, Rohs R, Bulyk ML. Genomic Regions Flanking E-Box Binding Sites Influence DNA Binding Specificity of bHLH Transcription Factors through DNA Shape. Cell Reports. 2013;3(4):1093–1104. doi: 10.1016/j.celrep.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annual Review of Biochemistry. 1988;57:159–197. doi: 10.1146/annurev.biochem.57.1.159. [DOI] [PubMed] [Google Scholar]
- Hanes SD, Brent R, Aggarwal AK, Rodgers DW, Drotter M, Ptashne M, Ptashne M. DNA specificity of the bicoid activator protein is determined by homeodomain recognition helix residue 9. Cell. 1989;57(7):1275–83. doi: 10.1016/0092-8674(89)90063-9. [DOI] [PubMed] [Google Scholar]
- Harrison SC. A structural taxonomy of DNA-binding domains. Nature. 1991;353:715–719. doi: 10.1038/353715a0. [DOI] [PubMed] [Google Scholar]
- Hoekstra HE, Coyne JA. The locus of evolution: Evo devo and the genetics of adaptation. Evolution. 2007;61(5):995–1016. doi: 10.1111/j.1558-5646.2007.00105.x. [DOI] [PubMed] [Google Scholar]
- Hong JW, Hendrix DA, Levine MS. Shadow enhancers as a source of evolutionary novelty. Science. 2008;321(5894):1314. doi: 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume MA, Barrera LA, Gisselbrecht SS, Bulyk ML. UniPROBE, update 2015: New tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Research. 2015;43(D1):D117–D122. doi: 10.1093/nar/gku1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inukai S, Kock KH, Bulyk ML. Transcription factor–DNA binding: beyond binding site motifs. Current Opinion in Genetics and Development. 2017;43:110–119. doi: 10.1016/j.gde.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-Wide Mapping of in Vivo Protein-DNA interactions. Science. 2007;316(5830):1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Taipale J. DNA-binding specificities of human transcription factors. Cell. 2013;152(1–2):327–339. doi: 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- Jolma A, Yin Y, Nitta KR, Dave K, Popov A, Taipale M, Taipale J. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature. 2015;527(7578):384–8. doi: 10.1038/nature15518. [DOI] [PubMed] [Google Scholar]
- Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Mathelier A. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Research. 2017;46(November 2017):260–266. doi: 10.1093/nar/gkx1126. [DOI] [Google Scholar]
- Kim S, Brostromer E, Xing D, Jin J, Chong S, Ge H, Xie XS. Probing Allostery Through DNA. Science. 2013;339(6121):816–19. doi: 10.1126/science.1229223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kribelbauer JF, Laptenko O, Chen S, Martini GD, Freed-Pastor WA, Prives C, Bussemaker HJ. Quantitative Analysis of the DNA Methylation Sensitivity of Transcription Factor Complexes. Cell Reports. 2017;19(11):2383–2395. doi: 10.1016/j.celrep.2017.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakovskiy IV, Vorontsov IE, Yevshin IS, Soboleva AV, Kasianov AS, Ashoor H, Makeev VJ. HOCOMOCO: Expansion and enhancement of the collection of transcription factor binding sites models. Nucleic Acids Research. 2016;44(D1):D116–D125. doi: 10.1093/nar/gkv1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe NM, Austin SE, Berman HM, Thornton JM. An overview of the structures of protein-DNA complexes. Genome Biology. 2000;1(1) doi: 10.1186/gb-2000-1-1-reviews001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Yang L, Rohs R, Noble WS. DNA sequence+shape kernel enables alignment-free modeling of transcription factor binding. Bioinformatics. 2017;33(19):3003–3010. doi: 10.1093/bioinformatics/btx336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maerkl SJ, Quake SR. A Systems Approach to Measuring the Binding Energy Landscapes of Transcription Factors. Science. 2007;315(5809):233–237. doi: 10.1126/science.1206034. [DOI] [PubMed] [Google Scholar]
- Majithia AR, Tsuda B, Agostini M, Gnanapradeepan K, Rice R, Peloso G, Altshuler D. Prospective functional classification of all possible missense variants in PPARG. Nature Genetics. 2016;48(12):1570–1575. doi: 10.1038/ng.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani L, Weinand K, Vedenko A, Barrera LA, Bulyk ML. Identification of Human Lineage-Specific Transcriptional Coregulators Enabled by a Glossary of Binding Modules and Tunable Genomic Backgrounds. Cell Systems. 2017;5(3):187–201.e7. doi: 10.1016/j.cels.2017.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown AN, Bridgham JT, Anderson DW, Murphy MN, Ortlund EA, Thornton JW. Evolution of DNA specificity in a transcription factor family produced a new gene regulatory module. Cell. 2014;159(1):58–68. doi: 10.1016/j.cell.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y, Vaessen B, Johnston K. Structure of the Elk-1 – DNA complex reveals how DNA- distal residues affect ETS domain recognition of DNA. Nature Structural Biology. 2000;7(4):3–8. doi: 10.1038/74055. [DOI] [PubMed] [Google Scholar]
- Morris Q, Bulyk ML, Hughes TR. Jury remains out on simple models of transcription factor specificity. Nature Biotechnology. 2011;29(6):483–484. doi: 10.1038/nbt0611-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Gisselbrecht SS, Rogers JM, Hartl DL, Bulyk ML. DNA-binding specificity changes in the evolution of forkhead transcription factors. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(30):12349–54. doi: 10.1073/pnas.1310430110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes MB, Christensen RG, Wakabayashi A, Stormo GD, Brodsky MH, Wolfe SA. Analysis of Homeodomain Specificities Allows the Family-wide Prediction of Preferred Recognition Sites. Cell. 2008;133(7):1277–1289. doi: 10.1016/j.cell.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder M, Barozzi I, Tissières V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, Pennacchio LA. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature. 2018;554(7691):239–243. doi: 10.1038/nature25461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Schevitz RW, Zhang RG, Lawson CL, Joachimiak A, Marmorstein RQ, Sigler PB. Crystal structure of trp repressor/operator complex at atomic resolution. Nature. 1988. [DOI] [PubMed]
- Parker DS, White MA, Ramos AI, Cohen BA, Barolo S. The cis-regulatory logic of Hedgehog gradient responses: key roles for gli binding affinity, competition, and cooperativity. Science Signaling. 2011;4(176):ra38. doi: 10.1126/scisignal.2002077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavletich NP, Pabo CO. Zinc finger–DNA recognition: crystal structure of a Zif268-DNA complex at 2.1Å. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- Pelossof R, Singh I, Yang JL, Weirauch MT, Hughes TR, Leslie CS. Affinity regression predicts the recognition code of nucleic acid–binding proteins. Nature Biotechnology. 2015;33(12):1242–1249. doi: 10.1038/nbt.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley TR, Lazarovici A, Mann RS, Bussemaker HJ. Building accurate sequence-to-affinity models from high-throughput in vitro protein-DNA binding data using featureREDUCE. eLife. 2015;4(e06397) doi: 10.7554/eLife.06397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. The role of DNA shape in protein-DNA recognition. Nature. 2009;461(7268):1248–1253. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan S, Siggers T, Lachke SA, Rowan S, Siggers T, Lachke SA, Maas RL. Precise temporal control of the eye regulatory gene Pax6 via enhancer-binding site affinity. Genes & Development. 2010;24:980–985. doi: 10.1101/gad.1890410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Xu J, Ragoczy T, Ippolito GC, Walkley CR, Maika SD, Orkin SH. Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009;460(7259):1093–1097. doi: 10.1038/nature08243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayou C, Monniaux M, Nanao MH, Moyroud E, Brockington SF, Thévenon E, Dumas R. A promiscuous intermediate underlies the evolution of LEAFY DNA binding specificity. Science. 2014;343(6171):645–8. doi: 10.1126/science.1248229. [DOI] [PubMed] [Google Scholar]
- Schlake T, Schorpp M, Nehls M, Boehm T. The nude gene encodes a sequence-specific DNA binding protein with homologs in organisms that lack an anticipatory immune system. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(8):3842–7. doi: 10.1073/pnas.94.8.3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz KN, Bondra ER, Moshe A, Villalta JE, Lieb JD, Kaplan T, Carolina N. Zelda is differentially required for chromatin accessibility, transcription factor binding, and gene expression in the early Drosophila embryo. 2015. pp. 1715–1726. [DOI] [PMC free article] [PubMed]
- Siggers T, Chang AB, Teixeira A, Wong D, Williams KJ, Ahmed B, Bulyk ML. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-κB family DNA binding. Nature Immunology. 2012;13(1):95–102. doi: 10.1038/ni.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggers T, Reddy J, Barron B, Bulyk ML. Diversification of transcription factor paralogs via noncanonical modularity in C2H2 Zinc finger DNA binding. Molecular Cell. 2014;55(4):640–648. doi: 10.1016/j.molcel.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery M, Riley T, Liu P, Abe N, Gomez-Alcala P, Dror I, Mann RS. Cofactor binding evokes latent differences in DNA binding specificity between hox proteins. Cell. 2011;147(6):1270–1282. doi: 10.1016/j.cell.2011.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EEM. Transcription factors: From enhancer binding to developmental control. Nature Reviews Genetics. 2012;13(9):613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- Stormo GD. Modeling the specificity of protein-DNA interactions. Quant Biol. 2013;1(2):115–130. doi: 10.1007/s40484-013-0012-4.Modeling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichmann Sa, Babu MM. Gene regulatory network growth by duplication. Nature Genetics. 2004;36(5):492–6. doi: 10.1038/ng1340. [DOI] [PubMed] [Google Scholar]
- Thornton JW. Resurrecting ancient genes: experimental analysis of extinct molecules. Nature Reviews. Genetics. 2004;5(5):366–75. doi: 10.1038/nrg1324. [DOI] [PubMed] [Google Scholar]
- Treisman J, Gönczy P, Vashishtha M, Harris E, Desplan C. A single amino acid can determine the DNA binding specificity of homeodomain proteins. Cell. 1989;59(3):553–62. doi: 10.1016/0092-8674(89)90038-X. [DOI] [PubMed] [Google Scholar]
- Tsong AE, Tuch BB, Li H, Johnson AD. Evolution of alternative transcriptional circuits with identical logic. Nature. 2006;443(7110):415–20. doi: 10.1038/nature05099. [DOI] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009;10(4):252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- Wagner GP, Lynch VJ. The gene regulatory logic of transcription factor evolution. Trends in Ecology and Evolution. 2008;23(7):377–385. doi: 10.1016/j.tree.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Hughes TR. Determination and Inference of Eukaryotic Transcription Factor Sequence Specificity. Cell. 2014;158(6):1431–1443. doi: 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Kwasnieski JC, Myers CA, Shen SQ, Corbo JC, Cohen BA. A Simple Grammar Defines Activating and Repressing cis-Regulatory Elements in Photoreceptors. Cell Reports. 2016;17(5):1247–1254. doi: 10.1016/j.celrep.2016.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White Ma, Myers Ca, Corbo JC, Cohen Ba. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proceedings of the National Academy of Sciences of the United States of America. 2013;109(29):11952–7. doi: 10.1073/pnas.1307449110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Barbosa-Morais NL, Schmidt D, Conboy CM, Vanes L, Tybulewicz VLJ, Odom DT. Species-Specific Transcription in Mice Carrying Human Chromosome 21. Science. 2008;322:434–438. doi: 10.1126/science.1163148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Odom DT. Evolution of transcriptional control in mammals. Current Opinion in Genetics and Development. 2009;19(6):579–585. doi: 10.1016/j.gde.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Wolfe SA, Nekludova L, Pabo CO. DNA Recognition by Cys2His2 Zinc Finger Proteins. Annu. Rev. Biophys. Biomol. Struct. 1999;3:183–212. doi: 10.1146/annurev.biophys.29.1.183. [DOI] [PubMed] [Google Scholar]
- Wunderlich Z, Bragdon MDJ, Vincent BJ, White JA, Estrada J, DePace AH. Krüppel Expression Levels Are Maintained through Compensatory Evolution of Shadow Enhancers. Cell Reports. 2015;12(11):1740–1747. doi: 10.1016/j.celrep.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Taipale J. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356(6337) doi: 10.1126/science.aaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Stormo GD. Quantitative analysis demonstrates most transcription factors require only simple models of specificity. Nature Biotechnology. 2011;29(6):480–483. doi: 10.1038/nbt0611-480b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Shen N, Yang L, Abe N, Horton J, Mann RS, Rohs R. Quantitative modeling of transcription factor binding specificities using DNA shape. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(15):4654–9. doi: 10.1073/pnas.1422023112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Byers K, McCord R, Shi Z, Berger M, Newburger D, Bulyk ML. High-resolution DNA binding specificity analysis of yeast transcription factors. Genome Res. 2009. pp. 556–566. [DOI] [PMC free article] [PubMed]