

Abstract
Sigma-1 receptor (sigma-1R), a 25-kDa integral membrane protein, is expressed at a high density in various tumor cell lines and its ligands mediate tumor cell proliferation. However, the effect of this receptor on proliferation and the associated intracellular molecules in tumors remains unclear. The present study aimed to investigate the effect of sigma-1R overexpression on MCF-7 cell proliferation and the associated intracellular molecules that serve a key role in this process. The sigma-1R proliferative function was examined by comparing the proliferation rates of a sigma-1R-overexpressing line, MCF-41 with a sigma-1R-defective line, MCF-7, in culture media with various serum concentrations. The results demonstrated that MCF-41 cells grew significantly faster compared with MCF-7 cells, indicating a proliferation-enhancing receptor function. This proliferation-enhancing effect was completely eliminated by adding a PKC inhibitor to the culture media for MCF-41 cells. To identify which PKC subtype affects the proliferative function of sigma-1R, five inhibitors of PKC subtypes or enzymes involved in the PKC signaling cascade were introduced to MCF-7 and MCF-41 cell culture media and their effects on cell proliferation were compared. It was revealed that only the classic PKC subtype inhibitor, GF109203×, significantly inhibited MCF-41 cell proliferation compared with the MCF-7 line. In conclusion, among PKC iso-enzymes only classic PKC subtype enzymes serve an important role in sigma-1R overexpression enhancing MCF-7 cell proliferation.
Keywords: sigma-1 receptor, protein kinase C, MCF-7, overexpression, proliferation
Introduction
Deregulated cell proliferation is one of the critical events that propel tumor cells and their progeny into uncontrolled expansion and invasion. Sigma-1 receptor (sigma-1R), a 25-kDa integral membrane protein, has been reported to play a role in tumor cell proliferation (1). This receptor was found to represent a unique, non-opioid, non-phencyclidine, haloperiodol-sensitive receptor family. Based on binding affinity, two subtypes of sigma receptors have been identified, namely sigma-1R and sigma-2R (2). Sigma-1R has been cloned from several species and various tissues, and the protein is 223 amino acids in length, with a molecular weight of 25 kDa (3,4). Sigma-1R is a distant homolog to the sterol isomerase in yeast and fungi, with a single putative transmembrane domain and a cytoplasmic reticulum retention sequence (3,5). Although lacking enzymatic activity, sigma-1R shares some pharmacological characteristics with this enzyme. The protein is considered to have two transmembrane-spanning helices (amino acids 9–28 and 81–101), with a 125-amino acid C-terminus (6,7). Sigma-1R is a chaperone protein binding with the chaperone binding immunoglobulin protein (BiP) in the lumen of the endoplasmic reticulum (ER). Agonists of the receptor stimulate its chaperone activity, which exerts a potent neuroprotective effect by ameliorating ER stress and reducing the levels of reactive oxygen radicals (5). Approaches using site-directed mutagenesis and photo-affinity probes identify that the ligand-binding region of sigma-1R is mainly located in the N-terminus of the receptor; however, the C-terminus facilitates the binding of the receptor with its ligands (8), has chaperone function and enhances bradykinin-stimulated calcium release (5,9). The sigma-1R chaperone function was found to be ligand-regulated in the intact protein; however, the ligand-binding and chaperone functions are separable (8).
Initial research on sigma-1R was focused on its role in the nervous system. Mounting evidence indicates that sigma-1R is involved in a number of nervous system diseases, such as methamphetamine or cocaine addiction, amnesia, pain depression, Alzheimer's disease, Parkinson's disease, stroke, Huntington's disease and HIV infection (10). Sigma-1R was later found to be expressed a thigh density in various tumor cells, suggesting the importance of this receptor in tumor cellular functions, as well as serving as a target binding site for tumor-imaging agents (11,12). Accumulating evidence shows that sigma-1R ligands affect tumor cell proliferation. Human mammary adenocarcinoma, colon carcinoma and melanoma cell proliferation was inhibited by sigma-1R ligands, such as haloperidol, DTG and SKF-10047, in vitro cell culture experiments (13). Similarly, sigma-1R ligands, including haloperidol, 2-IBP {N-[2-(piperidino) ethyl]-2-iodo-benzamide} and IPAB [2-piperidinyl-(aminoethyl)-4-iodobenzamide] were found to inhibit the growth of small-cell lung cancer cells (14). Some sigma-1R putative antagonists, but not putative agonists, inhibited tumor cell proliferation in vitro and in mouse tumor xenograft experiments (1,15). However, whether sigma-1R overexpression in cancer cells affects their proliferation has not yet been addressed.
Protein kinase C (PKC) is a family of serine/threonine kinases associated with tumor promotion. PKC play an important role in cell cycle regulation, cell survival, malignant transformation and apoptosis (16). PKC enzymes are stimulated by signals such as increase in the concentration of diacylglycerol (DAG) or calcium ions. Activated PKC phosphorylate hydroxyl groups of serine and threonine amino acid residues on various substrate proteins, depending on cell type, regulate various functions, including receptor desensitization, membrane structure events, gene transcription, immune response and cell growth. To date, at least 12 isoforms of PKC have been cloned, each displaying different enzymatic properties, tissue expression and intracellular localization (17,18). The PKC family comprises three groups: Classic PKCs (PKCα, PKCβ1, PKCβ2 and PKCγ), novel PKCs (PKCδ, PKCε, PKCη, PKCθ and PKCµ) and atypical PKCs (PKCζ, PKCλ and PKCι), which require different stimulators for their full activation.
It has been well-documented that the PKC family regulates cell proliferation and migration in breast tumor cell lines (19). PKCα plays a key role in controlling the proliferation of breast cancer cells through the activation of extracellular signal-regulated kinase (ERK) (20) and telomerase (21). Stimulating PKCβI and βII in MCF-7 cells enhance their growth by upregulating the expression of cyclin D1 (22). PKCη upregulation in malignant breast cells was found to be associated with cancer cell growth and survival via hormone-dependent cell growth pathways (23,24). PKCζ stimulates estrogen-mediated breast cancer cell growth by stabilizing steroid receptor coactivator-3 (25,26). Finally, PKCδ is a potential pro-proliferative factor in breast cancer cells, as TPA-mediated PKCδ activation leads to the activation and nuclear translocation of estrogen receptor (ER)α and enhances ER-dependent reporter gene expression (27). Although both sigma-1R and PKC are involved in cell proliferation, the possibility of a connection between them has not yet been addressed.
While evaluating the role of sigma-1R C-terminus in inositol 1,4,5-trisphosphate (IP3) receptor activation (9), we found that the sigma-1R-overexpressing cell line, MCF-41, proliferated significantly faster compared with the sigma-1R-defective line, MCF-7 (9,11). Based on these observations and the roles of sigma receptors and PKC isoenzymes in cell proliferation, the present study aimed to investigate how sigma-1R overexpression affects the proliferation rate of MCF-7 cells, and establish the connection via a signaling pathway between sigma-1R and PKC subtype enzymes in the pro-proliferative receptor function, which may provide potential molecular targets for anticancer treatment.
Materials and methods
Production of stably transfected cell lines and cell culture
Breast tumor cell lines stably transfected with pcDNA3.1 expression vectors harboring intact sigma-1R (MCF-41line) or its C-terminus (aa 102–223, sg101 line) were developed as previously described (9). MCF-7 (ATCC, Manassas, VA, USA), MCF-41 and sg101 cells were cultured in Dulbecco's minimal essential medium (DMEM) containing 1.5 g/l NaHCO3, 10% fetal bovine serum, insulin (10 mg/l) and penicillin/streptomycin (100 U/100 µg/ml). All cell cultures were performed in a humidified atmosphere of 5% CO2 and 95% air at 37°C. On the third day of the cell cultures, cells were observed from the culture flask under a phase-contrast inverted microscope. Images of the cells were captured using a Carl Zeiss Axiovert 200 microscope. No specific staining was performed.
Cell proliferation assays
Cells were seeded into 96-well plates at a density of 1×104 cells per well, and cultured in 5% CO2 at 37°C overnight. On the second day, the medium was replaced by fresh medium, with or without various concentrations of fetal bovine serum, and six parallel wells were arranged for one experiment. Media were refreshed every 3 days. CCK-8 reagent (10 µl) was added to each well at the indicated time points (days 0, 1, 2, 3, 4, 5, 6 and 7) and incubated for 1 h at 37°C. Optical absorbance was read by a Victor V plate reader at a wavelength of 450 nm. The readings were normalized to blank medium. Cell proliferation was calculated as the ratio of the readings of the selected day to the readings of day 0.
Proliferation of enzyme inhibiting assays
The protocol was similar to that described above for the proliferation assay. The main difference was that the cell medium was changed after 24 h and replaced by different concentrations of enzyme inhibitors, as described in the text, with complete serum or serum without insulin and fetal bovine serum. Measurements were performed as described in the proliferation assays and the readings were normalized to blank medium, either with or without serum and insulin.
Statistical analysis
Values are presented as mean ± standard deviation. Data were analyzed by the Student's t-test or one-way analysis of variance with post hoc comparisons using Student-Newman-Keuls test. The data were analyzed using SPSS software v20.0 (IBM Corp., Armonk, NY, USA). P<0.05 was considered to indicate statistically significant differences.
Results
Sigma-1R overexpression in MCF-7 cells was associated with higher proliferation rate and changed cell morphology
In order to investigate how serum differently affected the growth rates in the MCF-7, sg101 and MCF-41 cell lines, the cells were cultured in media containing various serum concentrations (0, 0.3 and 10%) and the proliferation rates were measured at indicated time points, as shown in Fig. 1A-C. As illustrated in Table I, MCF-41 cell proliferation was 4.5-, 3.5- and 3.4-fold during a 96-h cell culture in media containing 10, 0.3 and 0% serum, respectively. However, the proliferation of wild-type MCF-7 cells was only 1.8-, 1.5- and 1.3-fold during same period. Thus, MCF-41 cells exhibited a significantly higher proliferation rate compared with wild-type MCF-7 cells, even at low serum concentrations. Of note, sigma-1R overexpression enhanced cell proliferation in the absence of serum, i.e., absence of exogenous growth signals. The C-terminal segment of sigma-1R has been shown to enhance bradykinin-stimulated calcium signaling (9). We also determined the proliferation rate of the sg101 cell line in media with different serum concentrations. As shown in Fig. 1 and Table I, the proliferation rate of sg101 cells was 2.9-, 2.4- and 1.9-fold during a 96-h culture in media with varied concentration of serum, as mentioned above. The proliferation rates of sg101 cells were significantly higher compared with those of MCF-7 cells, but slightly lower compared with the MCF-41 cell line. The results demonstrated that sigma-1R played a key role in promoting the proliferation in breast tumor cells, and identified the C-terminus of sigma-1R as the functional domain driving cell proliferation. These results were consistent with previous findings supporting that the C-terminus of sigma-1R is the cellular function domain of the receptor (9). In addition to enhancing cell proliferation rate, overexpression of sigma-1R affected cell morphology. As shown in Fig. 2, the shape of MCF-7 cells was round-like, MCF-41 cells appeared more expanded and displayed pseudo-flagella, and the sg101 cell line displayed more prominent expansion compared with MCF-7 cells, but less prominent expansion compared with MCF-41 cells. Thus, sigma-1R expression was found to be associated with cell morphology, providing additional evidence supporting the cellular function of sigma-1R.
Figure 1.
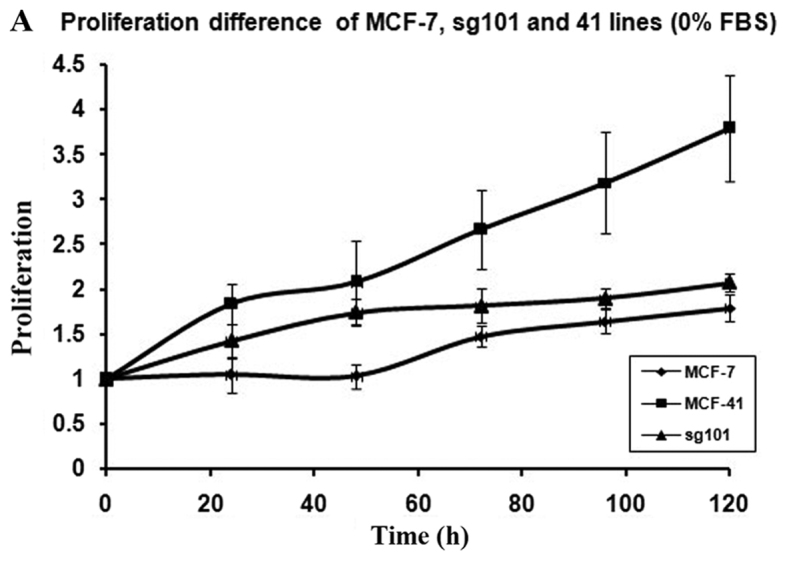
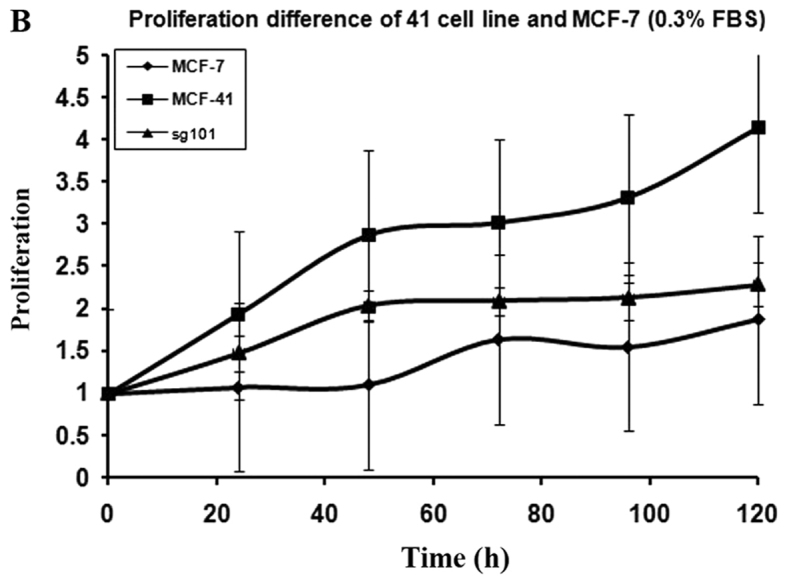
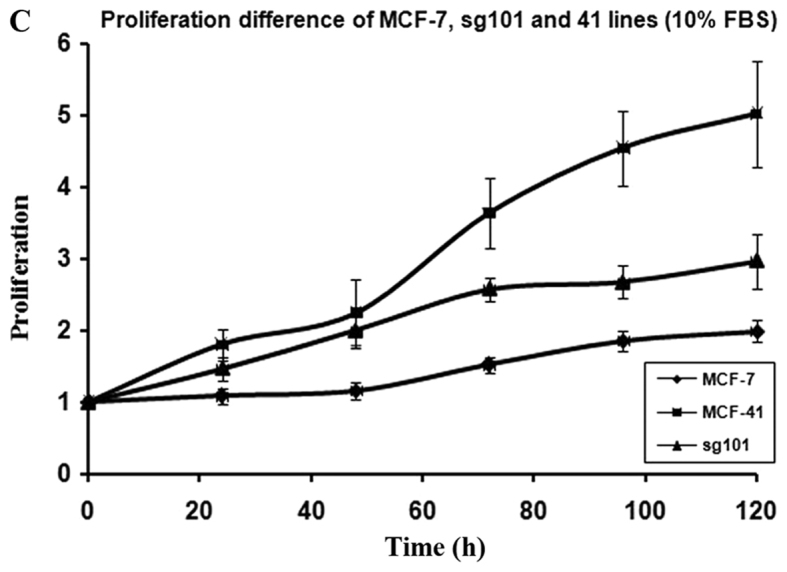
Proliferation curves for the MCF-41, sg101 and MCF-7 cell lines in different concentrations of serum. Cells were seeded into 96-well plates (1×104 cells per well) and cultured with media containing (A) 0, (B) 0.3 and (C) 10% fetal bovine serum, and cell proliferation rates were measured at specific time points (24, 48, 72, 96 and 120 h). The values are presented as the mean ± standard error of the mean.
Table I.
Summary proliferation rates of MCF-41, sg101 and MCF-7 after 96 h of cell culture in different media which contain 10, 0.3 and 0% fetal bovine serum.
| Cell lines | |||
|---|---|---|---|
| Serum concentration (%) | MCF-7 | MCF-41 | Sg101 |
| 10.0 | 1.8 | 4.5 | 2.9 |
| 0.3 | 1.5 | 3.5 | 2.4 |
| 0.0 | 1.3 | 3.4 | 1.9 |
Figure 2.
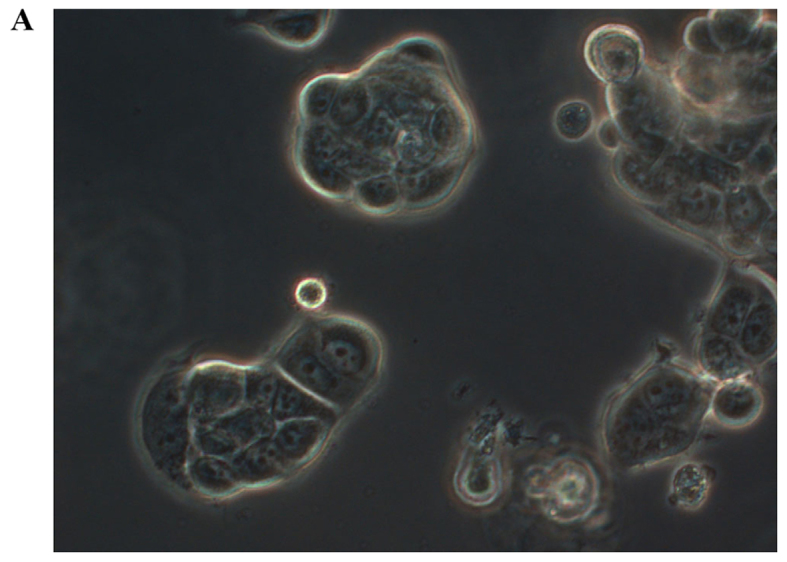
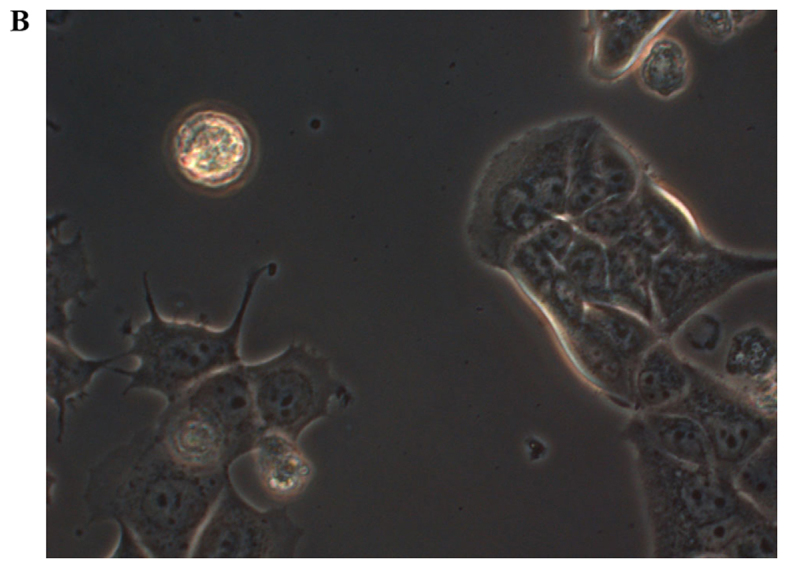
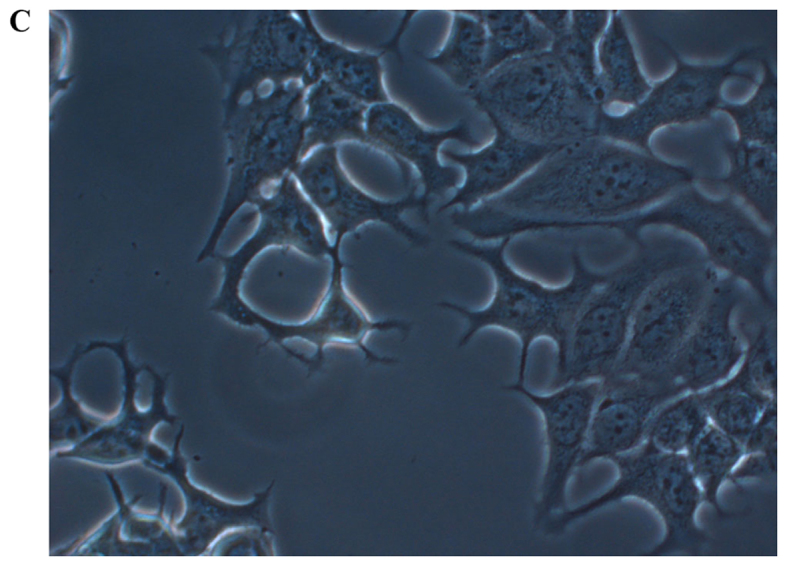
Live morphological observation of the MCF-7, sg101 and MCF-41 cell lines under a phase-contrast microscope at a magnification of ×200. The three cell lines, expressing different levels of sigma-1R or its truncated construct, displayed different morphologies. (A) MCF-7, (B) sg101 and (C) MCF-41 cells.
Sigma-1R overexpression increased MCF-7 cell proliferation rate via the PKC pathway
Tumors often progress from benign to malignant due to continuous mutations that enhance the sensitivity of cells to growth factor signals (28). The present study revealed that sigma-1R overexpression promotes tumor cell proliferation, which may be a part of this process. In order to identify the key molecules involved in the mechanism underlying the role of sigma-1R in proliferation, various enzyme inhibitors for 3 well-documented entities located upstream and downstream in the PKC signaling cascade, or PKC subclass isoenzymes associated with cell proliferation, were used in the CCK-8 assay and data were analyzed.
PI3K inhibition did not affect cell proliferation
To examine whether sigma-1R-overexpressing cells exhibited enhanced proliferation through the PI3K signaling pathway, the CCK-8 assay was performed with the PI3K inhibitor, LY294002. Cells were cultured in media containing various concentrations of LY294002, and proliferation rates were measured by CCK-8 as described in Materials and methods. The results are presented in Fig. 3A, and show that the PI3K inhibitor did not affect the proliferation rate of either MCF-7 or MCF-41 cells over a period of 48 h. These results indicated that the PI3K signaling pathway was not associated with sigma-1R overexpression enhancing cell proliferation. LY294002 and all inhibitors used in following studies were examined to confirm their effects and specification meet the research goals according to manufacturer's handbook when they were delivered from chemical companies (data not shown).
Figure 3.
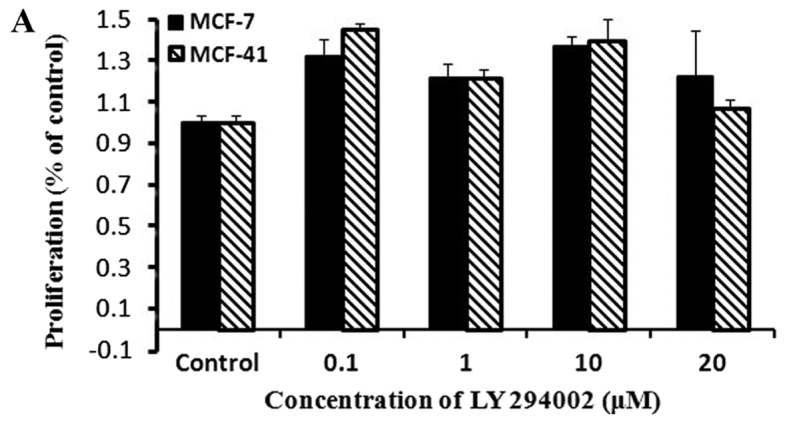
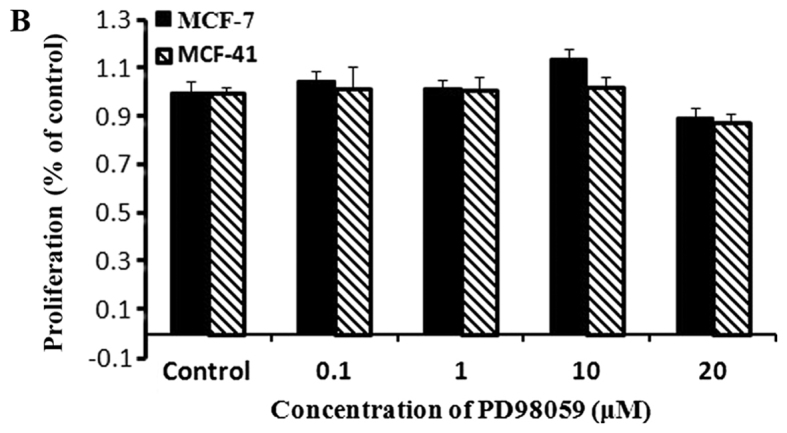
Neither the PI3K inhibitor LY294002, or the MEK1/2 inhibitor PD98059, affected cell proliferation. Cells were seeded into 96-well plates (1×104 cells per well) and cultured with regular media containing 0, 0.1, 1, 10 and 20 µΜ LY294002 or PD98059 for 48 h. (A) Cells cultured in medium containing various concentrations of LY294002. (B) Cells cultured in medium containing various concentrations of PD98059. Cell proliferation rates were measured at 48 h and normalized relative to the control (media without inhibitor). The experiment for each bar was repeated 6 times and each value represents the mean of six separate experiments. The values are presented as the mean ± standard error of the mean. PI3K, Phosphatidylinositol 3-kinase; MEK, mitogen-activated kinase.
MEK1/2 inhibition did not affect cell proliferation
We investigated whether the MEK1/2 signaling pathway was involved in sigma-1R enhancing cell proliferation. An experiment similar to that performed with the PI3K inhibitor was conducted using a MEK1/2 inhibitor, PD98059. Cells were cultured in media with various concentrations of PD98059, and CCK-8 assays were performed at the indicated time points, as described in Fig. 3B. The data revealed that PD98059 did not affect the proliferation of MCF-7 or MCF-41 cells within a period of 48 h. Thus, MEK1/2 is not involved in the signaling pathway of sigma-1R driving cell proliferation.
PKC inhibition affected cell proliferation
Based on the fact that PKC isozymes play critical roles in cell proliferation, we examined whether PKC were associated with sigma-1R overexpression driving cell growth. The effect of PKC inhibition on cell growth was examined by the CCK-8 assay with 10 µM sotrastaurin, a universal PKC inhibitor, at 24, 48 and 72 h. As shown in Fig. 4A, sotrastaurin inhibited MCF-41 cell proliferation more potently compared with the MCF-7 cell line. We investigated further to determine which PKC subtype was involved in this function. LY333531 and dimethoxy-substituted phenyl-thiophene (DSPT), inhibitors of nPKC and aPKC, respectively, were used in the CCK-8 assay, and the results revealed that neither inhibitor affected proliferation in the two cell lines (Fig. 4B and C). However, GF109203×, a classic PKC subtype enzyme inhibitor, inhibited cell proliferation (Fig. 4D). The results are shown in Fig. 4D: MCF-7 cell proliferation rates decreased slightly when GF109203× concentration was increased from 0 to 1 µM, but the proliferation rate mildly increased when the inhibitor concentration was further increased to 10 µM. In the MCF-41 cell line, however, the proliferation rates decreased significantly following an inhibitor concentration increase from 0 to 0.01 µM, and the rates decreased even more when the inhibitor concentration was increased from 1 to 10 µM. This experiment clearly indicated that sigma-1R overexpression driving MCF-41cell proliferation was associated with classic PKC subtype isoenzymes.
Figure 4.
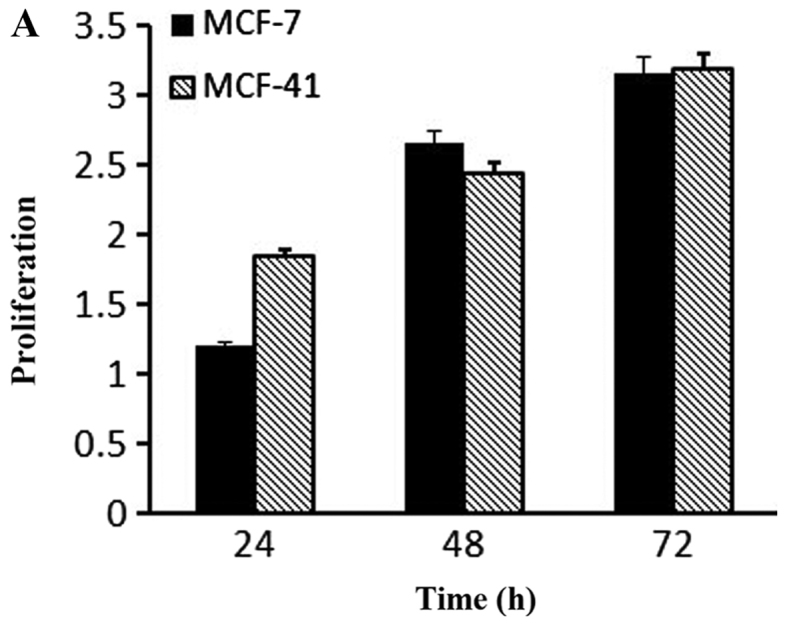
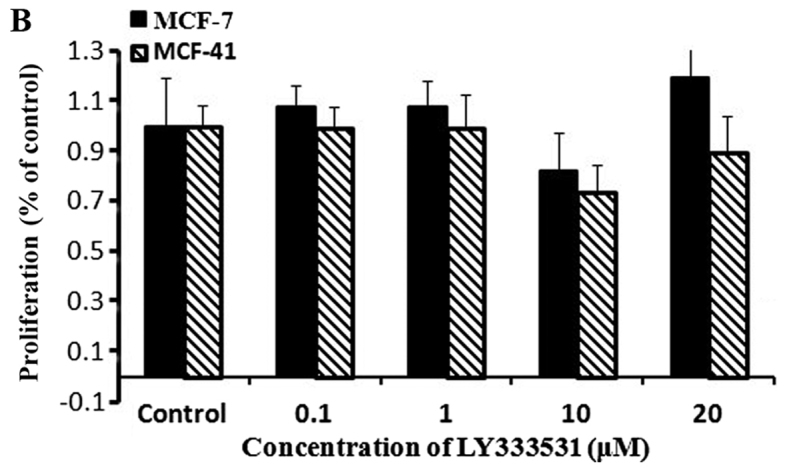
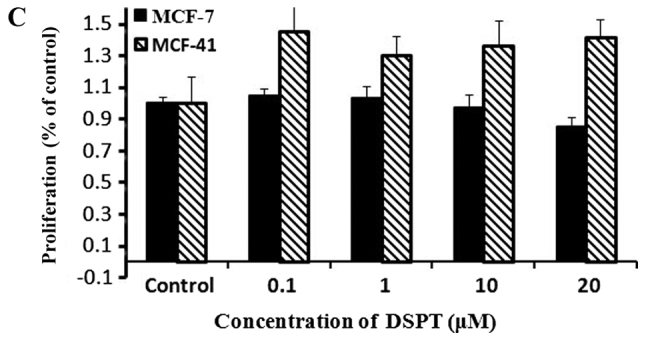
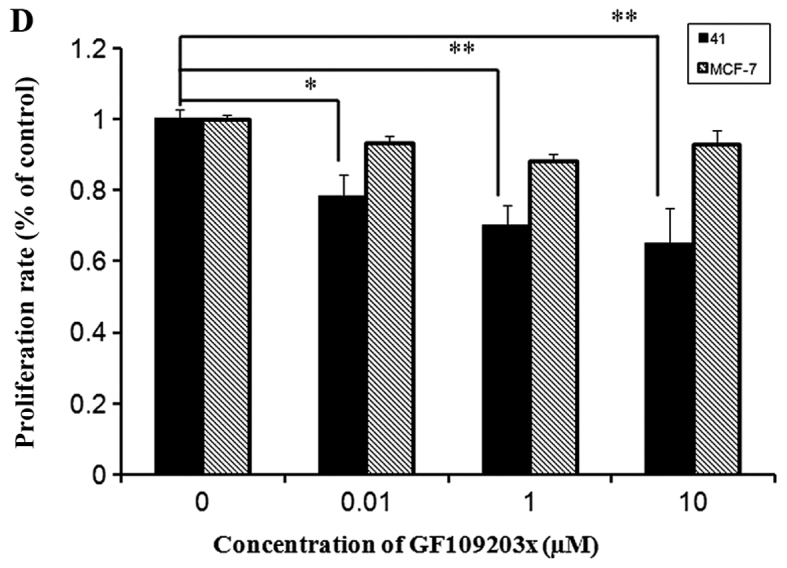
PKC enzyme inhibitor affects cell proliferation. Cells were seeded into 96-well plates (1×104 cells per well) and cultured with regular media containing (A) 10 µΜ sotrastaurin for 24, 48 and 72 h; 0.0, 0.1, 1.0, 10.0 and 20.0 µΜ of (B) LY333531or (C) DSPT; and (D) 0, 0.1, 1.0 and 10.0 µΜ GF109203× for 48 h. Cell proliferation rates were measured at the designated time points and normalized relative to the control (media without inhibitor). The experiment for each bar was repeated 6 times and each value represents the mean of six separate experiments. The values are presented as the mean ± standard error of the mean. *P<0.05; **P<0.01. PKC, protein kinase C.
Discussion
Overexpression of sigma receptors has been found in various types of human cancer cells (11,12). Previous studies revealed that sigma-1R ligands affected tumor cell proliferation (13–15). Overexpression of some receptors, such as TRIM29, was recently reported to facilitate cancer cell proliferation (29,30). However, little was known on whether the overexpression of sigma-1R in cancer cells regulated cell proliferation. In the present study, we demonstrated that overexpression of this receptor constitutively enhanced MCF-7 cell proliferation, even in absence of any of the receptor ligand, strongly indicating that sigma-1R is an oncoprotein. This result was consistent with a previous study reporting that sigma-1R-overexpressing cell lines exhibited constitutively enhanced bradykinin-induced calcium release, driving sigma agonist-independent dissociation of ANK 220 from IP3R-3, resulting in its activation (9). Thus, it is possible that the positive effect of sigma-1R on cell proliferation may be due to the receptor sensitizing cells to various growth signals associated with the IP3R-3/Ca2+ signaling pathway. In the present study, sigma-1R overexpression driving cell proliferation was inhibited by GF109203×, suggesting that the pro-proliferative effects of sigma-1R are closely associated with classic PKC subtype enzymes.
PKC isozymes are known to be involved in cell proliferation, survival, invasion and migration. Activation of PKC isozymes may be triggered by changes in intracellular cofactors of PKCs, which are activated by stimulation from receptor tyrosine kinases (RTKs), G-protein-coupled receptors (GPCR) and integrins (31). RTKs include receptors for epidermal growth factor (EGF), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), hepatocyte growth factor (Met), and insulin receptor family (IR) (32–34). Thus, various growth factors in the serum can stimulate RTKs on the cell surface. GPCRs recruit and activate PKC by releasing factor Gαq, which stimulates phospholipase C (PLC)-β to hydrolyze phosphatidylinositol biphosphate (PIP2) and produce IP3 and DAG, the two major secondary messengers required for stimulation of PKC (35). Integrins and other extracellular matrix protein clusters may also activate PKC via the PLC signaling cascade, which only acts in the formation of cellular focal adhesions (36). Thus, overexpression of sigma-1R regulating PKC activation to promote cell proliferation may be mediated only through the RTK or GPCR cascade rather than through the integrins pathway. The proliferation data revealed that sigma-1R overexpression drove cell proliferation in the absence of serum in the culture medium, indicating that sigma-1R regulates PKCs even when RTKs are silent. Therefore, our data indicated that sigma-1R regulating PKCs may be through the GPCR signaling pathway. A recent study confirmed that sigma-1R directly interacts with Rac1-GTPase in the brain mitochondria, providing more evidence supporting our hypothesis (37).
In breast cancer cells, several PKC isozymes, such as PKCα, β, δ, ε, ζ, η and θ, are involved in cell proliferation, differentiation, survival and apoptosis (19). In the present study, we first tested whether the Ras/Raf/MEK/ERK proliferation signaling pathway was associated with sigma-1R overexpression driving cell proliferation. The MEK1/2 inhibitor, PD98059, was employed in the cell proliferation assay and the data demonstrated it did not affect MCF-7 or MCF-41 cell proliferation, indicating that this signaling pathway is not involved in sigma-1R-mediated cell proliferation. Second, we tested whether the PI3K/Akt/mTOR proliferation signaling pathway was involved in sigma-1R overexpression driving cell proliferation. The PI3K inhibitor, LY294002, was used in the proliferation assay and it was found to exert no effect on cell proliferation. Therefore, this pathway was not associated with the effects of sigma-1R driving cell proliferation. Finally, a universal PKC inhibitor, sotrastaurin, was used in the cell proliferation assay, and the data revealed that the compound inhibited both MCF-7 and MCF-41 cell proliferation, suggesting that the PKC isoenzymes contribute to sigma-1R-induced cell proliferation. These findings were consistent with previous research reporting that PKC isozymes stimulate survival- or proliferation-associated signaling pathways in cancer (19). The PKC family can be classified into three subfamilies: Classic, novel and atypical PKCs. We investigated further to determine which PKC isoenzymes are involved in the pro-proliferative function of sigma-1R. The atypical PKC subtype inhibitor DSPT, the novel PKC subtype inhibitor LY333531, and the classic PKC subtype inhibitor GF109203× were used in MCF-41 and MCF-7 cell proliferation assays, and GF109203× inhibited cell proliferation to a significantly greater extent in the sigma-1R-overexpressing MCF-41 cell line compared with the sigma-1R-defective MCF-7 cell line. These results demonstrated that only classic PKC subtype enzymes are associated with sigma-1R-mediated cell proliferation. One signaling pathway for cell proliferation is the PKCζ-MEK-ERK (38), but our data demonstrated that MEK1/2 inhibition did not affect MCF-41 or MCF-7 cell proliferation, providing further evidence that atypical PKCs, such as PKCζ, are not involved insigma-1R-induced cell proliferation. It should be noted that in the experiments, the PI3K and MEK/ERK inhibitors exist in cell culture media up to 48 h, the cells still survive. The main reason may be that multiple signaling pathways are associated with cell growth and survival, and these signaling pathways usually serve as backup each other, when one or more pathways are blocked, the others can compensate them and this keep cells still survive.
GF109203× is a potent PKC inhibitor with an IC50 of 20, 17, 16 and 20 nM for PKCα, PKCβI, PKCβII and PKCγ, respectively, in cell-free assays. In the present study, the data revealed that GF109203× inhibited MCF-41 and MCF-7 cell proliferation, consistently with previous studies reporting that overexpression of PKCα may contribute to increased anchorage-independent growth, tumorigenicity and metastasis (39). The PKCβ isoform has been implicated in mammary tumorigenesis inhuman and rodent models, and is generally considered to be a growth-promoting kinase; in addition, the PKCβ-specific inhibitor LY379196 significantly reduces the growth of MCF-7, MDA-MB-231and BT-474 breast cancer cells (23). All these previous studies support that classic PKCs are key enzymes in tumor cell proliferation. Our data clearly indicated that sigma-1R-induced MCF-7 cell proliferation is mediated through the classic PKC subtype pathway, at least in part. However, the accurate PKC of classic PKC subtype enzymes is not identified due to short of specific chemical currently which inhibit one of classic PKC enzymes without affecting the other member in the group of PKC, and details on the mechanism underlying sigma-1R activation of classic PKC isoenzymes are unknown at present, and further investigation is required focus on these points in the future study.
In conclusion, our data demonstrated that the sigma-1R-overexpressing cell line, MCF-41, grew significantly faster compared with MCF-7, whereas this enhancement of cell proliferation was completely eliminated by the addition of PKC inhibitor to the culture media. Among PKC isoenzyme inhibitors, only the classic PKC subtype inhibitor, GF109203×, inhibited MCF-41 cell proliferation significantly compared with MCF-7 cells. In conclusion, sigma-1R overexpression driving MCF-7 cell proliferationis, at least in part, mediated through the classic PKC subtype signaling pathway. Due to sigma-1R highly expressing in various tumor cells (11,12), our finding provides new critical molecule targets for anti-tumor drug design in cancer therapy.
Acknowledgements
The authors would like to thank Dr Wayne D. Bowen at Brown University for his reviewing and valuable suggestions.
Funding
The present study was supported by the Scientific and Technological Development Funding of Zhejiang Province (grant no. 2016C33018).
Availability of data and materials
The datasets used in the present study are available from corresponding author on reasonable request.
Authors' contributions
ZW conceived and designed the experiments. YW, XB, XL and CZ performed the experiments. YW and ZW wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Aydar E, Palmer CP, Djamgoz MB. Sigma receptors and cancer: Possible involvement of ion channels. Cancer Res. 2004;64:5029–5035. doi: 10.1158/0008-5472.CAN-03-2329. [DOI] [PubMed] [Google Scholar]
- 2.Quirion R, Bowen WD, Itzhak Y, Junien JL, Musacchio JM, Rothman RB, Su TP, Tam SW, Taylor DP. A proposal for the classification of sigma binding sites. Trends Pharmacol Sci. 1992;13:85–86. doi: 10.1016/0165-6147(92)90030-A. [DOI] [PubMed] [Google Scholar]
- 3.Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E, Glossmann H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci USA. 1996;93:8072–8077. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hellewell SB, Bowen WD. A sigma-like binding site in rat pheochromocytoma (PC12) cells: Decreased affinity for (+)-benzomorphans and lower molecular weight suggest a different sigma receptor form from that of guinea pig brain. Brain Res. 1990;527:244–253. doi: 10.1016/0006-8993(90)91143-5. [DOI] [PubMed] [Google Scholar]
- 5.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 6.Aydar E, Palmer CP, Klyachko VA, Jackson MB. The receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron. 2002;34:399–410. doi: 10.1016/S0896-6273(02)00677-3. [DOI] [PubMed] [Google Scholar]
- 7.Palmer CP, Mahen R, Schnell E, Djamgoz BA, Aydar E. Sigma-1 receptors bind cholesterol and remodel lipid rafts in breast cancer cell lines. Cancer Res. 2007;67:11166–11175. doi: 10.1158/0008-5472.CAN-07-1771. [DOI] [PubMed] [Google Scholar]
- 8.Chu UB, Ruoho AE. Biochemical pharmacology of the sigma-1 receptor. Mol Pharmacol. 2016;89:142–153. doi: 10.1124/mol.115.101170. [DOI] [PubMed] [Google Scholar]
- 9.Wu Z, Bowen WD. Role of sigma-1 receptor C-terminal segment in inositol 1,4,5-trisphosphate receptor activation: Constitutive enhancement of calcium signaling in MCF-7 tumor cells. J Biol Chem. 2008;283:28198–28215. doi: 10.1074/jbc.M802099200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vilner BJ, John CS, Bowen WD. Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res. 1995;55:408–413. [PubMed] [Google Scholar]
- 12.van Waarde A, Rybczynska AA, Ramakrishnan NK, Ishiwata K, Elsinga PH, Dierckx RA. Potential applications for sigma receptor ligands in cancer diagnosis and therapy. Biochim Biophys Acta. 2015;1848:2703–2714. doi: 10.1016/j.bbamem.2014.08.022. [DOI] [PubMed] [Google Scholar]
- 13.Brent PJ, Pang GT. Sigma binding site ligands inhibit cell proliferation in mammary and colon carcinoma cell lines and melanoma cells in culture. Eur J Pharmacol. 1995;278:151–160. doi: 10.1016/0014-2999(95)00115-2. [DOI] [PubMed] [Google Scholar]
- 14.Moody TW, Leyton J, John C. Sigma ligands inhibit the growth of small cell lung cancer cells. Life Sci. 2000;66:1979–1986. doi: 10.1016/S0024-3205(00)00523-3. [DOI] [PubMed] [Google Scholar]
- 15.Kim FJ, Schrock JM, Spino CM, Marino JC, Pasternak GW. Inhibition of tumor cell growth by Sigma1 ligand mediated translational repression. Biochem Biophys Res Commun. 2012;426:177–182. doi: 10.1016/j.bbrc.2012.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erin M, Kazanietz GG. Protein kinase C and other diacylglycerol effector in cancer. Nat Rev Cancer. 2007;7:281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 17.Nishizuka Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature. 1988;334:661–665. doi: 10.1038/334661a0. [DOI] [PubMed] [Google Scholar]
- 18.Ono Y, Fujii T, Ogita K, Kikkawa U, Igarashi K, Nishizuka Y. The structure, expression, and properties of additional members of the protein kinase C family. J Biol Chem. 1988;263:6927–6932. [PubMed] [Google Scholar]
- 19.Kang JH. Protein kinase C (PKC) isozymes and cancer. New J Sci. 2014;2014:231418. doi: 10.1155/2014/231418. [DOI] [Google Scholar]
- 20.Gupta AK, Galoforo SS, Berns CM, Martinez AA, Corry PM, Guan KL, Lee YJ. Elevated levels of ERK2 in human breast carcinoma MCF-7 cells transfected with protein kinase C alpha. Cell Prolif. 1996;29:655–663. doi: 10.1111/j.1365-2184.1996.tb00979.x. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Zhao L, Yang Z, Funder JW, Liu JP. Telomerase is controlled by protein kinase Calpha in human breast cancer cells. J Biol Chem. 1998;273:33436–33442. doi: 10.1074/jbc.273.50.33436. [DOI] [PubMed] [Google Scholar]
- 22.Li H, Weinstein IB. Protein kinase C beta enhances growth and expression of cyclin D1 in human breast cancer cells. Cancer Res. 2006;66:11399–11408. doi: 10.1158/0008-5472.CAN-06-2386. [DOI] [PubMed] [Google Scholar]
- 23.Pal D, Outram SP, Basu A. Upregulation of PKCη by PKCε and PDK1 involves two distinct mechanisms and promotes breast cancer cell survival. Biochim Biophy Acta. 2013;1830:4040–4045. doi: 10.1016/j.bbagen.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karp G, Maissel A, Livneh E. Hormonal regulation of PKC: Estrogen up-regulates PKCeta expression in estrogen responsive breast cancer cells. Cancer Lett. 2007;246:173–181. doi: 10.1016/j.canlet.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Yi P, Feng Q, Amazit L, Lonard DM, Tsai SY, Tsai MJ, O'Malley BW. Atypical protein kinase C regulates dual pathways for degradation of the oncogenic coactivator SRC-3/AIB1. Mol Cell. 2008;29:465–476. doi: 10.1016/j.molcel.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allen-Petersen BL, Carter CJ, Ohm AM, Reyland ME. Protein kinase Cδ is required for ErbB2-driven mammary gland tumorigenesis and negatively correlates with prognosis in human breast cancer. Oncogene. 2014;33:1306–1315. doi: 10.1038/onc.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Servi B, Hermani A, Medunjanin S, Mayer D. Impact of PKCdelta on estrogen receptor localization and activity in breast cancer cells. Oncogene. 2005;24:4946–4955. doi: 10.1038/sj.onc.1208676. [DOI] [PubMed] [Google Scholar]
- 28.Gomperts B, Kramer IM, Tatham P. Signal transduction. 2nd edition. Elsevier Academic Press; Fourth printing: 2004. pp. 246–251. [Google Scholar]
- 29.Tan ST, Liu SY, Wu B. TRIM29 overexpression promotes proliferation and survival of bladder cancer cells through NF-KB signaling. Cancer Res Treat. 2016;48:1302–1312. doi: 10.4143/crt.2015.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Welm B, Boucher KM, Ebbert MT, Bernard PS. TRIM29 functions as a tumor suppressor in nontumorigenic breast cells and invasive ER+ breast cancer. Am J Pathol. 2012;180:839–847. doi: 10.1016/j.ajpath.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oliva JL, Griner EM, Kazanietz MG. PKC isozymes and diacylglycerol-regulated proteins as effectors of growth factor receptors. Growth Factors. 2005;23:245–252. doi: 10.1080/08977190500366043. [DOI] [PubMed] [Google Scholar]
- 32.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/S0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 34.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 35.Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- 36.Fogh BS, Multhaupt HA, Couchman JR. Protein kinase C, focal adhesions and the regulation of cell migration. J Histochem Cytochem. 2014;62:172–184. doi: 10.1369/0022155413517701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Natsvlishvili N, Goguadze N, Zhuravliova E, Mikeladze D. Sigma-1 receptor directly interacts with Rac1-GTPase in the brain mitochondria. BMC Biochem. 2015;16:11. doi: 10.1186/s12858-015-0040-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naranatt PP, Akula SM, Zien CA, Krishnan HH, Chandran B. Kaposi's sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: Implications for infectivity. J Virol. 2003;77:1524–1539. doi: 10.1128/JVI.77.2.1524-1539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ways DK, Kukoly CA, deVente J, Hooker JL, Bryant WO, Posekany KJ, Fletcher DJ, Cook PP, Parker PJ. MCF-7 breast cancer cells transfected with protein kinase C-alpha exhibit altered expression of other protein kinase C isoforms and display a more aggressive neoplastic phenotype. J Clin Invest. 1995;95:1906–1915. doi: 10.1172/JCI117872. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used in the present study are available from corresponding author on reasonable request.