

Abstract
Background:
Fine particulate matter (PM2.5) exacerbates airway inflammation and hyperreactivity in patients with asthma, but the mechanism remains unclear. The aim of this study was to observe the effects of prolonged exposure to high concentrations of PM2.5 on the pathology and airway hyperresponsiveness (AHR) of BALB/c mice undergoing sensitization and challenge with ovalbumin (OVA) and to observe the effects of apoptosis and T-cell immunoglobulin and mucin domain 1 (TIM-1) in this process.
Methods:
Forty female BALB/c mice were divided into four groups: control group, OVA group, OVA/PM group, and PM group (n = 10 in each group). Mice in the control group were exposed to filtered clean air. Mice in the OVA group were sensitized and challenged with OVA. Mice in the OVA/PM group were sensitized and challenged as in the OVA group and then exposed to PM2.5 for 4 h per day and 5 days per week for a total of 8 weeks using a nose-only “PM2.5 online enrichment system” in The Second Hospital of Hebei Medical University. Mice in the PM group were exposed to the PM2.5 online enrichment system only. AHR was detected. Bronchoalveolar lavage fluid (BALF) was collected for cell classification. The levels of interleukin-4 (IL-4), IL-5, and IL-33 in BALF were measured using enzyme-linked immunosorbent assay. Changes in histological structures were examined by light microscopy, and changes in ultramicrostructures were detected by electron microscopy. Apoptosis was determined by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay in the lung tissues. Western blotting and immunohistochemistry were utilized to analyze the expression of Bcl-2, Bax, and TIM-1 in the lungs.
Results:
The results showed that AHR in the OVA/PM group was significantly more severe than that in the OVA and PM groups (P < 0.05). AHR in the PM group was also considerably more severe than that in the control group (P < 0.05). The BALF of OVA/PM group (28.00 ± 6.08 vs. 12.33 ± 4.51, t = 4.631, P = 0.002) and PM group (29.00 ± 3.00 vs. 12.33 ± 4.51, t = 4.927, P = 0.001) had more lymphocytes than the BALF of the control group. The number of neutrophils in the BALF of the OVA/PM group (6.67 ± 1.53 vs. 3.33 ± 1.53, t = 2.886, P = 0.020) and PM group (6.67 ± 1.53 vs. 3.33 ± 1.53, t = 2.886, P = 0.020) was much higher than those in the BALF of OVA group (P < 0.05). TUNEL assays showed that the number of apoptotic cells in the OVA/PM group was significantly higher than that in the OVA group (Tunel immunohistochemical scores [IHS%], 1.20 ± 0.18 vs. 0.51 ± 0.03, t = 8.094, P < 0.001) and PM group (Tunel IHS%, 1.20 ± 0.18 vs. 0.51 ± 0.09, t = 8.094, P < 0.001), and that the number of apoptotic cells in the PM group was significantly higher than that in the control group (Tunel IHS%, 0.51 ± 0.09 vs. 0.26 ± 0.03, t = 2.894, P = 0.020). The concentrations of IL-4 (77.44 ± 11.19 vs. 48.02 ± 10.02 pg/ml, t = 4.595, P = 0.002) and IL-5 (15.65 ± 1.19 vs. 12.35 ± 0.95 pg/ml, t = 3.806, P = 0.005) and the Bax/Bcl-2 ratio (1.51 ± 0.18 vs. 0.48 ± 0.10, t = 9.654, P < 0.001) and TIM-1/β-actin ratio (0.78 ± 0.11 vs. 0.40 ± 0.06, t = 6.818, P < 0.001) in the OVA/PM group were increased compared to those in the OVA group. The concentrations of IL-4 (77.44 ± 11.19 vs. 41.47 ± 3.40 pg/ml, t = 5.617, P = 0.001) and IL-5 (15.65 ± 1.19 vs. 10.99 ± 1.40 pg/ml, t = 5.374, P = 0.001) and the Bax/Bcl-2 ratio (1.51 ± 0.18 vs. 0.97 ± 0.16, t = 5.000, P = 0.001) and TIM-1/β-actin ratio (0.78 ± 0.11 vs. 0.31 ± 0.06, t = 8.545, P < 0.001) in the OVA/PM group were increased compared to those in the PM group. The concentration of IL-4 (41.47 ± 3.40 vs. 25.46 ± 2.98 pg/ml, t = 2.501, P = 0.037) and the Bax/Bcl-2 ratio (0.97 ± 0.16 vs. 0.18 ± 0.03, t = 7.439, P < 0.001) and TIM-1/β-actin ratio (0.31 ± 0.06 vs. 0.02 ± 0.01, t = 5.109, P = 0.001) in the PM group were also higher than those in the control group.
Conclusions:
Exacerbated AHR associated with allergic asthma caused by PM2.5 is related to increased apoptosis and TIM-1 activation. These data might provide insights into therapeutic targets for the treatment of acute exacerbations of asthma induced by PM2.5.
Keywords: Apoptosis, Asthma, Fine Particulate Matter, T-cell Immunoglobulin and Mucin Domain 1
摘要
背景:
细颗粒物(PM2.5)会加剧哮喘患者的气道炎症和高反应性,但这种机制仍不确定。本研究的目的是观察长期暴露于高 浓度的PM2.5对于已经采用卵清蛋白(OVA)致敏的小鼠的病理、气道高反应性的影响,同时观察细胞凋亡和T细胞免疫球蛋白 域1(TIM-1)在这个过程中的作用。
方法:
四十只BALB/c小鼠被分为4组:对照组,OVA组,OVA/PM组,PM组(n = 10/组)。对照组吸入清洁空气,OVA组采用 OVA致敏和激发,OVA/PM组采用OVA同样的方法致敏和激发,同时将小鼠固定在PM2.5在线富集系统的口鼻暴露器上,每日 4小时,每周5天,连续8周,吸入富集的PM2.5,此项操作在中国河北省石家庄市河北医科大学第二医院进行。PM组仅仅给予 吸入富集的PM2.5。随后进行气道高反应性 (AHR)检测,收集肺泡灌洗液(BALF)进行细胞计数。采用酶联免疫法检测BALF 中的白细胞介素4 (interleukin-4, IL- 4),白细胞介素5 (interleukin-5, IL- 5)和白细胞介素33 (interleukin-33, IL- 33)的浓度。用光 学显微镜检查肺组织学结构,用电镜观察其超微结构。采用脱氧核苷酸末端转移酶介导的dUTP 缺口末端标记(TUNEL) 法检 测肺组织中的凋亡细胞。利用免疫印迹和免疫组织化学检测肺组织中的Bcl-2, Bax和TIM-1表达。
结果:
结果显示OVA/PM组小鼠气道反应性相对于OVA组和PM组明显升高(P < 0.05),而仅暴露PM2.5组小鼠气道高反应相对 于对照组也有明显升高(P < 0.05)。OVA/PM组(28.00 ± 6.08 vs 12.33 ± 4.51, t = 4.631, P = 0.002) 和PM组(29.00 ± 3.00 vs 12.33 ± 4.51, t = 4.927, P = 0.001)的BALF中淋巴细胞数量较对照组显著增高(P < 0.05). OVA/PM组 (6.67 ± 1.53 vs 3.33 ± 1.53, t = 2.886, P = 0.020) 和PM组 (6.67 ± 1.53 vs 3.33±1.53, t = 2.886, P = 0.020) 的BALF中中性粒细胞数量较OVA 组(P < 0.05)比较显 著升高。TUNEL检测显示OVA/PM组凋亡细胞显著高于OVA组(Tunel immunohistochemical scores [IHS%], 1.20 ± 0.18 vs 0.51 ± 0.03, t = 8.094, P < 0.001) 和 PM组 (Tunel IHS%, 1.20 ± 0.18 vs 0.51 ± 0.09, t = 8.094, P < 0.001),PM组凋亡细胞显著高于对照组 (Tunel IHS%, 0.51 ± 0.09 vs 0.26 ± 0.03, t = 2.894, P = 0.020)。IL-4(77.44 ± 11.19 vs 48.02 ± 10.02 pg/ml, t = 4.595, P = 0.002) 和 IL-5 (15.65 ± 1.19 vs 12.35 ± 0.95 pg/ml, t = 3.806, P = 0.005)的浓度和Bax/Bcl-2比(1.51 ± 0.18 vs 0.48 ± 0.10, t = 9.654, P < 0.001) 和TIM-1/IM-1) 4比 (0.78 ± 0.11 vs 0.40 ± 0.06, t = 6.818, P < 0.001) 在OVA/PM 组较OVA组比较显著增加。IL-4(77.44 ± 11.19 vs 41.47 ± 3.40 pg/ml, t = 5.617, P = 0.001)和IL-5 (15.65 ± 1.19 vs 10.99 ± 1.40 pg/ml, t = 5.374, P = 0.001) 的浓度和Bax/Bcl-2比(1.51 ± 0.18 vs 0.97 ± 0.16, t = 5.000, P = 0.001)和TIM-1/IM-1001比 (0.78 ± 0.11 vs 0.31 ± 0.06, t = 8.545, P < 0.001)在OVA/PM 组较PM 组比较显著增加。IL-4 (41.47 ± 3.40 vs 25.46 ± 2.98 pg/ml, t = 2.501, P = 0.037)浓度和Bax/Bcl-2比 (0.97 ± 0.16 vs 0.18 ± 0.03, t = 7.439, P < 0.001 )和TIM-1/IM-1 )9比(0.31 ± 0.06 vs 0.02 ± 0.01, t = 5.109, P = 0.001)在PM组较对照组比较显著增加。
结论:
PM2.5导致过敏性哮喘气道高反应性加剧与凋亡增加及T细胞免疫球蛋白域1(TIM-1)激活有关,这可能为治疗PM2.5诱发的哮喘急性加重提供治疗靶点。
INTRODUCTION
Air pollution from ambient particulate matter (PM), especially fine PM (PM2.5), has become one of the most serious environmental and public health challenges in many countries, particularly the northern regions of China. PM is a complex mixture of extremely small particles and liquid droplets in the atmosphere. Particles with diameters smaller than 2.5 μm are called PM2.5. PM2.5 can penetrate the bronchioles and alveoli, and thus, it is considered to be the most damaging particle to the lungs. Asthma is usually characterized by chronic airway inflammation and is associated with airway hyperresponsiveness (AHR). Asthma exacerbations usually occur in response to exposure to an external agent (e.g., viruses infecting the upper respiratory tract, pollen, and air pollutants). Epidemiological studies have shown that elevated concentrations of PM2.5 are correlated with an increased incidence and hospital admissions due to asthma.[1,2,3]
Many studies have confirmed that PM2.5 can enhance AHR,[4,5] but the mechanism is not yet clear. Research on the mechanisms of PM-induced toxicity has focused on inflammatory and oxidative stress responses, which have been considered important in the triggering of the cellular pathological process.[6,7,8] PM2.5 induces endoplasmic reticulum stress, mitochondrial swelling, autophagy, and apoptosis.[9,10,11] PM2.5 can induce apoptosis and AHR. However, whether the induction of apoptosis is the reason why PM2.5 aggravates the severity of asthma and how apoptosis induces AHR enhancement is unknown.
Recent studies have found that T-cell immunoglobulin and mucin domain 1 (TIM-1) is an important susceptibility gene for asthma and allergy. TIM-1 can be activated by the exposure of phosphatidylserine (PtdSer) on apoptotic cells, thus inducing asthma.[12,13,14] The aim of this study was to confirm whether PM2.5 can induce apoptosis and TIM-1 activation and thus exacerbate allergic asthma.
In previous studies, PM2.5 usually drips into the airway in the form of a suspension liquid or introduced through diesel engine exhaust,[4,5,15] but this is different from what happens in the real world. We treated mice with a nose-only “PM2.5 online enrichment system” to increase the concentration of PM2.5 in the air and simulate human exposure to pollutants in the atmosphere to observe the effects of PM2.5 on BALB/c mice undergoing sensitization and challenge with ovalbumin (OVA).
METHODS
Animals and ethics statement
Female BALB/c mice (specific pathogen-free grade, wild-type, 6 to 8 weeks old, 20 ± 2 g) were purchased from the Animal Center of Hebei Medical University and allowed to acclimatize to laboratory conditions for 1 week before being used in experiments. All animal experiments were approved by the Research Ethics Committee of The Second Hospital of Hebei Medical University (No. 2017-R023).
Animal groups and fine particulate matter exposure
BALB/c mice were housed in individually ventilated cages (IVCs) with filtered clean air, 12 h of light and 12 h of darkness at 24°C ± 2°C, and 50% ± 5% humidity in the presence of automatic water dispensers. Forty mice were acclimated for 1 week and randomly divided into the following four groups (n = 10 per group).
Control group
Mice were housed in IVCs and restrained in an animal restraining device for 4 h per day and 5 days per week for a total of 8 weeks.
Ovalbumin group
Mice were sensitized with chicken OVA (50 μg administered intraperitoneally, with 1 mg of alum in 0.2 ml of normal saline) on days 0 and 7, followed by repeated challenge with nebulized 3% OVA saline solution (30 min/day, 2 times/week).
Ovalbumin/fine particulate matter group
OVA treatment was performed in a similar manner as in the OVA group. In addition, the mice were restrained in an animal restraining device in a nose-only PM2.5 online enrichment system (Beijing HuiRongHe Technology Co., Ltd., China) for 4 h per day and 5 days per week regularly from February 14 to April 11, 2017, in Shijiazhuang City, Hebei Province, China, unless there was heavy rain or sandstorm, which can affect the enrichment effect. In the case of these inclement weather conditions, exposure was terminated and resumed during suitable weather while attempting to ensure exposure for an average of 5 days per week. The actual exposure duration was 38 days. The PM2.5 enrichment system can enrich for PM2.5 in the external environment to approximately 10-fold the concentration in the real world.
Particulate matter group
Mice were only exposed to the PM2.5 online enrichment system similarly to the OVA/PM group.
Lung mechanics, pulmonary functions, and airway hyperresponsiveness
Airway responsiveness was invasively determined based on lung resistance after challenge with aerosolized methacholine (Mch; Sigma-Aldrich, USA) as previously described.[16] Mice were anesthetized with 50 mg/kg pentobarbital and prepared for the measurement of lung mechanics (FlexiVent, SCIRESQ Scientific Respiratory Equipment Inc., Montreal, Canada). The mice were tracheostomized, intubated, and mechanically ventilated at a tidal volume of 10 ml/kg weight and a frequency of 150 breaths/min. Lung resistance was measured in response to increasing doses of aerosolized Mch (0, 3.125, 6.25, 12.5, 25, and 50 mg/ml) 24 h after the final exposure to the nose-only PM2.5 exposure system. Lung function parameters were calculated by tidal volume (10 ml/kg) dynamic PV loops using a single compartment model to determine the respiratory system resistance (Rrs), and low-volume (3 ml/kg) forced oscillation with prime frequencies was applied to the lungs. The resulting pressure and volume data were transformed into frequencies and fit to the constant phase model to calculate airway resistance (RN), tissue damping (related to tissue resistance; G), and tissue elasticity (H), and mean values were selected to indicate changes in lung functions.[17]
Collection of bronchoalveolar lavage fluid and cell classification
Mice were anesthetized and sacrificed for bronchoalveolar lavage fluid (BALF) collection 24 h after the final exposure. Then, 1 ml of normal saline was instilled into the mouse lung through tracheal intubation followed by several gentle compressions of the chest. This step was repeated twice, and up to 50% recovery was obtained. The recovered lavage fluid was centrifuged at 600 ×g for 10 min at 4°C. The number of eosinophils, neutrophils, lymphocytes, and macrophages were determined in a total of 200 cells using Wright staining under a microscope. The cell-free supernatant was frozen at −80°C for subsequent measurement of inflammatory cytokine level.
Enzyme-linked immunosorbent assay
Cytokine levels (interleukin-4 [IL-4], IL-5, and IL-33) in BALF supernatants were measured using individual enzyme-linked immunosorbent assay kits (R&D Systems, USA). The detailed procedures were performed as outlined in the instruction manuals.
Hematoxylin and eosin staining
After being fixed in 4% neutral formaldehyde solution for 72 h, the right lung tissues of mice were dehydrated, clarified, and embedded in paraffin. Tissue sections (4 μm thickness) were stained with hematoxylin and eosin (H and E) to evaluate general morphology.
Electron microscopy
After the mice were anesthetized, the left lungs were isolated as soon as possible and then divided into 1 mm × 1 mm × 1 mm sections and immersed in 4% glutaraldehyde solution at 4°C for 4 h. The samples were washed 3 times with 1/15 mol/L phosphate buffer, followed by 1% osmium tetroxide and fixed for 1–2 h, dehydrated in graded concentrations of acetone (50%, 70%, 80%, 90%, and 100%), and embedded in epoxy resin. The embedded samples were then cut into ultra-thin serial sections (50 nm, Ultramicrotome, Leica UC-7, Leica, Germany) and stained with lead citrate and uranyl acetate. The samples were subsequently visualized using an electron microscope (Hitachi, H-7500, Japan) at 80 kV at the Department of Electron Microscopy Center of Hebei Medical University. All slides were examined in a random and blinded fashion by two independent investigators.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling assays for the detection of apoptotic cells in lung
Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assays were conducted using the In situ Apoptosis Detection Kit (Roche, USA) and DAB Detection Kits (Roche, USA). Briefly, paraffin-embedded lung sections were deparaffinized and permeabilized. Then, 50 μ of TUNEL reaction mixture was added to the sections and incubated at 37°C in a humid box for 60 min. Subsequently, 50 μl of the enzyme reagent peroxidase (POD) was added to the sections and incubated at 37°C in a humid box for 30 min. The sections were washed 3 times with PBS, and 50–100 μl of DAB substrate solution was added to the sections and incubated at room temperature for 10 min. Finally, the nuclei were stained with hematoxylin, and the sections were sealed with coverslips. The negative control was parallelly incubated with 50 μl of reagent in place of the TUNEL reaction mixture. Analyses were performed using Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD, USA).
Immunohistochemistry
Bcl-2, Bax, and TIM-1 expression in lung was assessed by immunohistochemistry. Paraffin-embedded lung sections were cut into 4-μm sections, deparaffinized, hydrated in alcohol, and then subjected to antigen retrieval with citric acid. The sections were exposed to endogenous POD blockers for 10 min to quench endogenous POD activity. Lung tissue sections were incubated with antibodies against Bcl-2 (rabbit polyclonal antibody, 1:400, Proteintech Group Inc., USA), Bax (rabbit polyclonal mAb, 1:400, Cell Signaling, USA), and TIM-1 (rabbit polyclonal anti-TIM-1 antibody, 1:500, Abcam, UK) antibodies overnight at 4°C. After washing, the sections were incubated with a secondary antibody (goat polyclonal anti-rabbit IgG HRP, 1:8000, Affinity Biosciences, USA) for 20 min and exposed to a substrate chromogen mixture for 10 min. Finally, the sections were counterstained with hematoxylin and examined by light microscopy. Finally, signals were recorded by a chemiluminescence imaging analysis system (Bio-Rad ChemiDoc MP Imaging System, USA).
Western blotting
The expression of Bcl-2, Bax, and TIM-1 in the lung was assessed by Western blotting. Total protein was extracted from lung tissues with cold RIPA buffer (Beijing Solarbio Science and Technology Co., Ltd., China) containing protease and phosphatase inhibitors (Beijing Solarbio Science and Technology Co., Ltd, China). Lowry assay was conducted to quantify the protein concentration for each sample. Then, cell lysates were separated on 10% SDS-PAGE gels and transferred to PVDF membranes. The membranes were blocked with 5% nonfat milk, followed by incubation with primary antibodies against Bcl-2 (rabbit polyclonal antibody, 1:1000, Proteintech Group Inc., USA), Bax (rabbit polyclonal Bax mAb, 1:1000, Cell Signaling, USA), TIM-1 (rabbit polyclonal anti-TIM-1 antibody, 1:5000, Abcam, UK), or β-actin (1:2000, Cell Signaling, USA) overnight at 4°C. Subsequently, the membranes were incubated with an appropriate HRP-conjugated secondary antibody (goat polyclonal anti-rabbit IgG HRP, 1:8000, Affinity Biosciences, USA). Finally, protein bands were detected using SuperECL Plus detection reagents (LI-COR company, USA), and signals were recorded by a chemiluminescence imaging analysis system (Bio-Rad ChemiDoc MP Imaging System, USA). Densitometric analyses were performed using Photoshop CS6 (Adobe Systems Incorporated, USA). Protein expression levels are shown as fold changes relative to β-actin.
Statistical analysis
Data are expressed as the mean ± standard deviation (SD) and analyzed using SPSS software version 19.0 (SPSS Inc., Chicago, IL, USA). Differences among treatment groups were evaluated by one-way analysis of variance (ANOVA). A value of P < 0.05 was considered statistically significant.
RESULTS
Concentration and components of fine particulate matter
During the exposure period, the mean daily ambient PM2.5 concentration was 94 μg/m3(from the Shijiazhuang Environmental Protection Bureau). After considering the unexposed time and weekends (the actual exposure duration was 38 days), the calculated average daily PM2.5 concentration was 101 μg/m3. The mean daily ambient PM2.5 concentration in the exposure chamber was calculated. As shown in Figure 1, the maximum exposure concentration was 3016 μg/m3, the minimum exposure concentration was 109 μg/m3, and the mean daily concentration was 703 μg/m3.
Figure 1.
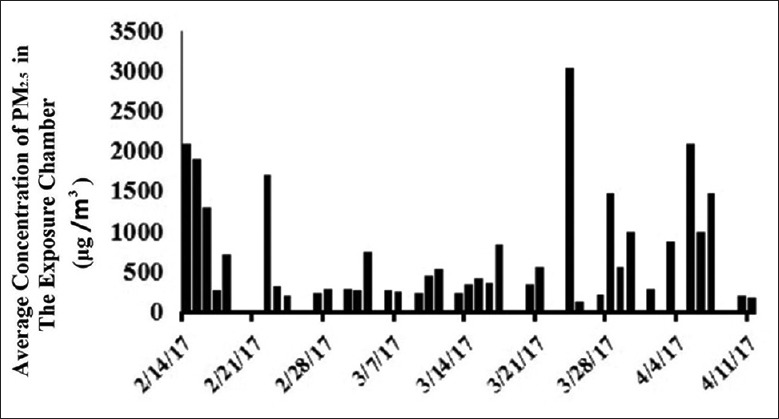
The mean concentration of PM2.5 in the exposure chamber from February 14 to April 11, 2017, in Shijiazhuang, Hebei, China. PM2.5: Fine particulate matter.
Animal behavior in different groups
The mice in the control group did not show any abnormal activities. The mice in the OVA/PM group displayed several typical characteristics of asthma such as fidgeting, staying silent, slow movement, shortness of breath, or nose grasping. The mice in the OVA group also showed behaviors associated with asthma such as fidgeting, staying silent, and slow movement, but the symptoms were milder than those in the OVA/PM group. The mice in the PM group also showed abnormal activities similar to the mice in the OVA group, but the degree was milder.
Lung mechanics, pulmonary functions, and airway hyperresponsiveness in different groups
To determine the effects of PM2.5 exposure on lung function, we tracheostomized, intubated, and mechanically ventilated the mice and then allowed them to inhale various concentrations of aerosolized Mch (0–50 mg/ml). Rrs, RN, G, and H were recorded as shown in Figure 2.
Figure 2.

Airway responsiveness in different groups. (a) Rrs in the control, OVA, OVA/PM, and PM groups at different concentrations of aerosolized Mch. (b) RN in the four groups. (c) G in the four groups. (d) H in the four groups. Values are represented as the mean ± SD. Based on one-way ANOVA, followed by LSD multiple range tests, comparing with control group, a significant difference is indicated by *P < 0.001, †P < 0.01, and ‡P < 0.05. Comparing with PM group, a significant difference is indicated by §P < 0.01 and ||P < 0.05. Comparing with OVA group, a significant difference is indicated by ¶P < 0.01 and **P < 0.05 (n = 6 per group). Rrs: Respiratory system resistance; RN: Airway resistance; G: Tissue damping; H: Tissue elasticity; Mch: Methacholine; PM: Particulate matter; OVA: Ovalbumin; SD: Standard deviation; ANOVA: Analysis of variance; LSD: Least significant difference.
The Rrs reflects the resistance of the entire respiratory system including the central airway, lung tissues, and thorax. As shown in Figure 2a, with 6.25 (1.64 ± 0.31 vs. 1.10 ± 0.06 cmH2O·ml−1·s−1, t = 4.115, P = 0.030), 12.5 (2.32 ± 0.59 vs. 1.15 ± 0.07 cmH2O·ml−1·s−1, t = 4.515, P = 0.002), 25 (2.68 ± 0.52 vs. 1.47 ± 0.26 cmH2O·ml−1·s−1, t = 4.911, P = 0.001), and 50 (2.83 ± 0.68 vs. 1.50 ± 0.23 cmH2O·ml−1·s−1, t = 4.463, P = 0.002) mg/ml Mch, the Rrs of the OVA/PM group was significantly higher than that of the control group. With 6.25 (1.64 ± 0.31 vs. 1.20 ± 0.01 cmH2O·ml−1·s−1, t = 3.290, P = 0.011) mg/ml Mch, the Rrs of the OVA/PM group was significantly higher than that of the OVA group. With 6.25 (1.64 ± 0.31 vs. 1.26 ± 0.06 cmH2O·ml−1·s−1, t = 2.832, P = 0.022) and 12.5 (2.32 ± 0.59 vs. 1.68 ± 0.25 cmH2O·ml−1·s−1, t = 2.416, P = 0.042) mg/ml Mch, the Rrs of the OVA/PM group was significantly higher than that of the PM group. With 25 (2.18 ± 0.16 vs. 1.47 ± 0.26 cmH2O·ml−1·s−1, t = 2.915, P = 0.019) and 50 (2.50 ± 0.07 vs. 1.50 ± 0.23 cmH2O·ml−1·s−1, t = 3.366, P = 0.010) mg/ml Mch, the Rrs of the PM group was significantly higher than that of the control group. With 12.5 (1.20 ± 0.01 vs. 1.15 ± 0.07 cmH2O·ml−1·s−1, t = 3.093, P = 0.015), 25 (1.94 ± 0.11 vs. 1.47 ± 0.26 cmH2O·ml−1·s−1, t = 3.814, P = 0.005), and 50 (2.49 ± 0.12 vs. 1.50 ± 0.23 cmH2O·ml−1·s−1, t = 3.332, P = 0.010) mg/ml Mch, the Rrs of the OVA group was significantly higher than that of the control group. With 12.5 mg/ml Mch, the Rrs of the OVA/PM group was 2.32 ± 0.59 cmH2O·ml−1·s−1, more than twice the baseline value (0.85 ± 0.05 cmH2O·ml−1·s−1), indicating the presence of AHR. With 25 mg/ml Mch, the Rrs of the PM group was 2.18 ± 0.16 cmH2O·ml−1·s−1, more than twice the baseline value (0.78 ± 0.02 cmH2O·ml−1·s−1). With 12.5 mg/ml Mch, the Rrs of the OVA group was 1.94 ± 0.17 cmH2O·ml−1·s−1, more than twice the baseline value (0.82 ± 0.03 cmH2O·ml−1·s−1).
The RN is based on the Newtonian resistance parameter of the constant-phase model and represents the resistance of the central airway. As shown in Figure 2b, with 6.25 (1.00 ± 0.35 vs. 0.52 ± 0.03 cmH2O·ml−1·s−1, t = 2.843, P = 0.022), 12.5 (1.17 ± 0.34 vs. 0.52 ± 0.02 cmH2O·ml−1·s−1, t = 4.479, P = 0.002), 25 (1.38 ± 0.45 vs. 0.63 ± 0.10 cmH2O·ml−1·s−1, t = 3.740, P = 0.006), and 50 (1.37 ± 0.18 vs. 0.71 ± 0.16 cmH2O·ml−1·s−1, t = 5.842, P = 0.001) mg/ml Mch, the RN of the OVA/PM group was significantly higher than that of the control group. With 50 (1.37 ± 0.18 vs. 1.07 ± 0.08 cmH2O·ml−1·s−1, t = 2.701, P = 0.036) mg/ml Mch, the RN of the OVA/PM group was significantly higher than that of the OVA group. With 50 (1.37 ± 0.18 vs. 1.02 ± 0.13 cmH2O·ml−1·s−1, t = 3.114, P = 0.034) mg/ml Mch, the RN of the OVA/PM group was significantly higher than that of the PM group. With 50 (1.02 ± 0.13 vs. 0.71 ± 0.16 cmH2O·ml−1·s−1, t = 2.737, P = 0.026) mg/ml Mch, the RN of the PM group was significantly higher than that of the control group. With 12.5 (0.95 ± 0.05 vs. 0.52 ± 0.02 cmH2O·ml−1·s−1, t = 2.925, P = 0.019) and 50 (1.07 ± 0.08 vs. 0.71 ± 0.16 cmH2O·ml−1·s−1, t = 3.149, P = 0.019) mg/ml Mch, the RN of the OVA group was significantly higher than that of the control group. In addition, with 6.25 mg/ml Mch, the RN of the OVA/PM group was 1.00 ± 0.35 cmH2O·ml−1·s−1, more than twice the baseline value (0.49 ± 0.09 cmH2O·ml−1·s−1). With 25 mg/ml Mch, the RN of the PM group was 0.94 ± 0.11 cmH2O·ml−1·s−1, more than twice the baseline value (0.44 ± 0.04 cmH2O·ml−1·s−1). With 12.5 mg/ml Mch, the RN of the OVA group was 0.95 ± 0.05 cmH2O·ml−1·s−1, more than twice the baseline value (0.49 ± 0.09 cmH2O·ml−1·s−1).
G represents tissue damping and is closely related to tissue resistance and reflects energy dissipation in lung tissues. As shown in Figure 2c, with 12.5 (14.56 ± 1.45 vs. 6.84 ± 0.82 cmH2O/ml, t = 5.493, P = 0.001), 25 (17.35 ± 3.26 vs. 7.65 ± 0.69 cmH2O/ml, t = 4.529, P = 0.002), and 50 (18.94 ± 4.50 vs. 9.51 ± 1.60 cmH2O/ml, t = 3.453, P = 0.009) mg/ml Mch, the G of the OVA/PM group was significantly higher than that of the control group. With 12.5 (14.56 ± 1.45 vs. 11.21 ± 2.21 cmH2O/ml, t = 2.381, P = 0.044) mg/ml Mch, the G of the OVA/PM group was significantly higher than that of the PM group. With 12.5 (11.21 ± 2.21 vs. 6.83 ± 0.82 cmH2O/ml, t = 3.112, P = 0.014) and 25 (12.76 ± 3.41 vs. 7.65 ± 0.69 cmH2O/ml, t = 2.386, P = 0.044) mg/ml Mch, the G of the PM group was significantly higher than that of the control group. With 12.5 (11.49 ± 2.50 vs. 6.83 ± 0.82 cmH2O/ml, t = 3.309, P = 0.011), 25 (14.26 ± 2.19 vs. 7.65 ± 0.69 cmH2O/ml, t = 3.085, P = 0.015), and 50 (15.75 ± 2.88 vs. 9.51 ± 1.60 cmH2O/ml, t = 2.284, P = 0.049) mg/ml Mch, the G of the OVA group was significantly higher than that of the control group. With 6.25 mg/ml Mch, the G of the OVA/PM group was 12.72 ± 1.00 cmH2O/ml, more than twice the baseline value (5.29 ± 0.06 cmH2O/ml). With 25 mg/ml Mch, the G of the PM group was 12.76 ± 3.41 cmH2O/ml, more than twice the baseline value (5.00 ± 0.34 cmH2O/ml). With 12.5 mg/ml Mch, the G of the OVA group was 11.49 ± 2.05 cmH2O/ml, more than twice the baseline value (5.50 ± 0.40 cmH2O/ml).
H represents tissue elasticity and reflects energy conservation in lung tissues. As shown in Figure 2d, with 6.25 (49.04 ± 11.31 vs. 23.38 ± 2.12 cmH2O/ml, t = 4.293, P = 0.003), 12.5 (59.42 ± 7.01 vs. 24.77 ± 2.96 cmH2O/ml, t = 6.367, P < 0.001), 25 (68.30 ± 12.81 vs. 27.80 ± 4.48 cmH2O/ml, t = 6.349, P < 0.001), and 50 (74.76 ± 9.49 vs. 31.25 ± 5.75 cmH2O/ml, t = 8.485, P < 0.001) mg/ml Mch, the H of the OVA/PM group was significantly higher than that of the control group. With 12.5 (59.42 ± 7.01 vs. 41.99 ± 9.48 cmH2O/ml, t = 3.204, P = 0.013), 25 (68.30 ± 12.81 vs. 48.85 ± 5.97 cmH2O/ml, t = 3.049, P = 0.016), and 50 (74.76 ± 9.49 vs. 56.87 ± 5.37 cmH2O/ml, t = 3.489, P = 0.008) mg/ml Mch, the H of the OVA/PM group was significantly higher than that of the OVA group. With 6.25 (49.04 ± 11.31 vs. 23.38 ± 2.12 cmH2O/ml, t = 2.851, P = 0.021), 12.5 (59.42 ± 7.01 vs. 39.32 ± 5.48 cmH2O/ml, t = 3.692, P = 0.006), 25 (68.30 ± 12.81 vs. 46.03 ± 4.92 cmH2O/ml, t = 3.492, P = 0.008), and 50 (74.76 ± 9.49 vs. 54.20 ± 2.41 cmH2O/ml, t = 4.009, P = 0.004) mg/ml Mch, the H of the OVA/PM group was significantly higher than that of the PM group. With 12.5 (39.32 ± 5.48 vs. 24.77 ± 2.96 cmH2O/ml, t = 2.675, P = 0.028), 25 (46.03 ± 4.92 vs. 27.80 ± 4.48 cmH2O/ml, t = 2.857, P = 0.021), and 50 (54.20 ± 2.41 vs. 31.25 ± 5.75 cmH2O/ml, t = 4.476, P = 0.002) mg/ml Mch, the H of the PM group was significantly higher than that of the control group. With 12.5 (41.99 ± 9.48 vs. 24.77 ± 2.96 cmH2O/ml, t = 3.164, P = 0.013), 25 (48.85 ± 5.97 vs. 27.80 ± 4.48 cmH2O/ml, t = 3.300, P = 0.011), and 50 (56.87 ± 5.37 vs. 31.25 ± 5.75 cmH2O/ml, t = 4.996, P = 0.001) mg/ml Mch, the H of the OVA group was significantly higher than that of the control group. With 6.25 mg/ml Mch, the H of the OVA/PM group was 49.04 ± 6.25 cmH2O/ml, more than twice the baseline value (23.07 ± 0.77 cmH2O/ml). With 25 mg/ml Mch, the H of the PM group was 46.03 ± 4.92 cmH2O/ml, more than twice the baseline value (20.57 ± 1.62 cmH2O/ml). With 25 mg/ml Mch, the H of the OVA group was 48.85 ± 5.97 cmH2O/ml, more than twice the baseline value (22.74 ± 0.84 cmH2O/ml).
The results showed that the AHR in the OVA/PM group was significantly more severe than that in the OVA group, and AHR in the PM group was also much more severe than that in the control group but less severe than that in the OVA/PM group.
Changes in inflammatory cells in bronchoalveolar lavage fluid
Eosinophils, lymphocytes, neutrophils, and macrophages in BALF were counted as shown in Figure 3. The number of eosinophils in the BALF of the OVA (21.33 ± 3.06 vs. 1.33 ± 0.58, t = 13.583, P < 0.001) and OVA/PM (18.67 ± 2.08 vs. 1.33 ± 0.58, t = 11.858, P < 0.001) groups was significantly higher than those in the BALF of the control group. The number of eosinophils in the BALF of the OVA (21.33 ± 3.06 vs. 1.33 ± 0.58, t = 12.937, P < 0.001) and OVA/PM (18.67 ± 2.08 vs. 1.33 ± 0.58, t = 11.212, P < 0.001) groups was significantly higher than those in the BALF of the PM group. The number of lymphocytes in the OVA/PM (28.00 ± 6.08 vs. 12.33 ± 4.51, t = 4.631, P = 0.002) and PM (29.00 ± 3.00 vs. 12.33 ± 4.51, t = 4.927, P = 0.001) groups was higher than those in the control group, and the number of lymphocytes in the OVA group (22.33 ± 1.53 vs. 12.33 ± 4.51, t = 2.956, P = 0.018) was higher than that in the control group. Furthermore, the number of neutrophils in the BALF of the OVA/PM (6.67 ± 1.53 vs. 3.33 ± 1.53, t = 2.886, P = 0.020) and PM group (6.67 ± 1.53 vs. 3.33 ± 1.53, t = 2.886, P = 0.020) was significantly higher than those in the BALF of OVA groups, and the number of neutrophils in the BALF of the OVA/PM (6.67 ± 1.53 vs. 2.00 ± 1.00, t = 4.041, P = 0.004) and PM groups (6.67 ± 1.53 vs. 2.00 ± 1.00, t = 4.041, P = 0.004) was significantly higher than those in the BALF of the control.
Figure 3.

Cell classification in BALF: number of eosinophils, lymphocytes, neutrophils, and macrophages based on a total of 200 cells in the BALF of the control, OVA, OVA/PM, and PM groups. Values are mean ± SD. Using one-way ANOVA, followed by LSD multiple range test, comparing with control group, a significant difference is indicated by *P < 0.001, †P < 0.01, and ‡P < 0.05. Comparing with PM group, significant difference is indicated by §P < 0.001, ||P < 0.01, and ¶P < 0.05. Comparing with OVA group, significant difference is indicated by **P < 0.05 (n = 6 per group). BALF: Bronchoalveolar lavage fluid; PM: Particulate matter; OVA: Ovalbumin; SD: Standard deviation; ANOVA: Analysis of variance; LSD: Least significant difference.
Changes in inflammatory cytokines in bronchoalveolar lavage fluid
As shown in Figure 4, IL-4 (77.44 ± 11.19 vs. 25.46 ± 2.98 pg/ml, t = 8.118, P < 0.001) and IL-5 (15.65 ± 1.19 vs. 9.75 ± 0.49 pg/ml, t = 6.804, P < 0.001) levels in the BALF of the OVA/PM group were significantly higher than those in the control group. IL-4 (77.44 ± 11.19 vs. 41.47 ± 3.40 pg/ml, t = 5.617, P = 0.001) and IL-5 (15.65 ± 1.19 vs. 10.99 ± 1.40 pg/ml, t = 5.374, P = 0.001) levels in the BALF of the OVA/PM group were also significantly higher than those in the PM group. IL-4 (77.44 ± 11.19 vs. 48.02 ± 10.02 pg/ml, t = 4.595, P = 0.002) and IL-5 (15.65 ± 1.19 vs. 12.35 ± 0.95 pg/ml, t = 3.806, P = 0.005) levels in the BALF of the OVA/PM group were also significantly higher than those in the OVA group. The IL-4 (41.47 ± 3.40 vs. 25.46 ± 2.98 pg/ml, t = 2.501, P = 0.037) level in the BALF of the PM group was significantly higher than that in the BALF of the control group. The IL-4 (48.02 ± 10.02 vs. 25.46 ± 2.98 pg/ml, t = 3.523, P = 0.008) and IL-5 (12.35 ± 0.95 vs. 9.75 ± 0.49 pg/ml, t = 2.998, P = 0.017) levels in the OVA group were significantly higher than those in the control group. IL-33 levels in the BALF of the OVA/PM group (151.27 ± 10.61 vs. 131.43 ± 8.71 pg/ml, t = 3.075, P = 0.015) and OVA group (151.36 ± 6.25 vs. 131.43 ± 8.71 pg/ml, t = 3.079, P = 0.015) were significantly higher than those in the BALF of the control groups. IL-33 levels in the BALF of the OVA/PM group (151.27 ± 10.61 vs. 136.21 ± 4.73 pg/ml, t = 2.335, P = 0.048) and OVA group (151.36 ± 6.25 vs. 136.21 ± 4.73 pg/ml, t = 2.349, P = 0.047) were significantly higher than those in the BALF of the PM groups.
Figure 4.

Cytokine analysis in BALF with ELISA: IL-4, IL-5, and IL-33 levels in the BALF of the control, OVA, OVA/PM, and PM groups. Values are mean ± SD. Using one-way ANOVA, followed by LSD multiple range test, comparing with control group, significant difference is indicated by *P < 0.001, †P < 0.01, and ‡P < 0.05. Comparing with PM group, a significant difference is indicated by §P < 0.01 and ||P < 0.05. Comparing with OVA group, a significant difference is indicated by ¶P < 0.01 (n = 6 per group). ELISA: Enzyme-linked immunosorbent assay; IL-4: Interleukin-4; IL-5: Interleukin-5; IL-33: Interleukin-33; PM: Particulate matter; OVA: Ovalbumin; SD: Standard deviation; ANOVA: Analysis of variance; LSD: Least significant difference.
Histopathological changes in the lung
Representative images of H and E-stained lung tissues are shown in Figure 5. The control group had intact terminal bronchioles and alveolar epithelia and showed no inflammation. The OVA group displayed hyperplasia of smooth muscles of the small bronchi, hyperplasia of lymphatic follicles, and infiltration of eosinophils. The OVA/PM group displayed mild loss of tracheal epithelial cells, widening of the alveolar septa, infiltration of inflammatory cells, and hyperplasia of smooth muscles of the small bronchi. The PM group displayed the presence of inflammatory cells in peribronchiolar regions and substantial alveolar epithelial hyperplasia.
Figure 5.
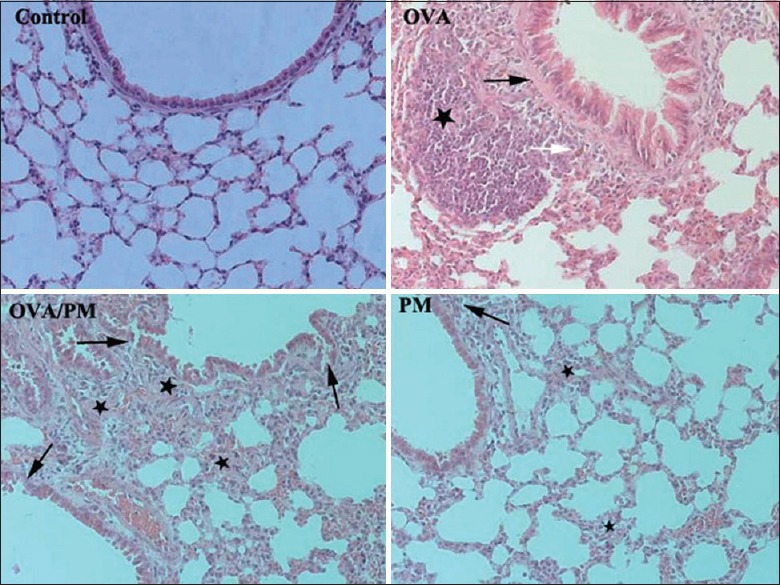
Comparison of the changes in lung tissues of mice in different groups (hematoxylin and eosin, ×200). The control group had intact terminal bronchioles and alveolar epithelia and showed no inflammation. The OVA group showed hyperplasia of smooth muscles of the small bronchi (black arrow), hyperplasia of lymphatic follicles (star), and infiltration of eosinophils (white arrow). The OVA/PM group displayed mild loss of tracheal epithelial cells, widening of the alveolar septa (black arrows), infiltration of inflammatory cells, and hyperplasia of smooth muscles of the small bronchi (stars). The PM group displayed the presence of inflammatory cells in peribronchiolar regions (black arrow) and substantial alveolar epithelial hyperplasia (stars). PM: Particulate matter; OVA: Ovalbumin.
Ultrastructural damage observed under electron microscopy
Normal lung ultramicrostructures were observed in the control group. As shown in Figure 6, the OVA/PM group displayed Type II alveolar epithelium with abnormal mitochondria and slight fusion and deterioration of the nuclear membrane and mitochondrial cristae and interstitial fibrosis. The PM group displayed Type II alveolar epithelium with abnormal mitochondria and slight fusion and deterioration of the nuclear membrane and mitochondrial cristae.
Figure 6.

Comparison of the ultrastructural changes in lung tissues of mice in different groups. The OVA group (both ×15,000) displayed eosinophilic hyperplasia. In the OVA/PM group, black arrow indicates interstitial fibrosis (upper ×8000) and white arrow indicates type II alveolar epithelium with abnormal mitochondria and slight fusion and deterioration of the nuclear membrane and mitochondrial cristae (lower ×5000). In the PM group, black arrows indicate type II alveolar epithelium with abnormal mitochondria and slight fusion and deterioration of the nuclear membrane and mitochondrial cristae (upper ×6000 and lower ×15,000). PM: Particulate matter; OVA: Ovalbumin.
Apoptosis analysis by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling assay
The TUNEL assay was used to determine lung cell apoptosis. As shown in Figure 7, multiple nuclei in the OVA/PM group showed obvious deep brown staining, which was significantly different from that in the OVA group (Tunel immunohistochemical scores [IHS%], 1.20 ± 0.18 vs. 0.51 ± 0.03, t = 8.094, P < 0.001), PM group (Tunel IHS%, 1.20 ± 0.18 vs. 0.51 ± 0.09, t = 8.094, P < 0.001), and control group (Tunel IHS%, 1.20 ± 0.18 vs. 0.26 ± 0.03, t = 11.000, P < 0.001). The OVA group (Tunel IHS%, 0.51 ± 0.03 vs. 0.26 ± 0.03, t = 2.894, P = 0.020) and PM group (Tunel IHS%, 0.51 ± 0.09 vs. 0.26 ± 0.03, t = 2.894, P = 0.020) displayed darker brown staining than did the control group. The OVA and PM groups demonstrated no significant difference.
Figure 7.

Apoptotic cells were determined by TUNEL staining in the lung tissues of mice in the control, OVA, OVA/PM, and PM groups (×200). TUNEL-positive cells were found in the bronchia and lung cells. Values are mean ± SD. Using one-way ANOVA, followed by LSD multiple range test, comparing with control group, a significant difference is indicated by *P < 0.001 and †P < 0.05. Comparing with PM group, a significant difference is indicated by ‡P < 0.001. Comparing with OVA group, a significant difference is indicated by §P < 0.001 (n = 6 per group). TUNEL: Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling; IHS: Immunohistochemical scores; PM: Particulate matter; OVA: Ovalbumin; SD: Standard deviation; ANOVA: Analysis of variance; LSD: Least significant difference.
Western blotting and immunohistochemistry
The protein levels of Bcl-2, Bax, and TIM-1 were measured by Western blotting as shown in Figure 8. The ratios of Bax/Bcl-2 (1.51 ± 0.18 vs. 0.18 ± 0.03, t = 12.448, P < 0.001) and TIM-1/β-actin (0.78 ± 0.11 vs. 0.02 ± 0.01, t = 13.655, P < 0.001) were significantly higher in the OVA/PM group than those in the control group. The ratios of Bax/Bcl-2 (1.51 ± 0.18 vs. 0.97 ± 0.16, t = 5.000, P = 0.001) and TIM-1/β-actin (0.78 ± 0.11 vs. 0.31 ± 0.06, t = 8.545, P < 0.001) were significantly higher in the OVA/PM group than those in the PM group. The ratios of Bax/Bcl-2 (1.51 ± 0.18 vs. 0.48 ± 0.10, t = 9.654, P < 0.001) and TIM-1/β-actin (0.78 ± 0.11 vs. 0.40 ± 0.06, t = 6.818, P < 0.001) were significantly higher in the OVA/PM group than those in the OVA group. The ratios of Bax/Bcl-2 (0.48 ± 0.10 vs. 0.18 ± 0.03, t = 2.785, P = 0.023) and TIM-1/β-actin (0.40 ± 0.06 vs. 0.02 ± 0.01, t = 6.818, P < 0.001) were significantly higher in the OVA group than those in the control group. The ratios of Bax/Bcl-2 (0.97 ± 0.16 vs. 0.18 ± 0.03, t = 7.439, P < 0.001) and TIM-1/β-actin (0.31 ± 0.06 vs. 0.02 ± 0.01, t = 5.109, P = 0.001) were significantly higher in the PM group than those in the control group. The Bax/Bcl-2 ratio was significantly higher in the PM group than that in the OVA group (0.97 ± 0.16 vs. 0.48 ± 0.10, t = 4.654, P = 0.002).
Figure 8.

Effects of PM2.5 on the expression of apoptosis-regulatory proteins and TIM-1 in mice in different groups determined by Western blotting. (a) Images of Western blotting for Bax and Bcl-2. (b) Comparisons of the Bax/Bcl-2 ratio. (c) Images of Western blotting for TIM-1. (d) Comparisons of the expression of TIM-1. Values are mean ± SD. Using one-way ANOVA, followed by LSD multiple range test, comparing with control group, comparing with control group, significant difference is indicated by *P < 0.001, †P < 0.01, and ‡P < 0.05. Comparing with PM group, significant difference is indicated by §P < 0.001 and ||P < 0.01. Comparing with OVA group, significant difference is indicated by ¶P < 0.001 (n = 6 per group). TIM-1: T-cell Immunoglobulin and Mucin Domain 1; PM: Particulate matter; OVA: Ovalbumin; SD: Standard deviation; ANOVA: Analysis of variance; LSD: Least significant difference.
The immunohistochemistry results showed the same tendency as Western blotting as demonstrated in Figure 9. The integral optical density (IOD) of Bax in OVA/PM group was significantly higher than that in the PM group (16,382.00 ± 1316.77 vs. 9386.00 ± 735.19, t = 10.572, P < 0.001), OVA group (16,382.00 ± 1316.77 vs. 5097.00 ± 511.87, t = 17.053, P < 0.001), and control group (16,382.00 ± 1316.77 vs. 2139.00 ± 302.16, t = 21.522, P < 0.001). The IOD of Bax in PM group was significantly higher than that in the OVA group (9386.00 ± 735.19 vs. 5097.00 ± 511.87, t = 6.481, P < 0.001) and control group (9386.00 ± 735.19 vs. 2139.00 ± 302.16, t = 10.951, P < 0.001). The IOD of Bax in OVA group was significantly higher than that in the control group (5097.00 ± 511.87 vs. 2139.00 ± 302.16, t = 4.319, P = 0.002).
Figure 9.

Effects of PM2.5 on the expression of apoptotic regulatory proteins and TIM-1 in mice in different groups with immunohistochemistry (×200). (a-d) Images of immunohistochemistry of Bax in control, OVA, OVA/PM, and PM groups; (e) IOD of Bax; (f-i) images of immunohistochemistry of Bcl-2 in four groups; (j) IOD of Bcl-2; (k-n) images of immunohistochemistry of TIM-1in four groups; (o) IHS% of TIM-1. Values are mean ± SD. Using one-way ANOVA, followed by LSD multiple range test, comparing with control group, significant difference is indicated by *P < 0.001, †P < 0.01, and ‡P < 0.05. Comparing with PM group, significant difference is indicated by §P < 0.001 and ||P < 0.01. Comparing with OVA group, significant difference is indicated by ¶P < 0.001, **P < 0.01, and ††P < 0.05 (n = 6 per group). IOD: Integral optical density; IHS: Immunohistochemical scores; PM: Particulate matter; OVA: Ovalbumin; SD: Standard deviation; ANOVA: Analysis of variance; LSD: Least significant difference.
The IOD of Bcl-2 in OVA/PM group was significantly lower than that in the PM group (20,221.00 ± 1380.52 vs. 34,941.67 ± 3998.41, t = −4.231, P = 0.003), OVA group (20,221.00 ± 1380.52 vs. 30,221.67 ± 2153.20, t = −2.875, P = 0.021), and control group (20,221.00 ± 1380.52 vs. 49,944.00 ± 7077.39, t = −8.544, P < 0.001). The IOD of Bcl-2 in PM group was significantly lower than that in the control group (34,941.67 ± 3998.41 vs. 49,944.00 ± 7077.39, t = −4.312, P = 0.003). The IOD of Bcl-2 in OVA group was significantly lower than that in the control group (30,221.67 ± 2153.20 vs. 49,944.00 ± 7077.39, t = −5.669, P < 0.001). The IOD of Bcl-2 was not significantly different between OVA and PM groups (P = 0.212).
The IHS (%) of TIM-1 in OVA/PM group was significantly higher than that in the OVA group (0.54 ± 0.12 vs. 0.23 ± 0.04, t = 5.508, P = 0.001), PM group (0.54 ± 0.12 vs. 0.26 ± 0.05, t = 5.000, P = 0.001), and control group (0.54 ± 0.12 vs. 0.07 ± 0.01, t = 8.281, P < 0.001). The IHS (%) of TIM-1 in PM group was significantly higher than that in the control group (0.26 ± 0.05 vs. 0.07 ± 0.01, t = 3.281, P = 0.012). The IHS (%) of TIM-1 in OVA group was significantly higher than that in the control group (0.23 ± 0.04 vs. 0.07 ± 0.01, t = 2.772, P = 0.025).
DISCUSSION
Shijiazhuang is located in Northern China and is one of the cities with heavy pollution in China. Shijiazhuang is the fifth contributor to pollution-related mortality, accounting for 2.02% of the total deaths caused by air pollution.[18] Asthma attack among all ages was the second disease-related hospital admissions due to PM pollution estimated from 2014 to 2015.[19] In Shijiazhuang, industrial emissions and secondary aerosols are the major sources of PM2.5, followed by vehicle emissions, and pollution caused by coal burning during the winter exacerbates this situation. Many studies have suggested that the effect of PM2.5 on asthma is mostly driven by total ambient pollutant exposure.[20,21] The mean daily PM2.5 concentration for the mice was 703 μg/m3, and the average daily PM2.5 concentration in Shijiazhuang was 101 μg/m3, which is much higher than the National Ambient Air Quality Standards set by the Ministry of Ecology and Environment of the People's Republic of China (15 μg/m3 for the annual mean and 35 μg/m3 for the 24 h mean) and the WHO Air Quality Guidelines (10 μg/m3 for the annual mean and 25 μg/m3 for the 24 h mean).
The common method of observing PM2.5-induced damage to the lungs involves the administration of PM2.5 through intranasal instillation in the form of a suspension liquid.[4,22] This type of treatment is suitable for the study of acute injury. For this method, the collection and recovery of PM2.5 involve the use of a multisolvent filter extraction technique that combines sonication in multiple solvents accompanied by microporous membrane filtration.[4] Some components may change or be lost during this process. We treated the mice using a nose-only exposure system on the “PM2.5 online enrichment system” for the purpose of observing the effects of PM2.5 on the lungs, thus maintaining the normal inhalation pathway and the characteristics of PM2.5 exposure in the real world. Therefore, our approach is better suited to study the effects of chronic exposure to PM2.5 and to reflect the situation in the real world.
PM2.5 can significantly aggravate AHR in OVA-exposed mice, and mice in the OVA/PM group display typical pathologic features of asthma. In the PM group, with 25 mg/ml Mch, the Rrs, Rn, G, and H were more than twice the baseline values, which means that AHR was established. In the OVA group, with 12.5 mg/ml Mch, the Rrs, Rn, and G were more than twice the baseline values, which means that AHR was established. For mice in the OVA/PM group, with 6.125 mg/ml to 12.5 mg/ml Mch, the Rrs, Rn, G, and H were greater than twice the baseline values and displayed significant differences compared with the mice in the OVA, PM, and control groups. These results indicated that 8 weeks of treatment with an average daily concentration of 703 μg/m3 PM2.5 combined with OVA stimulates AHR earlier than does OVA or PM alone.
Moreover, from the H and E staining results, we could see that the OVA/PM group showed mild loss of tracheal epithelial cells, widening of the alveolar septum, infiltration of inflammatory cells, and hyperplasia of smooth muscles of the small bronchi, indicative of the pathological features of asthma. The pathological changes in the OVA/PM group were significantly more severe than those in the PM and OVA groups. Moreover, no eosinophils were found in the PM group.
Previous studies have shown inconsistent results in PM2.5-associated Th1 or Th2 responses. Some studies have found that PM2.5 exposure drives a Th1-biased immune response in human or animal models,[23] and some studies have shown that the Th2 immune response is dominant.[24,25] Asthma induced by OVA is characterized by the presence of eosinophils in the airways. In the OVA/PM group, the number of eosinophils, lymphocytes, and neutrophils and the levels of IL-4 and IL-5 in BALF were significantly higher than those in the other three groups. However, in the PM group, the number of lymphocytes and neutrophils and the level of IL-4 were significantly higher than those in the control group. Thus, the results show that lymphocytes, neutrophils, and IL-4 contribute to airway inflammation, which is aggravated by PM2.5.
In this study, based on electron microscopy, mice in the OVA/PM group showed type II alveolar epithelium with abnormal mitochondria and slight fusion and deterioration of the nuclear membrane and mitochondrial cristae. The PM group has abnormal mitochondria. All of these indicate cellular damage, consistent with another study.[26]
Based on the TUNEL assay, the OVA/PM group had more apoptotic cells, which were significantly different from that in the OVA, PM, and control groups. Meanwhile, Bax/Bcl-2 levels increased, which confirmed that the extent of apoptosis was higher in mice in the OVA/PM group than in mice in the other three groups. PM2.5 might elicit oxidative stress and mitochondria-dependent apoptosis and autophagy,[6,27] and some studies on pollutants have confirmed that these changes can increase apoptosis induced by damage.[28,29] The results of these studies are consistent with ours.
However, in some previous studies, allergic asthma was characterized by higher Bcl-2 expression and low Bax expression, and the pathogenetic value of apoptotic disorders was established in persistent allergic inflammation.[30,31] Therefore, on the basis of increased apoptosis, mice in the OVA/PM group should show milder AHR than mice in the OVA group, but our results showed an enhancement of AHR.
TIM-1 is an important susceptibility gene for asthma and allergy. The protein encoded by the TIM-1 gene is a type I transmembrane glycoprotein, which is mainly expressed in CD4+ cells, induces Th2 cell activation, and functions as a potent costimulatory molecule for TH2 cells. The expression of TIM-1 results in a significant increase in the number of cells producing IL-4.[32,33,34]
TIM-1 is also a receptor for PtdSer. PtdSer is an important marker of cells undergoing programmed cell death or apoptosis.[12,35,36] PtdSer is normally localized to the inner leaflet of the plasma membrane but is redistributed and exposed on the outer membrane when a cell undergoes apoptosis. TIM-1, a receptor for PtdSer expressed by apoptotic cells, drives the development of asthma by sensing and responding to apoptotic airway epithelial cells. This result was further confirmed by a study with TIM-1(-/-) mice.[37]
We used Western blotting and immunohistochemistry to detect the levels of TIM-1 in lung tissues and found that TIM-1 levels were significantly higher in the PM group than in the control group. In addition, TIM-1 levels were considerably greater in the OVA/PM group than in the other three groups. TIM-1 expression was consistent with the trends in apoptosis and changes in lung functions.
Therefore, PM2.5 altered the airway, and the subsequent lung tissue damage induced cell apoptosis. PtdSer was exposed on the outer membrane and then activated TIM-1, thereby enhancing Th2 cell activation and leading to the AHR. The damage ultimately resulted in lung fibrosis after injury. Increased IL-4 levels were observed in the in BALF of the OVA/PM group, consistent with TIM-1-induced Th2 cell activation.[32,33,34]
There was a significant increase in IL-5 levels, but no significant increase in the number of eosinophils in the lungs of mice in the OVA/PM group. The reasons may be as follows: first, the low level of IL-5 is not sufficient to increase the number of eosinophils relative to that in the other groups. Second, the production of eosinophils is influenced by many factors other than IL-5.[38] Regrettably, due to the uncontrollable real-time changes in air pollution, we failed to implement antiapoptosis measures or other interventions after observing the possible effects of apoptosis. Therefore, no direct interventional experiments were performed in this study to prove a causal relationship between apoptosis, TIM-1, and AHR.
In conclusion, our results indicate that 8 weeks of treatment with a mean concentration of 703 μg/m3 PM2.5 exacerbates allergic asthma in previously sensitized BALB/c mice. Apoptosis was also significantly increased in mice in the OVA/PM group, consistent with TIM-1 expression. The findings show that increased AHR associated with allergic asthma caused by PM2.5 is related to increased apoptosis and TIM-1 activation and might thus provide therapeutic targets for the treatment of acute exacerbations of asthma induced by PM2.5.
Financial support and sponsorship
This study was supported by a grant from the National Natural Science Foundation of China (No. 81770020).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Peng Lyu
REFERENCES
- 1.Cheng MH, Chen CC, Chiu HF, Yang CY. Fine particulate air pollution and hospital admissions for asthma: A case-crossover study in Taipei. J Toxicol Environ Health A. 2014;77:1075–83. doi: 10.1080/15287394.2014.922387. doi: 10.1080/15287394.2014.922387. [DOI] [PubMed] [Google Scholar]
- 2.Tsai SS, Chiu HF, Liou SH, Yang CY. Short-term effects of fine particulate air pollution on hospital admissions for respiratory diseases: A case-crossover study in a tropical city. J Toxicol Environ Health A. 2014;77:1091–101. doi: 10.1080/15287394.2014.922388. doi: 10.1080/15287394.2014.922388. [DOI] [PubMed] [Google Scholar]
- 3.Liu Y, Wang HD, Yu ZX, Hua SC, Zhou LT, Peng LP, et al. Influence of air pollution on hospital admissions in adult asthma in northeast china. Chin Med J. 2018;131:1030–3. doi: 10.4103/0366-6999.230735. doi: 10.4103/0366-6999.230735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castañeda AR, Bein KJ, Smiley-Jewell S, Pinkerton KE. Fine particulate matter (PM2.5) enhances allergic sensitization in BALB/c mice. J Toxicol Environ Health A. 2017;80:197–207. doi: 10.1080/15287394.2016.1222920. doi: 10.1080/15287394.2016.1222920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang X, Zhong W, Meng Q, Lin Q, Fang C, Huang X, et al. Ambient PM2.5 exposure exacerbates severity of allergic asthma in previously sensitized mice. J Asthma. 2015;52:785–94. doi: 10.3109/02770903.2015.1036437. doi: 10.3109/02770903.2015.1036437. [DOI] [PubMed] [Google Scholar]
- 6.Hu R, Xie XY, Xu SK, Wang YN, Jiang M, Wen LR, et al. PM2.5 exposure elicits oxidative stress responses and mitochondrial apoptosis pathway activation in HaCaT keratinocytes. Chin Med J. 2017;130:2205–14. doi: 10.4103/0366-6999.212942. doi: 10.4103/0366-6999.212942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim HJ, Choi MG, Park MK, Seo YR. Predictive and prognostic biomarkers of respiratory diseases due to particulate matter exposure. J Cancer Prev. 2017;22:6–15. doi: 10.15430/JCP.2017.22.1.6. doi: 10.15430/JCP.2017.22.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazzoli-Rocha F, Fernandes S, Einicker-Lamas M, Zin WA. Roles of oxidative stress in signaling and inflammation induced by particulate matter. Cell Biol Toxicol. 2010;26:481–98. doi: 10.1007/s10565-010-9158-2. doi: 10.1007/s10565-010-9158-2. [DOI] [PubMed] [Google Scholar]
- 9.Ru Q, Xiong Q, Chen L, Tian X, Yue K, Ma B, et al. Lipopolysaccharide accelerates fine particulate matter-induced cell apoptosis in human lung bronchial epithelial cells. Int J Occup Med Environ Health. 2018;31:173–83. doi: 10.13075/ijomeh.1896.00527. doi: 10.13075/ijomeh.1896.00527. [DOI] [PubMed] [Google Scholar]
- 10.Piao MJ, Ahn MJ, Kang KA, Ryu YS, Hyun YJ, Shilnikova K, et al. Particulate matter 2.5 damages skin cells by inducing oxidative stress, subcellular organelle dysfunction, and apoptosis. Arch Toxicol. 2018;92:2077–91. doi: 10.1007/s00204-018-2197-9. doi: 10.1007/s00204-018-2197-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin X, Xue B, Zhou Q, Su R, Li Z. Mitochondrial damage mediated by ROS incurs bronchial epithelial cell apoptosis upon ambient PM2.5 exposure. J Toxicol Sci. 2018;43:101–11. doi: 10.2131/jts.43.101. doi: 10.2131/jts.43.101. [DOI] [PubMed] [Google Scholar]
- 12.DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, et al. Tcell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. 2010;184:1918–30. doi: 10.4049/jimmunol.0903059. doi: 10.4049/jimmunol.0903059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Encinas JA, Janssen EM, Weiner DB, Calarota SA, Nieto D, Moll T, et al. Anti-T-cell ig and mucin domain-containing protein 1 antibody decreases TH2 airway inflammation in a mouse model of asthma. J Allergy Clin Immunol. 2005;116:1343–9. doi: 10.1016/j.jaci.2005.08.031. doi: 10.1016/j.jaci.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 14.Umetsu DT, Dekruyff RH. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: Microbes, apoptosis and TIM-1 in the development of asthma. Clin Exp Immunol. 2010;160:125–9. doi: 10.1111/j.1365-2249.2010.04136.x. doi: 10.1111/j.1365-2249.2010.04136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamada K, Goldsmith CA, Suzaki Y, Goldman A, Kobzik L. Airway hyperresponsiveness caused by aerosol exposure to residual oil fly ash leachate in mice. J Toxicol Environ Health A. 2002;65:1351–65. doi: 10.1080/00984100290071586. doi: 10.1080/00984100290071586. [DOI] [PubMed] [Google Scholar]
- 16.Devos FC, Maaske A, Robichaud A, Pollaris L, Seys S, Lopez CA, et al. Forced expiration measurements in mouse models of obstructive and restrictive lung diseases. Respir Res. 2017;18:123. doi: 10.1186/s12931-017-0610-1. doi: 10.1186/s12931-017-0610-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips JE, Peng R, Burns L, Harris P, Garrido R, Tyagi G, et al. Bleomycin induced lung fibrosis increases work of breathing in the mouse. Pulm Pharmacol Ther. 2012;25:281–5. doi: 10.1016/j.pupt.2011.10.001. doi: 10.1016/j.pupt.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Yang J, Zhang P, Meng C, Su J, Wei Z, Zhang F, et al. Quantifying the sources of the severe haze over the southern Hebei using the CMAQ model. ScientificWorldJournal. 2013;2013:812469. doi: 10.1155/2013/812469. doi: 10.1155/2013/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maji KJ, Arora M, Dikshit AK. Burden of disease attributed to ambient PM2.5 and PM10 exposure in 190 cities in China. Environ Sci Pollut Res Int. 2017;24:11559–72. doi: 10.1007/s11356-017-8575-7. doi: 10.1007/s11356-017-8575-7. [DOI] [PubMed] [Google Scholar]
- 20.Pearce JL, Waller LA, Mulholland JA, Sarnat SE, Strickland MJ, Chang HH, et al. Exploring associations between multipollutant day types and asthma morbidity: Epidemiologic applications of self-organizing map ambient air quality classifications. Environ Health. 2015;14:55. doi: 10.1186/s12940-015-0041-8. doi: 10.1186/s12940-015-0041-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao Q, Liu Y, Mulholland JA, Russell AG, Darrow LA, Tolbert PE, et al. Pediatric emergency department visits and ambient air pollution in the U.S. State of Georgia: A case-crossover study. Environ Health. 2016;15:115. doi: 10.1186/s12940-016-0196-y. doi: 10.1186/s12940-016-0196-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurai J, Watanabe M, Sano H, Hantan D, Shimizu E. The effect of seasonal variations in airborne particulate matter on asthma-related airway inflammation in mice. Int J Environ Res Public Health. 2016;13:6. doi: 10.3390/ijerph13060579. doi: 10.3390/ijerph13060579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hosseini A, Hirota JA, Hackett TL, McNagny KM, Wilson SJ, Carlsten C, et al. Morphometric analysis of inflammation in bronchial biopsies following exposure to inhaled diesel exhaust and allergen challenge in atopic subjects. Part Fibre Toxicol. 2016;13:2. doi: 10.1186/s12989-016-0114-z. doi: 10.1186/s12989-016-0114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mei M, Song H, Chen L, Hu B, Bai R, Xu D, et al. Early-life exposure to three size-fractionated ultrafine and fine atmospheric particulates in Beijing exacerbates asthma development in mature mice. Part Fibre Toxicol. 2018;15:13. doi: 10.1186/s12989-018-0249-1. doi: 10.1186/s12989-018-0249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunea D, Iordache S, Liu HY, Bøhler T, Pohoata A, Radulescu C, et al. Quantifying the impact of PM2.5 and associated heavy metals on respiratory health of children near metallurgical facilities. Environ Sci Pollut Res Int. 2016;23:15395–406. doi: 10.1007/s11356-016-6734-x. doi: 10.1007/s11356-016-6734-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gai HF, An JX, Qian XY, Wei YJ, Williams JP, Gao GL, et al. Ovarian damages produced by aerosolized fine particulate matter (PM2.5) pollution in mice: Possible protective medications and mechanisms. Chin Med J. 2017;130:1400–10. doi: 10.4103/0366-6999.207472. doi: 10.4103/0366-6999.207472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng X, Zhang F, Rui W, Long F, Wang L, Feng Z, et al. PM2.5-induced oxidative stress triggers autophagy in human lung epithelial A549 cells. Toxicol In Vitro. 2013;27:1762–70. doi: 10.1016/j.tiv.2013.05.004. doi: 10.1016/j.tiv.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Kwon KY, Jang JH, Kwon SY, Cho CH, Oh HK, Kim SP, et al. Cadmium induced acute lung injury and TUNEL expression of apoptosis in respiratory cells. J Korean Med Sci. 2003;18:655–62. doi: 10.3346/jkms.2003.18.5.655. doi: 10.3346/jkms.2003.18.5.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weaver CV, Liu SP, Lu JF, Lin BS. The effects of benzene exposure on apoptosis in epithelial lung cells: Localization by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) and the immunocytochemical localization of apoptosis-related gene products. Cell Biol Toxicol. 2007;23:201–20. doi: 10.1007/s10565-006-0165-2. doi: 10.1007/s10565-006-0165-2. [DOI] [PubMed] [Google Scholar]
- 30.Mineev VN, Nesterovich II, Trofimov VI, Kashintseva TV, Rybakova MG, Grozov RV, et al. Evaluating the activity of the apoptosis-regulating genes from bcl-2, bax expression, and caspase-3 activity in bronchial epithelial cells in patients with asthma (in Russian) Arkh Patol. 2011;73:11–4. [PubMed] [Google Scholar]
- 31.Tian BP, Xia LX, Bao ZQ, Zhang H, Xu ZW, Mao YY, et al. Bcl-2 inhibitors reduce steroid-insensitive airway inflammation. J Allergy Clin Immunol. 2017;140:418–30. doi: 10.1016/j.jaci.2016.11.027. doi: 10.1016/j.jaci.2016.11.027. [DOI] [PubMed] [Google Scholar]
- 32.de Souza AJ, Oriss TB, O’malley KJ, Ray A, Kane LP. T cell Ig and mucin 1 (TIM-1) is expressed on in vivo-activated T cells and provides a costimulatory signal for T cell activation. Proc Natl Acad Sci U S A. 2005;102:17113–8. doi: 10.1073/pnas.0508643102. doi: 10.1073/pnas.0508643102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curtiss M, Colgan J. The role of the T-cell costimulatory molecule Tim-1 in the immune response. Immunol Res. 2007;39:52–61. doi: 10.1007/s12026-007-0063-6. doi: 10.1007/s12026-007-0063-6. [DOI] [PubMed] [Google Scholar]
- 34.Nakae S, Iikura M, Suto H, Akiba H, Umetsu DT, Dekruyff RH, et al. TIM-1 and TIM-3 enhancement of Th2 cytokine production by mast cells. Blood. 2007;110:2565–8. doi: 10.1182/blood-2006-11-058800. doi: 10.1182/blood-2006-11-058800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HH, Meyer EH, Goya S, Pichavant M, Kim HY, Bu X, et al. Apoptotic cells activate NKT cells through T cell Ig-like mucin-like-1 resulting in airway hyperreactivity. J Immunol. 2010;185:5225–35. doi: 10.4049/jimmunol.1001116. doi: 10.4049/jimmunol.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235:172–89. doi: 10.1111/j.0105-2896.2010.00903.x. doi: 10.1111/j.0105-2896.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim HY, Chang YJ, Chuang YT, Lee HH, Kasahara DI, Martin T, et al. T-cell immunoglobulin and mucin domain 1 deficiency eliminates airway hyperreactivity triggered by the recognition of airway cell death. J Allergy Clin Immunol. 2013;132:414–25. doi: 10.1016/j.jaci.2013.03.025. doi: 10.1016/j.jaci.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gandhi GR, Neta MT, Sathiyabama RG, Quintans JS, de Oliveira E Silva AM, Araújo AA, et al. Flavonoids as th1/Th2 cytokines immunomodulators: A systematic review of studies on animal models. Phytomedicine. 2018;44:74–84. doi: 10.1016/j.phymed.2018.03.057. doi: 10.1016/j.phymed.2018.03.057. [DOI] [PubMed] [Google Scholar]