

Abstract
Amyotrophic Lateral Sclerosis (ALS) is an incurable neurodegenerative disease primarily characterized by progressive degeneration of motor neurons in the motor cortex, brainstem and spinal cord. Due to relatively fast progression of ALS, early diagnosis is essential for possible therapeutic intervention and disease management. To identify potential diagnostic markers, we investigated age-dependent effects of disease onset and progression on regional neurochemistry in the SOD1G93A ALS mouse model using localized in vivo magnetic resonance spectroscopy (MRS). We focused mainly on the brainstem region since brainstem motor nuclei are the primarily affected regions in SOD1G93A mice and ALS patients. In addition, metabolite profiles of the motor cortex were also assessed. In the brainstem, a gradual decrease in creatine levels were detected starting from the pre-symptomatic age of 70 days postpartum. During the early symptomatic phase (day 90), a significant increase in the levels of the inhibitory neurotransmitter γ- aminobutyric acid (GABA) was measured. At later time points, alterations in the form of decreased NAA, glutamate, glutamine and increased myo-inositol were observed. Also, decreased glutamate, NAA and increased taurine levels were seen at late stages in the motor cortex. A proof-of-concept (PoC) study was conducted to assess the effects of coconut oil supplementation in SODG93A mice. The PoC revealed that the coconut oil supplementation together with the regular diet delayed disease symptoms, enhanced motor performance, and prolonged survival in the SOD1G93A mouse model. Furthermore, MRS data showed stable metabolic profile at day 120 in the coconut oil diet group compared to the group receiving a standard diet without coconut oil supplementation. In addition, a positive correlation between survival and the neuronal marker NAA was found. To the best of our knowledge, this is the first study that reports metabolic changes in the brainstem using in vivo MRS and effects of coconut oil supplementation as a prophylactic treatment in SOD1G93A mice.
Keywords: Amyotrophic lateral sclerosis, Magnetic resonance spectroscopy, SOD1G93A, Coconut oil, Brain metabolism, Creatine
Highlights
-
•
A gradual decrease in the energy marker creatine associated with the hypoglossal nucleus of SOD1G93A mice.
-
•
Significant changes in GABA, Glx, N-acetylaspartate, myo-inositol and taurine were observed.
-
•
Ketone body rich diet delayed the onset of symptoms, improved motor performance, extended survival and maintained normal metabolite profiles.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is an idiopathic, progressive and a fatal neurodegenerative disease characterized by selective death of motor neurons in the motor cortex, brainstem and spinal cord. The bulbar neurons regulate the swallowing and spinal motor neurons innervate limb muscles. ALS symptoms and signs include fasciculations, progressive atrophy of the skeletal muscles, weakness and paralysis of the upper and lower limbs (Cleveland and Rothstein, 2001). In addition, loss of speech and denervation of the respiratory muscles occurs with most ALS patients dying of respiratory failure.
The pathophysiological mechanisms leading to the degeneration of motor neurons are still largely unknown and possibly multifactorial. However, there are several hypotheses underlying the disease process including oxidative stress, glutamate excitotoxicity, impaired mitochondrial function, aberrant protein folding, disturbances in RNA metabolism and impaired axonal transport (Turner and Talbot, 2008; Wang et al., 2015; De Vos and Hafezparast, 2017; Liu and Tsai, 2017).
While approximately 10% of ALS cases have familial causes (fALS), in 90% the disease is considered sporadic (sALS). The etiology of sALS is largely unknown; however, about 20% of fALS is associated with a dominantly inherited mutation in the gene that encodes the Cu/Zn-superoxide dismutase 1 (SOD1). A key pathology observed in SOD1 mutation is the abnormal build-up of misfolded-dysfunctional mutant SOD1 protein aggregates in affected motor neurons (Bruijn et al., 1998). However, since most mutant SOD1 proteins are able to function normally, a toxic gain of function is more likely as a cause for pathogenesis of SOD-related ALS (Boillée et al., 2006; Sau et al., 2007; Bunton-Stasyshyn et al., 2015; Bastow et al., 2016).
The SOD1G93A transgenic mouse, which over-expresses a mutated form of the human SOD1 gene (Gurney et al., 1994) has been studied as an experimental model for fALS. This model presents with clinical symptoms and neuropathological features that mimic those characteristic of fALS (Rosen et al., 1993; Van Damme et al., 2017), such as severe hind limb paralysis with atrophy of skeletal muscles (Tu et al., 1996). In SOD1G93A mice, atrophy of the diaphragm muscles leads to respiratory failure and subsequent death (Tankersley et al., 2007). At the moment there is a strong urgency to find predictive biomarkers in the progression of ALS since early diagnosis is the key for possible disease management methods (Karitzky and Ludolph, 2001). Magnetic resonance imaging (MRI) and spectroscopy (MRS) have become essential in the diagnosis of various neurological diseases mainly because of their non-invasive nature, high soft tissue contrast and multi-parametric readouts. In human ALS patients, both T2-weighted and fluid-attenuated inversion recovery (FLAIR) MR images have shown non-specific hyperintense alterations in the primary motor cortex and corticospinal tract (Lee et al., 2003; Hecht et al., 2002). Also, it is well established that T2-weighted MRI is sensitive to tissue changes around the time of symptom onset in SOD1G93A mice (Zang et al., 2004; Niessen et al., 2006; Bucher et al., 2007). These studies reported that T2-weighted MRI revealed an early and progressive degeneration in the SOD1G93A hindbrain motor nuclei, including motor nuclei V (trigeminal), VII (facial), and XII (hypoglossal).
In vivo 1H-MRS) is a non-invasive method for measuring brain metabolites. It has been used to assess the progression of neurodegeneration in humans and mouse models of diseases including ALS, Alzheimer's, Huntington's and Parkinson's disease (Jenkins et al., 2000; Andreassen et al., 2001; Choi et al., 2003; Dedeoglu et al., 2004; Jenkins et al., 2005; Marjanska et al., 2005; Van der Perren et al., 2015). There are a number of neurometabolites that can be identified and quantified from in vivo MRS. N-acetyl aspartate (NAA), a known marker of neuronal health and function has been observed to decrease in motor cortex and then increase with riluzole therapy (Kalra et al., 1999). A longitudinal study showed a decline of NAA in the motor cortex but not in other cortical regions of ALS (Choi et al., 2009). Increases in the neurotransmitters glutamate and glutamine have also been observed in ALS medulla (Pioro et al., 1999). Other molecules such as taurine and myo-inositol, that are related to both osmotic regulation/ neurotransmitter modulator as well as glial cell proliferation can be measured in vivo. 1H-MRS also has the advantage of being able to measure changes in total creatine (the sum of creatine and phosphor-creatine), which is a marker for the energy metabolism in cells with dietary supplementation (Andreassen et al., 2001; Hersch et al., 2006; Choi et al., 2009).
There is considerable evidence to indicate that the ALS pathology arise due to impaired energy metabolism in motor neurons (Dupuis et al., 2011). In ALS, a lower body mass index (BMI) is linked with higher risk of developing the disease and poor prognosis. Therefore, maintaining a higher BMI is proven to be beneficial in ALS (Gallo et al., 2013; O'Reilly et al., 2013). Previous studies have shown that a ketogenic diet or caprylic triglyceride halts the impairment of motor function and reduces death of motor neurons in the spinal cord of SOD1G93A mice by restoring energy metabolism through ketone body utilization (Zhao et al., 2006; Zhao et al., 2012). In normal circumstances glucose acts as the main energy source for the cells. However, alternative energy sources such as ketone bodies or TCA cycle intermediates can potentially bypass the rate limiting steps associated with impaired neuronal glucose metabolism and restore mitochondrial adenosine triphosphate (ATP) production (Ari et al., 2014). Recently, metabolic-targeted therapies such as therapeutic ketosis have been used in the disease management process in several neurological disorders (Hartman, 2012; Hartman, 2013). In addition, coenzyme Q (Beal, 2002), creatine (Andreassen et al., 2001), high-fat diet and ketogenic-medium chain triglyceride diet (Zhao et al., 2006) were found to be effective against neuronal damage induced by excitotoxicity and mitochondrial inhibition. Furthermore, coconut oil which contains high levels of medium chain triglycerides (MCT) could act as a natural ketogenic supplement in ALS. However, we were unable to find any published clinical or preclinical trials of coconut oil, although sporadic reports exist on patient platforms (Gupta and Riis, 2011). Therefore, we designed a proof-of-concept (PoC) study to assess the effects of coconut oil in the SOD1G93A mouse model.
The goals in this study were to monitor the onset and progression of the disease by observing alterations in regional brain metabolism of SOD1G93A mice using in vivo MR spectroscopy and to examine the potential neuroprotective effects of coconut oil dietary supplement in this model.
2. Materials and methods
2.1. Transgenic mice
All animal experiments were performed according to the European Communities Council Directive of September 22nd 2010 (2010/63/EU) and approved by the local Animal Ethics Committee of the KU Leuven. Male transgenic mice carrying a high copy number of a mutant allele human superoxide dismutase 1 (SOD1) [B6SJL-TgN (SOD1-G93A) 1Gur, Stock #: 002726] were purchased and bred locally with female B6SJL mice (Jackson Laboratory, Bar Harbor, ME). The progeny were genotyped by tail DNA polymerase chain reaction (PCR).
In a first experiment, we used localized in vivo 1H-MRS to evaluate metabolic changes associated with disease onset and progression in SOD1G93A mice (n = 10). Metabolic profiles were assessed in the hindbrain and motor cortex in comparison to age-matched wild type (WT) mice (n = 10) of 30, 60, 70, 90, 110, 120 and 140 days postpartum (P30, P60… etc). In the second experiment, in vivo 1H-MRS was used to assess the effects of coconut oil supplementation versus standard diet, tested in additional cohorts of SOD1G93A (n = 6) and control mice (n = 6). Also, body weight, motor performance and survival was assessed. Sample sizes for both experiments were based on preliminary data and subsequent power analysis. For example, an 8% NAA reduction in a preliminary study resulted in a minimum group size of 6 for experiment 1 using an a priori Two tail test (αerror = 0.05, power = 0.8 effect size = 1.6: software G*Power 3.1.9.2).
Time-points were selected to reflect disease stages either long before the onset of clinical symptoms (P30), right before the disease onset (P60-P70), at the average disease onset (P90-P110) and at the terminal stage of ALS (P120 and P140). Since sex could possibly be a factor for variations in the survival in SOD1G93A model only males were used.
2.2. In vivo MRI and MR spectroscopy
All MR experiments were performed using a small animal 9.4 Tesla MR scanner (Biospec 94/20, Bruker Biospin, Ettlingen, Germany) with a horizontal bore of 20 cm and equipped with an actively shielded gradient set (600 mT m−1, inner diameter 11.7 cm). Single-voxel 1H-MR spectra were acquired using a linear polarized resonator (7 cm diameter) for transmission, combined with a mouse brain surface coil for receiving (both Bruker Biospin, Ettlingen, Germany).
2.2.1. Animal preparation and monitoring
Mice were fixed in an animal bed by placing the nose of the mouse in a nose cone, restraining it with a bite bar and custom made outer ear-inserts. The mouse head was lifted and gently stretched a few millimeters forward. By lining up the midline of the head and the spine produces an angle between the brainstem and the rest of the mouse body. This alignment decreases the distance between the surface coil and the brainstem region and allows acquisition of high quality data.
Throughout scanning, mice were anaesthetized with 1–2% isoflurane in 100% oxygen. Respiration and body temperature were continuously monitored and maintained at 60–100 min−1 and 37 ± 1 °C, respectively. Vidisic Eye Gel (Bausch & Lomb, Brussels, Belgium) was used to avoid the drying of eyes during scanning.
2.2.2. Magnetic resonance imaging
In order to visualize signal intensity changes, a T2-weighted 3D TurboRARE protocol was acquired with the following acquisition parameters: repetition time (TR) 1000 ms, echo time (TE) 12 ms, FOV 24 × 15 × 8.3 mm, matrix size 256 × 160 × 88, RARE factor 10, number of averages = 1. For the placement of the spectroscopy voxel, T2-weighted 2D RARE images (effective TE 50 ms; matrix 256 × 256; 300um slice thickness) were acquired. For T2 relaxometry, a multi-slice multi-echo (MSME) sequence was used with ten echoes collected at intervals of 12 ms from TE = 12 ms to TE = 120 ms, with TR = 3000 ms and with NA = 1, FOV 25 × 25 mm, 19 slices, slice thickness of 0.5 mm and a matrix of 256 × 256. T2 maps were produced using the imageJ software (NIH, USA). T2 values were calculated using ITKSNAP (Penn Image Computing and Science Laboratory, USA) and an in-house python scripts.
2.2.3. 1H-magnetic resonance spectroscopy
MR spectra were acquired for volumes in the motor cortex and the hindbrain with a voxel size of 2 × 2.5.x1.5 mm3 (see Fig. 2). The hindbrain voxel covered the following nuclei: hypoglossal, medial vestibular nucleus, and the dorsal motor nucleus of the vagus nerve. MR spectra were acquired with a PRESS pulse sequence using the following parameters: TR = 1800 ms, TE = 20 ms, and number of averages 320 for motor cortex and 640 for hindbrain. Water suppression was optimized using VAPOR (Griffey and Flamig, 1990). An unsuppressed water spectrum (TE = 20 ms, TR = 1400 ms, number of averages = 4) was acquired before each 1H-MRS spectrum (water suppressed) for quantification/ referencing. Shimming was performed using FASTMAP (Gruetter, 1993), resulting in a final water line width at half height < 16 Hz. Spectra were processed using jMRUI v6.0 (Stefan et al., 2009). Spectra were phase corrected and an HLSVD (Hankel Lanczos Singular Values Decomposition) filter was applied to remove the residual water signal (van den Boogaart et al., 1994). Only metabolites with a Cramer-Rao lower bound <25% were considered for quantification. Metabolites were quantified with the QUEST algorithm (Ratiney et al., 2004) in jMRUI using a simulated (NMRScopeB) basis set (Starčuk Jr. et al., 2008). Results are reported in reference to the non-suppressed water signal. A total of 16 brain metabolites (Alanine, Aspartic acid, total creatine, choline, GABA, glycine, glucose, glutamate, glutamine, NAA, myo-inositol, Scyllo-inositol, lactate, phosphoethanolamine, scyllo-inositol, taurine) was quantified.
Fig. 2.
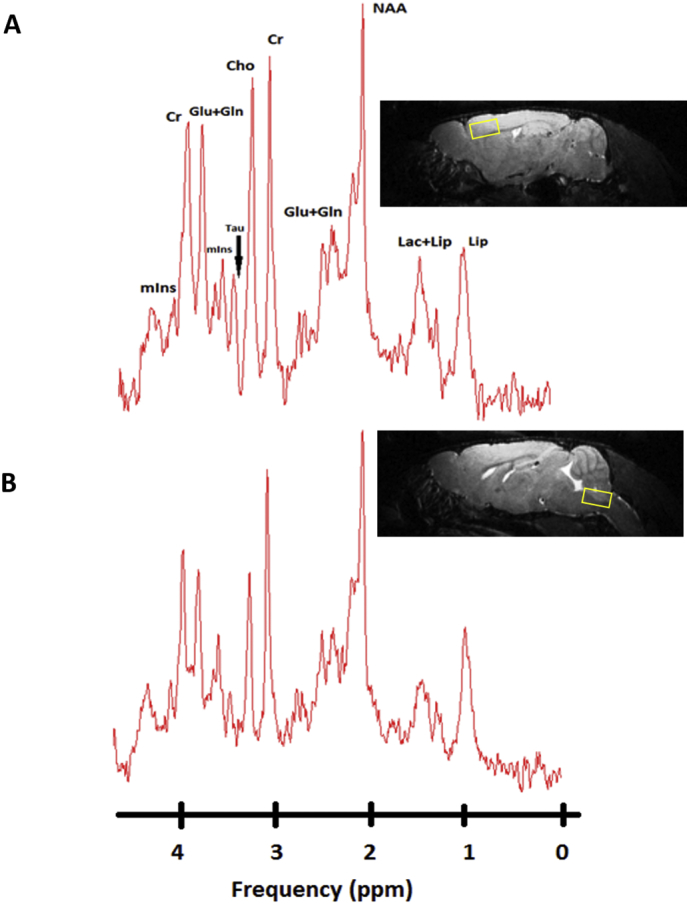
1H-MR spectra of the motor cortex and hindbrain of a SOD1G93A mouse 30 days postpartum acquired at 9.4 Tesla.
(A) In vivo1H-MR spectra aquired from the motor cortex and (B) hindbrain of a SOD1G93A mouse with labelled visible neurometabolite peaks. Right images show the voxel localization in both regions (2 × 2.5.x1.5 mm3). Abbreviations: Lip, lipd; Lac, lactate; NAA, N-acetylaspartate; Glu, glutamate; Gln, glutamine; Cr, creatine; Cho, choline; Tau, taurine; mIns, myo-inositol.
2.3. Diet preparation
Extra virgin coconut oil (supplementary Fig. S1 (A)), Royal Green, Frenchtop Natural Care Products BV, AL Hoornm Netherlands) was mixed with the standard rodent food (ssniff DIO D12450B, 4 ml with 15-20 g). Food was dispensed in a petri dish and placed inside the cage daily. Treatment started at 72 days after birth. At 70 days of age, animals underwent baseline MR spectroscopy, and the same animals were re-scanned at 120 days. Mice were followed up until end stage for behavior and survival studies. Control animals received standard rodent food (D12450B Rodent Diet with 10 kcal% fat from Ssniff laboratories).
Supplementary Fig. S1.

PoC study: Coconut oil dietary supplementation.
2.4. Behavioral assessments
Muscle strength and endurance was assessed by the paw grip endurance (PaGE) test (supplementary Fig. S1 (B)) (Weydt et al., 2003). Mice were placed on the center of a custom made metal grid and grid was turned upside down over a cage and the latency to fall (time in seconds) was recorded (Crawley, 2007). Longest hanging-time was recorded after two attempts were given for each mouse.
2.5. Survival and end-point
Survival endpoint criteria were defined as unable to return to the normal, upright position within 10s following push over, this was tested daily from P130 (Ari et al., 2014). When the mice reached the end-stage, mice were euthanized by IP injection of Sodium pentobarbital (50 mg/kg, Butler Schein, NDC #11695–4829-1).
2.6. Statistical analysis
All statistical tests were performed using GraphPad PRISM version 5 (GraphPad, San Diego). Two-way analysis of variance (ANOVA) tests were performed for each neurometabolite in hindbrain and motor cortex to evaluate the significance of the group (SOD1G93A and WT) and postpartum age (P30, P60, P70, P90, P110, P120 and P140) as fixed factors. Subsequently, those neurometabolites, which had a significant effect were further analyzed for age-dependent effects within each group with a one-way ANOVA. One-way ANOVA and t-tests were used to assess significant differences in mean concentrations within each group and between the two groups. Comparisons between coconut oil treated and standard diet animals were made using by t-tests or ANOVA with post hoc analyses and effect of coconut oil diet on survival was assessed by the Kaplan-Meier survival analysis. The threshold for statistical significance was set at P < .05. P-values were Bonferroni corrected for multiple comparisons. Pearson correlation coefficient, r, was used for correlation analysis.
3. Results
3.1. T2-weighted magnetic resonance imaging
T2-weighted MR images revealed increased hyperintense contrast in SOD1G93A brainstem motor nuclei V (trigeminal), VII (facial), and XII (hypoglossal) (Fig. 1). Contrast of the motor nuclei within the brainstem starts to increase relative to surrounding tissue around P90, parallel to the observation of first clinical symptoms such as tremors and gait abnormalities.
Fig. 1.
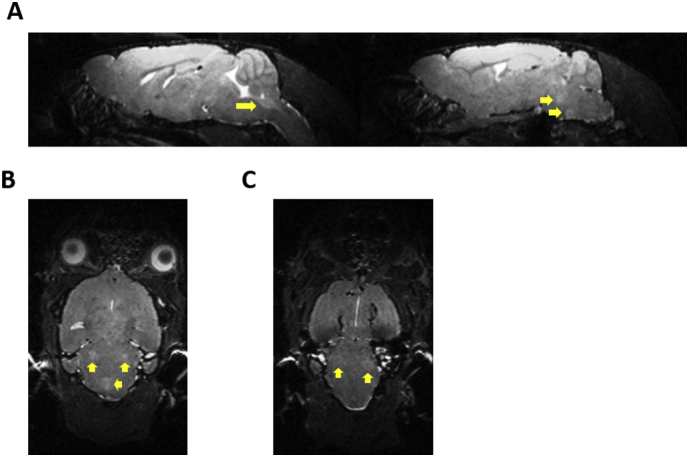
T2-weighted MRI Sections showing hyperintense regions in SOD1G93A mice.
T2-weighted MRI showing hyperintense contrast in the brainstem region compared to surrounding regions in a 140 day old SOD1G93A mouse. (A) Left: Sagittal sections: arrow shows hyperintensity in the brainstem nucleus Nc. XII (hypoglossal). Right: Sagittal sections: arrows show hyperintensities in the brainstem nuclei Nc V (trigeminal), Nc VII. (B) Coronal section: white arrows show bilateral hyperintensities in Nc V (trigeminal). Yellow arrow: hyperintensity in Nc XII (hypoglossal). (C) Coronal section: arrows show bilateral hyperintensities in Nc VII (facial).
3.2. Magnetic resonance spectroscopy
In order to assess neurometabolic alterations, we acquired MR spectra (motor cortex: signal to noise ratio > 14; line-width 9 ± 2 Hz, hindbrain: SNR > 10, line-width 12 ± 3 Hz, water/lipid/artifact suppressed). Typical MR spectra obtained from the motor cortex and hindbrain of a SOD1G93A mouse at P30 are shown in Fig. 2.
Two-way ANOVA tests for each 16 neurometabolites showed significant interactions in the motor cortex and the hindbrain of the SOD1G93A mice. The neurometabolic changes observed in SOD1G93A mice and the comparison to WT mice are shown in Fig. 3 (hindbrain) and Fig. 4 (motor cortex). In the motor cortex, significant interactions were found for glutamate (F = 3.0, P < .01), taurine (F = 4.7, P = .0002) and NAA (F = 6.4, P < .0001). As for the hindbrain, significant interaction of the two factors were found for creatine (F = 13.6, P < .001), GABA, (F = 3.3, P = .0044), glutamate (F = 4.1, P = .0009), glutamine (F = 4.2, P < .001), mIns (F = 4.1, P < .001), and NAA (F = 4.2, P = .0008). For the metabolites, which showed significant interaction, data were further analyzed by one-way ANOVAs separately in SOD1G93A mice and WT mice to evaluate influences of animal age for each brain region. In the motor cortex of SOD1G93A mice, age-dependent effects were found for the excitatory neurotransmitter glutamate, neuronal marker NAA and the neurotransmitter modulator taurine. Decreases in glutamate (P < .05) and NAA (P < .0001) concentrations around P120 as well as a continuous increase in taurine from P90 (P < .05) were found. The increase in taurine concentration was significant (P < .001) between the two groups at P110 (Fig. 4). In the hindbrain of SOD1G93A mice, a significant (P = .001) age dependent decrease in creatine levels were found at P110. However, the levels of creatine were significantly (P < .0001) different between SOD1G93A and WT around P70. A significant (P < .0001) decrease of glutamine was observed at early-symptomatic stage (P110). For the inhibitory neurotransmitter GABA, a significant (P < .0001) increase was found at P90 and a significant (P = .0110) decrease of NAA concentration was detected at P120. Also, a significant (P < .0001) decrease in the glutamate and an increase of the inflammation/glial marker myo-inositol (P < .0001) were found at P140. The mean concentrations of glutamate and taurine were significantly higher (41.2%, 118.5%, respectively) in the motor cortex than in the hindbrain for both groups (Fig. 5). A 3-time point summary of the significant neurometabolite concentrations in both regions for SOD1G93A and WT is presented in Table 1. To further analyze the dependence between the altered neurometabolite concentrations in the motor cortex and hindbrain, Pearson correlation coefficients (r) were calculated using all seven time points for SOD1G93A mice (Fig. 6 and Fig. 7). Strong correlations were observed for NAA and glutamate in the motor cortex and brainstem (r = 0.968, P = .0003; r = 0.915, P = .0038, respectively). Also in the motor cortex, significant negative correlations were found between NAA and taurine (r = −0.785, P = .036) and between glutamate and taurine (r = −0.758, P = .048). In the hindbrain, strong correlation was seen between NAA and creatine (r = 0.983, P < .0001), NAA-glutamate (r = 0.947, P = .0041), between NAA and myo-inositol (r = −0.905, P = .0050), and between glutamate and creatine (r = 0.889, P = .0074).
Fig. 3.

Absolute Concentrations of creatine, γ-aminobutyric acid (GABA), glutamate, glutamine, myo-inositol, and N-acetyl aspartate (NAA) in the hindbrain of SOD1G93A and WT mice.
Concentrations of creatine, γ-aminobutyric acid (GABA), glutamate, glutamine, myo-inositol, and N-acetyl aspartate (NAA) in the hindbrain of SOD1G93A (n = 10) and age-matched wild-type mice (n = 10) at 30, 60, 70, 90, 110, 120, 140 days postpartum were determined relative to the unsupressed water signal from the same voxel. Statistical significances between the two groups and mean concentrations within each group are shown. Concentration values are given as mean ± SD; *P < .05, **P < .01, ***P < .001.
Fig. 4.

Absolute Concentrations of NAA, taurine and glutamate in the motor cortex of SOD1G93A and WT mice.
Concentrations of NAA, taurine and glutamate in the motor cortex of SOD1G93A (n = 10) and age-matched wild-type mice (n = 10) at 30, 60, 70, 90, 110, 120, 140 days postpartum were determined relative to the unsupressed water signal from the same voxel. Statistical significances between the two groups and mean concentrations within each group are shown. Concentration values are given as mean ± SD; *P < .05, **P < .01, ***P < .001.
Fig. 5.

Absolute concentrations of glutamate, taurine in hindbrain and motor cortex of SODG93A mice.
Comparison of mean concentrations of glutamate, taurine in hindbrain and motor cortex of SOD1G93A (n = 10) at 30, 60, 70, 90, 110, 120, 140 days postpartum. Abbreviations: Mx. Motor cortex; Hb, hindbrain.
Table 1.
Summary of metabolite concentrations of SOD and WT mice.
| Metabolite Concentration (mean ± S.D in mmol/l) |
||||||||
|---|---|---|---|---|---|---|---|---|
| Model | Age | Location | NAA | tCr | Glu | Gln | mIns | Taurine |
| SOD | P30 | Hindbrain | 9.27 ± 0.79 | 7.45 ± 0.43 | 8.15 ± 0.73 | 3.87 ± 0.57 | 3.50 ± 0.39 | 2.23 ± 0.32 |
| Motor cortex | 8.77 ± 0.88 | 6.68 ± 0.79 | 12.38 ± 1.50 | 3.01 ± 0.44 | 2.02 ± 0.47 | 7.82 ± 0.65 | ||
| WT | Hindbrain | 9.42 ± 0.74 | 7.40 ± 0.20 | 8.30 ± 0.87 | 3.97 ± 0.47 | 3.04 ± 0.57 | 1.89 ± 0.85 | |
| Motor cortex | 9.21 ± 1.12 | 7.21 ± 0.72 | 13.14 ± 1.98 | 3.54 ± 0.32 | 2.15 ± 0.76 | 8.12 ± 0.57 | ||
| SOD | P110 | Hindbrain | 8.48 ± 0.81 | 6.47 ± 0.61 | 8.47 ± 1.05 | 2.59 ± 0.64 | 3.10 ± 1.03 | 2.50 ± 023 |
| Motor cortex | 8.93 ± 0.78 | 7.11 ± 0.65 | 13.05 ± 0.87 | 3.23 ± 0.35 | 2.24 ± 0.36 | 9.45 ± 0.70 | ||
| WT | Hindbrain | 9.43 ± 1.02 | 8.22 ± 0.59 | 9.13 ± 1.29 | 3.54 ± 0.36 | 3.24 ± 0.59 | 2.45 ± 0.14 | |
| Motor cortex | 8.84 ± 0.92 | 7.68 ± 0.53 | 11.68 ± 1.80 | 3.10 ± 0.42 | 1.97 ± 0.89 | 8.35 ± 0.84 | ||
| SOD | P140 | Hindbrain | 6.22 ± 0.49 | 5.23 ± 0.57 | 6.77 ± 0.61 | 2.91 ± 0.75 | 5.20 ± 0.55 | 2.36 ± 0.13 |
| Motor cortex | 6.29 ± 0.73 | 6.68 ± 0.79 | 9.62 ± 1.26 | 3.21 ± 0.33 | 2.32 ± 0.61 | 10.03 ± 0.63 | ||
| WT | Hindbrain | 10.05 ± 0.58 | 7.89 ± 0.78 | 9.24 ± 0.74 | 3.34 ± 0.42 | 3.45 ± 0.72 | 2.01 ± 0.35 | |
| Motor cortex | 9.21 ± 0.47 | 7.45 ± 0.87 | 12.85 ± 0.84 | 3.12 ± 0.25 | 2.04 ± 0.63 | 8.73 ± 0.65 | ||
Fig. 6.

Correlation between neurometabolite concentrations in SOD1G93A hindbrain.
Correlation between neurometabolite concentrations in the hindbrain of SOD1G93A mice for all time-points (30, 60, 70, 90, 110, 120, 140 days postpartum). (A) Correlation of regional glutamate and NAA concentrations across mouse age. The resulting slope in linear fit is represented by the dashed black line (R2 = 0.837). (B) Correlation between myo-Inositol versus NAA concentrations (R2 = 0.819). (C) Relation between glutamine and NAA concentrations (R2 = 0.898). (D) Correlation between creatine and NAA concentrations (R2 = 0.965). (E) Correlation between creatine and glutamate (R2 = 0.791) concentrations. Abbreviation: NAA, N-acetylaspartate; tCr, total creatine; mIns, myo-inositol;Glu, glutamate;
Fig. 7.

Correlation between metabolite concentrations in SOD1G93A motor cortex.
Correlation between neurometabolite concentrations in SOD1G93A motor cortex for all time-points (30, 60, 70, 90, 110, 120, 140 days postpartum). (A) Correlation between glutamate and NAA (R2 = 0.938). (B) Correlation between taurine and NAA (R2 = 0.616). (C) Correlation between taurine and glutamate (R2 = 0.575). Abbreviation: NAA, N-acetylaspartate; Glu, glutamate; Tau, taurine.
3.3. Development of symptoms
In this first experiment, SOD1G93A mice developed tremors in the hind-limbs between P90 and P100, and gait impairment around P115. Interestingly, in all animals an early and increasingly pronounced effect on left hind-limb tremors and dysfunction was detected. Total paralysis of both hind-limbs were detected between P140 and P150, reaching the endpoint of the disease.
3.4. Effect of coconut oil diet on motor performance in SOD1G93A mice
In a second experiment, animals were treated with a virgin coconut oil diet starting from 72 days of age. These animals were compared with animals receiving standard food. The diet had the following effects on SOD mice:
I. Body weight: Coconut oil diet had a significant net positive effect on retention of body weight at 90 (P < .001), 110 (P < .0001) and 130 (P < .0001) days of age compared to the mice fed with a normal diet (Fig. 8 A).
Fig. 8.

Effect of the coconut oil diet on SOD1G93A mice.
(A) The effect of the coconut oil diet on weight in SOD1G93A mice at the beginning of treatment (Day 70), day 90, day 110 and day 130 of the study. Coconut oil fed group had significant net positive effect on retention of body weight during P90 (P < 90), P110 (P < .001) and P130 (P < .0001). (B) The PaGE test showed better motor performance in the coconut oil diet group on week 14, 15 and 16 (P < .001, P < .0001, P < .0001, respectively), compared to the standard diet group. (C) Kaplan-Meier survival plot of the study groups. SOD1G93A mice treated with coconut oil diet showed significantly extended survival (13.6%) compared to control animals (p < .0001). Data = mean ± SD; Coconut oil diet group: n = 6, standard diet group: n = 6. **P < .01, ***P < .001, **** P < .0001.
II. Motor performance: Better motor performance was recorded in the treated group on week 14, 15 and 16 (P = .001, P = .0001, P = .0001) when compared to normal diet group (Fig. 8 B).
III. Survival: Coconut oil diet extended the mean survival by 11% (164.7 ± 5.4 days for coconut oil-fed versus 147.2 ± 3.3 days for normal fed, P < .0001, n = 6) (Fig. 8 C).
3.5. Effects of coconut oil diet on neurometabolites and MRI
MR spectra were acquired at baseline (P70) and P120 for both coconut oil and standard diet groups. We examined the changes in neurometabolite concentrations in the hindbrain within and among the two groups. Compared to the baseline, stable (no significant changes) metabolite levels were estimated at the late time point (P120) for the coconut oil fed group (Fig. 9 A). In contrast, the previously observed pattern of changes in neurometabolite levels (see first experiment) were observed for creatine (P < .05) and NAA (P < .05) in the standard diet group (Fig. 9 B). Even though the neurometabolite concentration for NAA was not significantly different when comparing the coconut oil treated group with the standard diet mice at P120 (Fig. 9 C), the levels were higher on average (8.6 ± 2.2 for coconut oil treated group vs. 7.6 ± 1.2 for standard diet group, respectively). Creatine concentrations for the treated group were significantly higher (P < .05) at P120 compared to the standard group (7.1 ± 1.3 vs. 5.60 ± 1.2 respectively).
Fig. 9.

The effects of coconut oil supplementation on hindbrain neurometabolite levels of SOD1G93A mice assessed with 1H-MRS.
(A–B) Neurometabolite profiles determined by in vivo MRS of the standard diet group and coconut oil diet group at day 70 and day 120. Significant decrease in creatine and NAA was found for the standard diet group between 70 and 120 days (P < .001, P < .005 respectively). No significant changes in neurometabolites were found for the coconut oil diet group between day 70 and day 120. (C) Comparison of the two groups at P120. Significant decrease in creatine was found for the standard diet group (P < .01) compared to the coconut oil diet group. (D) A highly significant (P < .0001) correlation (R2 = 0.922) between the survival and the hindbrain NAA levels was seen for the two groups. Dotted circles highlight the standard diet and coconut oil diet groups.
4. Discussion
MRI and MRS have been applied to monitor ALS progression and treatment response in both humans as well as transgenic ALS mouse models (Wang et al., 2011; Kumar et al., 2013; Verstraete et al. 2015). In the present study, we employed in vivo MRI and MRS to monitor the disease onset and progression in the SOD1G93A mouse model. Furthermore, we conducted a small-scale PoC study to assess the potential neuroprotective effect of coconut oil diet in SOD1G93A mice.
To the best of our knowledge, only three studies have examined the SOD1G93A neurometabolic changes utilizing in vivo MRS and/or ex vivo high resolution NMR spectroscopy (Andreassen et al., 2001; Choi et al., 2009; Niessen et al., 2007). Two of the studies also examined the possible beneficial effects of dietary creatine supplementation in the same mouse model. The set up and results of the above mentioned studies are summarized in the (Supplementary Table 1). Our study revealed significant changes in the hindbrain and motor cortex of SOD1G93A mice. While some observations are consistent with previous studies, our findings also indicate some dissimilar patterns.
4.1. T2-weighted MRI and SOD1G93A brainstem pathology
Previously it was shown that in SOD1G93A mice, spinal cord motor neurons and lower motor nuclei are the most vulnerable structures for degeneration (Niessen et al., 2006; Niessen et al., 2007). Consequently, it is reasonable to assume that neurometabolic changes occurring in those motor nuclei during the life-span of this mouse model (Kong et al., 1998). Using T2-weighted MR imaging Niessen et al. (2006) showed clear signal intensity enhancement compared with surrounding tissue in various motor nuclei within the brainstem of SOD1G93A mice, including the hypoglossal nucleus (Nc. Nv XII), nucleus ambiguus (Nc. Nv IX, X), facial nucleus (Nc. Nv VII), and trigeminal nucleus (Nc. Nv V). Using T2-weighted MR images, we observed similar increased contrast in the SOD1G93A brainstem motor nuclei V (trigeminal), VII (facial), and XII (hypoglossal) compared to WT mice. Trigeminal and hypoglossal nuclei are shown to be two key motor components involved in mastication (chewing) and licking behaviors of mice (Hillel et al., 1989; Zang et al., 2004). In our previous studies with this model, we observed impaired chewing and licking behavior through the progressive decline in body weight and food consumption of SOD1G93A mice. Caron et al. demonstrated that the hyperintensities in T2-weighted MRI of the brainstem region are associated with the formation of vacuoles rather than the loss of motor neurons (Caron et al., 2015). In fact, vacuolization is one of the initial signs of motor neuron changes in SOD1G93A (Higgins et al., 2003). Vacuolization, which is primarily due to abnormal and swollen mitochondria, has been observed as early as one month of age in dendrites and proximal axons of spinal motor neurons (Kong and Xu, 1998; Bendotti et al., 2001), before the loss of motor nuclei and the appearance of a glial reaction (Bendotti and Carri, 2004).
4.2. 1H-MR spectroscopy
Our study implies MR detectable involvement of the hindbrain and motor cortex in the course of the disease seen as significant changes in SOD1G93A neurometabolite concentrations. MR spectra of the time-evolution of neurometabolites in the hindbrain of SOD1G93A mice is presented in Fig. 10. Some of these significant changes in the hindbrain region of SOD1G93A mice are in accordance with previous observations by Niessen et al. (2007) and Choi et al. (2009) using the same mouse model. Niessen et al. observed a decrease in glutamine at P90, a decrease in NAA and glutamate at P120. Choi et al. also reported a decrease in NAA at P142 as well as a trend towards decreasing glutamate. Furthermore, they reported an increase in inflammation/glial marker myo-inositol between P110 and P142. We observed a decrease in glutamine at P110 and a decrease in NAA, glutamate at P120 and P140, respectively. Similar to Choi et al., we also observed an increase in myo-inositol around P140. However, our results show somewhat different patterns from those observed in those previous studies. For instance, unlike Andreassen et al. (2001) and Choi et al., 2009), we did not observe neither an increase in glutamate and glutamine at the early disease stages nor an increase in glutamine at end stage. Concerning the neuronal marker NAA, we were unable to confirm the early decrease of NAA in the brainstem at P75 as was reported by Niessen et al. Apart from the above mentioned differences, we were able to find significant neurometabolite changes in both the motor cortex and hindbrain regions that were not previously described in the SOD1G93A animal model.
Fig. 10.
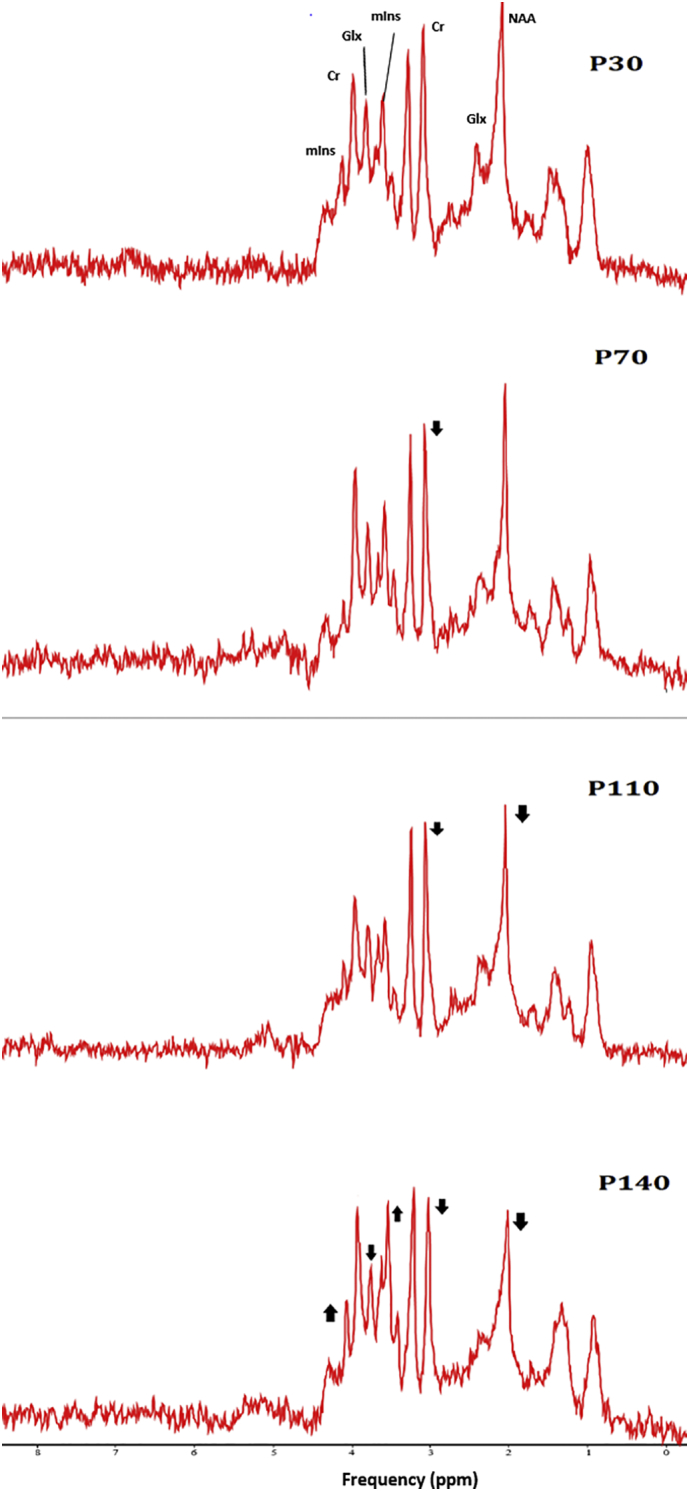
MR spectra illustrating the evolution of neurometabolites in the hindbrain of SOD1G93A mice.
Hindbrain 1H-MR spectra of postpartum day 30, 70, 110, and 140 of a single SOD1G93A mouse are shown. Arrows depict changes in signal intensities during the four time-points.
4.2.1. Decreased NAA and glutamate in the motor cortex of SOD1G93A mice
Reductions in the neuronal marker NAA have been reported in the motor cortex of individuals with ALS (Block et al., 1998; Bradley et al., 1999; Gredal et al., 1997). The most common finding reported in in vivo MRS studies of ALS patients is also the reduction of the neuronal marker NAA in the motor cortex, which is generally interpreted as neuronal loss (Agosta et al., 2010; Rule et al., 2004). Our results show decreased NAA levels at the end stages between P120 and P140. However, since NAA is synthesized in neuronal mitochondria, changes in NAA levels could also reveal mitochondrial dysfunction (Clark, 1998; Vielhaber et al., 2001). Furthermore, our study shows decreased glutamate levels in the motor cortex of SOD1G93A mice around P120 and P140, which was not previously reported for this model. Glutamate is the main excitatory neurotransmitter in the brain, though higher extracellular glutamate is associated with several neurodegenerative diseases including ALS. Previous MRS studies in patients, using low magnetic field strengths (1.5 or 3 Tesla) have reported variable results on glutamate concentrations in patients with ALS (Bradley et al., 1999; Bowen et al., 2000; Block et al., 1998; Han and Ma, 2010). However, a recent study by Atassi et al. (2017), using a higher magnetic field (7 Tesla) reported decreased glutamate levels in the motor cortex of ALS patients compared to healthy controls.
4.2.2. Increased taurine in the motor cortex of SOD1G93A mice
In the motor cortex, age-dependent concentration changes were found for taurine in SOD1G93A mice. An age-dependent increase in taurine started at around P90 and became significant at P110 compared to WT mice. Niessen et al. also reported increased taurine levels in the motor cortex at P90, however, the changes were similar in both SOD1G93A mice and control mice (Niessen et al., 2007). Our finding of increased taurine levels is in agreement with studies in postmortem ALS patients, where higher levels of taurine were detected in the brain and spinal cord (Yoshino et al., 1979; Perry et al., 1987; Malessa et al., 1991). Taurine is a non-essential amino acid whose exact function is unclear. However, it has been proposed as a modulator of neurotransmitter action, where it is shown to exert an inhibitory effect (Curtis and Watkins, 1965). Jung et al. reported that SOD1G93A mice have higher levels of brain taurine and higher expression of the taurine transporter, TauT (Jung et al., 2013). Evidently, expression of this transporter increases under oxidative stress and is controlled by the heat shock factor 1 protein. Increased expression of taurine transporters and uptake of taurine in motor neurons in ALS could be a compensatory mechanism (Jung et al., 2013).
4.2.3. Increased GABA in the hindbrain of SOD1G93A mice
Around P90, or in the early symptomatic stage we observed an increase in GABA levels in the hindbrain which normalized thereafter and remains stable. Imbalance of GABA has been implicated to play an important role in the pathogenesis of ALS. GABA is produced by decarboxylation of glutamic acid, catalyzed by the glutamic acid decarboxylase enzyme (Petroff, 2002). Therefore, an increase in GABA could have a pathological implication in reversing glutamate excitotoxicity and thereby act as a protective mechanism against cell damage. Along this mechanism, pharmacological agents that increase the levels of GABA such as baclofen and gabapentin have been suspected to be beneficial for treating ALS. The reason for our observation of GABA levels normalizing after P90 and remaining stable until P140 is currently unknown. Although the continuous reduction of glutamine and glutamate levels starting at around P110 may trigger a compensatory GABA level increase, which appears stable over time compared to other metabolites. In addition, in vivo GABA detection by 1H-MRS, even at high magnetic field strengths, presents significant challenges arising from the low brain concentration, overlap with more intense resonances and contamination by mobile macromolecule signals (Pfeuffer et al., 1999). Therefore, caution must be taken in interpreting MRS-GABA results.
4.2.4. Gradual decrease in total creatine levels in the hindbrain of pre-symptomatic SOD1G93A mice
1H-MRS detectable creatine constitutes of creatine and phosphocreatine (total creatine), which are indistinguishable at magnetic field strengths of 9.4 T and below (Sartorius et al., 2008). Creatine plays a major role in maintaining ATP levels constant in cells with high and unstable energy demands (Rae and Bröer, 2015; Gasparovic et al., 2009). In MRS, creatine is frequently used as an internal reference since it is considered stable even under most pathological conditions. However, clinical MR spectroscopic studies reported that early and progressive reduction of total creatine levels in the striatum is associated with the onset and progression of Huntington's disease (Sánchez-Pernaute et al., 1999; Reynolds Jr et al., 2005). In our study, we observed a gradual decrease in creatine levels in the SOD1G93A hindbrain starting between P60 and P90. The decrease was clearly evident in all ten SOD1G93A mice (supplementary Fig. S3). Our finding of decreased creatine level in the hindbrain of SOD1G93A mice may indicate a hyper-metabolic environment in the motor nuclei region. In a recent publication, Atassi et al. (2017) observed a decrease in creatine in the left precentral gyrus of ALS patients compared to healthy controls. In ALS, a hyper-metabolic state in the hindbrain may possibly occur due to the imbalance of glutamate-glutamine cycle. Increased glutamate/glutamine levels have been reported in the medulla of ALS patients (Pioro et al., 1999). Therefore, a possible pathway could be that enhanced excitatory neurotransmitter release leads to the hyper-metabolism of the hindbrain of SOD1G93A mice, which results in an overconsumption of creatine. An imbalance in supply-demand in creatine levels may threaten the viability of the neuronal cells and hence make them susceptible to degeneration. However, further investigations are needed to support this hypothesis. On the contrary, in vitro NMR studies using brainstem extracts from SOD1G93A mice, reported stable creatine levels (Niessen et al., 2007; Choi et al., 2009). One reason for this disagreement could be the brain extracts used in the ex vivo experiments covered a larger volume compared to the smaller voxel size (7.5 mm3) used in our in vivo MRS experiments. Since mainly the motor nuclei are affected in the brainstem, homogenization of brain extracts from a larger region may mask the detection of subtle changes in neurometabolites in those affected areas. In addition, biological variability, sample preparation technique and signal acquisition/ processing parameters might also influence the precision and accuracy of ex vivo measurement of neurometabolites.
Supplementary Fig. S3.

Evolution of creatine levels in the individual SODG93A and wild-type mice.
4.2.5. Increased myo-inositol in the hindbrain of SOD1G93A mice
We were able to detect a significant increase in the glial marker myo-inositols in the hindbrain of SOD1G93A mice from around P140. This finding is consistent with the observation of Choi et al. where they observed an increase in myo-inositol in the medulla between 110 and 142 days (Choi et al., 2009). Furthermore, similarly MRS studies with ALS patients have also shown increased myo-inositol (Block et al., 1998; Bowen et al., 2000; Kalra et al., 2006; Foerster et al., 2014; Atassi et al., 2017).
Correlation analyses between the neurometabolites in the hindbrain and motor cortex indicate that the factors regulating the levels of these metabolites may be interrelated. However, definite interpretations of the biological significance of these interrelations remain to be elucidated.
4.3. Coconut oil diet improved motor performance and increased survival in SOD1G93A mice
The influence of dietary factors in ALS progression has been a focus of previous research (Cacabelos et al., 2014). It is well accepted that malnutrition is indeed a negative prognostic factor in ALS (Greenwood, 2013). Furthermore, the ‘ALS Untangled study’ reported that saturated fatty acids, such as those present in coconut oil, could alleviate the disease progression by means of increasing energy bioavailability towards mitochondrial pathways (ALS Untangled 2012). Extracted from the meat of coconuts, coconut oil comprises 2.5% of world vegetable oil production (Hamid et al., 2011; ALS Untangled 2012). It is available in a basic “virgin” form, and also as an “RBD” (refined, bleached and deodorized) form, which has no coconut aroma or taste. Coconut oil can be fractionated into different medium-chain fatty acids; fractionated coconut oil is sometimes referred to as medium chain triglyceride (MCT) oil (ALS Untangled 2012).
4.3.1. Coconut oil as compensation for mitochondrial dysfunction in ALS
Mitochondrial dysfunction is likely to play an important role in ALS pathophysiology (Martin, 2011). Among the mitochondrial dysfunctions identified, cells extracted from patients with ALS show decreased complex-I activity, which contribute to impaired energy production (Swerdlow et al., 1998). MCT in coconut oil are converted in the liver into ketone bodies. In cultured neurons treated with drugs impairing complex 1 function, the addition of ketone bodies can restore complex 1 function (Tieu et al., 2003). Therefore, consumption of coconut oil and thereby raising ketone body levels may help compensate for mitochondrial dysfunction and impaired energy production in patients with ALS. Alternatively, there is growing evidence suggesting that nutritional status (Desport et al., 1999) and lipid metabolism (Dorst et al., 2011) are important prognostic factors in patients with ALS. In that context, coconut oil could possibly slow the disease progression by acting as a high caloric reservoir that increases circulating lipids.
4.3.2. Proof of concept study: coconut oil as a dietary treatment for SOD1G93A mice
One major finding of our study is that coconut oil supplemented diet demonstrated improved motor function in SOD mice on PaGE test compared to animals receiving standard diet during weeks 14 to 16. The coconut oil diet group showed 13.5% extended survival compared to the standard diet group. T2-weighted MR images acquired at P120 showed typical hyperintensities in the hypoglossal nucleus for animals receiving standard diet, which is less pronounced for animals of the coconut oil group (supplementary Fig. S2 A). T2-relaxometry maps from the hypoglossal nucleus confirmed increased T2-values for the standard diet group compared to the coconut oil group (supplementary Fig. S2 B, C). MRS data showed a stable neurometabolic profile in mice receiving a coconut oil supplemented diet compared to mice receiving standard diet at P120. Also we observed a significant (P < .0001) positive correlation (r = 0.88) between survival and NAA concentrations (Fig. 9 D), where the coconut oil group showed higher levels of NAA and extended survival. Kalra et al. showed that impaired neuronal integrity (decreased NAA) in the motor cortex negatively correlates with survival in ALS (Kalra et al., 2006). Therefore, improved NAA levels are a possible biological marker for vital prognosis in ALS. However, we cannot exclude the fact that a general improvement of the dietary state (instead of ketone bodies supplemented with the coconut oil) can also contribute to the improved survival and motor function.
Supplementary Fig. S2.

PoC study: Relaxometry.
5. Conclusion
In conclusion, MR images of SOD1G93A mice between 90 and 140 days revealed clear signal intensity enhancements compared to surrounding tissue in specific motor nuclei within the hindbrain, which are not detectable in age-matched WT mice. MRS evaluation of neurometabolic profiles revealed biochemical alterations in the motor cortex and hindbrain of transgenic ALS SOD1G93A mice. In a PoC study, we have demonstrated that coconut oil supplementation added to a standard rodent diet improved motor function, delayed neurological deficits, extended survival and was reflected by the absence of neurochemical changes otherwise seen in age matched animals of the SOD1G93A mouse model. Thus the study provides experimental evidence that prophylactic treatment with coconut oil diet may slow motor deterioration and protect motor neurons in SOD1G93A mice. Our initial results suggest that the neuroprotective effect by coconut oil may provide beneficial effects in ALS through a dietary intervention. Although promising, it is important to note that this was a pilot study, and more research is needed to validate these preliminary results. Finally, our study confirms that degeneration of neurons in the brainstem may indeed contribute significantly to the onset and overall disease progression in the SOD1G93A ALS model. Therefore, therapeutic trials in this mouse model should evaluate the beneficial effects to brainstem motor neurons. Furthermore, we confirm that 1H-MRS technique has the potential to be a used as diagnostic and a therapy monitoring tool in the SOD1G93A ALS mouse model.
The following are the supplementary data related to this article.
1H-MRS Studies of ALS SOD1G93A mice.
Disclosure/conflict of interest
The authors declared no potential conflicts of interest.
All experiments on rodents were approved by the local ethical committee of the University of Leuven, Belgium.
Funding
This work was partially funded by the European project EC-FP7 MC ITN ‘TRANSACT’ 2012 (no. 316679) and the KU Leuven program financing ‘IMIR’ (PF10/017).
Acknowledgements
Authors wish to thank Begga Schevenels, Nicole Hersmus and Caroline Eykens for the assistance with providing animals and information. We acknowledge the jMRUI team from the Czech Republic: Dr. Jana Starčuková, Dr. Zenon Starčuk jr and Michał Jabłonski for the technical support.
References
- Agosta F., Chiò A., Cosottini M., De Stefano N., Falini A., Mascalchi M., Rocca M.A., Silani V., Tedeschi G., Filippi M. The present and the future of neuroimaging in amyotrophic lateral sclerosis. AJNR Am. J. Neuroradiol. 2010;31(10):1769–1777. doi: 10.3174/ajnr.A2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALSUntangled Group. ALSUntangled 15 Coconut oil. Amyotr. Lateral Scler. 2012;13(3):328–330. doi: 10.3109/17482968.2012.671629. [DOI] [PubMed] [Google Scholar]
- Andreassen O.A., Jenkins B.G., Dedeoglu A., Ferrante K.L., Bogdanov M.B., Kaddurah-Daouk R., Beal M.F. Increases in cortical glutamate concentrations in transgenic amyotrophic lateral sclerosis mice are attenuated by creatine supplementation. J. Neurochem. 2001;77:383–390. doi: 10.1046/j.1471-4159.2001.00188.x. [DOI] [PubMed] [Google Scholar]
- Ari C., Poff A.M., Held H.E., Landon C.S., Goldhagen C.R., Mavromates N., D'Agostino D.P. Metabolic therapy with Deanna Protocol supplementation delays disease progression and extends survival in amyotrophic lateral sclerosis (ALS) mouse model. PLoS One. 2014;9(7) doi: 10.1371/journal.pone.0103526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atassi N., Xu M., Triantafyllou C., Keil B., Lawson R., Cernasov P., Ratti E., Long C.J., Paganoni S., Murphy A., Salibi N., Seethamraju R., Rosen B., Ratai E.M. Ultra high-field (7tesla) magnetic resonance spectroscopy in Amyotrophic Lateral Sclerosis. PLoS One. 2017;12(5) doi: 10.1371/journal.pone.0177680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastow E.L., Peswani A.R., Tarrant D.S., Pentland D.R., Chen X., Morgan A., Staniforth G.L., Tullet J.M., Rowe M.L., Howard M.J., Tuite M.F., Gourlay C.W. New links between SOD1 and metabolic dysfunction from a yeast model of amyotrophic lateral sclerosis. J. Cell Sci. 2016;129(21):4118–4129. doi: 10.1242/jcs.190298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal M.F. Coenzyme Q10 as a possible treatment for neurodegenerative diseases. Free Radic. Res. 2002;36(4):455–460. doi: 10.1080/10715760290021315. [DOI] [PubMed] [Google Scholar]
- Bendotti C., Carri M.T. Lessons from models of SOD1-linked familial ALS. Trends Mol. Med. 2004;10(8):393–400. doi: 10.1016/j.molmed.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Bendotti C., Calvaresi N., Chiveri L., Prelle A., Moggio M., Braga M., Silani V., De Beasi S. Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J. Neurol. Sci. 2001;191(1–2):25–33. doi: 10.1016/s0022-510x(01)00627-x. [DOI] [PubMed] [Google Scholar]
- Block W., Karitzky J., Träber F., Pohl C., Keller E., Mundegar R.R., Lamerichs R., Rink H., Ries F., Schild H.H., Jerusalem F. Proton magnetic resonance spectroscopy of the primary motor cortex in patients with motor neuron disease: subgroup analysis and follow-up measurements. Arch. Neurol. 1998;55(7):931–936. doi: 10.1001/archneur.55.7.931. [DOI] [PubMed] [Google Scholar]
- Boillée S., Vande Velde C., Cleveland D.W. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52(1):39–59. doi: 10.1016/j.neuron.2006.09.018. Review. [DOI] [PubMed] [Google Scholar]
- Bowen B.C., Pattany P.M., Bradley W.G., Murdoch J.B., Rotta F., Younis A.A., Duncan R.C., Quencer R.M. MR imaging and localized proton spectroscopy of the precentral gyrus in amyotrophic lateral sclerosis. Am. J. Neuroradiol. 2000;21(4):647–658. [PMC free article] [PubMed] [Google Scholar]
- Bradley W.G., Bowen B.C., Pattany P.M., Rotta F. 1H-magnetic resonance spectroscopy in amyotrophic lateral sclerosis. J. Neurol. Sci. 1999;169(1–2):84–86. doi: 10.1016/s0022-510x(99)00221-x. [DOI] [PubMed] [Google Scholar]
- Bruijn L.I., Houseweart M.K., Kato S., Anderson K.L., Anderson S.D., Ohama E., Reaume A.G., Scott R.W., Cleveland D.W. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Bucher S., Braunstein K.E., Niessen H.G., Kaulisch T., Neumaier M., Boeckers T.M., Stiller D., Ludolph A.D. Vacuolization correlates with spin-spin relaxation time in motor brainstem nuclei and behavioral tests in the transgenic G93A-SOD1 mouse model of ALS. Eur. J. Neurosci. 2007;26:1895–1901. doi: 10.1111/j.1460-9568.2007.05831.x. [DOI] [PubMed] [Google Scholar]
- Bunton-Stasyshyn R.K., Saccon R.A., Fratta P., Fisher E.M. SOD1 function and its implications for amyotrophic lateral sclerosis pathology: new and renascent themes. Neuroscientist. 2015;21(5):519–529. doi: 10.1177/1073858414561795. [DOI] [PubMed] [Google Scholar]
- Cacabelos D., Ayala V., Ramírez-Nunez O., Granado-Serrano A.B., Boada J., Serrano J.C., Cabré R., Nadal-Rey G., Bellmunt M.J., Ferrer I., Pamplona R., Portero-Otin M. Dietary lipid unsaturation influences survival and oxidative modifications of an amyotrophic lateral sclerosis model in a gender-specific manner. NeuroMolecular Med. 2014;16(4):669–685. doi: 10.1007/s12017-014-8317-7. [DOI] [PubMed] [Google Scholar]
- Caron I., Micotti E., Paladini A., Merlino G., Plebani L., Forloni G., Modo M., Bendotti C. Comparative magnetic resonance imaging and histopathological correlates in two SOD1 transgenic mouse models of amyotrophic lateral sclerosis. PLoS One. 2015;10(7) doi: 10.1371/journal.pone.0132159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I.Y., Lee S.P., Guilfoyle D.N., Helpern J.A. In vivo NMR studies of neurodegenerative diseases in transgenic and rodent models. Neurochem. Res. 2003;28:987–1001. doi: 10.1023/a:1023370104289. [DOI] [PubMed] [Google Scholar]
- Choi J., Kustermann E., Dedeoglu A., Jenkins B.G. Magnetic resonance spectroscopy of regional brain metabolite markers in FALS mice and the effects of dietary creatine supplementation. Eur. J. Neurosci. 2009;30(11):2143–2150. doi: 10.1111/j.1460-9568.2009.07015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J.B. N-acetyl aspartate: a marker for neuronal loss or mitochondrial dysfunction. Dev. Neurosci. 1998;20:271–276. doi: 10.1159/000017321. [DOI] [PubMed] [Google Scholar]
- Cleveland D.W., Rothstein J.D. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Crawley M.J. The R Book. John Wiley & Sons, Ltd; Chichester, UK: 2007. Data input. [Google Scholar]
- Curtis D.R., Watkins J.C. The pharmacology of amino acids related to gamma-aminobutyric acid. Pharmacol. Rev. 1965;17(4):347–391. [PubMed] [Google Scholar]
- De Vos K.J., Hafezparast M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017;105:283–299. doi: 10.1016/j.nbd.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedeoglu A., Choi J.K., Cormier K., Kowall N.W., Jenkins B.G. Magnetic resonance spectroscopic analysis of Alzheimer's disease mouse brain that express mutant human APP shows altered neurochemical profile. Brain Res. 2004;1012:60–65. doi: 10.1016/j.brainres.2004.02.079. [DOI] [PubMed] [Google Scholar]
- van den Boogaart A., van Ormondt D., Pijnappel W.W.F., de Beer R., Ala Korpel M. Mathematics of Signal Processing III. Clarendon Press; Oxford: 1994. Removal of the residual water resonance from 1H magnetic resonance spectra; pp. 175–195. [Google Scholar]
- Desport J.C., Preux P.M., Truong T.C., Vallat J.M., Sautereau D., Couratier P. Nutritional status is a prognostic factor for survival in ALS patients. Neurology. 1999;53(5):1059–1063. doi: 10.1212/wnl.53.5.1059. [DOI] [PubMed] [Google Scholar]
- Dorst J., Kühnlein P., Hendrich C., Kassubek J., Sperfeld A.D., Ludolph A.C. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survivalin amyotrophic lateral sclerosis. J. Neurol. 2011;258(4):613–617. doi: 10.1007/s00415-010-5805-z. [DOI] [PubMed] [Google Scholar]
- Dupuis L., Pradat P.F., Ludolph A.C., Loeffler J.P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011;10(1):75–82. doi: 10.1016/S1474-4422(10)70224-6. [DOI] [PubMed] [Google Scholar]
- Foerster B.R., Carlos R.C., Dwamena B.A., Callaghan B.C., Petrou M., Edden R.A.E.…Pomper M.G. Multimodal MRI as a diagnostic biomarker for amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2014;1(2):107–114. doi: 10.1002/acn3.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V., Wark P.A., Jenab M., Pearce N., Brayne C., Vermeulen R., Andersen P.M., Hallmans G., Kyrozis A., Vanacore N., Vahdaninia M., Grote V., Kaaks R., Mattiello A., Bueno-de-Mesquita H.B., Peeters P.H., Travis R.C., Petersson J., Hansson O., Arriola L., Jimenez-Martin J.M., Tjønneland A., Halkjær J., Agnoli C., Sacerdote C., Bonet C., Trichopoulou A., Gavrila D., Overvad K., Weiderpass E., Palli D., Quirós J.R., Tumino R., Khaw K.T., Wareham N., Barricante-Gurrea A., Fedirko V., Ferrari P., Clavel-Chapelon F., Boutron-Ruault M.C., Boeing H., Vigl M., Middleton L., Riboli E., Vineis P. Prediagnostic body fat and risk of death from amyotrophic lateral sclerosis: the EPIC cohort. Neurology. 2013;26(80(9)):829–838. doi: 10.1212/WNL.0b013e3182840689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparovic C., Yeo R., Mannell M., Ling J., Elgie R., Phillips J., Doezema D., Mayer A.R. Neurometabolite concentrations in gray and white matter in mild traumatic brain injury: an 1H-magnetic resonance spectroscopy study. J. Neurotrauma. 2009;26(10):1635–1643. doi: 10.1089/neu.2009.0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gredal O., Rosenbaum S., Topp S., Karlsborg M., Strange P., Werdelin L. Quantification of brain metabolites in amyotrophic lateral sclerosis by localized proton magnetic resonance spectroscopy. Neurology. 1997;48(4):878–881. doi: 10.1212/wnl.48.4.878. [DOI] [PubMed] [Google Scholar]
- Greenwood D.I. Nutrition management of amyotrophic lateral sclerosis. Nutr. Clin. Pract. 2013;28(3):392–399. doi: 10.1177/0884533613476554. [DOI] [PubMed] [Google Scholar]
- Griffey R.H., Flamig D.P. VAPOR for solvent -supressed, short-echo, volume-localized proton spectroscopy. J. Magn. Reson. 1990;88:161–166. [Google Scholar]
- Gruetter R. Automatic, localized in vivo adjustment of all first- and second-order shim coils. Magn. Reson. Med. 1993;29(6):804–811. doi: 10.1002/mrm.1910290613. [DOI] [PubMed] [Google Scholar]
- Gupta Sunil, Riis Jason. Harvard Business School; 2011. PatientsLikeMe: An Online Community of Patients. Case 511-03, February 2011. (Revised Novem 2012) [Google Scholar]
- Gurney M.E., Pu H., Chiu A.Y., Dal Canto M.C., Polchow C.Y., Alexander D.D., Caliendo J., Hentati A., Kwon Y.W., Deng H.X. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;17(264(5166)):1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hamid M.A., Sarmidi M.R., Mokhtar T.H., Sulaiman W.R.W., Azila R.A. Innovative integrated wet-process for virgin coconut oil production. J. Appl. Sci. 2011;11:2467–2469. [Google Scholar]
- Han J., Ma L. Study of the features of proton MR spectroscopy ((1) H-MRS) on amyotrophic lateral sclerosis. J. Magn. Reson. Imaging. 2010;31(2):305–308. doi: 10.1002/jmri.22053. [DOI] [PubMed] [Google Scholar]
- Hartman A.L. Neuroprotection in metabolism-based therapy. Epilepsy Res. 2012;100(3):286–294. doi: 10.1016/j.eplepsyres.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman A.L. Stafstrom CE. Harnessing the power of metabolism for seizure prevention: focus on dietary treatments. Epilepsy Behav. 2013;26(3):266–272. doi: 10.1016/j.yebeh.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht M.J., Fellner F., Fellner C., Hilz M.J., Neundorfer B., Heuss D. Hyperintense and hypointense MRI signals of the precentral gyrus and corticospinal tract in ALS: a follow-up examination including FLAIR images. J. Neurol. Sci. 2002;199:59–65. doi: 10.1016/s0022-510x(02)00104-1. [DOI] [PubMed] [Google Scholar]
- Hersch S.M., Gevorkian S., Marder K., Moskowitz C., Feigin A., Cox M., Como P., Zimmerman C., Lin M., Zhang L., Ulug A.M., Beal M.F., Matson W., Bogdanov M., Ebbel E., Zaleta A., Kaneko Y., Jenkins B., Hevelone N., Zhang H., Yu H., Schoenfeld D., Ferrante R., Rosas H.D. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2'dG. Neurology. 2006;66:250–252. doi: 10.1212/01.wnl.0000194318.74946.b6. [DOI] [PubMed] [Google Scholar]
- Higgins C.M., Jung C., Xu Z. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003;4:16. doi: 10.1186/1471-2202-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillel A.D., Miller R. Bulbar amyotrophic lateral sclerosis: patterns of progression and clinical management. Head Neck. 1989;11(1):51–59. doi: 10.1002/hed.2880110110. [DOI] [PubMed] [Google Scholar]
- Jenkins B.G., Klivenyi P., Kustermann E., Andreassen O.A., Ferrante R.J., Rosen B.R., Beal M.F. Nonlinear decrease over time in N-acetyl aspartate levels in the absence of neuronal loss and increases in glutamine and glucose in transgenic Huntington's disease mice. J. Neurochem. 2000;74:2108–2119. doi: 10.1046/j.1471-4159.2000.0742108.x. [DOI] [PubMed] [Google Scholar]
- Jenkins B.G., Andreassen O.A., Dedeoglu A., Leavitt B., Hayden M., Borchelt D., Ross C.A., Ferrante R.J., Beal M.F. Effects of CAG repeat length, HTT protein length and protein context on cerebral metabolism measured using magnetic resonance spectroscopy in transgenic mouse models of Huntington's disease. J. Neurochem. 2005;95:553–562. doi: 10.1111/j.1471-4159.2005.03411.x. [DOI] [PubMed] [Google Scholar]
- Jung M.K., Kim K.Y., Lee N.Y., Kang Y.S., Hwang Y.J., Kim Y., Sung J.J., McKee A., Kowall N., Lee J., Ryu H. Expression of taurine transporter (TauT) is modulated by heat shock factor 1 (HSF1) in motor neurons of ALS. Mol. Neurobiol. 2013;47(2):699–710. doi: 10.1007/s12035-012-8371-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra S., Arnold D.L., Cashman N.R. Biological markers in the diagnosis and treatment of ALS. J. Neurol. Sci. 1999;165:S27–S32. doi: 10.1016/s0022-510x(99)00023-4. [DOI] [PubMed] [Google Scholar]
- Kalra S., Vitale A., Cashman N.R., Genge A., Arnold D.L. Cerebral degeneration predicts survival in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry. 2006;77(11):1253–1255. doi: 10.1136/jnnp.2006.090696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karitzky J., Ludolph A.C. Imaging and neurochemical markers for diagnosis and disease progression in ALS. J. Neurol. Sci. 2001;191:35–41. doi: 10.1016/s0022-510x(01)00628-1. [DOI] [PubMed] [Google Scholar]
- Kong J., Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998;18(9):3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Ghosh D., Singh R.L. Amyotrophic lateral sclerosis and metabolomics: clinical implication and therapeutic approach. J. Biomark. 2013;2013:538765. doi: 10.1155/2013/538765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.C., Markus R., Hughes A. MRI in ALS: corticospinal tract hyperintensity. Neurology. 2003;61:1600. doi: 10.1212/01.wnl.0000096015.48322.2a. [DOI] [PubMed] [Google Scholar]
- Liu Y.J., Tsai P.Y., Chern Y. Energy homeostasis and abnormal rna metabolism in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2017;11:126. doi: 10.3389/fncel.2017.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malessa S., Leigh P.N., Bertel O., Sluga E., Hornykiewicz O. Amyotrophic lateral sclerosis: glutamate dehydrogenase and transmitter amino acids in the spinal cord. J. Neurol. Neurosurg. Psychiatry. 1991;54(11):984–988. doi: 10.1136/jnnp.54.11.984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjanska M., Curran G.L., Wengenack T.M., Henry P.G., Bliss R.L., Poduslo J.F., Jack C.R., Jr., Ugurbil K., Garwood M. Monitoring disease progression in transgenic mouse models of Alzheimer's disease with proton magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2005;102:11906–11910. doi: 10.1073/pnas.0505513102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L.J. Mitochondrial pathobiology in ALS. J. Bioenerg. Biomembr. 2011;43(6):569–579. doi: 10.1007/s10863-011-9395-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen H.G., Angenstein F., Sander K., Kunz W.S., Teuchert M., Ludolph A.C. In vivo quantification of spinal and bulbar motor neuron degeneration in the G93A-SOD1 transgenic mouse model of ALS by T2 relaxation time and apparent diffusion coefficient. Exp. Neurol. 2006;201:293–300. doi: 10.1016/j.expneurol.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Niessen H.G., Debska-Vielhaber G., Sander K., Angenstein F., Ludolph A.C., Hilfert L., Willker W., Leibfritz D., Heinze H.J., Kunz W.S., Vielhaber S. Metabolic progression markers of neurodegeneration in the transgenic G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 2007;25(6):1669–1677. doi: 10.1111/j.1460-9568.2007.05415.x. [DOI] [PubMed] [Google Scholar]
- O'Reilly É.J., Wang H., Weisskopf M.G., Fitzgerald K.C., Falcone G., McCullough M.L., Thun M., Park Y., Kolonel L.N., Ascherio A. Premorbid body mass index and risk of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemp. Degener. 2013;14(3):205–211. doi: 10.3109/21678421.2012.735240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry T.L., Hansen S., Jones K. Brain glutamate deficiency in amyotrophic lateral sclerosis. Neurology. 1987;37:1845–1848. doi: 10.1212/wnl.37.12.1845. [DOI] [PubMed] [Google Scholar]
- Petroff O.A. GABA and glutamate in the human brain. Neuroscientist. 2002;8(6):562–573. doi: 10.1177/1073858402238515. [DOI] [PubMed] [Google Scholar]
- Pfeuffer J., Tkác I., Provencher S.W., Gruetter R. Toward an in vivo neurochemical profile: quantification of 18 metabolites in short-echo-time (1)H NMR spectra of the rat brain. J. Magn. Reson. 1999;141(1):104–120. doi: 10.1006/jmre.1999.1895. [DOI] [PubMed] [Google Scholar]
- Pioro E.P., Majors A.W., Mitsumoto H., Nelson D.R., Ng T.C. 1H-MRS evidence of neurodegeneration and excess glutamate + glutamine in ALS medulla. Neurology. 1999;53:71–79. doi: 10.1212/wnl.53.1.71. [DOI] [PubMed] [Google Scholar]
- Rae C.D., Bröer S. Creatine as a booster for human brain function. How might it work? Neurochem. Int. 2015;89:249–259. doi: 10.1016/j.neuint.2015.08.010. [DOI] [PubMed] [Google Scholar]
- Ratiney H., Coenradie Y., Cavassila S., van Ormondt D., Graveron-Demilly D. Time-domain quantitation of 1H short echo-time signals: background accommodation. MAGMA. 2004;16(6):284–296. doi: 10.1007/s10334-004-0037-9. [DOI] [PubMed] [Google Scholar]
- Reynolds N.C., Jr., Prost R.W., Mark L.P. Heterogeneity in 1H-MRS profiles of presymptomatic and early manifest Huntington's disease. Brain Res. 2005;1031(1):82–89. doi: 10.1016/j.brainres.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Rosen D.R., Siddique T., Patterson D., Figlewicz D.A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J.P., Deng H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rule R.R., Suhy J., Schuff N., Gelinas D.F., Miller R.G., Weiner M.W. Reduced NAA in motor and non-motor brain regions in amyotrophic lateral sclerosis: a cross-sectional and longitudinal study. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2004:141–149. doi: 10.1080/14660820410017109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Pernaute R., García-Segura J.M., del Barrio Alba A., Viaño J., de Yébenes J.G. Clinical correlation of striatal 1H MRS changes in Huntington's disease. Neurology. 1999;11(53(4)):806–812. doi: 10.1212/wnl.53.4.806. [DOI] [PubMed] [Google Scholar]
- Sartorius A., Lugenbiel P., Mahlstedt M.M., Ende G., Schloss P., Vollmayr B. Proton magnetic resonance spectroscopic creatine correlates with creatine transporter protein density in rat brain. J. Neurosci. Methods. 2008;172(2):215–219. doi: 10.1016/j.jneumeth.2008.04.028. [DOI] [PubMed] [Google Scholar]
- Sau D., De Biasi S., Vitellaro-Zuccarello L., Riso P., Guarnieri S., Porrini M., Simeoni S., Crippa V., Onesto E., Palazzolo I., Rusmini P., Bolzoni E., Bendotti C., Poletti A. Mutation of SOD1 in ALS: a gain of a loss of function. Hum. Mol. Genet. 2007;1(16(13)):1604–1618. doi: 10.1093/hmg/ddm110. [DOI] [PubMed] [Google Scholar]
- Starčuk Z.O., Jr., Strbak J., Starcukova Graveron-Demilly. IEEE International Workshop on Imaging Systems and Techniques. 2008. Simulation of steady state free precession acquisition mode in coupled spin systems for fast MR spectroscopic imaging; pp. 302–306. [Google Scholar]
- Stefan D., Di Cesare F., Andrasescu A., Popa E., Lazariev A., Vescovo E., Strbak O., Williams S., Starcuk Z., Cabanas M., van Ormondt D., Graveron-Demilly D. Quantitation of magnetic resonance spectroscopy signals: the jMRUI software package. Meas. Sci. Technol. 2009;20:104035. (9 pp.) [Google Scholar]
- Swerdlow R.H., Parks J.K., Cassarino D.S., Trimmer P.A., Miller S.W., Maguire D.J., Sheehan J.P., Maguire R.S., Pattee G., Juel V.C., Phillips L.H., Tuttle J.B., Bennett J.P., Jr., Davi R.E., Parker W.D., Jr. Mitochondria in sporadic amyotrophic lateral sclerosis. Exp. Neurol. 1998;153(1):135–142. doi: 10.1006/exnr.1998.6866. [DOI] [PubMed] [Google Scholar]
- Tankersley C.G., Haenggeli C., Rothstein J.D. Respiratory impairment in a mouse model of amyotrophic lateral sclerosis. J. Appl. Physiol. 2007;102:926–932. doi: 10.1152/japplphysiol.00193.2006. [DOI] [PubMed] [Google Scholar]
- Tieu K., Perier C., Caspersen C., Teismann P., Wu D.C., Yan S.D., Naini A., Vila M., Jackson-Lewis V., Ramasamy R., Przedborski S. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Invest. 2003;112(6):892–901. doi: 10.1172/JCI18797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu P.H., Raju P., Robinson K.A., Gurney M.E., Trojanowski J.Q., Lee V.M. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc. Natl. Acad. Sci. U. S. A. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner B.J., Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 2008;85:94–134. doi: 10.1016/j.pneurobio.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Van Damme P., Robberecht W., Van Den Bosch L. Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis. Model. Mech. 2017;10(5):537–549. doi: 10.1242/dmm.029058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Perren A., Toelen J., Casteels C., Macchi F., Van Rompuy A.S., Sarre S., Casadei N., Nuber S., Himmelreich U., Osorio Garcia M.I., Michotte Y., D'Hooge R., Bormans G., Van Laere K., Gijsbers R., Van den Haute C., Debyser Z., Baekelandt V. Longitudinal follow-up and characterization of a robust rat model for Parkinson's disease based on overexpression of alpha-synuclein with adeno-associated viral vectors. Neurobiol. Aging. 2015;36(3):1543–1558. doi: 10.1016/j.neurobiolaging.2014.11.015. [DOI] [PubMed] [Google Scholar]
- Verstraete E., Foerster B.R. Neuroimaging as a New Diagnostic Modality in Amyotrophic Lateral Sclerosis. Neurotherapeutics. 2015;12(2):403–416. doi: 10.1007/s13311-015-0347-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vielhaber S., Kaufmann J., Kanowski M., Sailer M., Feistner H., Tempelmann C., Elger C.E., Heinze H.-J., Kunz W.S. Effect of creatine supplementation on metabolite levels in ALS motor cortices. Exp. Neurol. 2001;172:377–382. doi: 10.1006/exnr.2001.7797. [DOI] [PubMed] [Google Scholar]
- Wang S., Melhem E.R., Poptani H., Woo J.H. Neuroimaging in amyotrophic lateral sclerosis. Neurotherapeutics. 2011;8(1):63–71. doi: 10.1007/s13311-010-0011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Zhang F., Li L., Tang F., Siedlak S.L., Fujioka H., Liu Y., Su B., Pi Y., Wang X. MFN2 couples glutamate excitotoxicity and mitochondrial dysfunction in motorneurons. J. Biol. Chem. 2015;290(1):168–182. doi: 10.1074/jbc.M114.617167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P., Hong S.Y., Kliot M., Möller T. Assessing disease onset and progression in the SOD1 mouse model of ALS. Neuroreport. 2003;14(7):1051–1054. doi: 10.1097/01.wnr.0000073685.00308.89. [DOI] [PubMed] [Google Scholar]
- Yoshino Y., Koike H., Akai K. Free amino acids in motor cortex of amyotrophic lateral sclerosis. Experientia. 1979;35:219–220. doi: 10.1007/BF01920627. [DOI] [PubMed] [Google Scholar]
- Zang D.W., Yang Q., Wang H.X., Egan G., Lopes E.C., Cheema S.S. Magnetic resonance imaging reveals neuronal degeneration in the brainstem of the superoxide dismutase 1 transgenic mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 2004;20(7):1745–1751. doi: 10.1111/j.1460-9568.2004.03648.x. [DOI] [PubMed] [Google Scholar]
- Zhao Z., Lange D.J., Voustianiouk A., MacGrogan D., Ho L., Suh J., Humala N., Thiyagarajan M., Wang J., Pasinetti G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006;7:29. doi: 10.1186/1471-2202-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W., Varghese M., Vempati P., Dzhun A., Cheng A., Wang J., Lange D., Bilski A., Faravelli I., Pasinetti G.M. Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLoS One. 2012;7(11) doi: 10.1371/journal.pone.0049191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H-MRS Studies of ALS SOD1G93A mice.