

Abstract
Background
Human decidua basalis mesenchymal stem/multipotent stromal cells (DBMSCs) inhibit endothelial cell activation by inflammation induced by monocytes. This property makes them a promising candidate for cell-based therapy to treat inflammatory diseases, such as atherosclerosis. This study was performed to examine the ability of DBMSCs to protect endothelial cell functions from the damaging effects resulting from exposure to oxidatively stress environment induced by H2O2 and monocytes.
Methods
DBMSCs were co-cultured with endothelial cells isolated from human umbilical cord veins in the presence of H2O2 and monocytes, and various functions of endothelial cell were then determined. The effect of DBMSCs on monocyte adhesion to endothelial cells in the presence of H2O2 was also examined. In addition, the effect of DBMSCs on HUVEC gene expression under the influence of H2O2 was also determined.
Results
DBMSCs reversed the effect of H2O2 on endothelial cell functions. In addition, DBMSCs reduced monocyte adhesion to endothelial cells and also reduced the stimulatory effect of monocytes on endothelial cell proliferation in the presence of H2O2. Moreover, DBMSCs modified the expression of many genes mediating important endothelial cell functions. Finally, DBMSCs increased the activities of glutathione and thioredoxin reductases in H2O2-treated endothelial cells.
Conclusions
We conclude that DBMSCs have potential for therapeutic application in inflammatory diseases, such as atherosclerosis by protecting endothelial cells from oxidative stress damage. However, more studies are needed to elucidate this further.
Keywords: Placenta, Decidua basalis mesenchymal stem cells, Endothelial cells, H2O2, Proliferation, Adhesion, Migration, Monocytes
Background
Mesenchymal stem cells (MSCs) are adult multipotent stromal cells that can be isolated from many tissues, such as human placenta [1]. Recently, we isolated MSCs from the maternal decidua basalis tissue (DBMSCs) of human term placenta [2]. The tissue of decidua basalis is a main source of oxidative stress molecules, which are found in the maternal circulation due to pregnancy [3]. Therefore, DBMSCs in their niche (vascular microenvironment) are in direct contact with the maternal circulation, and therefore, they are exposed to high levels of inflammation and oxidative stress mediators [4]. In addition, we also isolated MSCs from the fetal tissue (chorionic villi) of the placenta [5]. These fetal chorionic MSCs are in direct contact with the fetal circulation and therefore exposed to lower levels of inflammation and oxidative stress molecules as compared to DBMSCs [5–7].
MSCs from placenta and other sources can differentiate into multiple cell lineages including adipocyte, osteoblast, and chondrocyte [1]. In addition, MSCs show low immunogenicity and anti-inflammatory properties [1]. Therefore, MSCs have been investigated as promising therapeutic agents in many inflammatory diseases, such as atherosclerosis [8].
Atherosclerosis is characterized by endothelial activation due to the accumulation of high amounts of low-density lipoprotein (LDL) and immune cells that lead to the production of high levels of oxidative stress mediators, such as hydrogen peroxide (H2O2) [9, 10].
H2O2 has several important effects on endothelial cell functions in physiological homeostasis and in inflammatory diseases [9, 10]. H2O2 alters the functional activities of proteins that cause the generation of more toxic radicals (i.e., peroxynitrite (ONOO−) and hydroxyl (·OH)), which induce oxidative damage in the cellular DNA and proteins [9, 10]. In addition, H2O2 can rapidly inactivate nitric oxide (NO) and this causes endothelial cell damage [9, 10].
Endothelial cell damage is usually associated with phenotypic changes (i.e., increased expression of inflammatory molecules), dysfunctional activities [i.e., increased endothelial cell proliferation, adhesion, migration, permeability, angiogenesis (blood vessel formational)], and also enhanced endothelial cell interaction with immune cells (i.e., enhanced monocyte adhesion to the endothelium and their infiltration into the tissues); these events are the typical characteristics of atherosclerosis [11]. In atherosclerosis, an inflammatory response is initiated at the injury site of endothelium that increases the expression of adhesion molecules (i.e., VCAM-1), which activates the recruitment and adhesion of immune cells (i.e., monocytes) to the injured site of endothelium [11]. This interaction between monocytes and endothelial cells will loosen up the tight junction between endothelial cells that increases the permeability of endothelium and subsequently monocytes and LDL will pass through the intima, where LDL undergoes oxidation while monocytes differentiate into macrophages, which take up oxidized LDL [11]. This lipid laden macrophages are known as “foam cells”, which eventually die by apoptosis, but the lipid content will accumulate in the intimal area leading to the formation of plaque [11].
Recently, we reported that DBMSCs can protect endothelial cells from activation by inflammation triggered by monocyte adhesion and increased endothelial cell proliferation [12]. These events are manifest in inflammatory diseases, such as atherosclerosis. These data make DBMSCs as a useful candidate to be employed in a therapeutic strategy for treating atherosclerosis. We performed this study to examine the ability of DBMSCs to protect endothelial cell functions from the damaging effects resulting from exposure to oxidatively stress environment induced by H2O2 and monocytes. We investigated the ability of DBMSCs to protect endothelial cell functions (adhesion, proliferation, and migration) from oxidative stress induced by H2O2. The effect of DBMSCs on the adhesion of monocytes to endothelial cells in oxidative stress environment was also examined. Finally, we investigated the effect of DBMSCs on endothelial cell expression of many genes under oxidative stress, and the mechanism underlying DBMSC protection of endothelial cells from oxidative stress was also determined. Our data suggest that DBMSCs have a protective effect on endothelial cells in oxidative stress environment and suggest that DBMSCs have the potential to treat inflammatory diseases, such as atherosclerosis by protecting endothelial cells from injury induced by oxidative stress and inflammatory cells. However, future studies are necessary to elucidate this further in vitro and in vivo.
Methods
Ethics and collection of human placentae and umbilical cords
The study was approved by the institutional review board (reference number IRBC/246/13) of KAIMRC (King Abdulla International Medical Research Centre, Saudi Arabia). Samples (placentae and umbilical cords of uncomplicated human pregnancies, 38–40 gestational weeks) were obtained and used immediately after signing consent forms. All clinical and experimental procedures were performed in compliance with KAIMRC research guidelines and regulations.
Isolation and culture of DBMSCs
MSCs were isolated from the decidua basalis (DBMSCs) of the maternal part of human term placenta as previously described by us [2]. Briefly, the decidual tissues were dissected and then digested using a sterile phosphate-buffered solution (PBS; pH 7.4) containing 0.3% collagenase type I (Life Technology, Grand Island, USA), 270 unit/mL DNase I (Life Technology), and antibiotics (100 μg/mL streptomycin and 100 U/mL penicillin). After 1-h incubation at 37 °C in a water bath, the cell mixture was filtered through a 100-μm nylon filter (Becton Dickinson, NJ, USA), and the red blood cells in the cell pellet were then removed as previously described [12]. Cells were then washed with sterile PBS and cultured in a complete DBMSC culture medium [DMEM-F12 medium containing 10% MSCFBS (mesenchymal stem cell-certified fetal bovine serum, catalogue number 12-662-011, Life Technology), and antibiotics described above] and then incubated at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air (a cell culture incubator). Prior to using DBMSCs in subsequent experiments, DBMSCs at passage 3 were characterized by flow cytometry using MSC and hematopoietic markers (Table 1) and then evaluated for differentiation into adipocytes, chondrocytes, and osteocytes as previously described by us [2]. DBMSCs (passage 3) of 30 placentae were used in this study.
Table 1.
Monoclonal antibodies used in this study
| Markers | Monoclonal antibodies | |||||||
|---|---|---|---|---|---|---|---|---|
| MSC markers | CD44 | CD90 | CD105 | CD146 | CD166 | HLA-ABC | ||
| Hematopoietic markers | CD14 | CD19 | CD40 | CD45 | CD80 | CD83 | CD86 | HLA-DR |
| Endothelial Cell Marker | CD31 | |||||||
| Adhesion Molecules | ICAM-1 | VCAM-1 | CD44 | |||||
Isolation and culture of human umbilical vein endothelial cells (HUVEC)
HUVEC were isolated from umbilical cord veins using our previously published method [12]. Following rinsing the cannulated umbilical veins with PBS for several times, veins were filled with a digestion PBS solution containing 6 mg/ml collagenase type II (catalogue number 17101-015, Life Technologies) and then incubated at 37 °C in a cell culture incubator. After 25 min, HUVEC were collected and then resuspended in a complete endothelial cell growth medium (catalogue number PCS-100-041™, ATCC, USA) and cultured at 37 °C in a cell culture incubator as previously described [12]. Prior to using HUVEC in subsequent experiments, they were characterized by flow cytometry using a CD31 endothelial cell marker (R and D Systems, Abingdon, UK) as previously described [12]. HUVEC (> 95% purity) from passages 3 to 5 of 30 umbilical cords were used in this study.
HUVEC proliferation in response to DBMSCs and H2O2
HUVEC (5 × 103) were seeded in wells of 96-well culture plates containing a complete endothelial cell growth medium and cultured at 37 °C in a cell culture incubator. Following 24 h, adherent HUVEC were incubated with different concentrations [1%, 5% and 25% (v/v) conditioned medium (CM) harvested from DBMSC culture (CMDBMSC) diluted in a complete DBMSC growth medium] of CMDBMSC and different ratios of 1:1, 5:1, and 10:1 HUVEC to DBMSC. Cells were then cultured in a complete endothelial cell growth medium with or without 100 μM H2O2 for 72 h at 37 °C in a cell culture incubator.
HUVEC proliferation was then evaluated after each indicated culture time points (24, 48, and 72 h) by a tetrazolium compound [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS] kit (catalogue number G5421, CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay, Promega, Germany), as previously described [12]. CMDBMSC was produced as previously described [12]. Before adding DBMSCs to HUVEC culture, DBMSCs were treated with 25 μg/ml Mitomycin C to inhibit their proliferation as previously described [12]. The blank was cells incubated in MTS solution in a complete endothelial cell growth medium alone. Results were presented as means (± standard errors). Each experiment was performed in triplicate and repeated with five independent HUVEC (passages 3–5) and DBMSC (passage 3) preparations.
Culture of HUVEC with different treatments of DBMSCs (conditioned medium, supernatant, and intercellular direct contact) and H2O2
HUVEC were cultured alone (Fig. 1a) or with 100 μM H2O2 (Fig. 1b) or with 25% CMDBMSC and 100 μM H2O2 (Fig. 1c) in a complete endothelial cell growth medium. For the coculture experiments (supernatant and intercellular direct contact), cells (HUVEC and DBMSCs) were separated by transwell chamber membrane culture system [catalogue number 657640, ThinCert™ Cell Culture Inserts, 0.4 μm, Greiner Bio-One, Germany]. For soluble factor experiments (SFDBMSC; Fig. 1d), DBMSCs were cultured on the upper compartments while HUVEC were cultured in the lower compartment. For intercellular direct contact experiments (ICDBMSC; Fig. 1e), DBMSCs were seeded on the reverse side of the membrane of the chamber, and HUVEC were seeded on the upper side of the membrane. In both culture systems, cells were cultured at 5HUVEC:1DBMSC ratio. Cells in the SFDBMSC and ICDBMSC culture systems were then cultured in a complete endothelial cell growth medium in the presence of 100 μM H2O2 and incubated as described above. HUVEC were also cultured with CMDBMSC, SFDBMSC, and ICDBMSC without H2O2. After 48 h in culture, HUVEC were harvested with TrypLE™ Express detachment solution (Life Technologies) and used in an adhesion, proliferation, and migration experiments as described below. HUVEC viability was determined using Trypan blue. Each experiment was performed and repeated as described above. HUVEC cultured in complete endothelial cell growth medium without DBMSCs were included as a negative control for all HUVEC cultured with different treatments of DBMSCs.
Fig. 1.

The culture system used in this study to culture HUVEC alone or with H2O2 in the presence or absence of different treatments of DBMSCs (CMDBMSC, SFDBMEC, and ICDBMSC). CMDBMSC culture system consisted of HUVEC seeded on a surface of 6-well culture plate in a complete endothelial cell growth culture medium (untreated HUVEC) (a) or with 100 μM H2O2 (b) or with 100 μM H2O2 and 25% CM obtained from unstimulated DBMSCs (c); SFDBMSC culture system consisted of DBMSCs seeded in the upper chamber while HUVEC seeded in the lower chamber of transwell membrane culture system (d); and ICDBMSC culture system consisted of DBMSCs seeded on the reverse side of the membrane of the chamber and HUVEC seeded on the upper side of the membrane (e). For SFDBMSC and ICDBMSC, 0.4-μm pore size transwell chamber membrane was used. HUVEC were incubated with different concentrations (1%, 5%, and 25% (v/v) CM diluted in complete DBMSC growth medium) of CMDBMSC and different ratios of 1:1, 5:1, and 10:1 HUVEC:DBMSC. Cells were then cultured in a complete endothelial cell growth medium with or without 100 μM H2O2 for 72 h at 37 °C in a cell culture incubator
HUVEC adhesion and proliferation using xCELLigence system
The xCELLigence system (RTCA-DP version; Roche Diagnostics, Mannheim, Germany) was used as we previously described [12, 13] to evaluate the adhesion and proliferation of HUVEC. The xCELLigence system is a real-time cell analyzer that constantly monitors and records the changes in electrical impedance, because of cellular events, and these changes are reported as an arbitrary cell index [12, 13]. Briefly, 100-μL complete endothelial cell growth medium was added to well in 16-well culture plates (catalogue number 05469813001, E-Plate 16, Roche Diagnostics), and the background impedance was then achieved as previously described [12, 13]. Then, 20 × 104 HUVEC (HUVEC were initially co-cultured with DBMSCs and 100 μM H2O2 or cultured alone as described above) were seeded in 100 μL of complete endothelial cell growth medium in quadruplicate wells, and equilibrium was achieved by leaving the culture plates for 30 min at RT before data recording. To record data, culture plates were placed in the xCELLigence system at 37 °C in a cell culture incubator. HUVEC cell index was then automatically monitored for 72 h. For data analysis, the xCELLigence software (version 1.2.1) was used. For cell adhesion, data was measured after 2 h and the value of cell index was then expressed as mean ± standard errors of the cell index. For cell proliferation, data was expressed as mean ± standard errors of the cell index normalized to the cell index recorded after 2 h (adhesion time point). The rate of cell proliferation was determined by calculating the normalized cell index at 24, 48, and 72 h. Each experiment was performed and repeated as described above.
HUVEC migration using xCELLigence system
The migration of HUVEC was evaluated using CIM migration plates (catalogue number 05665825001, Roche Diagnostics) in the xCELLigence system as previously described by us [12, 13]. The CIM plates have 16-migration wells that each consists of two chambers (upper and lower) separated by a membrane (polyethylene terephthalate) with a porous of 8 μm in size. The membrane is in contact with microelectrodes. Following the addition of 50-μl pre-warmed media to the wells of the upper chamber and 160-μl endothelial cell growth medium containing 30% FBS to the lower chamber, the plates were then locked in the RTCA DP device at 37 °C in a cell culture incubator for 1 h to obtain equilibrium, and a measurement step was then performed as previously described [12, 13]. The migration experiments were then initiated by seeding 20 × 103 HUVEC [HUVEC were initially co-cultured with DBMSCs and 100 μM H2O2 or cultured with DBMSCs (CMDBMSC, SFDBMSC and ICDBMSC) or cultured alone as described above] in the upper chamber containing 100-μL endothelial cell serum free medium and the plates were then incubated for 30 min at RT to allow the cells to settle onto the membrane as previously described [12, 13]. Experiments were performed in quadruplicate, and after equilibration, the impedance value of each well was automatically monitored every 15 min for 24 h by the xCELLigence system and then expressed as a cell index value. HUVEC migration observed in the presence and absence of 30% FBS served as positive and negative controls, respectively. Each experiment was performed and repeated as described above.
HUVEC proliferation in response to monocytes pretreated with DBMSCs and H2O2
To evaluate the effects of monocytes pre-treated with DBMSCs on the proliferation of endothelial cells, monocyte proliferation in response to DBMSCs was initially examined by adding DBMSCs to human monocytes (THP-1, catalogue number TIB-202™, ATCC, USA) in 96-well tissue culture plates at different THP-1:DBMSC ratios (2.5:1, 5:1, 10:1, and 20:1 THP-1:DBMSC) in the presence or absence of 100 μM H2O2 (Fig. 2). Cells were then cultured in a complete RPMI-1640 culture medium containing 10% FBS, 100 μg/mL l-glutamate, and antibiotics. After 24, 48, and 72 h incubation at 37 °C in a cell culture incubator, monocyte proliferation was examined using the MTS assay as previously described [12].
Fig. 2.

The culture systems used in this study to culture monocytes (THP-1) alone or with H2O2 or with DBMSCs and H2O2 or with HUVEC and H2O2. Monocytes (THP-1) cultured alone in RPMI-1640 culture medium (a) or with 100 μM H2O2 (b) or with DBMSCs (physical contact experiment) and 100 μM H2O2 (c), HUVEC cultured with THP-1 pretreated with DBMSCs and 100 μM H2O2 (Physical contact experiment) in a complete endothelial cell growth medium in the presence of 100 μM H2O2 (d). THP-1 were cultured with DBMSCs at different THP-1:DBMSC ratios (2.5:1, 5:1, 10:1 and 20:1 THP-1:DBMSC) in the presence or absence of 100 μM H2O2. HUVEC were cultured with THP-1- pre-treated with DBMSCs and H2O2 at different THP-1:HUVEC ratios (2.5:1, 5:1, 10:1 and 20:1 THP-1:HUVEC) in the presence or absence of 100 μM H2O2. Cells were then incubated for 96 h at 37 °C in a cell culture incubator
Next, endothelial cell proliferation in response to monocytes pre-cultured with DBMSCs and H2O2 at the indicated ratios (below) in the presence of 100 μM H2O2 was examined (Fig. 2). After 24-h culture with DBMSCs in the presence of 100 μM H2O2, THP-1 [THP-1 alone, THP-1+ H2O2 (THP-1 pretreated with H2O2), and THP-1/DBMSC+ H2O2 (THP-1 pretreated with DBMSCs and H2O2)] were harvested and then added to HUVEC at different THP-1:HUVEC ratios (2.5:1, 5:1, 10:1, and 20:1 THP-1:HUVEC) in the presence of 100 μM H2O2. Briefly, THP-1 were added to HUVEC that were initially seeded at a density of 5 × 103 per well in 96-well tissue culture plates. After 24-h culture in a complete HUVEC culture medium at 37 °C in a cell culture incubator, HUVEC proliferation was examined using the MTS assay as previously described [12]. Before using DBMSCs and THP-1 in the proliferation assays, cells were treated with 25 μg/ml Mitomycin C to inhibit their proliferation as previously described [12]. Results were presented as means (± standard errors). Each experiment was performed in triplicate and repeated for five times with five independent preparations of DBMSCs and HUVEC. DBMSCs and THP-1 cultured alone were included as negative controls.
Adhesion of monocyte to HUVEC
DBMSC effect on THP-1 adhesion to HUVECs was examined using our previously published method [12]. Briefly, H2O2-untreated THP-1 or pretreated with 100 μM H2O2 (TTHP-1) for 24 h were cocultured with H2O2-untreated DBMSCs (TTHP-1/UDBMSC) or with H2O2-treated DBMSCs (TTHP-1/TDBMSC) for 24 h at 5:1 THP-1:DBMSC ratio in a physical contact experiment by adding THP-1 to DBMSCs that were initially cultured on a plastic surface of 6-well culture plate for 24 h to allow cells to be fully adhered (Fig. 2). After 24-h incubation in a complete RPMI-1640 culture medium (above), THP-1 were harvested and then labelled with 5 μM green fluorescent cell tracker stain (5-chloromethylfluorescin diacetate; CMFDA; Molecular Probes, Life Technologies) for 4 h as previously described [12]. Following washing THP-1 with fresh RPMI-1640 culture medium, they were added to a monolayer layer of HUVEC at a ratio of 5THP-1:1HUVEC (HUVEC were initially cultured alone or with 100 μM H2O2 for 24 h). After incubation for 30 min, non-adherent THP-1 were gently removed by washing with PBS, and the fluorescence intensity of the THP-1 that had adhered to the monolayer of HUVEC was then measured at excitation 485 nm and emission 528 nm using a fluorescence microplate reader (Glomax Multi Detection System, Promega, Germany). Results were expressed as relative fluorescence intensity (RFI). Different ratios of HUVEC to THP-1 were evaluated. Experiments were performed in triplicate using HUVEC prepared from independent umbilical cord tissue and repeated three times.
Measurement of glutathione reductase activity
HUVEC (HUVEC were initially co-cultured with DBMSCs and 100 μM H2O2 or cultured alone as described above) were washed twice with cold PBS, and they were then lysed as previously described [12, 13]. Total protein in the supernatant was then determined by Bradford method [12, 13].
The activity of glutathione reductase was measured using OxiSelect™ Glutathione Reductase Assay Kit (catalogue number STA-812, Cell Biolabs, San Diego, USA) as previously described by us [13]. This assay is based on the reduction of glutathione disulfide (oxidized glutathione) (GSSG) to reduced glutathione (GSH) by glutathione reductase, using NADPH as a donor for H. Subsequently, the chromogen reacts with the thiol group of GSH to produce a colored compound that absorbs at 405 nm. The glutathione reductase content in HUVEC samples is determined by comparison with the predetermined glutathione reductase standard curve. The assay was performed using 100-μl aliquots of HUVEC supernatant protein immediately after preparation (30 μg protein) added to phosphate buffer containing excess GSSG and NADPH. The level of change was determined at 405 nm using a standard curve performed. Three experiments were performed in triplicate using HUVEC and DBMSCs as indicated above.
Measurement of thioredoxin reductase activity
Total protein was extracted from HUVEC (prepared as described above), and thioredoxin reductase (TrxR) activity (catalogue number 10007892, Cayman, Michigan, USA) was then evaluated as per the manufacturer’s instructions. This assay is based on the reduction of 5,5′-dithiobis (2-nitrobenzoic) acid (DTNB) with NADPH to 5-thio-2-nitrobenzoic acid, which generates a strong yellow color that can be measured at 412 nm. In the crude biological sample, glutathione reductase and glutathione peroxidase can also be reduced by DTNB. Therefore, TrxR specific inhibitor is used to determine the specific activity of TrxR. Therefore, the total DTNB reduction by the sample is initially estimated and the DTNB reduction by the sample in the presence of the TrxR specific inhibitor will then be estimated. The difference between the two results is the DTNB reduction due to TrxR activity. Three experiments were performed in triplicate using HUVEC and DBMSCs as indicated above.
RNA isolation, cDNA synthesis, and real-time polymerase chain reaction (RT-PCR) analysis
The expression of 84 genes related to endothelial cell biology (catalogue number PAHS-015ZD-24, Qiagen, Hilden, Germany) by HUVEC was determined using QuantiTect Primer Assay (Qiagen, Hilden, Germany) in a real-time polymerase chain reaction (RT-PCR) as previously published [2]. Briefly, total RNA from HUVEC pretreated with DBMSCs and 100 μM H2O2 for 48 h was isolated, and cDNA was then synthesized using FastLane Cell cDNA kit and RT Primer Mix (Qiagen) as previously published [2]. After quantifying mRNA using QuantiTect SYBR Green PCR Kit (Qiagen), the real-time PCR reaction was performed in triplicate on the CFX96 real-time PCR detection system (BIO-RAD) as previously published [2]. To analyze the data, the CFX manager software (Bio-Rad, CA, USA) was used. The results were exported to Microsoft Excel for further analysis. The results were expressed as fold change by calculating the ΔΔ−2 values. The relative expression of an internal house-keeping gene as a loading control was used as provided in the kit. Experiments were performed in triplicate using HUVEC prepared from independent umbilical cord tissue and repeated three times.
Flow cytometry
Cells were characterized by flow cytometry as previously described [12]. Briefly, cells (1 × 105) were stained with monoclonal antibodies (Table 1) for 30 min. Cells were then washed twice by adding cold PBS and centrifuged at 150×g for 5 min at 8 °C. Unstained and isotype controls were used. Immunoreactivity to cell surface antibody markers or intracellular proteins was assayed by a BD FACS CANTO II (Becton Dickinson, NJ, USA) flow cytometer.
Statistical analysis
Data were analyzed using the t test (unpaired t test, two tailed). These analyses were performed using GraphPad Prism 5. Results were considered to be statistically significant if P < 0.05.
Results
Isolation and characterization of DBMSCs
MSCs from decidua basalis of human term placenta were previously isolated and characterized by us [2]. DBMSCs at passage 3 were positive (> 95%) for MSC markers and negative for hematopoietic markers and were able to differentiate into adipocytes, chondrocytes, and osteocytes as previously report [2]. Subsequently, DBMSCs at passage 3 were used in all experiments.
DBMSCs and H2O2 modulated the proliferation of HUVEC
To evaluate the effects of DBMSCs on endothelial cell functions, the proliferation of HUVEC cultured with DBMSCs in the presence or absence of 100 μM H2O2 was examined using the MTS assay. The viability HUVEC exposed to 100 μM H2O2 was more than 90% at all culture time points (24, 48, and 72 h). This was consistent with our previous report [13]. The exposure of HUVEC to concentrations higher than 100 μM H2O2 reduced their viability to less than 50%, as we previously reported [13]. Consequently, 100 μM H2O2 was used in this study.
The effects of H2O2 on HUVEC proliferation maintained throughout the culture times (24, 48, and 72 h), but the addition of DBMSCs (CMDBMSC and DBMSCs) significantly (P < 0.05) induced the effect of H2O2 on HUVEC proliferation after 48 h in culture at all examined concentrations of CMDBMSC and ratios of DBMSCs, respectively, as compared to untreated HUVEC or H2O2-treated HUVEC, P < 0.05 (Fig. 3a, b), and had no significant changes on the effect of H2O2 on HUVEC proliferation after 24 and 72 h in culture, P > 0.05. Consequently, the culture time used in this study was 48 h. HUVEC proliferation was also significantly increased in response to at all examined concentrations of CMDBMSC, P < 0.05 (Fig. 3a), and only at a high ratio of DBMSCs to HUVEC (1:1), P < 0.05 (Fig. 3b) as compared to untreated HUVEC. As compared to HUVEC cultured with CMDBMSC, HUVEC proliferation did not significantly change in response to H2O2 and CMDBMSCs, P > 0.05 (Fig. 3a). As compared to HUVEC cultured with low ratios of DBMSCs to HUVEC (1:5 and 1:10), HUVEC proliferation significantly increased in response to H2O2 and DBMSCs, P < 0.05 (Fig. 3b).
Fig. 3.

Proliferation of HUVEC measured by MTS. As compared to untreated HUVEC, the proliferation of HUVEC significantly increased in response to H2O2 alone or with different concentrations (1%, 5%, and 25%) of CMDBMSC in the presence of 100 μM H2O2 (a) and with different ratios of DBMSC to HUVEC (1:1, 1:5, and 1:10) in the presence of 100 μM H2O2 (b). As compared to HUVEC treated with H2O2 (HUVEC + H2O2), the proliferation of HUVEC significantly increased in response to different concentrations (1%, 5% and 25%) of CMDBMSC in presence of 100 μM H2O2 (a) and with different ratios of DBMSC to HUVEC (1:1, 1:5, and 1:10) in presence of 100 μM H2O2 (b). As compared to untreated HUVEC, HUVEC proliferation significantly increased in response to different concentrations (1%, 5%, and 25%) of CMDBMSC (a), and at a high ratio of DBMSC to HUVEC (1:1) (b). As compared to HUVEC cultured with CMDBMSC, HUVEC proliferation did not significantly change in response to H2O2 and CMDBMSCs, P > 0.05 (a). As compared to HUVEC cultured with low ratios of DBMSCs to HUVEC (1:5 and 1:10), HUVEC proliferation significantly increased in response to H2O2 and DBMSCs (b). *P < 0.05. Bars represent standard errors. Each experiment was performed in triplicate and repeated for five times with five independent preparations of DBMSCs and HUVEC
The effects of DBMSCs and H2O2 on HUVEC proliferation are reversible
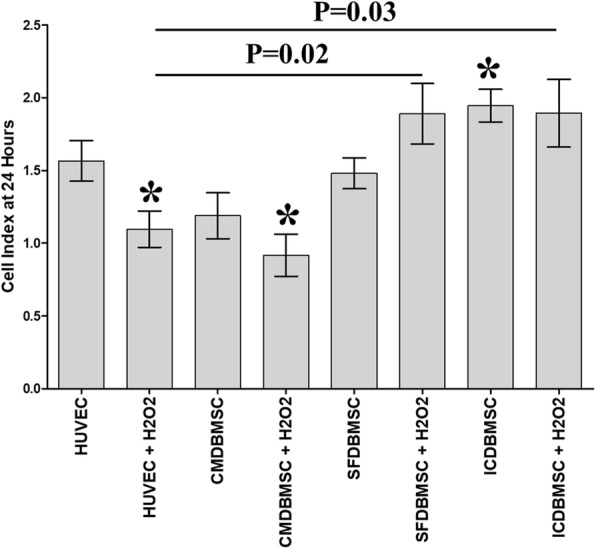
To evaluate the reversibility of DBMSC effects on the proliferation of H2O2-treated HUVEC, HUVEC were initially cultured with different treatments of DBMSCs in the presence of 100 μM H2O2 for 48 h and their proliferation was then determined using the xCELLigence system. After 24, 48, and 72 h, the proliferation of HUVEC pretreated with H2O2 (HUVEC + H2O2) and HUVEC pretreated with H2O2 and with different treatments of DBMSCs (CMDBMSC + H2O2, SFDBMSC + H2O2, and ICDBMSC + H2O2) significantly reduced as compared to untreated HUVEC, P < 0.05 (Fig. 4a–c). Similarly, after 24, 48, and 72 h, the proliferation of HUVEC pretreated with H2O2 and CMDBMSC (CMDBMSC + H2O2) significantly reduced as compared to HUVEC pretreated with H2O2, P < 0.05 (Fig. 4a–c). In contrast, after 48 and 72 h, and as compared to HUVEC pretreated with H2O2, the proliferation of HUVEC pretreated with H2O2 and SFDBMSC (SFDBMSC + H2O2) significantly increased (P < 0.05), but did not change significantly after 24 h, P > 0.05 (Fig. 4a–c). In addition, the culture with ICDBMSC (ICDBMSC + H2O2) did not significantly affect the proliferation of HUVEC pretreated with H2O2 as compared to HUVEC pretreated with H2O2 at all examined time points in culture, P > 0.05 (Fig. 4a–c).
Fig. 4.

The proliferation of HUVEC after removing the effects of DBMSCs and H2O2. HUVEC were initially cultured with DBMSCs and 100 μM H2O2 for 48 h and then used in a proliferation assay using the xCELLigence real-time cell analyzer. After 24 (a), 48 (b), and 72 (c) hours, the proliferation of HUVEC pretreated with H2O2 (HUVEC + H2O2) or with H2O2 and CMDBMSC (CMDBMSC + H2O2) or with H2O2 and SFDBMSC (SFDBMSC + H2O2) or with H2O2 and ICDBMSC (ICDBMSC + H2O2) significantly reduced as compared to untreated HUVEC. After 24, 48, and 72 h, the proliferation of HUVEC pretreated with H2O2 and CMDBMSC (CMDBMSC + H2O2) significantly reduced as compared to HUVEC pretreated with H2O2 (HUVEC + H2O2) (a–c). The proliferation of HUVEC pretreated with H2O2 and SFDBMSC (SFDBMSC + H2O2) significantly increased after 48 and 72 h as compared to H2O2-treated HUVEC (HUVEC + H2O2) (b, c), but did not change significantly after 24 h (P > 0.05) (a). The proliferation of HUVEC pretreated with H2O2 and ICDBMSC (ICDBMSC + H2O2) was not significantly changed (P > 0.05) as compared to H2O2-treated HUVEC (HUVEC + H2O2) (a–c). Each experiment was performed in triplicate and repeated with five independent HUVEC (passages 3–5) and DBMSC (passage 3) preparations. *P < 0.05. Bars represent standard errors
DBMSCs and H2O2 modulated HUVEC adhesion
To study the effects of DBMSCs on the adhesion of H2O2-treated HUVEC, HUVEC were initially cultured with different treatments of DBMSCs in the presence of 100 μM H2O2 for 48 h and their adhesion was then determined using the xCELLigence system. After 2 h, the adhesion of HUVEC pretreated with H2O2 (HUVEC + H2O2) and HUVEC pretreated with H2O2 and different treatments of DBMSCs (CMDBMSC + H2O2 and SFDBMSC+ H2O2) significantly increased as compared to untreated HUVEC, P < 0.05 (Fig. 5). In contrast, culturing with ICDBMSC (ICDBMSC + H2O2) decreased the adhesion of H2O2-treated HUVEC, but not significantly as compared to untreated HUVEC, P > 0.05 (Fig. 5). As compared to H2O2-treated HUVEC, the adhesion of H2O2-treated HUVEC cultured with SFDBMSC and ICDBMSC (SFDBMSC+ H2O2 and ICDBMSC+ H2O2) significantly increased and reduced, respectively (P < 0.05) (Fig. 5). In contrast, culturing with CMDBMSC (CMDBMSC + H2O2) had no significant changes on the adhesion of H2O2-treated HUVEC as compared to H2O2-treated HUVEC cultured alone, P > 0.05 (Fig. 5).
Fig. 5.
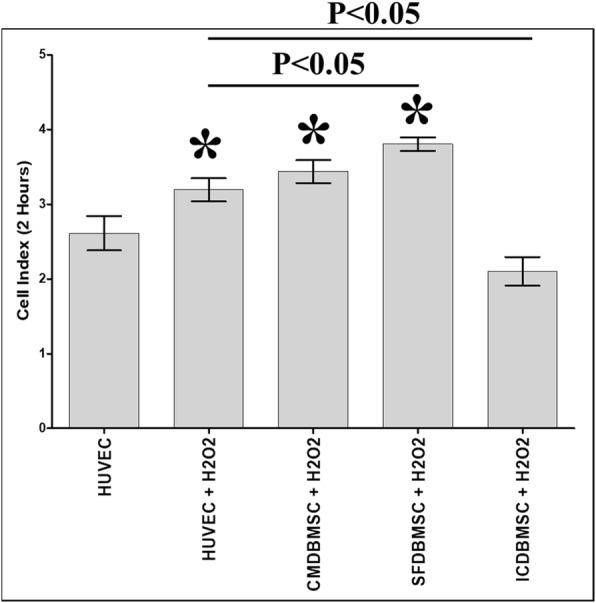
The adhesion of HUVEC after removing the effects of DBMSCs and H2O2. HUVEC were initially cultured with DBMSC with 100 μM H2O2 for 48 h and then used in an adhesion assay using the xCELLigence real-time cell analyzer. After 2 h, the adhesion of HUVEC pretreated with H2O2 alone (HUVEC + H2O2) or with H2O2 and CMDBMSC (CMDBMSC + H2O2) or with H2O2 and SFDBMSC (SFDBMSC + H2O2) significantly increased as compared to untreated HUVEC while the adhesion of HUVEC pretreated with H2O2 and ICDBMSC (ICDBMSC + H2O2) reduced, but not significantly (P > 0.05). After 2 h and as compared with HUVEC pretreated with H2O2 (HUVEC + H2O2), the adhesion of HUVEC pretreated with H2O2 and SFDBMSC (SFDBMSC + H2O2) or ICDBMSC (ICDBMSC + H2O2) significantly increased and decreased, respectively. Each experiment was performed in triplicate and repeated with five independent HUVEC (passages 3–5) and DBMSC (passage 3) preparations. *P < 0.05. Bars represent standard errors
DBMSCs and H2O2 modulated HUVEC migration
To further study the effect of DBMSCs and H2O2 on the migration of endothelial cells, HUVEC were initially cultured with different treatment of DBMSCs in the presence of 100 μM H2O2 (CMDBMSC + H2O2, SFDBMSC + H2O2, and ICDBMSC + H2O2) or in the absence of H2O2 (CMDBMSC, SFDBMSC, and ICDBMSC) for 48 h and then re-cultured in a 16-well migration culture plate and monitored using the xCELLigence system. After 24 h, the migration of H2O2-treated HUVEC (HUVEC + H2O2) cultured alone or with CMDBMSC (ICDBMSC + H2O2) significantly reduced as compared to untreated HUVEC, P < 0.05 (Fig. 6). In contrast, the migration of H2O2-treated HUVEC cultured with SFDBMSC and ICDBMSC (SFDBMSC + H2O2 and ICDBMSC + H2O2) did not change significantly as compared to untreated HUVEC (P > 0.05), but significantly increased as compared to H2O2-treated HUVEC, P < 0.05 (Fig. 6). The incubation with CMDBMSC (CMDBMSC+ H2O2) reduced the migration of H2O2-treated HUVEC, but not significantly (P > 0.05) as compared to H2O2-treated HUVEC (Fig. 6). After 24 h, the migration of HUVEC treated with CMDBMSC or SFDBMSC did not significantly change as compared to untreated HUVEC, P > 0.05 (Fig. 6). In contrast, the migration of HUVEC treated with ICDBMSC significantly increased as compared to untreated HUVEC, P < 0.05 (Fig. 6). As compared to HUVEC treated with CMDBMSC, SFDBMSC, or ICDBMSC, the migration of HUVEC treated with H2O2 in the presence of CMDBMSC or SFDBMSC or ICDBMSC did not significantly change, P > 0.05 (Fig. 6).
Fig. 6.
The migration of HUVEC after removing the effects of DBMSCs and H2O2. HUVEC were initially cultured with DBMSC with 100 μM H2O2 for 48 h and then used in a migration assay using the xCELLigence real-time cell analyzer. After 24 h, the migration of HUVEC pretreated with H2O2 alone (HUVEC + H2O2) or with H2O2 and CMDBMSC (CMDBMSC + H2O2) significantly reduced as compared to untreated HUVEC while the migration of HUVEC pretreated with H2O2 and with SFDBMSC (SFDBMSC + H2O2) or H2O2 and ICDBMSC (ICDBMSC + H2O2) increased, but not significantly (P > 0.05). After 24 h, the migration of HUVEC pretreated with H2O2 and with SFDBMSC (SFDBMSC + H2O2) or with ICDBMSC (ICDBMSC + H2O2) significantly increased as compared to HUVEC pretreated with H2O2 (HUVEC + H2O2). After 24 h and as compared to untreated HUVEC, the migration of HUVEC treated with CMDBMSC or SFDBMSC did not significantly change, P > 0.05. In contrast, the migration of HUVEC treated with ICDBMSC significantly increased as compared to untreated HUVEC after 24 h. After 24 h and as compared to HUVEC treated with CMDBMSC, SFDBMSC, or ICDBMSC, the migration of HUVEC treated with H2O2 in the presence of CMDBMSC or SFDBMSC or ICDBMSC did not significantly change, P > 0.05. Each experiment was performed in triplicate and repeated with five independent HUVEC (passages 3–5) and DBMSC (passage 3) preparations. *P < 0.05. Bars represent standard errors
DBMSCs reduced the stimulatory effect of monocytes and H2O2 on HUVEC proliferation
To study the effect of DBMSCs on the interaction between monocytes and H2O2 (THP-1 + H2O2) on the proliferation of HUVEC, the effect of DBMSCs on the proliferation of monocytes in the presence of H2O2 (THP-1:DBMSC + H2O2) was first examined using the MTS assay. Then, the proliferation of HUVEC in response to monocytes pre-cultured with DBMSCs and H2O2 (HUVEC:THP-1/DBMSC + H2O2) was also evaluated. After 24-h culture with DBMSCs, the proliferation of monocytes cultured with H2O2 and DBMSCs (THP-1:DBMSC + H2O2) at all examined ratios of monocytes and DBMSCs significantly increased, P < 0.05 (Fig. 7a) as compared to monocytes cultured alone or with H2O2 (THP-1 + H2O2). This stimulatory effect of DBMSCs on the proliferation of monocytes is reversible and time dependent (data not shown). The effect of DBMSCs on the proliferation of monocytes (THP-1) alone was also evaluated. After 24-h culture with DBMSCs, THP-1 proliferation at all examined ratios of monocytes and DBMSCs significantly increased, P < 0.05 (Fig. 7a) as compared to monocytes cultured alone. Following the addition of H2O2 to monocyte cultured with DBMSCs, monocyte proliferation did not significantly change as compared to monocytes cultured with H2O2 alone, P > 0.05 (Fig. 7a).
Fig. 7.

The proliferation of monocytes (THP-1) and HUVEC evaluated by the MTS assay. After 24 h and as compared to untreated THP-1, the proliferation of THP-1 in the presence of H2O2 and DBMSCs (THP-1/DBMSC + H2O2) significantly increased at different THP-1 and DBMSC ratios (20:1, 10:1, 5:1, and 2.5:1) (a). After 24 h and as compared with untreated HUVEC, the proliferation of H2O2-treated HUVEC (HUVEC + H2O2) and H2O2-treated HUVEC in the presence of THP-1 pretreated with H2O2 (THP-1 + H2O2) at 1:10 HUVEC:THP-1 ratio significantly increased (b). The proliferation of H2O2-treated HUVEC in the presence of THP-1 pretreated with H2O2 and DBMSCs (THP-1/DBMSC + H2O2) significantly reduced as compared with H2O2-treated HUVEC cultured with THP-1 pretreated with H2O2 (b). After 24 h culture with DBMSCs and as compared to monocytes cultured alone, the proliferation of THP-1 significantly increased at all examined ratios of monocytes and DBMSCs (a). As compared to monocytes cultured with H2O2 alone, the proliferation of monocyte cultured with DBMSCs did not significantly change, P > 0.05 (a). Each experiment was performed in triplicate and repeated for five times with five independent preparations of DBMSCs and HUVEC. *P < 0.05. Bars represent standard errors
After 24 h, the proliferation of HUVEC cultured in H2O2 significantly increased after the addition of a high ratio of monocytes pretreated with H2O2 (THP-1 + H2O2) to HUVEC (1HUVEC:10THP-1 + H2O2), and this stimulatory effect of monocytes pretreated with H2O2 on HUVEC proliferation was significantly reduced (P < 0.05) by monocytes pretreated with DBMSCs and H2O2 (THP-1/DBMSC + H2O2), P < 0.05 (Fig. 7b). This stimulatory effect of monocytes pretreated with H2O2 (THP-1 + H2O2) on the proliferation of HUVEC cultured in H2O2 and the inhibitory effect of DBMSCs on monocytes pretreated with H2O2 (THP-1/DBMSC + H2O2) inducing the proliferation of HUVEC cultured in H2O2 are reversible and time dependent (data not shown). The effects of DBMSCs on the proliferative responses of HUVEC cultured with or without H2O2 and monocytes pretreated with or without H2O2 were not significantly changed, P > 0.05 (data not shown).
DBMSCs reduced the adhesion of H2O2-treated monocytes to HUVEC
To evaluate the effect of DBMSCs on the adhesion of monocytes to endothelial cells, monocytes pretreated with H2O2 (TTHP-1) were cultured with H2O2-untreated DBMSCs (UDBMSC) or DBMSC pretreated with H2O2 (TDBMSC) for 24 h, and THP-1 were then harvested and labelled with the green fluorescent dye CMFDA and added to endothelial cells (HUVEC were initially cultured alone or with 100 μM H2O2 for 24 h) in an adhesion assay. Results showed that the adhesion of monocytes pretreated with H2O2 to endothelial cells (HUVEC were initially cultured alone or with H2O2) was significantly reduced after culturing with UDBMSC or TDBMSC, P < 0.05 (Fig. 8a, b). There was no difference in the adhesion of H2O2-untreated monocytes and H2O2-treated monocytes to endothelial cells by DBMSCs (data not shown).
Fig. 8.

The adhesion of monocytes (THP-1) to HUVEC was evaluated by measuring THP-1 fluorescence intensity using a fluorescence microplate reader. THP-1 were initially cultured alone (THP-1) or with 100 μM H2O2 (TTHP-1) for 24 h and then cultured with DBMSCs (5:1 THP-1:DBMSC ratio) that were initially cultured alone (UDBMSC) or with 100 μM H2O2 (TDBMSC) for 24 h. After 24 h culture, THP-1 were labelled with 5 μM green fluorescent cell tracker stain CMFDA and added to HUVEC monolayer (HUVEC were initially cultured with or without 100 μM H2O2 for 24 h). As compared to untreated THP-1, the adhesion of TTHP-1 to H2O2 untreated HUVEC significantly increased while the adhesion of TTHP-1/UDBMSCs and TTHP-1/TDBMSC to H2O2 untreated HUVEC significantly reduced after 30 min (a). As compared to TTHP-1, the adhesion of TTHP-1/UDBMSCs and TTHP-1/TDBMSC to H2O2 untreated HUVEC significantly reduced after 30 min (a). As compared to untreated THP-1, the adhesion of TTHP-1 to H2O2 pretreated HUVEC was not significantly changed (P > 0.05) after 30 min while the adhesion of TTHP-1/UDBMSCs and TTHP-1/TDBMSC to H2O2 pretreated HUVEC significantly reduced after 30 min as compared to untreated THP-1 and TTHP-1 (b). Each experiment was performed in triplicate and repeated for five times with five independent preparations of DBMSCs and HUVEC. *P < 0.05. Bars represent standard errors
Next, we studied that the inhibitory effects of DBMSCs on the adhesion of monocytes pretreated with H2O2 to endothelial cells were mediated by modulating the expression of adhesion molecules by monocytes pretreated with H2O2. A range of adhesion molecules were studied by flow cytometry and expression recorded as median fluorescence intensity or as a percentage of cells. After 24 h, the expression of ICAM-1 in monocytes pretreated with H2O2 (TTHP-1) was significantly increased as compared to H2O2-untreated monocytes (THP-1), and culturing with DBMSCs (TTHP-1 + DBMSC) significantly increased ICAM-1 expression in TTHP-1, P < 0.05 (Fig. 9a). Similarly, the expression of CD44 in TTHP-1 and TTHP-1 + DBMSC was significantly increased as compared to THP-1 (P < 0.05), and there was no difference in CD44 expression between TTHP-1 and TTHP-1 + DBMSC (Fig. 9c). In contrast, the expression of VCAM-1 in TTHP-1was not significantly changed as compared to THP-1 (P < 0.05), but the culture with DBMSCs (TTHP-1 + DBMSC) significantly decreased VECAM-1 expression in TTHP-1 (Fig. 9b).
Fig. 9.

The flow cytometric analysis of monocytes (THP-1) expression of ICAM-1, VCAM-1, and CD44. THP-1 were initially cultured with 100 μM H2O2 (TTHP-1) for 24 h and then cultured with DBMSCs at 5:1 THP-1:DBMSC ratio (TTHP-1/DBMSC). The flow cytometry was then evaluated after 24 h culture. The expression of ICAM-1 by TTHP-1 and by TTHP-1/DBMSC significantly increased as compared with H2O2-untreated THP-1 (a). As compared to TTHP-1, the expression of ICAM-1 by TTHP-1/DBMSC significantly increased (a). As compared with untreated THP-1, TTHP-1 expression of VCAM-1 was not significantly changed (P > 0.05) while significantly increased by TTHP-1/DBMSC (b). As compared with TTHP-1, TTHP-1/ DBMSC expression of VCAM-1 significantly increased (b). TTHP-1 and TTHP-1/DBMSC expression of CD44 significantly increased as compared to untreated THP-1 while TTHP-1/DBMSC expression of CD44 was not significantly changed as compared with TTHP-1 (c). Each experiment was performed in triplicate and repeated for five times with five independent preparations of DBMSCs and HUVEC. *P < 0.05. Bars represent standard errors
DBMSCs increased the activities of glutathione and thioredoxin reductases in H2O2-treated HUVEC
Cells are protected from injury induced by oxidative stress by employing several antioxidant defense mechanisms [14]. To address the possibility that DBMSCs can protect endothelial cells from injury induced by H2O2, we examined the effect of DBMSCs on the activities of glutathione and thioredoxin reductases (an antioxidant enzyme) in H2O2-treated endothelial cells.
Glutathione reductase activity in H2O2-treated HUVEC cultured with CMDMSC, SFDBMSC, and ICDBMSC is significantly higher than H2O2-treated HUVEC (P < 0.05). At baseline, the level of glutathione reductase in HUVEC was 77.5 mU/mL ± 4.87 mU/mL. Exposure of HUVEC to 100 μM H2O2 for 48 h, the level of glutathione reductase was 29.50 ± 2.55. The level of glutathione reductase levels in H2O2-treated endothelial cell cultured with CMDBMSC, SFDBMSC, and ICDBMSC were 56.90 mU/mL ± 3.11 mU/mL, 62.38 mU/mL ± 3.01 mU/mL, and 65.49 mU/mL ± 3.95 mU/mL, respectively. As compared to H2O2-treated HUVEC, there were an approximately 1.92-, 2.11-, and 2.22-fold increase in the levels of glutathione reductase in H2O2-treated HUVEC cultured with CMDMSC, SFDBMSC, and ICDBMSC, respectively, P < 0.05.
Thioredoxin reductase activity in H2O2-treated HUVEC cultured with CMDMSC, SFDBMSC, and ICDBMSC is significantly higher than H2O2-treated HUVEC (P < 0.05). At baseline, the activity of thioredoxin reductase was 91.67 ± 10.14 mU/106 cell. Exposure of HUVEC to 100 μM H2O2 for 48 h, the activity of the enzyme was reduced to 43.33 ± 10.73 mU/106 cell. The activity of thioredoxin reductase in H2O2-treated endothelial cell cultured with CMDBMSC, SFDBMSC, and ICDBMSC was 79.33 ± 7.21 mU/106 cell, 88.33 ± 9.28 mU/106 cell, and 83.33 ± 9.28 mU/106 cell, respectively. As compared to H2O2-treated HUVEC, there were an approximately 1.83-, 2.03-, and 1.92-fold increase in the activity of thioredoxin reductase in H2O2-treated HUVEC cultured with CMDBMSC, SFDBMSC, and ICDBMSC, respectively. These data suggest that culturing H2O2-treated HUVEC with DBMSCs can protect endothelial cells from oxidative stress induced by H2O2.
DBMSCs modulated the effect of H2O2 on the expression of genes important in endothelial cell functions
The expression of genes mediating endothelial cell functions was studied after culturing endothelial cells with H2O2 in the presence or absence of DBMSCs for 48 h and then analyzed and assessed using the real-time PCR assay. Results show that DBMSCs modulated H2O2 effects on endothelial cell expression of genes underlying many of endothelial cell functional activities including survival, apoptosis, injury, fibrosis formation, inflammation, angiogenesis, permeability, thrombus formation, and leukocyte adhesion as well as infiltration as compared to untreated endothelial cells (Tables 2, 3, 4, 5, and 6).
Table 2.
DBMSCs modulate the expression of genes involved in endothelial cell (EC) survival, apoptosis, injury, fibrosis formation, and inflammation. THUVEC (HUVEC were cultured with 100 μM H2O2 for 48 h). TDBMSC (HUVEC were cultured with DBMSC and 100 μM H2O2 for 48 h)
| # | Gene symbol | Gene full name | THUVEC mean ΔΔ−2 values | TDBMSC Mean ΔΔ−2 values | Fold change (TDBMSC Vs. THUVEC) P < 0.05 |
Biological activities |
|---|---|---|---|---|---|---|
| 1 | BCL2 | B-cell Lymphoma 2 | 2 | 23 | 11.5-fold ↑ | Induce EC survival |
| 2 | EDN1 | Endothelin-1 | 12 | 82 | > 6.64-fold ↑ | |
| 3 | EDNRA | Endothelin-1 (ET-1) Receptor A | 3 | 9 | 3-fold ↑ | |
| 4 | HMOX1 | Heme Oxygenase-1 | 7 | 158 | 20-fold ↑ | |
| 5 | KDR | Vascular Endothelial Growth Factor Receptor 3 (VEGFR3) | 2 | 52 | 22.57-fold ↑ | |
| 6 | MMP2 | Matrix Metallopeptidase 2 | 9 | 36 | 4-fold ↑ | |
| 7 | SPHK1 | Sphingosine Kinase 1 | 44 | 840,958 | 19112.68-fold ↑ | |
| 8 | MMP9 | Matrix Metallopeptidase 9 | 1 | 6 | 6-fold ↑ | |
| 9 | PECAM1 | Platelet endothelial cell adhesion molecule | 1090 | 7242 | 6.64-fold ↑ | |
| 10 | TNFSF10 | TNF-related apoptosis-inducing ligand (TRAIL) | 7 | 30 | > 4.20-fold ↑ | |
| 11 | TYMP | Thymidine Phosphorylase | 5 | 27 | > 5-fold ↑ | |
| 12 | FAS | Fas cell surface death receptor | 3 | 0.76 | > 3-fold ↓ | Induce EC apoptosis |
| 13 | PF4 | Platelet Factor 4 | 21 | 0.69 | > 30-fold ↓ | |
| 14 | HIF1α | Hypoxia-Inducible Factor-1α | 159 | 11 | > 14-fold ↓ | Induce EC injury |
| 15 | SERPINE1 (PAI-1) | Type 1 plasminogen activator inhibitor | 997 | 248 | 4-fold ↓ | |
| 16 | TIMP1 | TIMP Metallopeptidase Inhibitor 1 | 3 | 1 | 3-fold ↓ | |
| 17 | ACE | Angiotensin I Converting Enzyme | 15,765 | 5 | 3153-fold ↓ | |
| 18 | F2R | Coagulation Factor II Thrombin Receptor | 20 | 1 | 20-fold ↓ | Induce EC inflammation |
| 19 | ADAM17 | A disintegrin and Metalloprotease 17 | 256 | 4 | 64-fold ↓ | |
| 20 | FLT1 | Vascular Endothelial Growth Factor Receptor 1 | 23 | 12 | 1.91-fold ↓ | |
| 21 | TNF-α | Tumor Necrosis Factor-α | 591 | 2 | > 295-fold ↓ | |
| 22 | PTK2 | PTK2 protein tyrosine kinase 2 (PTK2) or focal adhesion kinase (FAK), | 8 | 2 | 4-fold ↓ | Inhibits inflammation and fibrosis |
| 23 | PLG | Plasminogen | 8 | 90 | > 11-fold ↑ | Inhibits inflammation and, promotes fibrin clearance |
| 24 | F3 | Coagulation Factor III, Tissue Factor | 30 | 0.06 | 500-fold ↓ | Induces EC injury by induction of fibrin and thrombus formation |
| 25 | THBD | Thrombomodulin | 1013 | 1 | 1013-fold ↓ | |
| 26 | THBS1 | Thrombospondin 1 (TSP-1) | 1097 | 3 | > 365-fold ↓ | Induces EC inflammation and injury |
| 27 | TFPI | Tissue Factor Pathway Inhibitor | 11 | 23 | ~ 2-fold ↑ | Inhibits EC injury by reducing thrombus formation |
Table 3.
DBMSCs modulate the expression of genes mediating endothelial cell (EC) angiogenesis and migration. THUVEC (HUVEC were cultured with 100 μM H2O2 for 48 h). TDBMSC (HUVEC were cultured with DBMSC and 100 μM H2O2 for 48 h)
| # | Gene symbol | Gene full name | THUVEC mean ΔΔ−2 values | TDBMSC mean ΔΔ−2 values | Fold change (TDBMSC Vs. THUVEC) P < 0.05 |
Biological activities |
|---|---|---|---|---|---|---|
| 1 | CAV1 | Caveolin-1 | 12 | 1472 | > 122-fold ↑ | Inhibit EC angiogenesis |
| 2 | VWF | von Willebrand Factor | 4 | 693 | > 173-fold ↑ | |
| 3 | AGT | Angiotensinogen | 1 | 1018 | 1018-fold ↑ | |
| 4 | CASP3 | Caspases 3 | 24 | 177 | > 7-fold ↑ | |
| 5 | BAX | Bcl-2-associated X | 4 | 27 | 6.75-fold ↑ | |
| 6 | F2R | Coagulation Factor II Thrombin Receptor | 20 | 1 | 20-fold ↓ | Induce EC angiogenesis |
| 7 | FGF1 | Fibroblast Growth Factor 1 | 3384 | 0.12 | 28,200-fold ↓ | |
| 8 | FGF2 | Fibroblast Growth Factor 2 | 14 | 4 | 3.5-fold ↓ | |
| 9 | KIT | Tyrosine Protein Kinase Kit or CD117 | 122,521 | 0.74 | > 165568-fold ↓ | |
| 10 | PTGIS | Prostacyclin Synthase | 7 | 2 | 3.5-fold ↓ | |
| 11 | PTGS2 | Cyclooxygenase (COX) | 15 | 2 | 7.5-fold ↓ | |
| 12 | SELPLG | P-selectin glycoprotein ligand-1 | 16 | 4 | 4-fold ↓ | |
| 13 | TEK | EK Receptor Tyrosine Kinase (TIE-2) | 67 | 20 | 3.35-fold ↓ | |
| 14 | VEGFA | Vascular Endothelial Growth Factor A | 2919 | 0.52 | > 5613-fold ↓ | |
| 15 | CFLAR | CASP8 and FADD like apoptosis regulator | 530 | 85 | > 6-fold ↓ | |
| 16 | EDN1 | Endothelin-1 | 12 | 82 | 6.83-fold ↑ | Induce EC migration |
| 17 | SPHK1 | Sphingosine Kinase 1 | 44 | 840,958 | 19112.68-fold ↑ | |
| 18 | HMOX1 | Heme Oxygenase-1 | 7 | 158 | 22.57-fold ↑ |
Table 4.
DBMSCs modulate the expression of genes mediating endothelial cell (EC) permeability. THUVEC (HUVEC were cultured with 100 μM H2O2 for 48 h). TDBMSC (HUVEC were cultured with DBMSC and 100 μM H2O2 for 48 h)
| # | Gene Symbol | Gene full name | THUVEC Mean ΔΔ−2 values | TDBMSC Mean ΔΔ−2 values | Fold change (TDBMSC Vs. THUVEC) P < 0.05 |
Biological activities |
|---|---|---|---|---|---|---|
| 1 | ACE | Angiotensin I Converting Enzyme | 15,765 | 5 | 3153-fold ↓ | Induce EC permeability |
| 2 | ADAM17 | A disintegrin and Metalloprotease 17 | 256 | 4 | 64-fold ↓ | |
| 3 | IL1β | Interleukin 1 beta | 4 | 0.46 | 8.69-fold ↓ | |
| 4 | IL6 | Interleukin 6 | 194 | 59 | > 3-fold ↓ | |
| 5 | VEGFA | Vascular Endothelial Growth Factor A | 2919 | 0.52 | > 5613-fold ↓ | |
| 6 | CAV1 | Caveolin-1 | 12 | 1472 | > 122-fold ↑ | Inhibit EC permeability |
| 7 | NPR1 | Natriuretic Peptide Receptor A/ Guanylate Cyclase A (Atrionatriuretic Peptide Receptor A) | 1 | 12 | 12-fold ↑ |
Table 5.
DBMSCs modulate the expression of genes mediating leukocyte infiltration of endothelial cells (EC), adhesion of inflammatory cells and monocyte adhesion and transmigration. THUVEC (HUVEC were cultured with 100 μM H2O2 for 48 h). TDBMSC (HUVEC were cultured with DBMSC and 100 μM H2O2 for 48 h)
| # | Gene symbol | Gene full name | THUVEC mean ΔΔ−2 values | TDBMSC mean ΔΔ−2 values | Fold change (TDBMSC Vs. THUVEC) P < 0.05 |
Biological activities |
|---|---|---|---|---|---|---|
| 1 | CDH5 | VE-Cadherin (Vascular Endothelial Cadherin) | 61 | 3 | ~ 20-fold ↓ | Induce leukocyte infiltration |
| 2 | SELE | E- selectin | 21 | 1 | 21-fold ↓ | |
| 3 | VCAM1 | Vascular Cell Adhesion Molecule 1 | 7 | 0.26 | 26.92-fold ↓ | |
| 4 | PLG | Plasminogen | 8 | 90 | ~ 11-fold ↑ | Inhibits adhesion of inflammatory cells |
| 5 | THBS1 | Thrombospondin 1 (TSP-1) | 1097 | 3 | > 365-fold ↓ | Induces monocyte adhesion and transmigration |
Table 6.
DBMSCs effects on genes involved in endothelial cell (EC) biology. THUVEC (HUVEC were cultured with 100 μM H2O2 for 48 h). TDBMSC (HUVEC were cultured with DBMSC and 100 μM H2O2 for 48 h)
| # | Gene symbol | Gene full name | THUVEC mean ΔΔ−2 values | TDBMSC mean ΔΔ−2 values | Fold change (TDBMSC Vs. THUVEC) P < 0.05 |
|---|---|---|---|---|---|
| 1 | ANGPT1 | Angiopoietin 1 | 1.35 | 0.64 | Fold change is not statically significant, P > 0.05 |
| 2 | AGTR1 | Angiotensin II Receptor Type 1 | 0.13 | 0.05 | |
| 3 | ALOX5 | Arachidonate 5-Lipoxygenase | 0.61 | 0.36 | |
| 4 | ANXA5 | Annexin A5 | 9 | 5 | |
| 5 | APOE | Apolipoprotein E | 0.01 | 0.01 | |
| 6 | BCL2L1 | BCL2L1 | 11 | 10 | |
| 7 | CALCA | Calcitonin Related Polypeptide Alpha | 0.71 | 0.70 | |
| 8 | CCL2 | C-C motif chemokine ligand 2 | 1 | 0.39 | |
| 9 | CX3CL1 | C-X3-C Motif Chemokine Ligand 1 | 0.50 | 0.34 | |
| 10 | EDN2 | Endothelin 2 | 0.01 | 0.01 | |
| 11 | FASLG | Fas Ligand | 0.39 | 2.54 | |
| 12 | FN1 | Fibronectin 1 | 0.06 | 0.04 | |
| 13 | ICAM1 | Intercellular Adhesion Molecule 1 | 1 | 0.30 | |
| 14 | IL3 | Interleukin 3 | 0.66 | 1.47 | |
| 15 | IL7 | Interleukin 7 | 0.10 | 2.81 | |
| 16 | KLK3 | Kallikrein Related Peptidase 3 | 1 | 2 | |
| 17 | MMP1 | Matrix Metallopeptidase 1 | 3 | 3 | |
| 18 | NOS3 | Nitric Oxide Synthase 3 | 1 | 0.92 | |
| 19 | NPPB | Natriuretic Peptide B | 0.80 | 0.32 | |
| 20 | OCLN | Occludin | 4.99 | 1.48 | |
| 21 | PDGFRA | Platelet Derived Growth Factor Receptor Alpha | 1.06 | 0.26 | |
| 22 | PGF | Placental Growth Factor | 1.40 | 2.96 | |
| 23 | PLAT | Plasminogen Activator, Tissue Type | 3.73 | 0.34 | |
| 24 | PLAU | Plasminogen Activator, Urokinase | 1.59 | 2 | |
| 25 | SELL | Selectin L | 0.16 | 0.09 | |
| 26 | SOD1 | Superoxide Dismutase 1 | 17 | 17 | |
| 27 | TGFB1 | Transforming Growth Factor Beta 1 | 9 | 7 | |
| 28 | ENG | Endoglin | 19 | 16 |
Discussion
We previously reported that DBMSCs protect endothelial cell activation by reducing the adhesion of monocytes to endothelial cells and their stimulatory effect on the proliferation of endothelial cells [2, 12]. These two events are the basis of endothelial cell injury in inflammatory diseases, such as atherosclerosis [12]. Inflammatory diseases are also associated with high level of oxidative stress mediators, such as H2O2 [15–19]. Recently, we reported the ability of DBMSCs to survive and function under the stress of H2O2 [20]. In addition, DBMSCs inhibit the angiogenesis of endothelial cells in H2O2 environment [20]. Therefore, DBMSCs have the potential to be used as a cell-based therapy for the treatment of inflammatory diseases. In this study, we investigated the ability of DBMSCs to protect endothelial cell functions from stress induced by both H2O2 and monocytes.
First, we determined the effect of DBMSCs on endothelial cell function under H2O2. DBMSCs significantly induced the stimulatory effect of H2O2 on endothelial cell proliferation (Fig. 3a, b). This contrasts with our recent finding that MSCs from the chorionic villi of human placentae (pMSCs) reverse the proliferative effect of H2O2 on endothelial cells [13]. This discrepancy can be attributed to the niche of both DBMSCs and pMSCs. During normal pregnancy, DBMSCs are located in the decidua where they are in a continuous exposure to high levels of oxidative stress mediators, because they are in a closed proximity to the maternal vessels [21, 22] while pMSCs are usually exposed to a lower levels of oxidative stress mediators because they are in a continuous contact with the fetal circulation [6, 7]. Therefore, DBMSCs have possibly acquired characteristics similar to H2O2 on the functions of endothelial cells.
Next, we demonstrated that the stimulatory effects of DBMSCs and H2O2 on endothelial cell proliferation are reversible (Fig. 4). However, the paracrine communication between DBMSCs and endothelial cells in the presence of H2O2 showed more stimulatory effect on endothelial cell proliferation than CMDBMSC (molecules produced by unstimulated DBMSCs) and ICDBMSC (intercellular direct contact), Fig. 4b, c. We also showed that DBMSCs may protect endothelial cells from oxidative stress by the finding that DBMSCs can induce the expression of many genes mediating the survival of endothelial cells and can also reduce the expression of genes that trigger apoptosis, injury, and inflammation in endothelial cells (Table 2) [23–48]. This protective role for DBMSCs on endothelial cells from stress induced by H2O2 is further confirmed by the ability of DBMSCs to increase the activities of glutathione and thioredoxin reductases (antioxidant enzymes) in H2O2-treated endothelial cells. Therefore, DBMSCs can protect endothelial cells from stress induced by H2O2, therefore suggesting a therapeutic potential for DBMSCs in inflammatory diseases.
We also found that ICDBMSC can reduce the stimulatory effect of H2O2 on endothelial cell adhesion (Fig. 5). In contrast, CMDBMSC and SFDBMSC could not reverse the stimulatory effect of H2O2 on endothelial cell adhesion (Fig. 5). Instead, SFDBMSC induced the stimulatory effect of H2O2 on the adhesiveness of endothelial cells. H2O2 is known to increase the adhesiveness property of endothelial cells through ICAM-1 and VCAM-1 [49–52]. DBMSCs secrete IL-1β and IL-10 [2]. IL-1β is a proinflammatory cytokine that induces endothelial cell production of H2O2 while IL-10 is an anti-inflammatory cytokine that reduces endothelial cell production of H2O2 [53, 54]. This may explain why DBMSCs exert dual functions on endothelial cell adhesion depending on the nature of DBMSC treatment. Therefore, paracrine communication with endothelial cells may stimulate DBMSC production of IL1-β while the intercellular direct contact with endothelial cells may stimulate DBMSC production of IL-10. The interaction with DBMSCs (ICDBMSC) reduced endothelial cell expression of VCAM (Table 5), thus suggesting that VCAM may mediate the anti-adhesive effect of DBMSC on endothelial cells. Our data highlight that DBMSCs have dual effects “a double-edged sword” on endothelial cells as it was previously reported for the immunomodulatory properties of bone marrow-derived MSCs [55]. However, a future study is essential to reveal this mechanism.
DBMSCs show also dual effects on endothelial cell migration. DBMSCs (SFDBMSC and ICDBMSC) reversed the inhibitory effect of H2O2 on endothelial cell migration while CMDBMSC enhanced the inhibitory effect of H2O2 on the migration of endothelial cells (Fig. 6). In this study, we found that DBMSCs induced the expression of a number of genes (e.g., endothelin-1, sphingosine kinase 1, and heme oxygenase-1) by H2O2-treated endothelial cells. These genes mediate the migration of endothelial cells [56–58], thus suggesting that these genes may mediate the stimulatory effect of DBMSCs on endothelial cell migration.
Adhesion and migration are the early steps towards endothelial cell angiogenesis [12]. Recently, we showed that DBMSCs inhibit H2O2-treated endothelial cell angiogenesis [20]. In this study, we found that DBMSCs increased and decreased H2O2-treated endothelial cell expression of various antiangiogenic [25, 59–63] and proangiogenic [24, 39, 64–70] genes, respectively (Table 3). These data suggest that these genes may mediate DBMSC inhibitory effect on endothelial cell angiogenesis. However, future functional studies are essential to elucidate the roles of these genes in the antiangiogenic properties of DBMSCs.
We previously reported that DBMSCs have an inhibitory effect on monocyte induction of endothelial cell proliferation and on their adhesion to endothelial cells [12]. In this study, we also show that in the presence of H2O2, DBMSCs inhibit the adhesion of monocytes to endothelial cells (Fig. 8) and also inhibit endothelial cell proliferation (Fig. 7b). This further confirms the protective role of DBMSCs on endothelial cells proliferation (discussed above) from oxidative stress. These data indicate that DBMSCs have the ability to reduced endothelial cell proliferation, a pathological phenomenon that is known to contribute to the formation of atheroma plaque in atherosclerosis [71].
DBMSCs reduced monocyte expression of VCAM-1 (Fig. 9b), thus indicating that this adhesion molecule may mediate monocyte adhesion to endothelial cells. We also found that DBMSCs modulated H2O2-treated endothelial cell expression of various genes mediating endothelial cell proliferation, adhesion [44, 45], and permeability [38, 40, 59, 69, 72–74] as well as monocyte infiltration of endothelial cells [44, 75, 76]. Together, these data demonstrate the protective roles that DBMSCs may exert on endothelial cells via mechanisms may involve genes listed in Tables 2, 3, 4, and 5. However, functional studies are necessary to elucidate the effects of these genes in mediating DBMCs protective activities on endothelial cells.
Conclusions
This is the first comprehensive study to demonstrate the protective role of DBMSCs on endothelial cells in harsh oxidative stress environment. DBMSCs can protect endothelial cells from injury induced by oxidative stress or immune cells. Endothelial cell injury is a hallmark of inflammatory diseases, such as atherosclerosis where endothelial cells show increased functional activities (proliferation, adhesion, migration, angiogenesis, and permeability) and increased fibrin and thrombus formation as well as increased adhesion to immune cells, such as monocytes and their infiltration. These functional activities could be therapeutical targets for DBMSCs to repair endothelial cell injury and treat atherosclerosis.
Acknowledgements
We appreciate the staff and patients of the Delivery Unit, King Abdul Aziz Medical City for giving us placentae.
Funding
This study was supported by grants from KAIMRC (Grant No. RC12/133).
Availability of data and materials
All data generated during this study are included in this published article.
Authors’ contributions
MHA proposed and supervised the project. MHA designed the experiments. MAA performed the experiments. MHA, MAA, and TK analyzed the data. MHA wrote the manuscript. MHA, FMA, TK, BK, SAM, RK, AOA, and ASA contributed to the data analysis and interpretation of results. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The institutional review board (IRB) at King Abdulla International Medical Research Centre (KAIMRC), Saudi Arabia, approved this study. Samples (placentae and umbilical cords of uncomplicated human pregnancies, 38–40 gestational weeks) were obtained and used immediately after signing consent forms.
Consent for publication
“Not applicable”. All authors agree to publish this manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
M. A. Alshabibi, Email: Malshabibi@kacst.edu.sa
T. Khatlani, Email: khatlanita@NGHA.MED.SA
F. M. Abomaray, Email: fawaz.abomaray@ki.se
A. S. AlAskar, Email: askaras@ngha.med.sa
B. Kalionis, Email: bill.kalionis@thewomens.org.au
S. A. Messaoudi, Email: safia.massoudi@nauss.edu.sa
R. Khanabdali, Email: rkhanabdali@student.unimelb.edu.au
A. O. Alawad, Email: alawad@kacst.edu.sa
M. H. Abumaree, Email: mohamedabumaree@hotmail.com, Email: abumareem@ksau-hs.edu.sa
References
- 1.Abumaree MH, Abomaray FM, Alshabibi MA, AlAskar AS, Kalionis B. Immunomodulatory properties of human placental mesenchymal stem/stromal cells. Placenta. 2017;59:87–95. doi: 10.1016/j.placenta.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Abomaray FM, Al Jumah MA, Alsaad KO, Jawdat D, Al Khaldi A, AlAskar AS, et al. Phenotypic and functional characterization of mesenchymal stem/multipotent stromal cells from decidua basalis of human term placenta. Stem Cells Int. 2016;2016:5184601. doi: 10.1155/2016/5184601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kanasaki K, Kalluri R. The biology of preeclampsia. Kidney Int. 2009;76(8):831–837. doi: 10.1038/ki.2009.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang YC, Yang ZM, Chen XH, Tan MY, Wang J, Li XQ, et al. Isolation of mesenchymal stem cells from human placental decidua basalis and resistance to hypoxia and serum deprivation. Stem Cell Rev. 2009;5(3):247–255. doi: 10.1007/s12015-009-9069-x. [DOI] [PubMed] [Google Scholar]
- 5.Abumaree MH, Al Jumah MA, Kalionis B, Jawdat D, Al Khaldi A, AlTalabani AA, et al. Phenotypic and functional characterization of mesenchymal stem cells from chorionic villi of human term placenta. Stem Cell Rev. 2013;9(1):16–31. doi: 10.1007/s12015-012-9385-4. [DOI] [PubMed] [Google Scholar]
- 6.Braekke K, Harsem NK, Staff AC Oxidative stress and antioxidant status in fetal circulation in preeclampsia. Pediatr Res. 2006;60(5):560–564. doi: 10.1203/01.pdr.0000242299.01219.6a. [DOI] [PubMed] [Google Scholar]
- 7.Kusuma Gina D., Abumaree Mohamed H., Pertile Mark D., Perkins Anthony V., Brennecke Shaun P., Kalionis Bill. Mesenchymal Stem/Stromal Cells Derived From a Reproductive Tissue Niche Under Oxidative Stress Have High Aldehyde Dehydrogenase Activity. Stem Cell Reviews and Reports. 2016;12(3):285–297. doi: 10.1007/s12015-016-9649-5. [DOI] [PubMed] [Google Scholar]
- 8.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109(23 Suppl 1):III27–III32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 9.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192(1):1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 10.Shono T, Ono M, Izumi H, Jimi SI, Matsushima K, Okamoto T, et al. Involvement of the transcription factor NF-kappaB in tubular morphogenesis of human microvascular endothelial cells by oxidative stress. Mol Cell Biol. 1996;16(8):4231–4239. doi: 10.1128/MCB.16.8.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 12.Alshabibi MA, Al Huqail AJ, Khatlani T, Abomaray FM, Alaskar AS, Alawad AO, et al. Mesenchymal stem/multipotent stromal cells from human decidua basalis reduce endothelial cell activation. Stem Cells Dev. 2017;26(18):1355–1373. doi: 10.1089/scd.2017.0096. [DOI] [PubMed] [Google Scholar]
- 13.Abumaree MH, Hakami M, Abomaray FM, Alshabibi MA, Kalionis B, Al Jumah MA, et al. Human chorionic villous mesenchymal stem/stromal cells modify the effects of oxidative stress on endothelial cell functions. Placenta. 2017;59:74–86. doi: 10.1016/j.placenta.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Sies H. Oxidative stress: from basic research to clinical application. Am J Med 1991;91(3C):31S–38S. PubMed PMID: 1928209. [DOI] [PubMed]
- 15.Valle-Prieto A, Conget PA. Human mesenchymal stem cells efficiently manage oxidative stress. Stem Cells Dev. 2010;19(12):1885–1893. doi: 10.1089/scd.2010.0093. [DOI] [PubMed] [Google Scholar]
- 16.Lodi Daniele, Iannitti Tommaso, Palmieri Beniamino. Stem cells in clinical practice: applications and warnings. Journal of Experimental & Clinical Cancer Research. 2011;30(1):9. doi: 10.1186/1756-9966-30-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mimeault M, Batra SK. Concise review: recent advances on the significance of stem cells in tissue regeneration and cancer therapies. Stem Cells. 2006;24(11):2319–2345. doi: 10.1634/stemcells.2006-0066. [DOI] [PubMed] [Google Scholar]
- 18.Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840(8):2506–2519. doi: 10.1016/j.bbagen.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Auletta JJ, Cooke KR, Solchaga LA, Deans RJ, van't Hof W. Regenerative stromal cell therapy in allogeneic hematopoietic stem cell transplantation: current impact and future directions. Biol Blood Marrow Transplant. 2010;16(7):891–906. doi: 10.1016/j.bbmt.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khatlani T., Algudiri D., Alenzi R., Al Subayyil A. M., Abomaray F. M., Bahattab E., AlAskar A. S., Kalionis B., El-Muzaini M. F., Abumaree M. H. Preconditioning by Hydrogen Peroxide Enhances Multiple Properties of Human Decidua Basalis Mesenchymal Stem/Multipotent Stromal Cells. Stem Cells International. 2018;2018:1–13. doi: 10.1155/2018/6480793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raijmakers MT, Roes EM, Poston L, Steegers EA, Peters WH. The transient increase of oxidative stress during normal pregnancy is higher and persists after delivery in women with pre-eclampsia. Eur J Obstet Gynecol Reprod Biol. 2008;138(1):39–44. doi: 10.1016/j.ejogrb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 22.Burton GJ, Jauniaux E. Oxidative stress. Best practice & research Clinical obstetrics & gynaecology. 2011;25(3):287–299. doi: 10.1016/j.bpobgyn.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong Feng, Zhang Xiaochun, Wold Loren E, Ren Qun, Zhang Zhaojie, Ren Jun. Endothelin-1 enhances oxidative stress, cell proliferation and reduces apoptosis in human umbilical vein endothelial cells: role of ETBreceptor, NADPH oxidase and caveolin-1. British Journal of Pharmacology. 2005;145(3):323–333. doi: 10.1038/sj.bjp.0706193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao C, Sun W, Christofidou-Solomidou M, Sawada M, Newman DK, Bergom C, et al. PECAM-1 functions as a specific and potent inhibitor of mitochondrial-dependent apoptosis. Blood. 2003;102(1):169–179. doi: 10.1182/blood-2003-01-0003. [DOI] [PubMed] [Google Scholar]
- 25.Kern TS, Du Y, Miller CM, Hatala DA, Levin LA. Overexpression of Bcl-2 in vascular endothelium inhibits the microvascular lesions of diabetic retinopathy. Am J Pathol. 2010;176(5):2550–2558. doi: 10.2353/ajpath.2010.091062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinderlerer A. R., Pombo Gregoire I., Hamdulay S. S., Ali F., Steinberg R., Silva G., Ali N., Wang B., Haskard D. O., Soares M. P., Mason J. C. Heme oxygenase-1 expression enhances vascular endothelial resistance to complement-mediated injury through induction of decay-accelerating factor: a role for increased bilirubin and ferritin. Blood. 2008;113(7):1598–1607. doi: 10.1182/blood-2008-04-152934. [DOI] [PubMed] [Google Scholar]
- 27.Kwon YG, Min JK, Kim KM, Lee DJ, Billiar TR, Kim YM. Sphingosine 1-phosphate protects human umbilical vein endothelial cells from serum-deprived apoptosis by nitric oxide production. J Biol Chem. 2001;276(14):10627–10633. doi: 10.1074/jbc.M011449200. [DOI] [PubMed] [Google Scholar]
- 28.Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170(6):3369–3376. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- 29.Liang P, Cheng SH, Cheng CK, Lau KM, Lin SY, Chow EY, et al. Platelet factor 4 induces cell apoptosis by inhibition of STAT3 via up-regulation of SOCS3 expression in multiple myeloma. Haematologica. 2013;98(2):288–295. doi: 10.3324/haematol.2012.065607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mikhail M, Vachon PH, D'Orleans-Juste P, Jacques D, Bkaily G. Role of endothelin-1 and its receptors, ETA and ETB, in the survival of human vascular endothelial cells. Can J Physiol Pharmacol. 2017;95(10):1298–1305. doi: 10.1139/cjpp-2017-0412. [DOI] [PubMed] [Google Scholar]
- 31.Toi M, Atiqur Rahman M, Bando H, Chow LW. Thymidine phosphorylase (platelet-derived endothelial-cell growth factor) in cancer biology and treatment. Lancet Oncol. 2005;6(3):158–166. doi: 10.1016/S1470-2045(05)01766-3. [DOI] [PubMed] [Google Scholar]
- 32.Wang JF, Zhang X, Groopman JE. Activation of vascular endothelial growth factor receptor-3 and its downstream signaling promote cell survival under oxidative stress. J Biol Chem. 2004;279(26):27088–27097. doi: 10.1074/jbc.M314015200. [DOI] [PubMed] [Google Scholar]
- 33.Secchiero P, Gonelli A, Carnevale E, Milani D, Pandolfi A, Zella D, et al. TRAIL promotes the survival and proliferation of primary human vascular endothelial cells by activating the Akt and ERK pathways. Circulation. 2003;107(17):2250–2256. doi: 10.1161/01.CIR.0000062702.60708.C4. [DOI] [PubMed] [Google Scholar]
- 34.Suhara T, Fukuo K, Sugimoto T, Morimoto S, Nakahashi T, Hata S, et al. Hydrogen peroxide induces up-regulation of Fas in human endothelial cells. J Immunol. 1998;160(8):4042–4047. [PubMed] [Google Scholar]
- 35.Lee S, Chung J, Ha IS, Yi K, Lee JE, Kang HG, et al. Hydrogen peroxide increases human leukocyte adhesion to porcine aortic endothelial cells via NFkappaB-dependent up-regulation of VCAM-1. Int Immunol. 2007;19(12):1349–1359. doi: 10.1093/intimm/dxm104. [DOI] [PubMed] [Google Scholar]
- 36.Brodsky SV, Malinowski K, Golightly M, Jesty J, Goligorsky MS. Plasminogen activator inhibitor-1 promotes formation of endothelial microparticles with procoagulant potential. Circulation. 2002;106(18):2372–2378. doi: 10.1161/01.CIR.0000033972.90653.AF. [DOI] [PubMed] [Google Scholar]
- 37.Moore R, Hawley A, Sigler R, Farris D, Wrobleski S, Ramacciotti E, et al. Tissue inhibitor of metalloproteinase-1 is an early marker of acute endothelial dysfunction in a rodent model of venous oxidative injury. Ann Vasc Surg. 2009;23(4):498–505. doi: 10.1016/j.avsg.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 38.Mu X, He K, Sun H, Zhou X, Chang L, Li X, et al. Hydrogen peroxide induces overexpression of angiotensin-converting enzyme in human umbilical vein endothelial cells. Free Radic Res. 2013;47(2):116–122. doi: 10.3109/10715762.2012.749987. [DOI] [PubMed] [Google Scholar]
- 39.Kaplanski G, Fabrigoule M, Boulay V, Dinarello CA, Bongrand P, Kaplanski S, et al. Thrombin induces endothelial type II activation in vitro: IL-1 and TNF-alpha-independent IL-8 secretion and E-selectin expression. J Immunol. 1997;158(11):5435–41. PubMed PMID: 9164965. [PubMed]
- 40.Dreymueller D, Martin C, Kogel T, Pruessmeyer J, Hess FM, Horiuchi K, et al. Lung endothelial ADAM17 regulates the acute inflammatory response to lipopolysaccharide. EMBO Mol Med. 2012;4(5):412–423. doi: 10.1002/emmm.201200217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shibuya Masaubmi. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): a dual regulator for angiogenesis. Angiogenesis. 2006;9(4):225–230. doi: 10.1007/s10456-006-9055-8. [DOI] [PubMed] [Google Scholar]
- 42.Valen G, Erl W, Eriksson P, Wuttge D, Paulsson G, Hansson GK. Hydrogen peroxide induces mRNA for tumour necrosis factor alpha in human endothelial cells. Free Radic Res. 1999;31(6):503–512. doi: 10.1080/10715769900301071. [DOI] [PubMed] [Google Scholar]
- 43.Sharma R, Colarusso P, Zhang H, Stevens KM, Patel KD. FRNK negatively regulates IL-4-mediated inflammation. J Cell Sci. 2015;128(4):695–705. doi: 10.1242/jcs.156588. [DOI] [PubMed] [Google Scholar]
- 44.Liu Z, Morgan S, Ren J, Wang Q, Annis DS, Mosher DF, et al. Thrombospondin-1 (TSP1) contributes to the development of vascular inflammation by regulating monocytic cell motility in mouse models of abdominal aortic aneurysm. Circ Res. 2015;117(2):129–141. doi: 10.1161/CIRCRESAHA.117.305262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao Q, Danton MJ, Witte DP, Kowala MC, Valentine MT, Bugge TH, et al. Plasminogen deficiency accelerates vessel wall disease in mice predisposed to atherosclerosis. Proc Natl Acad Sci U S A. 1997;94(19):10335–10340. doi: 10.1073/pnas.94.19.10335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steffel J, Luscher TF, Tanner FC. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. 2006;113(5):722–731. doi: 10.1161/CIRCULATIONAHA.105.567297. [DOI] [PubMed] [Google Scholar]
- 47.Boehme MW, Galle P, Stremmel W. Kinetics of thrombomodulin release and endothelial cell injury by neutrophil-derived proteases and oxygen radicals. Immunology. 2002;107(3):340–349. doi: 10.1046/j.1365-2567.2002.01469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Westrick RJ, Bodary PF, Xu Z, Shen YC, Broze GJ, Eitzman DT. Deficiency of tissue factor pathway inhibitor promotes atherosclerosis and thrombosis in mice. Circulation. 2001;103(25):3044–3046. doi: 10.1161/hc2501.092492. [DOI] [PubMed] [Google Scholar]
- 49.Bradley JR, Johnson DR, Pober JS. Endothelial activation by hydrogen peroxide. Selective increases of intercellular adhesion molecule-1 and major histocompatibility complex class I. Am J Pathol. 1993;142(5):1598–1609. [PMC free article] [PubMed] [Google Scholar]
- 50.Lo S. K., Janakidevi K., Lai L., Malik A. B. Hydrogen peroxide-induced increase in endothelial adhesiveness is dependent on ICAM-1 activation. American Journal of Physiology-Lung Cellular and Molecular Physiology. 1993;264(4):L406–L412. doi: 10.1152/ajplung.1993.264.4.L406. [DOI] [PubMed] [Google Scholar]
- 51.Lakshminarayanan V, Beno DW, Costa RH, Roebuck KA. Differential regulation of interleukin-8 and intercellular adhesion molecule-1 by H2O2 and tumor necrosis factor-alpha in endothelial and epithelial cells. J Biol Chem. 1997;272(52):32910–32918. doi: 10.1074/jbc.272.52.32910. [DOI] [PubMed] [Google Scholar]
- 52.Mendoza L, Carrascal T, De Luca M, Fuentes AM, Salado C, Blanco J, et al. Hydrogen peroxide mediates vascular cell adhesion molecule-1 expression from interleukin-18-activated hepatic sinusoidal endothelium: implications for circulating cancer cell arrest in the murine liver. Hepatology. 2001;34(2):298–310. doi: 10.1053/jhep.2001.26629. [DOI] [PubMed] [Google Scholar]
- 53.Anasagasti MJ, Alvarez A, Avivi C, Vidal-Vanaclocha F. Interleukin-1-mediated H2O2 production by hepatic sinusoidal endothelium in response to B16 melanoma cell adhesion. J Cell Physiol. 1996;167(2):314–323. doi: 10.1002/(SICI)1097-4652(199605)167:2<314::AID-JCP16>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 54.Shao Y, Cheng Z, Li X, Chernaya V, Wang H, Yang XF. Immunosuppressive/anti-inflammatory cytokines directly and indirectly inhibit endothelial dysfunction--a novel mechanism for maintaining vascular function. J Hematol Oncol. 2014;7:80. doi: 10.1186/s13045-014-0080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li W, Ren G, Huang Y, Su J, Han Y, Li J, et al. Mesenchymal stem cells: a double-edged sword in regulating immune responses. Cell Death Differ. 2012;19(9):1505–1513. doi: 10.1038/cdd.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paik JH, Chae S, Lee MJ, Thangada S, Hla T. Sphingosine 1-phosphate-induced endothelial cell migration requires the expression of EDG-1 and EDG-3 receptors and Rho-dependent activation of alpha vbeta3- and beta1-containing integrins. J Biol Chem. 2001;276(15):11830–11837. doi: 10.1074/jbc.M009422200. [DOI] [PubMed] [Google Scholar]
- 57.Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Li Calzi S, et al. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J Exp Med. 2007;204(3):605–618. doi: 10.1084/jem.20061609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daher Z, Noel J, Claing A. Endothelin-1 promotes migration of endothelial cells through the activation of ARF6 and the regulation of FAK activity. Cell Signal. 2008;20(12):2256–2265. doi: 10.1016/j.cellsig.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 59.Bauer PM, Yu J, Chen Y, Hickey R, Bernatchez PN, Looft-Wilson R, et al. Endothelial-specific expression of caveolin-1 impairs microvascular permeability and angiogenesis. Proc Natl Acad Sci U S A. 2005;102(1):204–209. doi: 10.1073/pnas.0406092102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Starke RD, Ferraro F, Paschalaki KE, Dryden NH, McKinnon TA, Sutton RE, et al. Endothelial von Willebrand factor regulates angiogenesis. Blood. 2011;117(3):1071–1080. doi: 10.1182/blood-2010-01-264507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Celerier J, Cruz A, Lamande N, Gasc JM, Corvol P. Angiotensinogen and its cleaved derivatives inhibit angiogenesis. Hypertension. 2002;39(2):224–228. doi: 10.1161/hy0202.103441. [DOI] [PubMed] [Google Scholar]
- 62.Nor JE, Mitra RS, Sutorik MM, Mooney DJ, Castle VP, Polverini PJ. Thrombospondin-1 induces endothelial cell apoptosis and inhibits angiogenesis by activating the caspase death pathway. J Vasc Res. 2000;37(3):209–218. doi: 10.1159/000025733. [DOI] [PubMed] [Google Scholar]
- 63.Rehn M, Veikkola T, Kukk-Valdre E, Nakamura H, Ilmonen M, Lombardo C, et al. Interaction of endostatin with integrins implicated in angiogenesis. Proc Natl Acad Sci U S A. 2001;98(3):1024–1029. doi: 10.1073/pnas.031564998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang X, Liaw L, Prudovsky I, Brooks PC, Vary C, Oxburgh L, et al. Fibroblast growth factor signaling in the vasculature. Curr Atheroscler Rep 2015;17(6):509. 10.1007/s11883-015-0509-6. PubMed PMID: 25813213; PubMed Central PMCID: PMCPMC4593313. [DOI] [PMC free article] [PubMed]
- 65.Kim JY, Choi JS, Song SH, Im JE, Kim JM, Kim K, et al. Stem cell factor is a potent endothelial permeability factor. Arterioscler Thromb Vasc Biol. 2014;34(7):1459–1467. doi: 10.1161/ATVBAHA.114.303575. [DOI] [PubMed] [Google Scholar]
- 66.He T, Lu T, d'Uscio LV, Lam CF, Lee HC, Katusic ZS. Angiogenic function of prostacyclin biosynthesis in human endothelial progenitor cells. Circ Res. 2008;103(1):80–88. doi: 10.1161/CIRCRESAHA.108.176057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Foubert P, Silvestre JS, Souttou B, Barateau V, Martin C, Ebrahimian TG, et al. PSGL-1-mediated activation of EphB4 increases the proangiogenic potential of endothelial progenitor cells. J Clin Invest. 2007;117(6):1527–1537. doi: 10.1172/JCI28338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim YM, Kim KE, Koh GY, Ho YS, Lee KJ. Hydrogen peroxide produced by angiopoietin-1 mediates angiogenesis. Cancer Res. 2006;66(12):6167–6174. doi: 10.1158/0008-5472.CAN-05-3640. [DOI] [PubMed] [Google Scholar]
- 69.Chua CC, Hamdy RC, Chua BH. Upregulation of vascular endothelial growth factor by H2O2 in rat heart endothelial cells. Free Radic Biol Med. 1998;25(8):891–897. doi: 10.1016/S0891-5849(98)00115-4. [DOI] [PubMed] [Google Scholar]
- 70.Scharner Dörte, Rössig Lothar, Carmona Guillaume, Chavakis Emmanouil, Urbich Carmen, Fischer Ariane, Kang Tae-Bong, Wallach David, Chiang Yungping Jeffrey, Deribe Yonathan Lissanu, Dikic Ivan, Zeiher Andreas M., Dimmeler Stefanie. Caspase-8 Is Involved in Neovascularization-Promoting Progenitor Cell Functions. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29(4):571–578. doi: 10.1161/ATVBAHA.108.182006. [DOI] [PubMed] [Google Scholar]
- 71.Fuster JJ, Fernandez P, Gonzalez-Navarro H, Silvestre C, Nabah YN, Andres V. Control of cell proliferation in atherosclerosis: insights from animal models and human studies. Cardiovasc Res. 2010;86(2):254–264. doi: 10.1093/cvr/cvp363. [DOI] [PubMed] [Google Scholar]
- 72.Puhlmann Markus, Weinreich David M, Farma Jeffrey M, Carroll Nancy M, Turner Ewa M, Alexander H Richard. Journal of Translational Medicine. 2005;3(1):37. doi: 10.1186/1479-5876-3-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wassmann S, Stumpf M, Strehlow K, Schmid A, Schieffer B, Bohm M, et al. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ Res. 2004;94(4):534–541. doi: 10.1161/01.RES.0000115557.25127.8D. [DOI] [PubMed] [Google Scholar]
- 74.Furst R, Bubik MF, Bihari P, Mayer BA, Khandoga AG, Hoffmann F, et al. Atrial natriuretic peptide protects against histamine-induced endothelial barrier dysfunction in vivo. Mol Pharmacol. 2008;74(1):1–8. doi: 10.1124/mol.108.045773. [DOI] [PubMed] [Google Scholar]
- 75.Sigala F, Vourliotakis G, Georgopoulos S, Kavantzas N, Papalambros E, Agapitos M, et al. Vascular endothelial cadherin expression in human carotid atherosclerotic plaque and its relationship with plaque morphology and clinical data. Eur J Vasc Endovasc Surg. 2003;26(5):523–528. doi: 10.1016/S1078-5884(03)00342-3. [DOI] [PubMed] [Google Scholar]
- 76.Foster CA. VCAM-1/alpha 4-integrin adhesion pathway: therapeutic target for allergic inflammatory disorders. J Allergy Clin Immunol. 1996;98(6 Pt 2):S270–S277. doi: 10.1016/S0091-6749(96)70075-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated during this study are included in this published article.