

Transparent polymers that can match the optical and thermal expansion properties of glass were synthesized for flexible devices.
Abstract
The key component currently missing for the next generation of transparent and flexible displays is a high-performance polymer material that is flexible, while showing optical and thermal properties of glass. It must be transparent to visible light and show a low coefficient of thermal expansion (CTE). While specialty plastics such as aromatic polyimides are promising, reducing their CTE and improving transparency simultaneously proved challenging, with increasing coloration the main problem to be resolved. We report a new poly(amide-imide) material that is flexible and displays glass-like behavior with a CTE value of 4 parts per million/°C. This novel polymer was successfully used as a substrate to fabricate transparent and flexible indium-gallium-zinc oxide thin-film transistors.
INTRODUCTION
The worldwide proliferation of handheld devices and smartphones has created an urgent need for light, flexible, energy-efficient, and nonbrittle display materials that can be used in the next generation of electronic devices (1) to create, for instance, flexible and transparent displays. Whereas polymer materials are uniquely suited to meet the required technical demands, transparency to visible light and low coefficient of thermal expansion (CTE) (2) proved difficult to realize. Metals and ceramics typically show low CTE values, in the range of 0.5 to 25 parts per million (ppm)/°C, and maintain their physical dimensions over a wide range of temperatures. Non–cross-linked organic polymers, however, experience relatively large thermal expansion upon heating and show large CTE values ranging from 30 to 400 ppm/°C. This behavior is easy to understand. Because the interactions between individual polymer chains are weak, they experience substantial thermal expansions upon heating (3).
To lower the CTE values of thermoplastic polymers, researchers have typically added filler materials (4–7) or introduced rigid rod-like backbones into the polymer (8–10). However, the filled polymer composites often suffer from poor toughness and are optically hazy and difficult to process, and the reduction in CTE is often modest. Polymers with rigid backbones and strong secondary interchain interactions, such as aromatic polyimides and polyamides, have inherently low CTE values and even become negative in the case of Kevlar yarns (11) and the rigid polyimide from pyromellitic dianhydride (PMDA) (8, 12). Effective chain packing aided by secondary interchain interactions can greatly suppress thermal expansion, especially when the polymer chains are aligned. However, these polymers are extremely difficult to process into films due to their low solubility. In addition, aromatic polyimides, such as Kapton, are well known to have a yellow hue due to the presence of aromatic rings in the backbone and the formation of charge-transfer complex between the electron-deficient dianhydride and electron-rich diamine units. The challenge has been to develop polymeric materials with low CTE while simultaneously retaining high transparency, good processability, and high thermal stability.
To overcome these fundamental challenges, we examined aromatic poly(amide-imide) as a new platform. We chose a rigid and strongly interacting aromatic poly(amide-imide) backbone, and we decorated the two diamine groups with sterically bulky trifluoromethyl groups capable of forming an intricate hydrogen-bonding network in two different directions. This approach achieved polymers with ~90% transparency with CTE values as low as 4 ppm/°C. We found that the CTE values can be precisely tuned in the range of 4 to 20 ppm/°C by controlling the fraction of asymmetrically positioned trifluoromethyl groups in the polymer backbone, with no sacrifice in high transparency. Structural analysis of the chain packing combined with density functional theory (DFT) calculations indicates that the asymmetrically positioned trifluoromethyl groups increase the interchain distance and the possible number of interchain hydrogen bonds, which contribute to both the low CTE value and high transparency without forming charge-transfer complexes. We demonstrate how the composition of the monomer can be optimized rationally for chain packing to change the CTE of the amorphous polymers, and this approach can be applied not only to polymers but also to organic materials.
RESULTS AND DISCUSSION
The novel poly(amide-imide) materials are based on a 4,4′-biphenyldiamine monomer, where two trifluoromethyl groups are introduced at the 2 and 6 positions on one of the aromatic rings (Fig. 1A). As a result, the two aromatic rings are not equivalent, and the synthesis requires a cross-coupling reaction, as shown in figs. S1 to S4. This diamine monomer is designated uDA, in contrast to the symmetric biphenyldiamine monomer (sDA) (13) that carries trifluoromethyl groups at the 2 position of both arenes. The sDA monomer was used previously to prepare polyimides (10) and poly(amide-imide)s (14) to increase their processability and transparency while retaining a high glass transition temperature (Tg). This is accomplished by frustrating the compact overlap between polymer chains while hindering rotation around the C–C bond between the aromatic rings.
Fig. 1. Transparent and thermostable poly(amide-imide)s with low CTE.
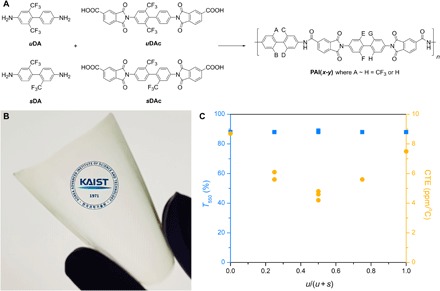
(A) Synthetic route to PAIs. (B) Photo of PAI(s-u) freestanding film. Photo credit: Byungyong Lee. (C) Relationship of transmittance and CTE as a function of u diamine content in the PAI.
Using both uDA and sDA as monomers, a series of poly(amide-imide) materials were synthesized as outlined in Fig. 1A. First, two different diacid monomers were synthesized by the reaction of uDA and sDA with trimellitic anhydride to form two internal imide linkages, as shown in figs. S5 to S8. Then, the diamine and the diacid monomers were polymerized in the presence of triphenyl phosphite and pyridine to produce the target poly(amide-imide)s with various combinations of the diamine repeating units in the backbone. The resulting polymers were labeled PAI(x-y), where x and y indicate the diamine repeating units between the amide and imide linkages, respectively. Here, su is used to indicate that an equimolar mixture of the diamine monomers was used in the synthesis, for example, as PAI(su-u). The position of the trifluoromethyl groups in the polymer originating from uDA is not regular, since uDA and uDAc are not symmetric. Characterization details of the synthesized polymers are summarized in Table 1.
Table 1. Characterization of PAIs.
| Polymer code | u/(u + s)* |
ηinh (dl g−1)† |
λco (nm)‡ | T550 (%)§ | Td5 (°C)║ | CTE (ppm/°C)¶ | ||
| In N2 | In air | Second run | Third run | |||||
| PAI(s-s) | 0.00 | 3.76 | 371 | 88 | 457 | 432 | 8.7 | 9.5 |
| PAI(s-su) | 0.25 | 3.90 | 371 | 88 | 468 | 474 | 6.1 | 7.4 |
| PAI(su-s) | 0.25 | 2.90 | 364 | 88 | 497 | 479 | 5.6 | 5.2 |
| PAI(s-u) | 0.50 | 3.98 | 371 | 88 | 478 | 484 | 4.2 | 4.4 |
| PAI(u-s) | 0.50 | 3.02 | 371 | 89 | 485 | 486 | 4.6 | 5.7 |
| PAI(su-su) | 0.50 | 3.40 | 371 | 88 | 482 | 492 | 4.8 | 4.8 |
| PAI(su-u) | 0.75 | 3.62 | 371 | 88 | 511 | 496 | 5.6 | 6.1 |
| PAI(u-u) | 1.00 | 3.50 | 371 | 88 | 465 | 476 | 7.5 | 7.6 |
*Molar fraction of uDA-originated repeating units in the total diamine content.
†Inherent viscosity measured in NMP at 0.5 g dl−1 concentration at 30°C.
‡Cutoff wavelength determined by UV-vis spectra of freestanding polymer films 70 to 80 μm thick.
§Transmittance at 550 nm.
║5% weight loss temperature measured by TGA at a heating rate of 10°C/min.
¶Calculated from the mean coefficient of linear thermal expansion between 50° and 250°C in the second and third heating runs, respectively.
All polymers were characterized by 1H–nuclear magnetic resonance (NMR) spectroscopy (fig. S9) and showed good solubility in various organic solvents (table S1). They also showed a high viscosity in N-methyl-2-pyrrolidone (NMP), indicating that polymers with high molar mass were successfully synthesized, although the two amine groups in uDA have different reactivities due to the electron-withdrawing trifluoromethyl groups. This allowed for processing all polymers into transparent, flexible, and tough films by casting the polymer solutions in N,N-dimethylacetamide (DMAc) on a glass support and evaporating the solvent (fig. S10). A representative example of a 70-μm-thick PAI(s-u) film is shown in Fig. 1B, highlighting its high transparency. The cutoff wavelengths (λco) were determined to be ~370 nm by ultraviolet-visible (UV-vis) spectroscopy for most of the polymer films, except for PAI(su-s), which showed a λco of 364 nm, indicating that the films only show a slight yellow hue (fig. S11). At 550 nm, the transmittance was ≥88% for all the polymers tested (Fig. 1C). The high transparency is attributed to the presence of the bulky and electron-withdrawing trifluoromethyl groups on the polymer backbone, which prohibits the formation of charge-transfer complexes by sterically repelling adjacent chains, and lowers the electron density in the diamine moiety. A prism-coupling analysis indicates that the birefringence of the films is related to the polymers having higher nTE values than nTM, suggesting that the chains are oriented in-plane (table S2).
These new polymers are remarkably stable at high temperatures. PAI(su-u) showed a 5% weight loss at above 510°C in nitrogen, and even in air, all polymers maintained their weight within 5% up to 470°C with the notable exception of PAI(s-s) (fig. S12). Whereas the glass transition temperatures (Tg) of the polymers were not detected by differential scanning calorimetry (DSC) analysis in the range of room temperature to 400°C (fig. S13), dynamic mechanical analysis (DMA) of the films exhibited α, β, and γ transitions, presumably corresponding to glass transition, and rotation-related relaxation processes (fig. S14 and table S3). The storage modulus of the PAIs was notably higher than conventional polyimides such as Kapton, indicating significant improvement of the mechanical properties in these new materials. The CTE values of the polymer films were determined by thermomechanical analysis (TMA) (fig. S15). Subjected to three cycles of heating from room temperature to 300°C, the mean coefficient of linear thermal expansion was estimated in the temperature range between 50° and 250°C in the second and the third heating scans, respectively, and the CTE values were calculated (Table 1). Only slight changes were observed in the CTE values between the second and the third heating cycles. During the second cycle, the CTE value of PAI(u-u) was determined to be 7.5 ppm/°C, which is slightly lower than the CTE value of PAI(s-s) previously reported (15). The PAIs containing both sDA and uDA repeating units exhibited even lower CTE values than PAI(u-u). Remarkably, the lowest CTE value of 4.2 ppm/°C, which is comparable to that of Pyrex glass, was obtained with PAI(s-u). Surprised by this finding, we produced different batches of PAI(s-u)s and confirmed that it is reproducible.
Figure 1C shows a plot of CTE values as a function of uDA repeating units in the total diamine content, regardless of its position, denoted as u/(u + s). It appears that the thermal expansion is more suppressed as the ratio of uDA and sDA repeating units approaches unity. This trend suggests that incorporation of both uDA and sDA synergistically reduces the CTE value beyond the straightforward steric and electronic effects exerted by the trifluoromethyl functionalities. To understand the origin of the unprecedentedly low CTE and relatively high transparency of the PAIs, the chain packing structure of the PAIs was investigated by grazing incidence wide-angle x-ray scattering (GIWAXS) experiments (15). Thin films of ~100-nm PAIs were prepared for the scattering experiments by spin coating the DMAc solution on a silicon wafer. Representative two-dimensional (2D) GIWAXS data are shown in Fig. 2A (see fig. S16 for the whole data), revealing an intense scattering arc along the out-of-plane (qz) direction with relatively weak scattering along the in-plane (qx) direction in all cases. These scattering profiles indicate that the PAIs are amorphous but have anisotropic packing, as observed in the prism-coupling analysis, where the polymer chains are preferentially stacked parallel to the substrate.
Fig. 2. Analysis of thermal expansion behavior of poly(amide-imide)s.
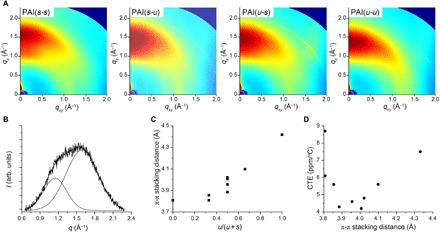
(A) Representative 2D GIWAXS data of the PAI(s-s), PAI(s-u), PAI(u-s), and PAI(u-u). (B) Azimuthal profile in the qz direction, which was deconvoluted assuming Gaussian functions. arb. units, arbitrary units. (C) A plot of π-π stacking distance versus u(u + s). (D) A plot of CTE versus π-π stacking distance.
On the basis of the liquid crystalline–like model suggested previously, we interpreted the scattering peaks by deconvoluting the scattering intensities assuming a Gaussian peak shape (Fig. 2B and figs. S17 and S18). This analysis suggests that the scattering peaks originate from π-π stacking between the biphenyl units and chain packing between the polymer chains in the liquid crystalline domains (16). The peak positions as a function of scattering vector (q), calculated domain spacings (d = 2π/q), and their assignment are given in table S4. The most discernible scattering peak from π-π stacking in the qz direction shows that the π-π stacking distance is almost linearly proportional to u/(u + s). This indicates that the asymmetrically introduced trifluoromethyl groups effectively increase the interchain distance as the population of the uDA fragments is increased in the polymer backbone (Fig. 2C). However, when the CTE values were plotted against the π-π stacking distance, CTE reached a minimum and then increased as the π-π stacking distance increased further (Fig. 2D). This behavior is in contrast to the conventional notion that high packing coefficient and small free volume lead to low CTE and suggests that the interchain interaction is not simply governed by distance. This is supported by the β-relaxation temperature, which is related to the rotational motion of the polymer backbone (16, 17) and is measured by DMA. It roughly increases with decreasing CTE but does not show a noticeable correlation with the π-π stacking distance (fig. S19).
To gain additional insight into the influence of uDA on the chain packing in the PAI materials, we prepared model compounds corresponding to the amide-linked uDA (amide-uDA) and imide-linked uDA (imide-uDA) (figs. S20 to S23). Their crystal structures, as revealed by x-ray diffraction (XRD) studies, indicate that the biphenyls in the amide-uDA are aligned antiparallel, to reduce steric congestion between the trifluoromethyl groups, but form hydrogen bonds between the amides (fig. S24 and tables S5 and S6), as envisioned in our original design strategy. The angle between two aromatic rings was significantly distorted at 78.6°. Whereas the imide-uDA exhibited a similar distortion of 76.4°, a parallel stacking of the biphenyls was observed, suggesting that the packing was predominantly driven by stacking of the imide rings. The possible packing structures of PAIs were further investigated by DFT calculations. Figure 3 shows the optimized structures of the PAI(s-s), PAI(s-u), PAI(u-s), and PAI(u-u) repeating units, along with their interaction energies between the chains and the optimized distances between the phenyl rings (figs. S25 to S28 and table S7). While the phenyl-phenyl distances increased with increasing u content, consistent with the GIWAXS data, the interaction energies of PAI(s-u) and PAI(u-s) were larger than those of PAI(s-s) and PAI(u-u), which explain the trend in the CTE values, since the interchain interaction energy is inversely proportional to the CTE. PAI(s-s) favored a parallel arrangement, whereas the others preferred antiparallel, as observed for amide-uDA, indicating the decisive influence of the uDA on molecular packing.
Fig. 3. Optimized structures of poly(amide-imide)s’ repeating units.
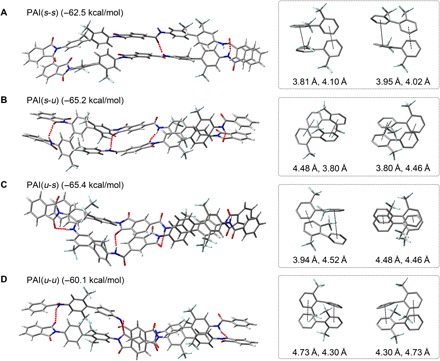
Optimized structures of PAI(s-s) (A), PAI(s-u) (B), PAI(u-s) (C), and PAI(u-u) (D) repeating units by DFT calculations. Their corresponding binding energies are also given. The red dotted lines indicate hydrogen bonds. The π-π stacking structures between two biphenyl units along with the distances between two phenyl rings are separately shown on the right side.
To exploit the excellent mechanical properties, high transparency, and the remarkable operational temperature range of the PAIs, we used the PAI(s-u) material as a substrate to fabricate high-performance electronic devices that are transparent and flexible. PAI(s-u) was chosen because of its thermal stability and ultralow CTE, allowing us to anneal the fabricated device to increase its performance. These unique advantages are well illustrated in the transparent and flexible indium-gallium-zinc oxide (IGZO) thin-film transistors (TFTs) fabricated using PAI(s-u) and shown in Fig. 4A. The PAI(s-u) successfully withstood radio frequency (RF) sputtering damage during deposition of the indium-zinc oxide (IZO) transparent electrodes and IGZO channels, thermal stress during the atomic layer deposition of gate insulator (Al2O3, 170 nm thick) at 150°C, and thermal annealing at 250°C in ambient air. With the channel length (L), width (W), and thickness of 200 μm, 980 μm, and 50 nm, respectively, the IGZO TFTs on the 40-μm-thick PAI(s-u) substrate exhibited a large on/off ratio of 3 × 107, a subthreshold swing of 201 mV/decade, and a saturation mobility (μsat) of 12 cm2/V·s at a drain bias (VD) of 10 V, as shown in Fig. 4B. As can be seen from the characteristics obtained in the forward and reverse scan directions (red and blue traces in Fig. 4B), hysteresis was relatively low with ΔV of ca. 0.5 V at an ID (drain current) of 1 nA. This level of performance and near hysteresis-free characteristics are not expected in IGZO TFTs, unless they are annealed to a significant degree (18, 19). The IGZO TFTs prepared with the new material are and stay transparent even after in-air thermal annealing (250°C at 1 hour), showing 83% transmittance at 550 nm (Fig. 4C). This is a significant breakthrough compared to conventional polyimides and PAIs that typically show significant yellowing, presumably because charge-transfer complex is formed, and further oxidation occurs during the thermal annealing process. The substrates in this work did not go through yellowing even with the prolonged annealing as long as it was done at temperature within ca. 250°C in air. The performance of the IGZO TFTs was further evaluated as a function of bending radius to investigate their potential as flexible devices (Fig. 4D). When measured flat after being repeatedly bent at 10 times at a given bending radius, the TFTs under study exhibited consistent performance with virtually no change in characteristics down to a bending radius of 4 mm. Although permanent degradation was observed in TFT characteristics at a lower bending radius than 4 mm (fig. S29), the devices were functional even at a bending radius of 0.9 mm, with only a slight decrease in on-current. All these results show that PAI(s-u) is a promising platform for high-performance electronic devices with transparent and/or flexible form factors.
Fig. 4. TFT device fabricated on poly(amide-imide) film.
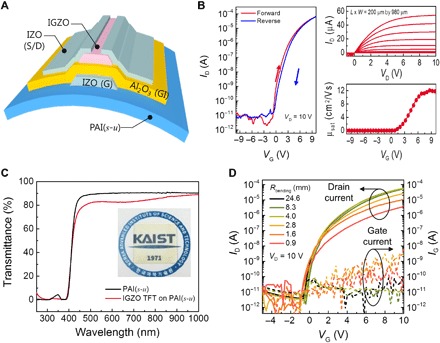
(A) Schematic structure of the transparent and flexible IGZO TFT fabricated on PAI(s-u). (B and C) Electrical characteristics (B) and transmittance spectra (C) of the IZGO TFTs. (D) TFT performance as a function of bending radius.
CONCLUSION
We developed novel poly(amide-imide) materials with a CTE value as low as 4 ppm/°C, which is exceptionally low for a non–cross-linked, amorphous polymer, while retaining high transparency and excellent thermal and mechanical properties. This breakthrough was possible because of careful and rational engineering of the chain packing structure of the polymers that allowed for controlling the position of the trifluoromethyl substituent on the polymer backbone. Specifically, tweaking the packing modes, the packing distances, and the intermolecular interactions gave rise to a remarkably different thermal behaviors of the polymers compared to conventional non–cross-linked polymers. The poly(amide-imide) materials developed in this study were tested as a substrate for transparent IGZO TFTs, and we found exceptional performance, demonstrating the potential of this new material as a versatile platform for electronic devices that require flexible and transparent form factors.
MATERIALS AND METHODS
Materials
2-Bromo-5-nitro-1,3-bis(trifluoromethyl)benzene (1) was synthesized, as reported in our previous paper (20). 4-Aminophenylboronic acid hydrochloride (Small Molecules), tetrakis(triphenylphosphine)palladium(0) (TCI), palladium 10% on carbon (Pd/C; Sigma-Aldrich), glacial acetic acid (Daejung), triphenyl phosphite (Sigma-Aldrich), pyridine (Sigma-Aldrich), benzyl chloride (Sigma-Aldrich), and trimethylamine (Sigma-Aldrich) were used as received. Reagent-grade calcium chloride was dried under vacuum at 180°C before use. Trimellitic anhydride (TCI) and 2,2′-bis(trifluoromethyl)benzidine (sDA; TCI) were purified by sublimation. NMP was purified by distillation under reduced pressure over calcium hydride before polymerization. Other commercially available reagent-grade chemicals were used without further purification.
Instrumentation
The Fourier transform infrared (FTIR) spectra of the compounds were obtained with a Bruker Equinox 55 spectrophotometer using a KBr pellet or film. The NMR spectra of the synthesized compounds were recorded on a Bruker Fourier Transform Avance 400 spectrometer. The chemical shift of the NMR was reported in parts per million using tetramethylsilane as an internal reference. Splitting patterns were designated as s (singlet), d (doublet), dd (doublets of doublet), t (triplet), q (quartet), or m (multiplet). Mass spectra were obtained on a Bruker Daltonik microTOF-QII mass spectrometer using the electrospray ionization method. Elemental analyses of the synthesized compounds were carried out with a FLASH 2000 Series. The inherent viscosities of polymers were measured using an Ubbelohde viscometer. Thermogravimetric analysis (TGA) and DSC were performed on a TA Instruments TGA Q500 and a DSC Q100, respectively. The TGA measurements were conducted at a heating rate of 10°C/min in N2 and air. The melting points (m.p.) of the synthesized compounds and the glass transition temperature (Tg) values of the polymers were obtained by DSC at a heating rate of 10°C/min in N2. No distinct Tg values were detected on the DSC measurements in the range between 0° and 400°C. DMA was performed on a TA Instruments Q800 with a tension film mode, using 10 mN preload, 15-μm amplitude, 125% force tracking, and 1-Hz frequency. Temperature ramp was conducted at a rate of 5°C min−1 from room temperature to 450°C. The in-plane linear CTEs of polymer films were measured by TMA using a TA Instruments TMA 2940 thermomechanical analyzer. To assess the expansion behavior over a large temperature range, polymer film specimens (10 mm long, 5 mm wide, and typically 70 μm thick) were heated at a heating rate of 5°C/min up to 300°C three times. CTE values were calculated from the mean coefficient of linear thermal expansion over a specific temperature range between 50° and 250°C in the second and third heating scans, respectively. Raman spectroscopy was performed on the specimens using an ARAMIS Raman spectrometer (Horiba Jobin Yvon, France) with a HeNe laser (λ = 633 nm) as an excitation source. UV-vis spectra were recorded on an Optizen POP spectrophotometer in the transmittance mode. The refractive indices nTE and nTM for the transverse electric (TE) and transverse magnetic (TM) modes of the polymer films were measured with a Sairon SPA-4000 prism coupler with a gadolinium gallium garnet prism at a wavelength of 633 nm at room temperature. The birefringence values (Δn) were calculated as the difference between nTE and nTM.
Monomer syntheses
4′-Nitro-2′,6′-bis(trifluoromethyl)-[1,1′-biphenyl]-4-amine (2)
4-Aminophenylboronic acid hydrochloride (3.47 g, 20.0 mmol) was reacted with 1 (3.48 g, 10.3 mmol) in the presence of Pd(PPh3)4 (0.60 g). The reaction mixture of 20 ml of toluene, 10 ml of ethanol, and 9.60 g of K2CO3 dissolved in 20 ml of water was refluxed for 20.5 hours. The product was extracted with ethyl acetate and passed through a silica column using CH2Cl2/hexane (v/v = 1:2) as an eluent to give solid product (2) (2.63 g, 7.51 mmol; yield, 72.9%): m.p. 106° and 107°C. FTIR (KBr): 3499 and 3401 cm−1 (NH2); 1620 cm−1 (aromatic C=C); 1533, 1359, and 1333 cm−1 (NO2); and 1126 to 1295 cm−1 (C–F). 1H-NMR (DMSO-d6, 400 MHz): 8.68 ppm (s, 2H), 6.84 ppm (d, J = 8.2 Hz, 2H), 6.56 ppm (d, J = 8.5Hz, 2H), and 5.35 ppm (s, NH2). 13C-NMR (DMSO-d6, 100 MHz): 149.39, 147.64, 146.36, and 132.11 ppm (q, J = 30.2 Hz); 129.70 and 124.63 ppm (q, J = 5.8 Hz); 122.25 ppm (q, J = 275.2Hz); 118.29 and 112.30 ppm. Anal. calcd. (Analytically calculated) for C14H8F6N2O2: C, 48.01; H, 2.30; N, 8.00. Found: C, 49.05; H, 2.56; N, 7.71.
2,6-Bis(trifluoromethyl)benzidine (uDA)
The Schlenk tube was charged with 2 (5.01 g, 14.3 mmol), 10% Pd/C, and 60 ml of the mixture of ethanol/ethyl acetate (v/v = 1:1). The reaction mixture was degassed with hydrogen and stirred under an atmosphere of H2 at room temperature for 20.5 hours. Quantitative hydrogenation was confirmed by thin-layer chromatography. The catalyst was filtered out through celite, and the solvent was removed in an evaporator. The product was passed through a silica column using ethyl acetate/hexane (v/v = 1:2) as an eluent to give pale yellow solid, which was further purified by sublimation to obtain white crystal (4.53 g, 14.1 mmol; yield, 98.9%). m.p. 127° and 128°C. FTIR (KBr): 3486, 3335, and 3221 cm−1 (NH2); 1640 and 1475 cm−1 (aromatic C=C); and 1119 to 1279 cm−1 (C–F). 1H-NMR (DMSO-d6, 400 MHz): 7.17 ppm (s, 2H), 6.77 ppm (d, J = 8.0 Hz, 2H), 6.50 ppm (d, J = 8.0 Hz, 2H), 5.95 ppm (s, NH2), and 5.08 ppm (s, NH2). 13C-NMR (DMSO-d6, 100 MHz): 148.19, 147.92, 131.13, and 131.02 ppm (q, J = 27.5 Hz); 126.00 and 123.70 ppm (q, J = 275.0 Hz); 121.35 and 113.23 ppm (q, J = 6.2 Hz); 112.40 ppm. Liquid chromatography–mass spectrometry (LC-MS): m/z (mass/charge ratio): calcd.: 320.07; found: 321.08 [M]. Anal. calcd. for C14H10F6N2: C, 52.51; H, 3.15; N, 8.75. Found: C, 53.71; H, 3.04; N, 8.74.
2,6-Bis(trifluoromethyl)-4,4′-bis(trimellitimido)biphenyl (uDAc)
A flask was charged with a mixture of 1.50 g (4.68 mmol) of uDA, 1.81 g (9.44 mmol) of trimellitic anhydride, and 20 ml of glacial acetic acid. The heterogeneous mixture was refluxed for 21.5 hours. The reaction mixture was precipitated in methanol and filtered to yield a white solid, which was rinsed with methanol several times (2.74 g, 4.10 mmol; yield, 87.6%). m.p. 361° and 362°C. FTIR (KBr): 2885 to 3436 cm−1 (COOH); 1785 cm−1 [asymmetrical (asym) C=O stretch (str)]; 1728 cm−1 [symmetrical (sym) C=O str]; 1475 cm−1 (aromatic C=C); 1383 cm−1 (C–N str); 1217, 1191, and 1132 cm−1 (C–F in CF3); and 728 cm−1 (imide ring deformation). 1H-NMR (DMSO-d6, 400 MHz): 13.80 ppm (broad, COOH), 8.46 ppm (dd, J = 7.8, 1.4 Hz, 1H), 8.43 ppm (dd, J = 7.8, 1.4 Hz, 1H), 8.37 ppm (dd, J = 1.4, 0.8 Hz, 1H), 8.37 ppm (s, 2H), 8.33 ppm (dd, J = 1.4, 0.7 Hz, 1H), 8.16 ppm (dd, J = 7.7, 0.7 Hz, 1H), 8.11 ppm (dd, J = 7.7, 0.7 Hz, 1H), and 7.57 ppm (s, 4H). 13C-NMR (DMSO-d6, 100 MHz): 166.08, 166.07, 165.76, 165.72, 165.71, 165.68, 138.24, 136.81, 136.51, 135.74, 135.47, 134.85, 134.74, 132.43, 132.29, 132.15, 132.01, 131.94, and 130.61 ppm (q, J = 29.7 Hz); 130.34 and 128.35 ppm (q, J = 5.7 Hz); 125.45, 124.10, 123.83, 123.58, 123.38, and 122.78 ppm (q, J = 274.9 Hz). LC-MS: m/z: Calcd.: 668.07; found: 691.05 [M + Na+]. Anal. calcd. for C32H14F6N2O8: C, 57.50; H, 2.11; N, 4.19. Found: C, 57.36; H, 1.91; N, 4.26.
2,2′-Bis(trifluoromethyl)-4,4′-bis(trimellitimido)biphenyl (sDAc)
This compound was prepared according to the same procedure with that of uDAc. A flask was charged with a mixture of 1.51 g (4.72 mmol) of sDA, 1.87 g (9.72 mmol) of trimellitic anhydride, and 35 ml of glacial acetic acid. The heterogeneous mixture was refluxed for 23.5 hours. The reaction mixture was precipitated in methanol and filtered to yield a white solid, which was rinsed with methanol several times (1.24 g, 1.86 mmol; yield, 39.4%). m.p. 340° and 341°C. 1H-NMR (DMSO-d6, 400 MHz): 13.82 ppm (broad, COOH), 8.45 ppm (dd, J = 7.8, 1.4 Hz, 2H), 8.35 ppm (dd, J = 1.4, 0.7 Hz, 2H), 8.14 ppm (dd, J = 7.7, 0.7 Hz, 2H), 8.07 ppm (d, J = 2.1 Hz, 2H), 7.88 ppm (dd, J = 8.2, 2.1 Hz, 2H), and 7.70 ppm (d, J = 8.4 Hz, 2H). 13C-NMR (DMSO-d6, 100 MHz): 166.38 ppm (d, J = 1.7 Hz); 166.21, 137.10, 136.06, 135.88, 135.30, 132.96, 132.83, 132.48, 130.47, and 128.26 ppm (q, J = 31.0 Hz); 124.98, 124.44, 123.95, and 123.87 ppm (q, J = 274.4 Hz). LC-MS: m/z: calcd.: 668.07; found: 691.06 [M + Na+]. Anal. calcd. for C32H14F6N2O8: C, 57.50; H, 2.11; N, 4.19. Found: C, 57.12; H, 2.41; N, 4.29.
Polymerization
PAI(u-u)
A typical polymerization procedure is as follows: 25-ml three-necked round-bottom flask (RBF) equipped with a nitrogen inlet, a Dean-Stark trap, and a mechanical stirrer was charged with uDA (0.1923 g, 0.600 mmol), uDAc (0.4015 g, 0.600 mmol), 0.3 g of calcium chloride, 1 ml of triphenyl phosphite, 1 ml of pyridine, and 6 ml of NMP. The reaction mixture was heated and stirred at 100°C for 8 hours, under nitrogen atmosphere. As the polycondensation proceeded, the reaction mixture became viscous gradually, and small amount of NMP (total additional volume, 9 ml) was added periodically during this time to maintain the viscosity of the reaction mixture. At the end of the reaction, the polymer solution was poured slowly into 300 ml of methanol under rapid stirring. The product was filtered, washed thoroughly with methanol, and dried under vacuum at 180°C overnight. The yield was 100%. The inherent viscosity of the polymer in NMP was 3.50 dl g−1, measured at a concentration of 0.5 g dl−1 at 30°C. FTIR (thin film): 3344 cm−1 (NH str); 1784 cm−1 (asym C=O str of imide); 1732 cm−1 (sym C=O str of imide); 1686 cm−1 (C=O str of amide); 1475 to 1599 cm−1 (aromatic C=C); 1373 cm−1 (C–N str); 1190 and 1134 cm−1 (C–F in CF3); and 725 cm−1 (imide ring deformation). 1H-NMR (DMSO-d6, 400 MHz, 100°C): 11.02 ppm (s, 1H), 10.57 to 10.56 ppm (m, 1H), 8.74 to 8.46 ppm (m, 6H), 8.41 ppm (s, 2H), 8.19 ppm (m, 2H), 7.91 ppm (d, J = 8.0 Hz, 2H), 7.65 ppm (d, J = 7.2 Hz, 2H), 7.57 ppm (d, J = 8.0 Hz, 2H), and 7.32 ppm (d, J = 8.0 Hz, 2H). Anal. calcd. for C46H20F12N4O6: C, 57.99; H, 2.12; N, 5.88. Found: C, 57.63; H, 2.36; N, 5.61.
PAI(s-s)
The same procedure used for PAI(u-u) was repeated with sDA (0.1890 g, 0.590 mmol), sDAc (0.3945 g, 0.590 mmol), 0.3 g of calcium chloride, 1 ml of triphenyl phosphite, 1 ml of pyridine, and 6 ml of NMP. As the polycondensation proceeded, small amount of NMP (total additional volume, 8 ml) was added periodically to maintain the viscosity of the reaction mixture. The yield was 100%. The inherent viscosity of the polymer in NMP was 3.76 dl g−1, measured at a concentration of 0.5 g dl−1 at 30°C. FTIR (thin film): 3356 cm−1 (NH str), 1783 cm−1 (asym C=O str of imide), 1730 cm−1 (sym C=O str of imide), 1689 cm−1 (C=O str of amide), 1491 to 1594 cm−1 (aromatic C=C), 1370 cm−1 (C–N str), 1173 and 1135 cm−1 (C–F in CF3), and 727 cm−1 (imide ring deformation). 1H-NMR (DMSO-d6, 400 MHz, 100°C): 10.72 ppm (s, 2H), 8.64 ppm (s, 2H), 8.56 ppm (d, J = 7.6 Hz, 2H), 8.36 ppm (s, 2H), 8.24 to 8.13 ppm (m, 4H), 8.12 ppm (s, 2H), 7.95 ppm (d, J = 8.7 Hz, 2H), 7.67 ppm (d, J = 8.7 Hz, 2H), and 7.42 ppm (d, J = 8.6 Hz, 2H). Anal. calcd. for C46H20F12N4O6: C, 57.99; H, 2.12; N, 5.88. Found: C, 57.52; H, 2.29; N, 6.05.
PAI(s-u)
The same procedure used for PAI(u-u) was repeated with sDA (0.1909 g, 0.596 mmol), uDAc (0.3984 g, 0.596 mmol), 0.3 g of calcium chloride, 1 ml of triphenyl phosphite, 1 ml of pyridine, and 15 ml of NMP. In this case, because the reaction mixture showed proper viscosity, no additional amount of NMP was added into the reaction mixture. The yield was 100%. The inherent viscosity of the polymer in NMP was 3.98 dl g−1, measured at a concentration of 0.5 g dl−1 at 30°C. FTIR (thin film): 3352 cm−1 (NH str), 1784 cm−1 (asym C=O str of imide), 1732 cm−1 (sym C=O str of imide), 1686 cm−1 (C=O str of amide), 1476 to 1595 cm−1 (aromatic C=C), 1377 cm−1 (C–N str), 1174 and 1131 cm−1 (C–F in CF3), and 725 cm−1 (imide ring deformation). 1H-NMR (DMSO-d6, 400 MHz, 100°C): 10.73 ppm (s, 2H), 8.74 to 8.49 ppm (m, 4H), 8.41 ppm (s, 2H), 8.36 ppm (s, 2H), 8.18 ppm (m, 4H), 7.66 ppm (d, J = 8.3 Hz, 2H), 7.56 ppm (d, J = 8.6 Hz, 2H), and 7.42 ppm (d, J = 8.4 Hz, 2H). Anal. calcd. for C46H20F12N4O6: C, 57.99; H, 2.12; N, 5.88. Found: C, 57.27; H, 2.09; N, 5.76.
PAI(u-s)
The same procedure used for PAI(u-u) was repeated with uDA (0.1906 g, 0.595 mmol), sDAc (0.3980 g, 0.595 mmol), 0.3 g of calcium chloride, 1 ml of triphenyl phosphite, 1 ml of pyridine, and 6 ml of NMP. As the polycondensation proceeded, the reaction mixture became viscous gradually, and small amount of NMP (total additional volume, 6 ml) was added periodically to maintain the viscosity of the reaction mixture. The yield was 100%. The inherent viscosity of the polymer in NMP was 3.02 dl g−1, measured at a concentration of 0.5 g dl−1 at 30°C. FTIR (thin film): 3353 cm−1 (NH str), 1784 cm−1 (asym C=O str of imide), 1731 cm−1 (sym C=O str of imide), 1689 cm−1 (C=O str of amide), 1476 to 1598 cm−1 (aromatic C=C), 1366 cm−1 (C–N str), 1179 and 1136 cm−1 (C–F in CF3), and 725 cm−1 (imide ring deformation). 1H-NMR (DMSO-d6, 400 MHz, 100°C): 10.96 ppm (s, 1H), 10.48 ppm (s, 1H), 8.76 to 8.43 ppm (m, 6H), 8.29 to 8.05 ppm (m, 4H), 7.92 ppm (dd, J = 20.8, 8.6 Hz, 4H), 7.67 ppm (d, J = 8.8 Hz, 2H), and 7.32 ppm (d, J = 8.6 Hz, 2H). Anal. calcd. for C46H20F12N4O6: C, 57.99; H, 2.12; N, 5.88. Found: C, 57.03; H, 2.03; N, 5.79.
PAI(s-su)
The same procedure used for PAI(u-u) was repeated with sDA (0.1906 g, 0.595 mmol), uDAc (0.1989 g, 0.298 mmol), sDAc (0.1992 g, 0.298 mmol), 0.3 g of calcium chloride, 1 ml of triphenyl phosphite, 1 ml of pyridine, and 6 ml of NMP. As the polycondensation proceeded, small amount of NMP (total additional volume, 10 ml) was added periodically to maintain the viscosity of the reaction mixture. The yield was 100%. The inherent viscosity of the polymer in NMP was 3.90 dl g−1, measured at a concentration of 0.5 g dl−1 at 30°C. FTIR (thin film): 3354 cm−1 (NH str), 1784 cm−1 (asym C=O str of imide), 1731 cm−1 (sym C=O str of imide), 1689 cm−1 (C=O str of amide), 1490 to 1594 cm−1 (aromatic C=C), 1373 cm−1 (C–N str), 1174 and 1134 cm−1 (C–F in CF3), and 726 cm−1 (imide ring deformation). 1H-NMR (DMSO-d6, 400 MHz, 100°C, ppm): 10.72 (s, 4H), 8.65 (d, J = 7.9 Hz, 4H), 8.56 (q, J = 6.8, 6.1 Hz, 4H), 8.41 (s, 2H), 8.36 (s, 4H), 8.21 to 8.14 (m, 8H), 8.11 (s, 2H), 7.95 (d, J = 8.5 Hz, 2H), 7.66 (d, J = 8.8 Hz, 4H), 7.56 (d, J = 8.3 Hz, 2H), 7.42 (d, J = 8.7 Hz, 4H). Anal. Calcd for C46H20F12N4O6: C, 57.99; H, 2.12; N, 5.88. Found: C, 57.46; H, 2.29; N, 5.82.
PAI(su-s)
The same procedure used for PAI(u-u) was repeated with uDA (0.0958 g, 0.299 mmol), sDA (0.0963 g, 0.301 mmol), sDAc (0.4009 g, 0.600 mmol), 0.3 g of calcium chloride, 1 ml of triphenyl phosphite, 1 ml of pyridine, and 6 ml of NMP. As the polycondensation proceeded, small amount of NMP (total additional volume, 6 ml) was added periodically to maintain the viscosity of the reaction mixture. The yield was 100%. The inherent viscosity of the polymer in NMP was 2.90 dl g−1, measured at a concentration of 0.5 g dl−1 at 30°C. FTIR (thin film): 3353 cm−1 (NH str), 1784 cm−1 (asym C=O str of imide), 1730 cm−1 (sym C=O str of imide), 1687 cm−1 (C=O str of amide), 1491 to 1596 cm−1 (aromatic C=C), 1368 cm−1 (C–N str), 1175 and 1134 cm−1 (C–F in CF3), and 726 cm−1 (imide ring deformation). 1H-NMR (DMSO-d6, 400 MHz, 100°C): 10.94 ppm (s, 1H), 10.72 ppm (s, 2H), 10.47 ppm (s, 1H), 8.70 to 8.59 ppm (m, 6H), 8.56 ppm (d, J = 7.1 Hz, 4H), 8.36 ppm (s, 2H), 8.22 to 8.14 ppm (m, 6H), 8.11 ppm (s, 4H), 7.95 ppm (d, J = 8.3 Hz, 4H), 7.89 ppm (d, J = 8.5 Hz, 2H), 7.66 ppm (d, J = 8.7 Hz, 4H), 7.41 ppm (d, J = 8.5 Hz, 2H), and 7.32 ppm (d, J = 8.8 Hz, 2H). Anal. calcd. for C46H20F12N4O6: C, 57.99; H, 2.12; N, 5.88. Found: C, 56.26; H, 2.24; N, 5.66.
PAI(su-u)
The same procedure used for PAI(u-u) was repeated with uDA (0.0958 g, 0.299 mmol), sDA (0.0958 g, 0.299 mmol), uDAc (0.4001 g, 0.599 mmol), 0.3 g of calcium chloride, 1 ml of triphenyl phosphite, 1 ml of pyridine, and 6 ml of NMP. As the polycondensation proceeded, small amount of NMP (total additional volume, 8 ml) was added periodically to maintain the viscosity of the reaction mixture. The yield was 100%. The inherent viscosity of the polymer in NMP was 3.62 dl g−1, measured at a concentration of 0.5 g dl−1 at 30°C. FTIR (thin film): 3353 cm−1 (NH str), 1784 cm−1 (asym C=O str of imide), 1732 cm−1 (sym C=O str of imide), 1691 cm−1 (C=O str of amide), 1476 to 1597 cm−1 (aromatic C=C), 1376 cm−1 (C–N str), 1189 and 1135 cm−1 (C–F in CF3), and 725 cm−1 (imide ring deformation). 1H-NMR (DMSO-d6, 400 MHz, 100°C): 10.94 ppm (s, 1H), 10.71 ppm (s, 2H), 10.46 ppm (s, 1H), 8.64 ppm (d, J = 10.7 Hz, 6H), 8.62 to 8.49 ppm (m, 4H), 8.41 ppm (s, 4H), 8.36 ppm (s, 2H), 8.27 to 8.08 ppm (m, 6H), 7.89 ppm (d, J = 8.3 Hz, 2H), 7.66 ppm (d, J = 7.5 Hz, 4H), 7.56 ppm (d, J = 8.6 Hz, 4H), 7.41 ppm (d, J = 8.3 Hz, 2H), and 7.32 ppm (d, J = 8.4 Hz, 2H). Anal. calcd. for C46H20F12N4O6: C, 57.99; H, 2.12; N, 5.88. Found: C, 57.48; H, 2.19; N, 5.62.
PAI(su-su)
The same procedure used for PAI(u-u) was repeated with uDA (0.0961 g, 0.300 mmol), sDA (0.0961 g, 0.300 mmol), uDAc (0.2002 g, 0.300 mmol), sDAc (0.2010 g, 0.301 mmol), 0.3 g of calcium chloride, 1 ml of triphenyl phosphite, 1 ml of pyridine, and 6 ml of NMP. As the polycondensation proceeded, small amount of NMP (total additional volume, 7 ml) was added periodically to maintain the viscosity of the reaction mixture. The yield was 100%. The inherent viscosity of the polymer in NMP was 3.40 dl g−1, measured at a concentration of 0.5 g dl−1 at 30°C. FTIR (thin film): 3354 cm−1 (NH str), 1784 cm−1 (asym C=O str of imide), 1731 cm−1 (sym C=O str of imide), 1689 cm−1 (C=O str of amide), 1476 to 1597 cm−1 (aromatic C=C), 1371 cm−1 (C–N str), 1177 and 1135 cm−1 (C–F in CF3), and 725 cm−1 (imide ring deformation). 1H-NMR (DMSO-d6, 400 MHz, 100°C): 10.94 ppm (s, 1H), 10.72 ppm (s, 2H), 10.47 ppm (s, 1H), 8.65 ppm (s, 6H), 8.56 ppm (d, J = 6.9 Hz, 4H), 8.41 ppm (s, 2H), 8.36 ppm (s, 2H), 8.24 to 8.13 ppm (m, 6H), 8.11 ppm (s, 2H), 7.95 ppm (d, J = 8.7 Hz, 2H), 7.89 ppm (d, J = 8.4 Hz, 2H), 7.66 ppm (d, J = 8.6 Hz, 4H), 7.56 ppm (d, J = 8.5 Hz, 2H), 7.41 ppm (d, J = 8.6 Hz, 2H), and 7.32 ppm (d, J = 8.7 Hz, 2H). Anal. calcd. for C46H20F12N4O6: C, 57.99; H, 2.12; N, 5.88. Found: C, 57.46; H, 2.23; N, 5.60.
Preparation of polymer films
DMAc solution of the polymers [7.5 weight % (wt %)] was prepared at room temperature. The DMAc solution was filtered and cast onto a glass plate. The solvent was evaporated in a vacuum oven at room temperature for 5 hours and then heated to 180°C for 10 hours to remove the residual solvent and yield a freestanding film with thicknesses of 70 to 80 μm. For refractive index measurements, the polymer films with thicknesses of 3 to 15 μm were prepared from 2.5 wt % DMAc solution.
Grazing incidence wide-angle x-ray scattering
GIWAXS experiments were performed at the 3C beamline in Pohang Accelerator Laboratory. A monochromatized x-ray radiation source of 10.2499 keV with a sample-to-detector distance of 210.5 mm was used. Scattering intensity was monitored by a Mar 165-mm-diameter CCD detector with 2048 by 2048 pixels. X-ray beam was set at 0.12 or 0.13 typically, which are between the critical angle of the PAI film and the silicon substrate. Scattering angles were corrected according to the positions of the x-ray beams with respect to a precalibrated sucrose. The scattering intensity profiles of in-plane and out-of-plane directions were extracted from the 2D scattering pattern. The magnitude of scattering vector was calculated with q = 4π/λsinθ with wavelength of x-ray (λ) and scattering angle (θ). Domain spacing (d) was estimated from the position of the specific peaks at q following the relationship d = 2/q.
Model compound syntheses and crystallographic characterization
Amide-uDA
A mixture of 3,5-bis(trifluoromethyl)benzidine (0.1988 g, 0.6211 mmol), triethylamine (0.1649 g, 1.630 mmol), and tetrahydrofuran (THF) (2 ml) was prepared in a 10-ml RBF. After stirring the mixture under nitrogen for 30 min, benzoyl chloride (0.2200 g, 1.5651 mmol) diluted with THF and was dropwise added to the RBF at 0°C. The reaction mixture kept stirring for overnight at room temperature. Then, the reaction mixture was poured into water, and the crude product was obtained by extraction with ethyl acetate and evaporation of the organic solvent. The product was further purified by recrystallization in methanol (0.2733 g, 0.517 mmol; yield, 82.7%). m.p. 309° and 310°C. FTIR (powder): 3350 and 3181 cm−1 (N–H str of amide), 1647 to 1649 cm−1 (C=O str of amide), 1594 and 1580 cm−1 (N–H rot of amide), 1466 to 1507 cm−1 (aromatic C=C), and 1133 to 1184 cm−1 (C–F in CF3). 1H-NMR (DMSO-d6, 400 MHz, 40°C): 10.87 ppm (s, 1H), 10.39 ppm (s, 1H), 8.65 ppm (s, 2H), 8.05 ppm (d, J = 9.6 Hz, 2H), 7.99 ppm (d, J = 8.8 Hz, 2H), 7.85 ppm (d, J = 11.2 Hz, 2H), 7.69 to 7.53 ppm (m, 6H), and 7.28 ppm (d, J = 11.2 Hz, 2H). LC-MS: m/z: calcd.: 528.13; found: 551.12 [M + Na+]. Anal. calcd. for C28H18F6N2O2: C, 63.64; H, 3.43; F, 21.57; N, 5.30. Found: C, 63.61; H, 4.05; N, 5.21.
Imide-uDA
A 5-ml of RBF was charged with 2,6-bis(trifluoromethyl)benzidine (50.4 mg, 0.157 mmol), phthalic anhydride (46.7 mg, 0.315 mmol), isoquinoline (one drop), and 1 ml of m-cresol. The reaction mixture was stirred at room temperature for 0.5 hours and at 190°C for 0.5 hours. The reaction mixture was poured into ethanol and then filtered (90.1 mg; yield, 98.9%). FTIR (KBr): 1711 cm−1 (C=O str), 1478 cm−1 (aromatic C=C), 1380 cm−1 (C–N str), 1123 to 1194 cm−1 (C–F in CF3), and 722 cm−1 (imide ring deformation). 1H-NMR (DMSO-d6, 400 MHz, 25°C): 8.36 ppm (s, 2H), 8.04 ppm (dd, J = 5.5, 3.0 Hz, 2H), 8.00 ppm (dd, J = 5.4, 3.1 Hz, 2H), 7.96 ppm (dd, J = 5.5, 3.1 Hz, 2H), 7.92 ppm (dd, J = 5.5, 3.1 Hz, 2H), and 7.56 ppm (d, J = 1.9 Hz, 4H). 1H-NMR (DMSO-d6, 400 MHz, 100°C): 8.36 ppm (s, 2H), 8.07 to 7.83 ppm (m, 8H), 7.59 ppm (d, J = 8.3 Hz, 2H), and 7.51 ppm (d, J = 8.1 Hz, 2H). 13C-NMR (DMSO-d6, 100 MHz, 25°C): 167.27, 166.92, 138.51, 135.46, 135.20, 133.10, 132.75, 132.63, 131.97, and 131.01 ppm (q, J = 29.7 Hz); 130.77 and 128.87 ppm (q, J = 5.5 Hz); and 125.96, 124.16, 123.92, and 123.28 (q, J = 274.9 Hz). LC-MS: m/z: calcd.: 580.09; found: 603.08 [M + Na+]. Anal. calcd. for C30H14F6N2O4: C, 62.08; H, 2.43; N, 4.83. Found: C, 61.89; H, 2.52; N, 4.62.
X-ray single crystallography
The single-crystal diffraction data of the model compounds were collected on a Bruker SMART 1000. A suitable size and quality of crystal was coated with Paratone-N oil and mounted on a MiTeGen MicroLoop. The data were collected with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 120 K. Cell parameters were determined and refined by the SMART program. Data reduction was performed using the SAINT software. An empirical absorption correction was applied using the SADABS program. The structure was solved by direct methods, and all nonhydrogen atoms were subjected to anisotropic refinement by full-matrix least squares on F2 by using the SHELXTL/PC package. Full crystallographic details can be obtained free of charge from the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif (CCDC 1587696 and 1587708).
Computational details
We searched various stacking structures between two monomers using the Monte Carlo basin-hopping (MCBH) sampling method (21) combined with density functional tight-binding (DFTB) calculations (22). The 3ob-3-1 pair potential parameters (23) were used for DFTB calculations. For each of PAI(s-s), PAI(s-u), PAI(u-s), and PAI(u-u), 600 stacking structures were first sampled using the MCBH method with constraints to keep stacking structures, and 10 most stable ones out of them were further optimized at the DFT level with the ωb97xd functional (24) and the 6-31g(d) basis set as implemented in Gaussian 09 package (25). The binding energy evaluation was also done by using the same functional and the 6-31+g(d,p) basis set, together with the Boys’ basis set superposition error correction scheme (26). We refer to figs. S25 to S28 and table S7 for the details of computational results. Figures S25 to S28 show the 10 most stable structures of PAI(s-s), PAI(s-u), PAI(u-s), and PAI(u-u), respectively, and the corresponding bonding energies are summarized in table S7. We chose PAI(s-s)-5, PAI(s-u)-1, PAI(u-s)-2, and PAI(u-u)-2 among them as the representatives shown in Fig. 3, because more stable ones in table S7 have intertwined structures and thus are inappropriate for the formation of layer-like packing of polymer chains proposed by the GIWAXS data.
Fabrication and characterization of IGZO TFTs
IGZO TFTs were fabricated on the PAI(s-u) as follows: 50-nm-thick IZO layers were first deposited as a transparent gate electrodes via RF magnetron sputtering. The substrates were held at 50°C during deposition. On top of the IZO layers, 170-nm-thick Al2O3 layers were grown by atomic layer deposition at 150°C. Then, 40-nm-thick IGZO film with the atomic composition of 1:1:2.5 (In/Ga/Zn) was deposited in the RF magnetron sputtering chamber at O2/(Ar + O2) ratio of 50%. Subsequently, IZO layers were deposited onto the active layer as transparent source/drain electrodes with a channel length of 200 μm and a width of 980 μm defined with a shadow mask. Last, the devices were passivated by fluoropolymer passivation layer known as CYTOP (Asahi Glass Co.) using spin coating (27). To get the high-quality oxide semiconductors, the IGZO TFTs on polymer substrates were annealed at 250°C in ambient air. Output and transfer electrical characteristics of IGZO TFTs were analyzed using a semiconductor parameter analyzer (HP4155A, Agilent Inc.) in a nitrogen-filled glove box. The flexural strain measurement of IGZO TFTs was tested after repeated bending cycles (10 times) at several bending radii with a custom-built bending tester. Then, the electrical characteristics of the IGZO TFTs were evaluated in the flat state.
Supplementary Material
Acknowledgments
We thank S. H. Kim for DMA. Funding: This work was supported by Samsung Research Funding and Incubation Center for Future Technology (SFRC) through contract number SRFC-MA1401-09. This work was also supported by National Research Foundation (NRF) of Korea (NRF-2015R1A2A1A10055222). Author contributions: S.D.K. and B.L. are coauthors. S.D.K., B.L., and T.B. synthesized and characterized all the monomers, polymers, and model compounds. I.S.C. prepared the asymmetric diamine monomer. I.S., N.Y.A., and M.S. did GIWAX experiments. Y.L. did XRD analysis. Y.K. and W.Y.K. did DFT calculations. H.K., H.M., and S.Y. fabricated and characterized the IGZO TFT device. M.S. prepared the manuscript, and S.Y.K. revised the manuscript. S.Y.K. proposed the work and led the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplementary materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaau1956/DC1
Fig. S1. Synthetic route to uDA.
Fig. S2. 1H NMR spectra of the nitro intermediate 2 and uDA.
Fig. S3. 13C NMR spectra of the nitro intermediate 2 and uDA.
Fig. S4. FTIR spectra of the nitro intermediate 2 and uDA.
Fig. S5. Synthetic route to diacids.
Fig. S6. 1H NMR spectra of the diacids uDAc and sDAc.
Fig. S7. 13C NMR spectra of the diacids uDAc and sDAc.
Fig. S8. FTIR spectrum of uDAc.
Fig. S9. 1H-NMR spectra of PAIs.
Fig. S10. Photos of PAI films.
Fig. S11. UV-vis spectra of PAIs.
Fig. S12. TGA data of PAIs.
Fig. S13. DSC data of representative PAIs.
Fig. S14. DMA data of PAIs.
Fig. S15. TMA data of PAIs.
Fig. S16. 2D GIWAXS data of PAIs.
Fig. S17. 1D deconvoluted plot of the GIWAXS data in out-of-plane direction.
Fig. S18. 1D deconvoluted plot of the GIWAXS data in in-plane direction.
Fig. S19. A plot of CTE and π-π stacking distance versus β-relaxation temperature determined by the DMA.
Fig. S20. Synthetic route to model compound amide-uDA and imide-uDA.
Fig. S21. 1H NMR spectra of model compounds amide-uDA and imide-uDA.
Fig. S22. 13C NMR spectra of model compound amide-uDA and imide-uDA.
Fig. S23. FTIR spectra of model compound amide-uDA and imide-uDA.
Fig. S24. Chemical and crystal structures of model compound amide-uDA and imide-uDA.
Fig. S25. Ten most stable structures of PAI(s-s).
Fig. S26. Ten most stable structures of PAI(s-u).
Fig. S27. Ten most stable structures of PAI(u-s).
Fig. S28. Ten most stable structures of PAI(u-u).
Fig. S29. Transfer characteristics of the IGZO TFTs under study.
Table S1. Solubility of PAIs
Table S2. Birefringence of PAIs
Table S3. Transition temperatures of PAIs identified by the DMA
Table S4. Summary of the GIWAXS peak positions
Tables S5. Crystal data and structure refinement for amide-uDA
Tables S6. Crystal data and structure refinement for imide-uDA
Table S7. Bonding energies and geometric features of the 10 most stable dimeric structures for each of PAI(s-s), PAI(s-u), PAI(u-s), and PAI(u-u)
REFERENCES AND NOTES
- 1.MacDonald W. A., Looney M. K., MacKerron D., Eveson R., Adam R., Hashimoto K., Rakos K., Latest advances in substrates for flexible electronics. J. Soc. Inf. Disp. 15, 1075–1083 (2007). [Google Scholar]
- 2.D. R. Askeland, P. P. Fulay, Essentials of Materials Science and Engineering (Cengage Learning, ed. 2, 2009). [Google Scholar]
- 3.N. W. Ashcroft, N. D. Mermin, Solid State Physics (Holt, Rinehart & Winston, 1976). [Google Scholar]
- 4.Jin J., Ko J.-H., Yang S., Bae B.-S., Rollable transparent glass-fabric reinforced composite substrate for flexible devices. Adv. Mater. 22, 4510–4515 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Kim J.-S., Yang S., Bae B.-S., Thermally stable transparent sol–gel based siloxane hybrid material with high refractive index for light emitting diode (LED) encapsulation. Chem. Mater. 22, 3549–3555 (2010). [Google Scholar]
- 6.Zhi C., Bando Y., Terao T., Tang C., Kuwahara H., Golberg D., Towards thermoconductive, electrically insulating polymeric composites with boron nitride nanotubes as fillers. Adv. Funct. Mater. 19, 1857–1862 (2009). [Google Scholar]
- 7.Wang S., Liang Z., Gonnet P., Liao Y. H., Wang B., Zhang C., Effect of nanotube functionalization on the coefficient of thermal expansion of nanocomposites. Adv. Funct. Mater. 17, 87–92 (2007). [Google Scholar]
- 8.Matsuura T., Hasuda Y., Nishi S., Yamada N., Polyimide derived from 2,2′-bis(trifluoromethyl)-4,4′-diaminobiphenyl. 1. Synthesis and characterization of polyimides prepared with 2,2′-bis(3,4-dicarboxyphenyl)hexafluoropropane dianhydride or pyromellitic dianhydride. Macromolecules 24, 5001–5005 (1991). [Google Scholar]
- 9.Feiring A. E., Auman B. C., Wonchoba E. R., Synthesis and properties of fluorinated polyimides from novel 2,2′-bis(fluoroalkoxy)benzidines. Macromolecules 26, 2779–2784 (1993). [Google Scholar]
- 10.Lin S.-H., Li F., Cheng S. Z. D., Harris F. W., Organo-soluble polyimides: Synthesis and polymerization of 2,2′-bis(trifluoromethyl)-4,4′,5,5′-biphenyltetracarboxylic dianhydride. Macromolecules 31, 2080–2086 (1998). [Google Scholar]
- 11.Ii T., Tashiro K., Kobayashi M., Tadokoro H., Thermomechanical and ultrasonic properties of high-modulus aromatic polyamide fibers. Macromolecules 19, 1809–1814 (1986). [Google Scholar]
- 12.Ando S., Sekiguchi K., Mizoroki M., Okada T., Ishige R., Anisotropic linear and volumetric thermal-expansion behaviors of self-standing polyimide films analyzed by thermomechanical analysis (TMA) and optical interferometry. Macromol. Chem. Phys. 219, 1700354–1700363 (2018). [Google Scholar]
- 13.Rogers H. G., Gaudiana R. A., Hollinsed W. C., Kalyanaraman P. S., Manello J. S., McGowan C., Minns R. A., Sahatjian R., Highly amorphous, birefringent, para-linked aromatic polyamides. Macromolecules 18, 1058–1068 (1985). [Google Scholar]
- 14.Hasegawa M., Ishigami T., Ishii J., Sugiura K., Fujii M., Solution-processable transparent polyimides with low coefficients of thermal expansion and self-orientation behavior induced by solution casting. Eur. Polym. J. 49, 3657–3672 (2013). [Google Scholar]
- 15.Wakita J., Jin S., Shin T. J., Ree M., Ando S., Analysis of molecular aggregation structures of fully aromatic and semialiphatic polyimide films with synchrotron grazing incidence wide-angle x-ray scattering. Macromolecules 43, 1930–1941 (2010). [Google Scholar]
- 16.Kochi M., Chen C., Yokota R., Hasegawa M., Hergenrother P., Isomeric biphenyl polyimides. (II) Glass transitions and secondary processes. High Perform. Polym. 17, 335–347 (2005). [Google Scholar]
- 17.Sekiguchi K., Takizawa K., Ando S., Thermal expansion behavior of the ordered domain in polyimide films investigated by variable temperature WAXD measurements. J. Photopolym. Sci. Technol. 26, 327–332 (2013). [Google Scholar]
- 18.Nomura K., Ohta H., Takagi A., Kamiya T., Hirano M., Hosono H., Room-temperature fabrication of transparent flexible thin-film transistors using amorphous oxide semiconductors. Nature 432, 488–492 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Mativenga M., Choi M. H., Choi J. W., Jang J., Transparent flexible circuits based on amorphous-indium–gallium–zinc–oxide thin-film transistors. IEEE Electron Device Lett. 32, 170–172 (2011). [Google Scholar]
- 20.Kim S. D., Ka D., Chung I. S., Kim S. Y., Poly(arylene ether)s with low refractive indices: Poly(biphenylene oxide)s with trifluoromethyl pendant groups via a meta-activated nitro displacement reaction. Macromolecules 45, 3023–3031 (2012). [Google Scholar]
- 21.Kim Y., Choi S., Kim W. Y., Efficient basin-hopping sampling of reaction intermediates through molecular fragmentation and graph theory. J. Chem. Theory Comput. 10, 2419–2426 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Aradi B., Hourahine B., Frauenheim T., DFTB+, a sparse matrix-based implementation of the DFTB method. J. Phys. Chem. A 111, 5678–5684 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Kubillus M., Kubař T., Gaus M., Řezáč J., Elstner M., Parameterization of the DFTB3 method for Br, Ca, Cl, F, I, K, and Na in organic and biological systems. J. Chem. Theory Comput. 11, 332–342 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Chai J.-D., Head-Gordon M., Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008). [DOI] [PubMed] [Google Scholar]
- 25.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09 Revision D.01 (Gaussian Inc., Wallingford, CT, 2009).
- 26.Boys S. F., Bernardi F., The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 19, 553–566 (1970). [Google Scholar]
- 27.Choi S.-H., Kang J.-H., Kim J.-J., Han M.-K., Low-temperature organic (CYTOP) passivation for improvement of electric characteristics and reliability in IGZO TFTs. IEEE Electron Device Lett. 33, 381–383 (2012). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaau1956/DC1
Fig. S1. Synthetic route to uDA.
Fig. S2. 1H NMR spectra of the nitro intermediate 2 and uDA.
Fig. S3. 13C NMR spectra of the nitro intermediate 2 and uDA.
Fig. S4. FTIR spectra of the nitro intermediate 2 and uDA.
Fig. S5. Synthetic route to diacids.
Fig. S6. 1H NMR spectra of the diacids uDAc and sDAc.
Fig. S7. 13C NMR spectra of the diacids uDAc and sDAc.
Fig. S8. FTIR spectrum of uDAc.
Fig. S9. 1H-NMR spectra of PAIs.
Fig. S10. Photos of PAI films.
Fig. S11. UV-vis spectra of PAIs.
Fig. S12. TGA data of PAIs.
Fig. S13. DSC data of representative PAIs.
Fig. S14. DMA data of PAIs.
Fig. S15. TMA data of PAIs.
Fig. S16. 2D GIWAXS data of PAIs.
Fig. S17. 1D deconvoluted plot of the GIWAXS data in out-of-plane direction.
Fig. S18. 1D deconvoluted plot of the GIWAXS data in in-plane direction.
Fig. S19. A plot of CTE and π-π stacking distance versus β-relaxation temperature determined by the DMA.
Fig. S20. Synthetic route to model compound amide-uDA and imide-uDA.
Fig. S21. 1H NMR spectra of model compounds amide-uDA and imide-uDA.
Fig. S22. 13C NMR spectra of model compound amide-uDA and imide-uDA.
Fig. S23. FTIR spectra of model compound amide-uDA and imide-uDA.
Fig. S24. Chemical and crystal structures of model compound amide-uDA and imide-uDA.
Fig. S25. Ten most stable structures of PAI(s-s).
Fig. S26. Ten most stable structures of PAI(s-u).
Fig. S27. Ten most stable structures of PAI(u-s).
Fig. S28. Ten most stable structures of PAI(u-u).
Fig. S29. Transfer characteristics of the IGZO TFTs under study.
Table S1. Solubility of PAIs
Table S2. Birefringence of PAIs
Table S3. Transition temperatures of PAIs identified by the DMA
Table S4. Summary of the GIWAXS peak positions
Tables S5. Crystal data and structure refinement for amide-uDA
Tables S6. Crystal data and structure refinement for imide-uDA
Table S7. Bonding energies and geometric features of the 10 most stable dimeric structures for each of PAI(s-s), PAI(s-u), PAI(u-s), and PAI(u-u)