

Abstract
Balanced general anesthesia, the most common management strategy used in anesthesia care, entails the administration of different drugs together to create the anesthetic state. Anesthesiologists developed this approach to avoid sole reliance on ether for general anesthesia maintenance. Balanced general anesthesia uses less of each drug than if the drug were administered alone, thereby increasing the likelihood of its desired effects and reducing the likelihood of its side effects. To manage nociception intraoperatively and pain postoperatively, the current practice of balanced general anesthesia relies almost exclusively on opioids. While opioids are the most effective antinociceptive agents, they have undesirable side effects. Moreover, overreliance on opioids has contributed to the opioid epidemic in the United States. Spurred by concern of opioid overuse, balanced general anesthesia strategies are now using more agents to create the anesthetic state. Under these approaches, called “multimodal general anesthesia,” the additional drugs may include agents with specific central nervous system targets such as dexmedetomidine and ones with less specific targets, such as magnesium. It is postulated that use of more agents at smaller doses further maximizes desired effects while minimizing side effects. Although this approach appears to maximize the benefit-to-side effect ratio, no rational strategy has been provided for choosing the drug combinations. Nociception induced by surgery is the primary reason for placing a patient in a state of general anesthesia. Hence, any rational strategy should focus on nociception control intraoperatively and pain control postoperatively. In this Special Article, we review the anatomy and physiology of the nociceptive and arousal circuits, and the mechanisms through which commonly used anesthetics and anesthetic adjuncts act in these systems. We propose a rational strategy for multimodal general anesthesia predicated on choosing a combination of agents that act at different targets in the nociceptive system to control nociception intraoperatively and pain postoperatively. Because these agents also decrease arousal, the doses of hypnotics and/or inhaled ethers needed to control unconsciousness are reduced. Effective use of this strategy requires simultaneous monitoring of antinociception and level of unconsciousness. We illustrate the application of this strategy by summarizing anesthetic management for 4 representative surgeries.
General anesthesia is a drug-induced reversible state consisting of unconsciousness, amnesia, antinociception, and immobility, with maintenance of physiological stability.1 Balanced general anesthesia, the most common management strategy used in anesthesia care, entails administering a combination of different agents to create the anesthetic state. Anesthesiologists developed this approach to avoid sole reliance on ether for maintenance of general anesthesia.2 There is evidence that balanced general anesthesia uses less of each drug than if the drug were administered alone.3 This approach is believed to increase the likelihood of a drug’s desired effects and reduce the likelihood of its side effects.
Current practice of balanced general anesthesia relies on a hypnotic, such as propofol, for induction and on an inhaled ether or on a hypnotic infusion to maintain unconsciousness. Although midazolam is often administered before induction to treat anxiety, amnesia is managed implicitly by rendering the patient unconscious. And while muscle relaxants are administered to produce immobility, administration of propofol and of inhaled ethers also contributes to muscle relaxation. To date, balanced general anesthesia has relied almost exclusively on opioids administered as intermittent boluses or as continuous infusions to manage nociception intraoperatively and pain postoperatively.
We distinguish here between nociception and pain. Nociception is the propagation through the sensory system of potentially noxious and harmful stimuli, whereas pain is the conscious perception of nociceptive information.4 For example, if a patient is unconscious after receiving only propofol and has an increase in heart rate and blood pressure in response to the surgical incision, then this is an example of nociception. If a surgeon makes an incision to create a dialysis fistula after an inadequate administration of local anesthesia for a field block and the patient says, “Ouch,” then this is pain. The heart rate and blood pressure most certainly would go up as a physiological response. Someone monitoring the vital signs but unable to hear the patient would appreciate the patient’s nociceptive response.
Nociception induced by surgery, due to tearing of tissue and inflammation, is the primary reason for placing a patient in a state of general anesthesia.5 If not controlled, nociceptive perturbations are also the primary source of hemodynamic and stress responses intraoperatively and of chronic pain syndromes postoperatively. While opioids are the most effective antinociceptive agents, they have undesirable side effects, including respiratory depression, nausea, vomiting, urinary retention, constipation, ileus, and pruritus.6 Overreliance on opioids has certainly contributed to the opioid epidemic in the United States.7 Propofol and the inhaled ethers also contribute to antinociception by maintaining unconsciousness, and thereby altering perception of nociceptive stimuli.
Spurred by concerns about opioid overuse and their undesirable side effects, strategies for balanced general anesthesia are now using multiple agents in addition to or in lieu of opioids to manage the nociceptive component of the anesthetic state. Under this approach, called “multimodal general anesthesia,” the additional drugs may include agents with specific central nervous system targets such as dexmedetomidine8 as well as ones whose targets are less specific such as lidocaine.9 It is postulated that the use of more agents at smaller doses further maximizes desired effects while minimizing side effects.10 And while the multimodal approach appears to maximize the benefit-to-side effect ratio, no rational strategy for choosing the drug combinations has been proposed.
We propose that a rational strategy for multimodal general anesthesia should: (1) administer combinations of antinociceptive agents chosen so that each one targets a different circuit in the nociceptive system; (2) monitor continuously levels of antinociception and unconsciousness; (3) use explicitly the sedative effects of the antinociceptive agents to reduce the doses of hypnotic agents and inhaled anesthetics administered to maintain unconsciousness; and (4) continue multimodal pain control during the in-hospital postoperative period and after discharge.
We review the anatomy and physiology of the parts of nociceptive and arousal systems, and the mechanisms through which commonly used anesthetic and nonanesthetic drugs act in these systems. We show that understanding these systems can be used to formulate a rational strategy for multimodal general anesthesia management. We illustrate the new strategy by summarizing anesthetic management for 4 representative surgeries.
MULTIMODAL GENERAL ANESTHESIA: THEORY
The nociceptive system of the body consists of the nociceptors, the ascending nociceptive pathways, and the descending nociceptive pathways (Figure 1).4,11 Nociceptors are unspecialized, bare nerve cell endings located in peripheral tissue and in the viscera that initiate nociception or pain (Figure 1A). The cell bodies arise in the dorsal horn of the spinal cord and send one axonal process to the periphery and the other to the spinal cord or brainstem. The ascending pathways transmit nociceptive stimuli from the periphery to the spinal cord to the brainstem (medulla and midbrain), the amygdala, the thalamus, and on to the primary and secondary sensory cortices. The descending nociceptive pathways begin in the sensory cortex and project to the hypothalamus and amygdala. The projections from the hypothalamus and amygdala synapse in the periaqueductal gray in the midbrain, and the nucleus of the tracts solitarius and the rostral ventral medulla in the medulla. The periaqueductal gray projects to the spinal cord primarily through the rostra ventral medulla. The descending pathways are activated immediately by the ascending nociceptive ascending pathways and modulate (upregulate and downregulate) nociceptive information transmission (Figure 1B).
Figure 1.
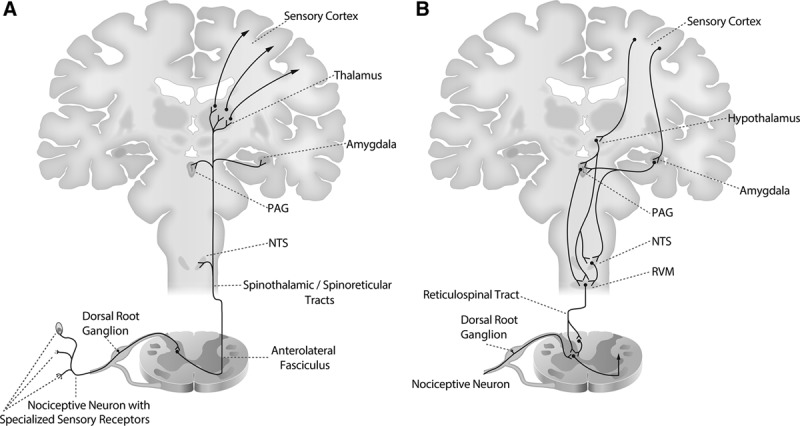
Ascending and descending nociception pathways. A, Nociceptive signals enter the spinal cord through nociceptive neurons that have specialized sensory receptors which lie in the tissue and cell bodies which lie in the dorsal root ganglia. These neurons synapse in the dorsal horn of the spinal cord onto primary projection neurons that travel in the anterolateral fasciculus through the spinal reticular tract (to the NTS and the amygdala) and spinal thalamic tract (to the thalamus). Projections from the thalamus continue to primary sensory cortex. B, The descending pathways begin in the sensory cortex and project to the hypothalamus and amygdala. Projections from the hypothalamus and amygdala synapse in the PAG, NTS, and RVM. Projections from the RVM carried in the reticular spinal tract modulate incoming nociceptive information by synapsing onto inputs to nociceptive neurons at the level of the dorsal horn. NTS indicates nucleus of the tractus solitarius; PAG, periaqueductal gray; RVM, rostral ventral medulla.
Because there are multiple different neurotransmitters and neural relays in the ascending and descending pathways, there are multiple targets at which antinociceptive agents can act to disrupt nociceptive information processing. Targeting simultaneously multiple targets in the nociceptive system is the key concept underlying the design of a multimodal strategy for nociceptive control, and hence, multimodal general anesthesia. We focus our discussion of antinociceptive agents on opioids, ketamine, magnesium, dexmedetomidine, nonsteroidal anti-inflammatory drugs (NSAIDs), and the local anesthetic lidocaine. The nociceptive pathways have strong connections with the arousal pathways, which is why administering antinociceptive agents decreases arousal. We focus our discussion of hypnotic agents on propofol and sevoflurane.
Antinociceptive Agents
Opioids.
Opioids, the primary class of drugs used as antinociceptive agents, target multiple classes of opioid receptors in the periaqueductal gray, the spinal cord, amygdala, rostral ventral medulla, and cortex (Figure 2).12–15 Binding to opioid receptors disrupts information trans mission in the nociceptive circuits by decreasing conductance of voltage-gated calcium channels and opening of inward-rectifying potassium channels.16 Activation of the opioid receptors has 2 principal effects on nociceptive information transmission: blocking afferent nociceptive inputs into the spinal cord; and enhancing descending inhibition of nociceptive inputs beginning at the level of the periaqueductal gray. These descending projections originate in the periaqueductal gray and project to the spinal cord through synapses in the rostral ventral medulla. These 2 effects of opioids decrease nociceptive information processing. In contrast, action of the opioids in the amygdala decreases nociceptive perception and the emotional effect of nociceptive and pain stimulation.17,18 These multisite antinociceptive effects are also one of the mechanisms through which opioids decrease arousal. Opioids also decrease arousal through their inhibitory actions on brainstem cholinergic circuits at the level of the lateral dorsal tegmental nucleus, the pedunculopontine tegmental nucleus, the median pontine reticular formation, and the thalamus.19,20 Opioids enhance cholinergic input to the sinoatrial node, induce bradycardia, and thereby, mitigate sympathetic responses associated with nociception.21
Figure 2.
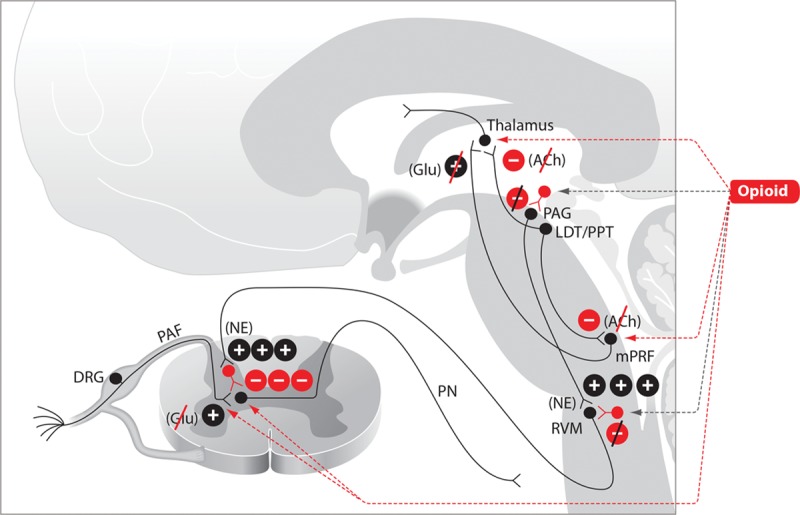
Opioids. The mechanisms of opioid-induced antinociception are produced by opioid binding to opioid receptors in the brainstem and spinal cord. Opioid-induced decrease in arousal is produced by blockade of cholinergic arousal projections from the brainstem to the thalamus and cortex. The symbol  denotes an excitatory effect. The symbol
denotes an excitatory effect. The symbol  denotes an inhibitory effect. The symbols
denotes an inhibitory effect. The symbols  and
and  denote inhibition of the indicated effects. ACh indicates acetylcholine; DRG, dorsal root ganglia; Glu, glutamate; LDT, laterodorsal tegmental area; mPRF, medial pontine reticular formation; NE, norepinephrine; PAF, peripheral afferent fiber; PAG, periaqueductal gray; PN, projection neuron; PPT, pedunculopontine tegmental area; RVM, rostral ventral medulla.
denote inhibition of the indicated effects. ACh indicates acetylcholine; DRG, dorsal root ganglia; Glu, glutamate; LDT, laterodorsal tegmental area; mPRF, medial pontine reticular formation; NE, norepinephrine; PAF, peripheral afferent fiber; PAG, periaqueductal gray; PN, projection neuron; PPT, pedunculopontine tegmental area; RVM, rostral ventral medulla.
Ketamine.
The principal antinociceptive effects of ketamine are due to its targeting of N-methyl-d-aspartate (NMDA) glutamate receptors located on peripheral afferent nociceptive neurons that synapse in the dorsal horn of the spinal cord (Figure 3).22 Glutamate is the primary excitatory neurotransmitter in the nervous system. Blocking nociceptive inputs at this juncture impedes the entrance of these signals into the spinal cord. The actions of ketamine at NMDA receptors in the cortex and other parts of the arousal system contribute both to its antinociceptive effects and to its capacity to decrease arousal.8 At low doses, ketamine binds preferentially to NMDA receptors on γ-aminobutyric acid-ergic (GABAergic) inhibitory interneurons, leading to general disinhibition of pyramidal neurons and diffuse excitatory cortical activity.23,24 This activity is marked by gamma oscillations (25–30 Hz) in the electroencephalogram and an altered state of arousal that commonly includes hallucination.1,25 At higher doses, ketamine also blocks NMDA receptors on excitatory pyramidal neurons. Pyramidal neurons with NMDA receptors can be found in many locations throughout the brain and central nervous system. However, key targets for altering arousal are the glutamatergic projections from the parabrachial nucleus and from the medial pontine reticular formation in the brainstem to the thalamus and to the basal forebrain.26–28 These glutamatergic projections are very potent, excitatory arousal pathways. At higher ketamine doses, inactivation of these arousal pathways leads most likely to profound unconsciousness marked by electroencephalogram patterns, which show profound slow-delta oscillations (0.1–4 Hz) alternating with gamma oscillations.29
Figure 3.
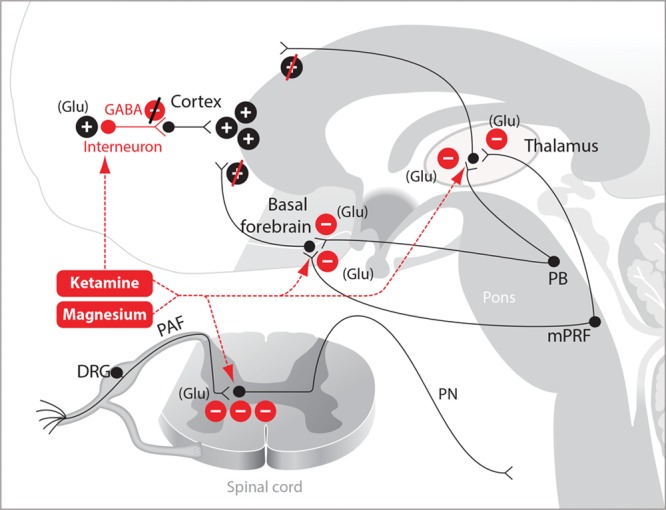
Ketamine and magnesium. The mechanisms of ketamine- and magnesium-induced antinociception are produced primarily by blockade of glutamatergic receptors in the spinal cord and in arousal projections emanating from the brainstem. Ketamine at low doses blocks GABAergic interneurons. DRG indicates dorsal root ganglia; GABA, γ-aminobutyric acid; Glu, glutamate; mPRF, medial pontine reticular formation; PAF, peripheral afferent fiber; PB, parabrachial nucleus; PN, projection neuron.
Magnesium.
Magnesium, the fourth most common cation in the body, plays a critical role in numerous physiological processes (Figure 3).30 In obstetrical anesthesia care, magnesium is used as an antihypertensive and a muscle relaxant to treat preeclampsia.31 Several of these therapeutic effects are mediated through its blockade of calcium channels.32 In addition, magnesium blocks NMDA receptors.33 Because of its inhibitory effects at glutamatergic synapses, a magnesium infusion can be used as an adjunct to reduce the dose of antinociceptive agents being administered as part of a multimodal general anesthesia regimen. Magnesium also potentiates the effects of the hypnotics on arousal.34 The antinociceptive and hypnotic targets of magnesium are most likely the same as ketamine. However, because magnesium is a widely prevalent ion, it most certainly contributes to these states as well through its wide range of nonspecific targets. Magnesium’s profound effects on blood pressure and muscle relaxation must be taken into account when this agent is administered as part of a multimodal general anesthesia regimen.31 Similarly, it is important to be cognizant that high doses of magnesium can lead to decreased atrioventricular conduction, heart block, and possibly cardiac arrest.30
Dexmedetomidine.
Dexmedetomidine, the α-2 adrenergic receptor agonist, like clonidine, exerts its antinociceptive effects in 2 primary locations (Figure 4). It enhances descending inhibition of nociceptive transmission by activating inhibitory interneurons that synapse onto projection neurons in the dorsal horn of the spinal cord.35 Dexmedetomidine also exerts a key antinociceptive effect by decreasing arousal. In this regard, dexmedetomidine acts presynaptically to decrease release of norepinephrine from locus coeruleus neurons projecting to the basal forebrain, intralaminar nucleus of the thalamus, the preoptic area of the hypothalamus, and diffusely to the cortex.8 Decreased norepinephrine release in the preoptic area of the hypothalamus disinhibits GABAergic and galanergic inhibitory projections to the major arousal nuclei in the midbrain and pons.36,37 The actions of dexmedetomidine at these 4 principal targets lead to a substantial decrease in arousal due to decreased noradrenergic excitatory inputs to the preoptic area of the hypothalamus, the thalamus, basal forebrain, and the cortex. Under dexmedetomidine, the electroencephalogram shows spindle (intermittent 9–15 Hz) oscillations and slow-delta oscillations at low-to-moderate doses, and only slow-delta oscillations at high doses.38,39
Figure 4.
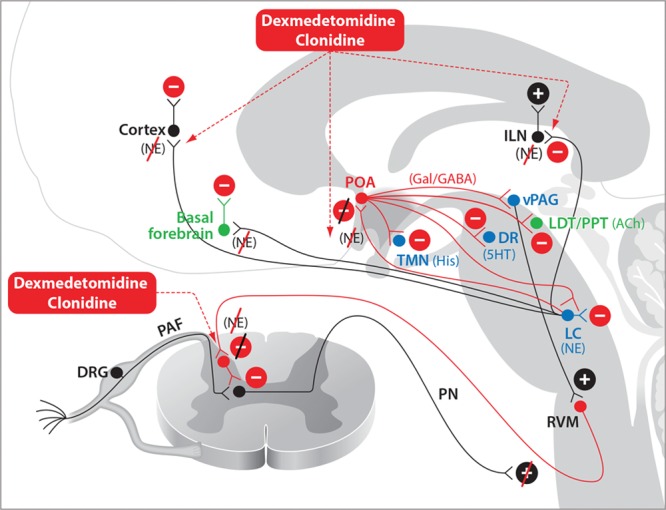
Dexmedetomidine and clonidine. Dexmedetomidine- and clonidine-induced antinociception occur primarily through enhanced inhibitory activity in the descending nociceptive pathways. Sedation induced by dexmedetomidine or clonidine and loss of consciousness induced by dexmedetomidine occur through NE-mediated disinhibition of the POA of the hypothalamus and decreased noradrenergic signaling in the thalamus and cortex. 5HT indicates serotonin; ACh, acetylcholine; DA, dopamine; DR, dorsal raphé; DRG, dorsal root ganglia; GABAA, γ-aminobutyric acid receptor subtype A; Gal, galanin; His, histamine; ILN, intralaminar nucleus of the thalamus; LC, locus coeruleus; LDT, laterodorsal tegmental area; NE, norepinephrine; PAF, peripheral afferent fiber; PN, projection neuron; POA, preoptic area; PPT, pedunculopontine tegmental area; RVM, rostral ventral medulla; TMN, tuberomammillary nucleus; vPAG, ventral periaqueductal gray.
Nonsteroidal Anti-Inflammatory Drugs.
Each surgical insult induces local release of numerous inflammatory mediators, which enter the circulation and spread systemically.40,41 These inflammatory mediators are major enhancers of the nociceptive response to the insult.4 One of the most prevalent of these mediators are the prostaglandins. Prostaglandins activate the nociceptive system by binding to tissue nociceptors. Suppression of prostaglandin synthesis at sites of inflammation is the primary mechanism underlying the antinociceptive effects of NSAIDs (Figure 5).42 The antinociceptive and anti-inflammatory effects of NSAIDs result from their ability to inhibit the activity of the cyclooxygenase isoforms 1 and 2.43 Inhibition of the cyclooxygenase enzymes impairs the 2-step transformation of arachidonic acid into prostaglandins, and with this, the production of these key inflammatory mediators and nociceptive agents.44
Figure 5.
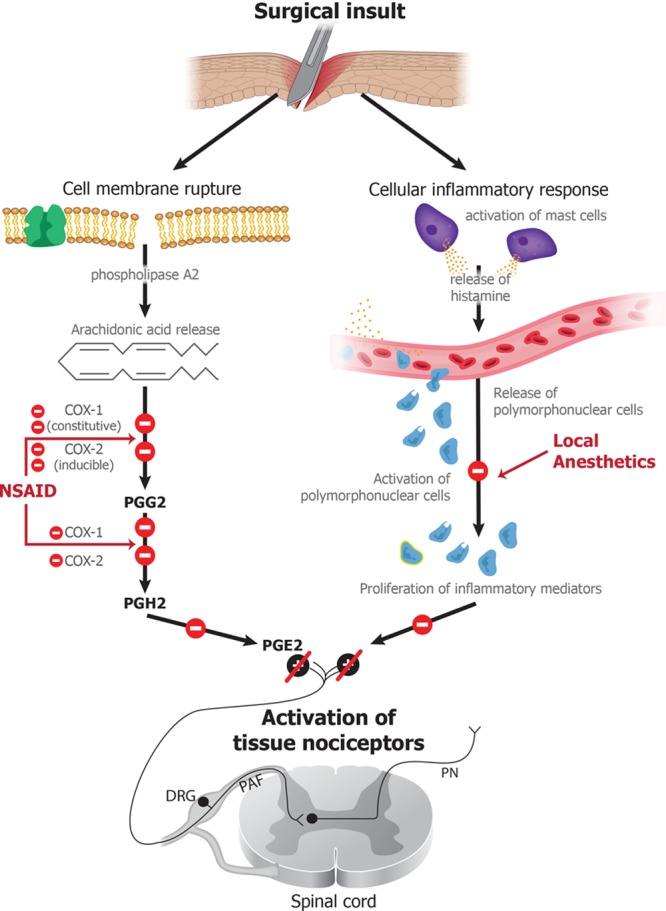
NSAIDs and lidocaine. Surgical insults induce rupture of cell membranes, leading to release of arachidonic acid, which, through the action of COX-1 and COX-2, is converted into prostaglandins, which are potent inflammatory and nociceptive mediators. NSAIDs modulate the nociceptive response by blocking the actions of COX-1 and COX-2, and lidocaine exerts their nociceptive effects by inactivating sodium channels, thus inhibiting excitation of nerve endings and blocking conduction of action potentials in peripheral nerves. Lidocaine also impedes neutrophil degranulation, thereby impeding the amplification of the inflammatory response. COX indicates cyclooxygenase; DRG, dorsal root ganglion; NSAID, nonsteroidal anti-inflammatory drug; PAF, peripheral afferent fiber; PGE2, prostaglandin E2; PGH2, prostaglandin H2; PGG2, prostaglandin G2; PN, projection neuron.
Lidocaine.
An infusion of the local anesthetic lidocaine is commonly used as an adjunct to control intraoperative nociception and postoperative pain. When used in nerve blocks or regional anesthesia, local anesthetics produce antinociception by either inhibiting excitation of nerve endings or by blocking conduction of action potentials in peripheral nerves.45 Local anesthetics accomplish these effects by binding reversibly to sodium channels and thus, blocking the sodium influx required to sustain action potentials.46 The primary target of local anesthetics is the sodium channel open state, and the effectiveness of the local anesthetic in blocking action potential propagation depends critically on how frequently the neuron is depolarized.46 Blocking of sodium channels is not likely to be the primary mechanism through which local anesthetic infusions contribute to antinociception because these effects are achieved at relatively low blood concentrations.9 A more likely mechanism is by their ability to block neutrophil priming (Figure 5).47 In response to tissue injury such as a surgical incision, primed neutrophils degranulate and amplify the inflammatory response. Lidocaine likely downregulates neutrophil degranulation.48 This G-protein–mediated effect presumably impedes the ability of neutrophils to amplify the inflammatory response and hence, the nociceptive signaling created by a surgical insult.49 Alternative mechanisms through which lidocaine could likely enhance antinociception are through blockade of sodium channels, NMDA receptors, and/or glycine receptors on neurons in brainstem arousal circuits and in the amygdala, thus diminishing nociceptive transmission and enhancing sedation.50–52
Hypnotic Agents
Propofol and Sevoflurane.
The intravenous anesthetic propofol and the inhaled ether anesthetic sevoflurane are the principal hypnotics administered in current anesthesiology practice. These agents do not act directly in the nociceptive pathways but rather decrease perception of nociceptive stimuli by rendering the patient unconscious. The primary targets of these anesthetics are γ-aminobutyric acid subtype A (GABAA) receptor53 synapses of inhibitory interneurons on to pyramidal neurons in the cortex, thalamus, brainstem, striatum, and spinal cord (Figure 6).54,55 In addition to GABAA receptors, sevoflurane and the other inhaled ethers also block 2-pore potassium channels, hyperpolarizing-activated cyclic nucleotide-gated channels, and NMDA receptors.54
Figure 6.
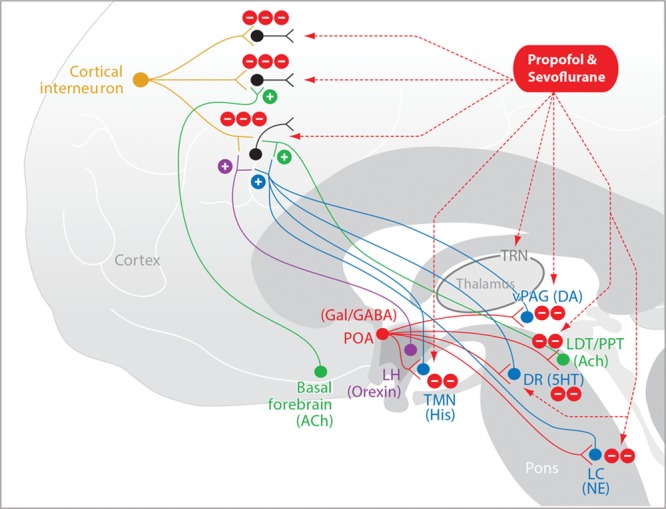
Propofol and sevoflurane. Propofol and sevoflurane induce unconsciousness by enhancing inhibitory GABAergic activity of inhibitory interneurons in the cortex, in the thalamus, and at the inhibitory GABAergic projections from the POA of the hypothalamus onto the arousal centers in the brainstem. 5HT indicates serotonin; ACh, acetylcholine; DA, dopamine; DR, dorsal raphé; GABA, γ-aminobutyric acid; Gal, galanin; His, histamine; LC, locus coeruleus; LDT, laterodorsal tegmental area; LH, lateral hypothalamus; NE, norepinephrine; POA, preoptic area; PPT, pedunculopontine tegmental area; TMN, tuberomammillary nucleus; TRN, thalamic reticular nucleus; vPAG, ventral periaqueductal gray.
The effects of these anesthetics in the brain are readily visible in the electroencephalogram of young adults as characteristic slow-delta oscillations and alpha oscillations (8–12 Hz). The slow-delta oscillations most likely represent hyperpolarization of the thalamus and cortex due to direct inhibition of pyramidal neurons by the anesthetics in cortex and in the thalamic reticular nucleus. The anesthetics also contribute to these slow-delta oscillations by decreasing excitatory brainstem inputs to the thalamus and cortex due to their actions at the GABAergic synapses from the preoptic area of the hypothalamus onto the major arousal centers in the midbrain and pons (Figure 6).36,37 The slow oscillations are incoherent across the cortex and are associated with highly phase-limited spiking activity across the cortex.56,57 The alpha oscillations are coherent across the front of the head and most likely represent coherent activity between the thalamus and the cortex.58,59 The presence of the phase-limited spiking activity maintained by the slow oscillations and the coherent alpha oscillations most likely maintain unconsciousness by impairing neuronal communication within and between brain regions. Above 1 minimal alveolar concentration, sevoflurane and the other inhaled ether anesthetics show a downward shift in the alpha power and an upward shift in the delta power that gives the appearance of a theta oscillation (4–8 Hz).25 These shifts in power in these 2 frequency bands are empirically associated with more profound states of unconsciousness.25
Multimodal General Anesthesia: Rational Design of a Practice Strategy
Using this analysis of the antinociceptive and hypnotic agents, we summarize in Table 1 a rational strategy for implementing multimodal general anesthesia. For each of the principal behavioral states of general anesthesia, our strategy defines drugs that are administered to explicitly maintain that state and those that contribute implicitly to maintaining the state. For example, remifentanil is administered explicitly to maintain antinociception, yet its sedative effects contribute implicitly to unconsciousness. The key features of this strategy are to make control of nociception a primary objective of the choice of anesthetics used for maintenance by using combinations of mechanistically distinct agents; take advantage of the fact that each anesthetic has an explicit and an implicit effect when choosing drug combinations, particularly the effect of the antinociceptive agents on unconsciousness; and make multimodal pain control a key objective in the postoperative period (Table 2).
Table 1.
Multimodal General Anesthesia: Explicit and Implicit Maintenance of the Anesthetic State

Table 2.
Examples of Multimodal Anesthesia Care Practice
Antinociception.
For maintenance of antinociception during general anesthesia, we use simultaneously multiple antinociceptive agents, including opioids (Table 1, column II, row A). Use of multiple antinociceptive agents in addition to an opioid creates the opioid-sparing effect of these agents. Each agent targets a different component of the nociceptive system so that together, they suppress more completely nociceptive transmission. The hypnotic agents reduce the ability to perceive pain, and thereby, contribute implicitly to antinociception (Table 1, column III, row A).
Unconsciousness.
During general anesthesia, uncon sciousness is maintained primarily by using a single titratable agent such as propofol or sevoflurane (Table 1, column II, row B). The antinociceptive agents profoundly contribute to unconsciousness by arresting nociceptive-induced arousal (Table 1, column III, row B). Because each of the antinociceptive agents decreases arousal, their combination reduces appreciably the hypnotic dose required to maintain unconsciousness. When considering the use of an inhaled ether to maintain unconsciousness, the fact that less is required when multiple antinociceptive agents are administered is the commonly cited special case of the minimal alveolar concentration–sparing effect of the antinociceptive drugs. Amnesia is maintained by ensuring unconsciousness because a patient who is truly unconscious and not simply unresponsive is also amnestic.
Immobility.
A single muscle relaxant (nicotinic anticholinergic agent) can be used to maintain immobility (Table 1, column II, row C). The GABAergic hypnotic agents contribute to muscle relaxation by blocking α motor neurons at the level of the spinal cord (Table 1, column III, row C). Magnesium, administered as part of an antinociceptive regimen (Table 1, column II, row A), also enhances muscle relaxation significantly (Table 1, column III, row C). In this case, the muscle relaxant dose has to be reduced accordingly.
Unconsciousness and Antinociception Monitoring.
In addition to standard monitors required for tracking the physiological state during general anesthesia, electroencephalogram monitoring is essential to track level of unconsciousness and to guide hypnotic dosing. The ability to apply our multimodal strategy would be substantially enhanced by a monitor to track level of antinociception and guide dosing of the antinociceptive agents.25 Such monitors are becoming commercially available.60–63 At present, we use heart rate and blood pressure changes as a measure of the nociceptive medullary adrenergic circuit response to nociceptive stimuli.64
MULTIMODAL GENERAL ANESTHESIA IN PRACTICE
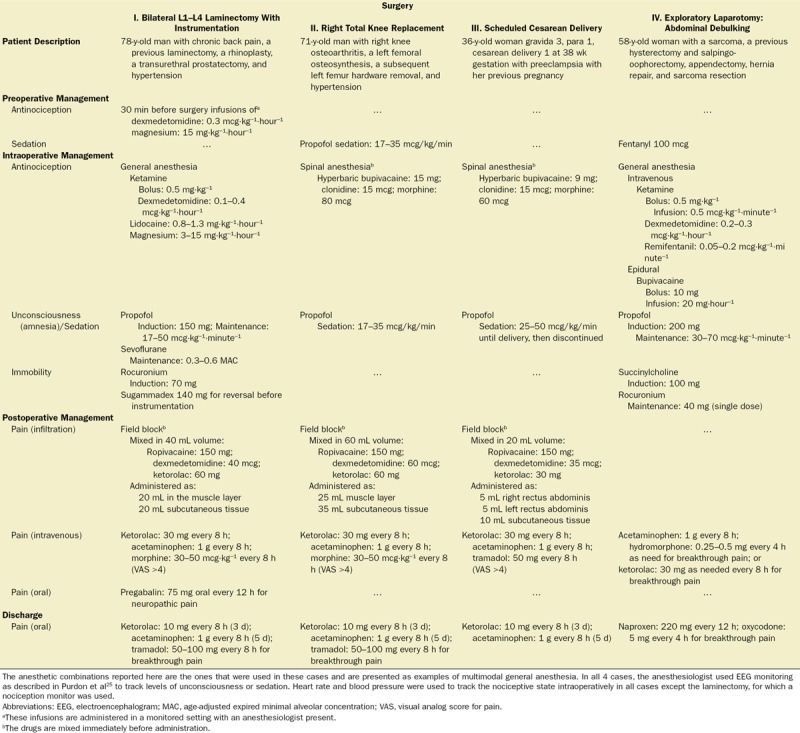
To illustrate our multimodal general anesthesia strategy, we summarize the perioperative management of 4 surgeries: laminectomy, total knee replacement, cesarean delivery, and exploratory laparotomy (Table 2). The strategy requires an explicit plan for preoperative, intraoperative, postoperative, and discharge antinociception/pain management. The drug doses and combinations are ones the anesthesiologist chose for the particular case. They are intended solely as examples. The management strategy, anesthetic choices, and anesthetic doses must be adapted to the needs of the individual patient.
Lumbar Laminectomy With Instrumentation
The preoperative anesthetic management for the lumbar laminectomy with instrumentation (Table 2, column I) started 30 minutes before the surgery by administering low-dose infusions of dexmedetomidine and magnesium. These infusions, which initiate the patient’s antinociceptive regimen, commonly induce substantial sedation and muscle relaxation. Unconsciousness was induced with propofol and maintained with a low-rate propofol infusion and a low concentration of sevoflurane. After a ketamine bolus, ketamine and lidocaine infusions were added to complete simultaneous multimodal targeting of nociception. Muscle relaxation was maintained with rocuronium, which was reversed with sugammadex before the start of the instrumentation. The magnesium, lidocaine, and dexmedetomidine infusions were stopped 15–30 minutes before the projected end of the surgery to avoid prolonged recovery of consciousness. For postoperative antinociceptive management, a field block was performed by combining ropivacaine, dexmedetomidine, and ketorolac. Half of the volume was administered in the muscle layer along the wound, and the remaining half was administered in the subcutaneous tissue. This field block provided postoperative analgesia for approximately 24 hours.
We (M.N.) used the visual analog scale to allow the patient to describe their level of postoperative pain. Intravenous ketorolac and acetaminophen were the primary agents used for pain control, and morphine was administered to treat breakthrough pain. Before discharge, we counseled the patient on the benefits of taking pain medication on a set schedule and on the potential adverse effects of opioids. Our (M.N.) goal is to discharge patients using just NSAIDs, such as either ketorolac or acetaminophen to control pain.65 This patient was also discharged on pregabalin as he had been taking it for neuropathic pain before his surgery.
Total Knee Replacement and Cesarean Delivery
The multimodal strategy also applies to surgeries such as total knee replacements (Table 2, column II) and cesarean deliveries (Table 2, column III), which were conducted using regional anesthetic techniques. For the total knee replacement, a low-dose propofol infusion was used for sedation during placement of the spinal and during the surgery. During the cesarean delivery, a low-dose propofol infusion was used for sedation until delivery then discontinued at the end of the surgery. The spinal anesthetics for both cases used a combination of bupivacaine, clonidine, and morphine to achieve multimodal antinociception and muscle relaxation. Both patients received field blocks using a combination of ropivacaine, dexmedetomidine, and ketorolac at the completing of their surgeries before closing the skin incisions. For the field block, the anesthetic solution is injected in a systematic way around the length of the incision. Postoperative pain management relied on ketorolac and acetaminophen, with morphine as the rescue agent after the total knee replacement and tramadol as the rescue agent after the cesarean delivery. Both patients were discharged home on ketorolac 10 mg every 8 hours for 3 days and acetaminophen 1 g every 8 hours for 5 days. In addition to the ketorolac and acetaminophen, the patient who had the total knee replacement was given tramadol 50 mg every 8 hours for breakthrough pain.
Exploratory Laparotomy
For the exploratory laparotomy (Table 2, column IV), the patient received 100 mg of fentanyl as a preinduction sedative. Intraoperative antinociception management included infusions of ketamine, dexmedetomidine, and remifentanil, along with a continuous epidural bupivacaine infusion. The patient received a bolus of ketamine (0.5 mg·kg−1) before starting the ketamine infusion. Induction was with propofol and succinylcholine, and a low-dose propofol infusion (30–70 µg·kg−1·minute−1) was used to maintain unconsciousness. Muscle relaxation was maintained with rocuronium and reversed at the end of the surgery with neostigmine and glycopyrrolate. The epidural was continued for 2 days postoperatively for pain control using a bupivacaine/hydromorphone infusion. Acetaminophen and 1 dose of hydromorphone were administered for breakthrough pain. On postoperative day 3, the patient received acetaminophen and was discharged home on naproxen 220 mg every 12 hours and oxycodone 5 mg every 4 hours as needed for breakthrough pain.
CONCLUSIONS
For many years after the initial use of ether as the first anesthetic in the 1840s, anesthesiologists relied almost exclusively on this single agent for anesthetic management. With time, anesthesiologists learned that using balanced general anesthesia to create the anesthetic state offered a greater likelihood of achieving the beneficial effects while minimizing side effects. The several undesirable side effects of the opioids and the recent opioid epidemic have catalyzed efforts to develop new balanced anesthesia paradigms, which reduce or eliminate opioid use.10,66
For example, a recent review proposes an opioid-free multimodal balanced general anesthesia strategy that provides unconsciousness with amnesia and muscle relaxation while maintaining appropriate tissue perfusion and sympathetic stability to protect organs.10 This strategy emphasizes use of medications other than opioids to create stress-free intraoperative conditions and asserts that analgesia is only important postoperatively and can be achieved with medications other than opioids. In contrast, we believe that nociception should be maintained intraoperatively and postoperatively using multiple antinociceptive agents.
A recent report has summarized the modalities (nonanesthetic and anesthetic adjuncts, and regional techniques) that can be used to reduce opioid use perioperatively.67 Our framework offers a principled approach for designing and implementing multimodal strategies for use in anesthesiology practice. The fundamental feature of our strategy is administration of multiple antinociceptive agents simultaneously to suppress nociceptive trafficking during both general and regional anesthesia (Table 2). Each agent targets a different component of the nociceptive system. Our neural circuit analyses provide a neurophysiologically based approach for understanding the effects of each anesthetic and for choosing the anesthetic combinations (Figures 2–6). As stated in the “Introduction,” surgically induced nociception is the primary reason for administering general anesthesia and the primary source of the patient’s hemodynamic and stress responses. If nociceptive control is adequate, the stress responses will be minimized and sympathetic stability will be achieved. Moreover, our approach also takes account of the implicit effects of the anesthetics being administered (Table 1). Suppression of nociceptive transmission has the significant added benefit of decreasing arousal, which appreciably reduces the hypnotic doses required to maintain unconsciousness and amnesia. We postulate that reduction in hypnotic use may facilitate faster recovery and help reduce the contribution of general anesthesia to postoperative cognitive dysfunction. Similarly, muscle relaxants decrease arousal by decreasing proprioceptive feedback. Under our strategy, opioid use need not be eliminated. Instead, other agents are used along with opioids to achieve antinociception control intraoperatively and pain control postoperatively (Tables 1 and 2).
To achieve adequate postoperative pain control, multimodal pain management has to be continued in the immediate postoperative period and after discharge (Table 2). This formal planning that provides an explicit way to reduce postoperative opioid use requires coordinated management during the perioperative period among the anesthesiology, surgical, and nursing teams. Our experience suggests that this multimodal strategy has important potential. We will test our strategy further in clinical practice and in clinical trial comparisons with existing approaches.
DISCLOSURES
Name: Emery N. Brown, MD, PhD.
Contribution: This author helped conceive and write major sections of the manuscript.
Conflicts of Interest: Masimo Corporation has licensed and paid royalties on intellectual property to Massachusetts General Hospital created by E. N. Brown. This author is also a cofounder of PASCALL, a company developing closed loop physiological control systems for anesthesiology.
Name: Kara J. Pavone, BS, BSN, RN.
Contribution: This author helped write major sections of the manuscript and design the figures.
Conflicts of Interest: None.
Name: Marusa Naranjo, MD.
Contribution: This author helped write major sections of the manuscript and prepare the tables.
Conflicts of Interest: M. Naranjo has served as a paid speaker for Masimo Corporation and Medtronics.
This manuscript was handled by: Jean-Francois Pittet, MD.
APPENDIX
Glossary of Terms
α-2 adrenergic receptor is a subtype of G-protein–coupled, presynaptic receptor through which catecholamines like norepinephrine and epinephrine signal in the central and peripheral nervous systems.
Arousal system (arousal pathways) is (are) a collection of nuclei located primarily in the brainstem and their ascending neuronal projections to the thalamus and cortex that is (are) responsible for creating the awake component of consciousness. Inactivation of these nuclei and/or their pathways is a mechanism for producing unconsciousness.
Basal forebrain (BF) is an area of the cortex located in at the base of the frontal cortex that is a major source of excitatory cholinergic projections to the thalamus and cortex.
Cyclooxygenase (COX) is an important enzyme for biosynthesis of prostaglandins that promote inflammation and fever. COX-1 and COX-2 are 2 important types COX enzymes, the actions of which are inhibited by nonsteroidal anti-inflammatory drugs (NSAIDs).
Dorsal raphé is a brainstem area located in the central pons that sends primarily excitatory serotonergic projections to the cortex.
Frequency bands are electroencephalogram frequency ranges that have been established by convention. The commonly used bands, characterized in cycles per second or Hertz (Hz), are as follows: slow-delta, 0.1–4 Hz; theta, 4–7 Hz; alpha, 8–12 Hz; beta, 13–25 Hz; gamma, >25 Hz.
Galaninergic pathways are inhibitory pathways that project from the preoptic area of the hypothalamus onto nearly each one of the major arousal nuclei in the pons and midbrain. The neuropeptide galanin is the neurotransmitter in these pathways.
γ-amino butyric acid is the primary inhibitory neurotransmitter in the central nervous system.
Laterodorsal tegmentum is a brainstem area located in the superior posterior region of the midbrain that is a major source of excitatory cholinergic projections to the thalamus and cortex.
Locus ceruleus is a brainstem area located in the central pons that sends primarily noradrenergic projections to the cortex, central thalamus, BF, and the preoptic area of the hypothalamus.
Neutrophil priming is the process by which polymorphonuclear lymphocytes are activated, and as a consequence, readily degranulate inducing amplification of an inflammatory response.
N-methyl-d-aspartate receptors are a pharmacologically identified subset of glutamate receptors and ion channel proteins that are primarily excitatory.
Nociception is the propagation through the sensory system of potentially noxious and harmful chemical, mechanical, or thermal stimuli. The nociceptive system or pathways consist of the nociceptors in the periphery and in the viscera, the ascending nociceptive pathways and the descending nociceptive pathways. The ascending pathways transmit nociceptive stimuli from the periphery to the spinal cord to the brainstem (medulla and midbrain), the amygdala, the thalamus, and on to the primary and secondary sensory cortices. The brainstem descending component of the nociceptive pathway begins in the periaqueductal gray located in the midbrain, and projects through the rostral ventral medulla in the medulla to the spinal cord. The descending pathways are activated immediately by the nociceptive inputs from the ascending pathways and modulate (upregulate and downregulate) the nociceptive information.
Nociceptors are unspecialized bare nerve cell endings that initiate nociception or pain. Their cell bodies arise in the dorsal horn of the spinal cord and send one axonal process to the periphery and the other to the spinal cord or brainstem through the spinothalamic tract.
NSAIDs are pharmacological agents that block specifically the COX-1 and COX-2 enzymes that play a major role in prostaglandin synthesis. Because prostaglandins are primary mediators of inflammation, NSAIDs are key agents for blocking inflammation and thereby reducing inflammation-induced nociception and pain.
Pain is the conscious perception of nociceptive information.
Parabrachial nucleus is a brainstem area located in the dorsolateral pons that surrounds the superior cerebellar peduncle as it enters the brainstem from the cerebellum. The parabrachial nucleus provides important glutamatergic projections to the central thalamus and the BF.
Pedunculopontine tegmentum is brainstem area located in the superior posterior region of the midbrain that is a major source of excitatory cholinergic projections to the thalamus and to the cortex.
Periaqueductal gray is the midbrain relay of the descending pathways for modulating nociceptive inputs into the central nervous system.
Pyramidal neurons are multipolar, commonly teardrop-shaped excitatory neurons that are located primarily in the amygdala, the hippocampus, and the cortex.
Rostral ventral medulla is a brainstem area located in the upper ventral part of the medulla that relays descending modulation of nociceptive information from the periaqueductal gray to the spinal cord.
Rostral and caudal ventral lateral medulla are brainstem areas located respectively in the upper and lower ventral lateral parts of the medulla. These areas relay sympathetic signals from the nucleus of the tractus solitarius—also in the medulla—to the sympathetic ganglia in the thoracolumbar trucks.
Spindles are waxing and waning oscillations in the 9–15 Hz range that are a defining feature of stage 2 nonrapid eye movement sleep. These oscillations are also observed in the EEG of patients receiving low-dose dexmedetomidine.
Striatum is a general term used to denote the putamen and caudate in the basal ganglia. It plays a critical role in motor control.
Thalamic reticular nucleus is a network of inhibitory neurons that surround the thalamus and modulate nearly all thalamic output, particularly to the cortex.
Footnotes
Funding: This work was supported by the National Institutes of Health (Bethesda, MD): R01 GM104948 (to E.N.B.) and P01GM118269 (to E.N.B.); and by the Department of Anesthesia, Critical Care and Pain Medicine, Massachusetts General Hospital, Boston, MA.
Conflicts of Interest: See Disclosures at the end of the article.
A glossary of terms is available in the Appendix.
Reprints will not be available from the authors.
REFERENCES
- 1.Brown EN, Lydic R, Schiff ND. General anesthesia, sleep, and coma. N Engl J Med. 2010;363:2638–2650.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lundy JS. Balanced anesthesia. Minn Med. 1926;9:399–404.. [Google Scholar]
- 3.Hendrickx JF, Eger EI, II, Sonner JM, Shafer SL. Is synergy the rule? A review of anesthetic interactions producing hypnosis and immobility. Anesth Analg. 2008;107:494–506.. [DOI] [PubMed] [Google Scholar]
- 4.Purves D, Augustine GJ, Fitzpatrick D, Hall WC, Lamantia A, White LE. Purves D, Augustine GJ, Fitzpatrick D, Hall WC, Lamantia A, White LE. Pain. In: Neuroscience. 2012:5th ed Sunderland, MA: Sinauer Associates, Inc; 209–228.. [Google Scholar]
- 5.Lake APJ. Balanced anaesthesia 2005: avoiding the transition from acute to chronic pain. South Afr J Anaesth Analg. 2005;11:14–18.. [Google Scholar]
- 6.McNicol E, Horowicz-Mehler N, Fisk RA, et al. ; American Pain Society. Management of opioid side effects in cancer-related and chronic noncancer pain: a systematic review. J Pain. 2003;4:231–256.. [DOI] [PubMed] [Google Scholar]
- 7.Volkow ND, Collins FS. The role of science in the opioid crisis. N Engl J Med. 2017;377:1798. [DOI] [PubMed] [Google Scholar]
- 8.Brown EN, Purdon PL, Van Dort CJ. General anesthesia and altered states of arousal: a systems neuroscience analysis. Annu Rev Neurosci. 2011;34:601–628.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn LK, Durieux ME. Perioperative use of intravenous lidocaine. Anesthesiology. 2017;126:729–737.. [DOI] [PubMed] [Google Scholar]
- 10.Mulier J. Opioid free general anesthesia: a paradigm shift? Rev Esp Anestesiol Reanim. 2017;64:427–430.. [DOI] [PubMed] [Google Scholar]
- 11.Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474.. [DOI] [PubMed] [Google Scholar]
- 12.Rabiner EA, Beaver J, Makwana A. Pharmacological differentiation of opioid receptor antagonists by molecular and functional imaging of target occupancy and food reward-related brain activation in humans. Mol Psychiatry. 2011;16:826–835.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burn DJ, Rinne JO, Quinn NP, Lees AJ, Marsden CD, Brooks DJ. Striatal opioid receptor binding in Parkinson’s disease, striatonigral degeneration and Steele-Richardson-Olszewski syndrome, A [11C]diprenorphine PET study. Brain. 1995;118(pt 4):951–958.. [DOI] [PubMed] [Google Scholar]
- 14.Waldhoer M, Bartlett SE, Whistler JL. Opioid receptors. Annu Rev Biochem. 2004;73:953–990.. [DOI] [PubMed] [Google Scholar]
- 15.Stein C. The control of pain in peripheral tissue by opioids. N Engl J Med. 1995;332:1685–1690.. [DOI] [PubMed] [Google Scholar]
- 16.Fukuda K. Opioids. 20097th ed New York, NY: Churchill Livingstone. [Google Scholar]
- 17.Veinante P, Yalcin I, Barrot M. The amygdala between sensation and affect: a role in pain. J Mol Psychiatry. 2013;1:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becerra L, Harter K, Gonzalez RG, Borsook D. Functional magnetic resonance imaging measures of the effects of morphine on central nervous system circuitry in opioid-naive healthy volunteers. Anesth Analg. 2006;103:208–216.. [DOI] [PubMed] [Google Scholar]
- 19.Lydic R, Baghdoyan HA. Sleep, anesthesiology, and the neurobiology of arousal state control. Anesthesiology. 2005;103:1268–1295.. [DOI] [PubMed] [Google Scholar]
- 20.Mortazavi S, Thompson J, Baghdoyan HA, Lydic R. Fentanyl and morphine, but not remifentanil, inhibit acetylcholine release in pontine regions modulating arousal. Anesthesiology. 1999;90:1070–1077.. [DOI] [PubMed] [Google Scholar]
- 21.Griffioen KJ, Venkatesan P, Huang ZG. Fentanyl inhibits GABAergic neurotransmission to cardiac vagal neurons in the nucleus ambiguus. Brain Res. 2004;1007:109–115.. [DOI] [PubMed] [Google Scholar]
- 22.Sinner B, Graf BM. Ketamine. Handb Exp Pharmacol. 2008:313–333.. [DOI] [PubMed] [Google Scholar]
- 23.Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–533.. [DOI] [PubMed] [Google Scholar]
- 24.Seamans J. Losing inhibition with ketamine. Nat Chem Biol. 2008;4:91–93.. [DOI] [PubMed] [Google Scholar]
- 25.Purdon PL, Sampson A, Pavone KJ, Brown EN. Clinical electroencephalography for anesthesiologists: part I: background and basic signatures. Anesthesiology. 2015;123:937–960.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boon JA, Milsom WK. NMDA receptor-mediated processes in the parabrachial/Kölliker fuse complex influence respiratory responses directly and indirectly via changes in cortical activation state. Respir Physiol Neurobiol. 2008;162:63–72.. [DOI] [PubMed] [Google Scholar]
- 27.Fuller PM, Fuller P, Sherman D, Pedersen NP, Saper CB, Lu J. Reassessment of the structural basis of the ascending arousal system. J Comp Neurol. 2011;519:933–956.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fulwiler CE, Saper CB. Subnuclear organization of the efferent connections of the parabrachial nucleus in the rat. Brain Research. 1984;319:229–259.. [DOI] [PubMed] [Google Scholar]
- 29.Akeju O, Song AH, Hamilos AE. Electroencephalogram signatures of ketamine anesthesia-induced unconsciousness. Clin Neurophysiol. 2016;127:2414–2422.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Do SH. Magnesium: a versatile drug for anesthesiologists. Korean J Anesthesiol. 2013;65:4–8.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pairu J, Triveni GS, Manohar A. The study of serum calcium and serum magnesium in pregnancy induced hypertension and normal pregnancy. Int J Reprod Contracept Obstet Gynecol. 2015;4:30–34.. [Google Scholar]
- 32.Gourgoulianis KI, Chatziparasidis G, Chatziefthimiou A, Molyvdas PA. Magnesium as a relaxing factor of airway smooth muscles. J Aerosol Med. 2001;14:301–307.. [DOI] [PubMed] [Google Scholar]
- 33.Ruppersberg JP, Kitzing E, Schoepfer R. The mechanism of magnesium block of NMDA receptors. Semin Neurosci. 1994;6:87–96.. [Google Scholar]
- 34.Seyhan TO, Tugrul M, Sungur MO. Effects of three different dose regimens of magnesium on propofol requirements, haemodynamic variables and postoperative pain relief in gynaecological surgery. Br J Anaesth. 2006;96:247–252.. [DOI] [PubMed] [Google Scholar]
- 35.Andrieu G, Roth B, Ousmane L. The efficacy of intrathecal morphine with or without clonidine for postoperative analgesia after radical prostatectomy. Anesth Analg. 2009;108:1954–1957.. [DOI] [PubMed] [Google Scholar]
- 36.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263.. [DOI] [PubMed] [Google Scholar]
- 37.Sherin JE, Elmquist JK, Torrealba F, Saper CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998;18:4705–4721.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akeju O, Kim SE, Vazquez R. Spatiotemporal dynamics of dexmedetomidine-induced electroencephalogram oscillations. PLoS One. 2016;11:e0163431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akeju O, Pavone KJ, Westover MB. A comparison of propofol- and dexmedetomidine-induced electroencephalogram dynamics using spectral and coherence analysis. Anesthesiology. 2014;121:978–989.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bautmans I, Njemini R, De Backer J, De Waele E, Mets T. Surgery-induced inflammation in relation to age, muscle endurance, and self-perceived fatigue. J Gerontol A Biol Sci Med Sci. 2010;65:266–273.. [DOI] [PubMed] [Google Scholar]
- 41.Arias J, Aller M-A, Arias J-I. Surgical Inflammation. 2013Madrid, Spain: Bentham Science Publishers. [Google Scholar]
- 42.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ajmone-Cat MA, Bernardo A, Greco A, Minghetti L. Non-steroidal anti-inflammatory drugs and brain inflammation: effects on microglial functions. Pharmaceuticals (Basel). 2010;3:1949–1965.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231:232–235.. [DOI] [PubMed] [Google Scholar]
- 45.Berde C, Strichartz GR. Miller R, Eriksson L, Fleisher L, Wiener-Kronish J, Cohen N, Young W. Local Anesthetics. In: Miller's Anesthesia. 2015:8th ed Philadelphia, PA: Elsevier; 1028–1053.. [Google Scholar]
- 46.Wang GK, Strichartz GR. State-dependent inhibition of sodium channels by local anesthetics: a 40-year evolution. Biochem (Mosc) Suppl Ser A Membr Cell Biol. 2012;6:120–127.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hollmann MW, Herroeder S, Kurz KS. Time-dependent inhibition of G protein-coupled receptor signaling by local anesthetics. Anesthesiology. 2004;100:852–860.. [DOI] [PubMed] [Google Scholar]
- 48.Miralda I, Uriarte SM, McLeish KR. Multiple phenotypic changes define neutrophil priming. Front Cell Infect Microbiol. 2017;7:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hollmann MW, McIntire WE, Garrison JC, Durieux ME. Inhibition of mammalian Gq protein function by local anesthetics. Anesthesiology. 2002;97:1451–1457.. [DOI] [PubMed] [Google Scholar]
- 50.Wagman IH, De Jong RH, Prince DA. Effects of lidocaine on the central nervous system. Anesthesiology. 1967;28:155–172.. [DOI] [PubMed] [Google Scholar]
- 51.Muth-Selbach U, Hermanns H, Stegmann JU. Antinociceptive effects of systemic lidocaine: involvement of the spinal glycinergic system. Eur J Pharmacol. 2009;613:68–73.. [DOI] [PubMed] [Google Scholar]
- 52.Cummins TR, Sheets PL, Waxman SG. The roles of sodium channels in nociception: implications for mechanisms of pain. Pain. 2007;131:243–257.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bowery NG, Hudson AL, Price GW. GABAA and GABAB receptor site distribution in the rat central nervous system. Neuroscience. 1987;20:365–383.. [DOI] [PubMed] [Google Scholar]
- 54.Hemmings HC, Jr, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci. 2005;26:503–510.. [DOI] [PubMed] [Google Scholar]
- 55.Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–386.. [DOI] [PubMed] [Google Scholar]
- 56.Purdon PL, Pierce ET, Mukamel EA, et al. Electroencephalogram signatures of loss and recovery of consciousness from propofol. Proc Natl Acad Sci U S A. 2013;110:E1142–E11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lewis LD, Weiner VS, Mukamel EA, et al. Rapid fragmentation of neuronal networks at the onset of propofol-induced unconsciousness. Proc Natl Acad Sci U S A. 2012;109:E3377–E33.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ching S, Cimenser A, Purdon PL, Brown EN, Kopell NJ. Thalamocortical model for a propofol-induced alpha-rhythm associated with loss of consciousness. Proc Natl Acad Sci U S A. 2010;107:22665–22670.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flores FJ, Hartnack KE, Fath AB, et al. Thalamocortical synchronization during induction and emergence from propofol-induced unconsciousness. Proc Natl Acad Sci U S A. 2017;114:E6660–E6668.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dolosys GmbH. The Dolosys Paintracker. 2017. Available at: http://www.dolosys.de/Products-EN.htm. Accessed June 29, 2017.
- 61.ANI (Analgesia Nociception Index). Available at: https://www.mdoloris.com/en/technologies/ani-analgesia-nociception-index/. Accessed August 19, 2018.
- 62.Storm H. Med-Storm. 2016PainMonitor™:Oslo, Norway. [Google Scholar]
- 63.Huiku M, Kamppari L, Viertio-Oja H. Surgical Plethysmographic Index (SPI) in Anesthesia Practice. 2014Helsinki, Finland: General Electric Healthcare. [Google Scholar]
- 64.Brown EN, Solt K, Purdon PL, Akeju O. Miller R, Eriksson L, Fleisher L, Wiener-Kronish J, Cohen N, Young W. Monitoring brain state during general anesthesia and sedation. In: Miller’s Anesthesia. 2015:8th ed Philadelphia, PA: Elsevier; 1524–1540.. [Google Scholar]
- 65.Clarke R, Derry S, Moore RA. Single dose oral etoricoxib for acute postoperative pain in adults. Cochrane Database Syst Rev. 2014:CD004309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ljungqvist O, Scott M, Fearon KC. Enhanced recovery after surgery: a review. JAMA Surg. 2017;152:292–298.. [DOI] [PubMed] [Google Scholar]
- 67.Kumar K, Kirksey MA, Duong S, Wu CL. A review of opioid-sparing modalities in perioperative pain management: methods to decrease opioid use postoperatively. Anesth Analg. 2017;125:1749–1760.. [DOI] [PubMed] [Google Scholar]