

Abstract
Rationale:
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a disorder characterized by a symmetrical, sensorimotor involvement and slowly progressive onset peripheral neuropathy. Peripheral neuropathies have been reported in some central demyelination patients. However, the central nervous system focus in the CIDP patient can mimic neuromyelitis optica have not been recognized by most of us.
Patient concerns:
The numbness and weakness of limbs about eight weeks.
Diagnoses:
Chronic inflammatory demyelinating polyradiculoneuropathy.
Interventions:
Immunotherapy with intravenous immunoglobulins was applied to this patient.
Outcomes:
After 1 year follow-up, the results showed there was still slight numbness of all limbs, and he could walk slowly without help. Gastrocnemius muscle atrophy did not aggravate.
Lessons:
So It is suggest that CIDP can combine with central lesions mimicking neuromyelitis optica. We should take the diagnosis of CIDP into consideration when we find focus in central nervous system.
Keywords: central nervous system involvement, chronic inflammatory demyelinating polyradiculoneuropathy, neuromyelitis optica
1. Introduction
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a disorder characterized by a symmetrical, sensorimotor involvement and slowly progressive onset peripheral neuropathy.[1] Neuromyelitis optica (NMO, Devic syndrome) is an autoimmune chronic inflammatory disease of the central nervous system (CNS) characterized by transverse myelitis and optic neuritis. Magnetic resonance imaging (MRI) typically presents longitudinally extensive lesions spanning 3 or more vertebral segment, and highly showed in CNS such as subcortical and aquaporin-4 antibodies (AQP4-IgG) high-expressed area.[2,3] Peripheral neuropathies have been reported in some patients with central demyelination. However, the CNS focus in the CIDP patient can mimic NMO have not been recognized by most of us. Herein we described a novel case of CIDP with thalamus and periaqueductal gray involvement, the imaging of which was initially suggestive NMO.
2. Case Presentation
A 53-year-old male patient came for numbness and weakness of all limbs. Eight weeks before the visit, he had numbness and weakness of the lower limbs. The numbness appeared most obvious on the toes. And the weakness was most obvious when going up and down the stairs. With time progressed, the weakness aggravated, which led to difficulty in walking. The symptoms spread to upper limbs 2 weeks later, which gave priority to distal parts. The patient could not make a fist, but could lift his arms over the shoulder. The MRI of the brain showed abnormal signal around lateral ventricle, midbrain aqueduct, and IV ventricle (Fig. 1). The cerebrospinal fluid pressure was 150 mm H2O (reference is 80–180 mm H2O), protein was 450 mg/L (reference is 150–450 mg/L), and glucose was 5.16 mmol/L (reference is 2.4–4.4 mmol/L). However, several days later, not only the numbness but also weakness accelerated. The patient presented with weak voice and difficulty in breathing. Examination revealed that voice was weak. Decline of superficial sensation of 4 limbs, proprioceptive sensation of the lower limbs. As for the muscle strength, the distal of 4 limbs was 1/5 (MRCS, grade 0–5), and the proximal of upper was 4/5 but the lower was 2/5 only. There was mild atrophy of gastrocnemius muscle. The biceps reflex, triceps reflex, and knee jerk were weak, and the pathological sign was negative. We reperformed lumbar puncture. The results showed that the protein of cerebrospinal fluid was 542.82 mg/L (reference is 150–450 mg/L), and the white blood cell was 5 × 106/L. The laboratory examination displayed folic acid was 2.23 ng/mL (reference values was >5.38 ng/mL), and vitamin B12 was >2000 pg/mL. Based on the findings at present, NMO was suspected at first. To rule out other possible diseases, cervical and thoracic vertebra MRI was performed, and the result was normal. The serum AQP4 antibodies were negative, but the anti-GM1 IgM was positive. The somatosensory-evoked potential showed abnormalities in the lower limbs (damage of period conduction pathway). Electromyography (EMG) showed axonal demyelinating polyradiculoneuropathy, abnormal distal latency with very low amplitude, disappearance of F waves, and numerous spontaneous potential (Table 1). According to the diagnostic criteria brought up by Wingerchuk et al, NMO was not considered any more, and according to the CIDP diagnosis criteria, the patient was finally diagnosed with CIDP. Immunotherapy with intravenous immunoglobulins was applied. Brain MRI was reperformed on the 12th day after treatments, the results showed lateral ventricle, midbrain aqueduct, and IV ventricle abnormal signal diminished obviously (Fig. 2). The numbness and weakness improved and the patient could walk with the help of others; the atrophy did not aggravate any more when the patient was discharged from hospital on the 17th day after treatments. After the patient was discharged from the hospital, we conducted a 1-year follow-up to him by telephone. We telephoned the patient every 60 days. The recent results showed there was still slight numbness of all limbs, and he could walk slowly without help. Gastrocnemius muscle atrophy did not aggravate. Unfortunately, he rejected to perform brain magnetic resonance and electrophysiological examinations any more.
Figure 1.
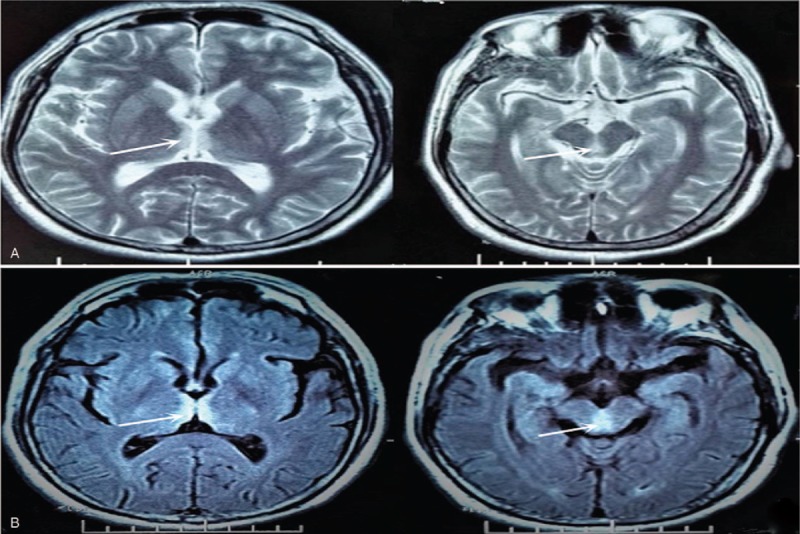
Axial T2-weighted imaging and T2 flair magnetic resonance showing high signal around lateral ventricle, midbrain aqueduct, and IV ventricle (A, axi-T2WI; B, axi-T2FLAIR).
Table 1.
The results of nerve conductions.
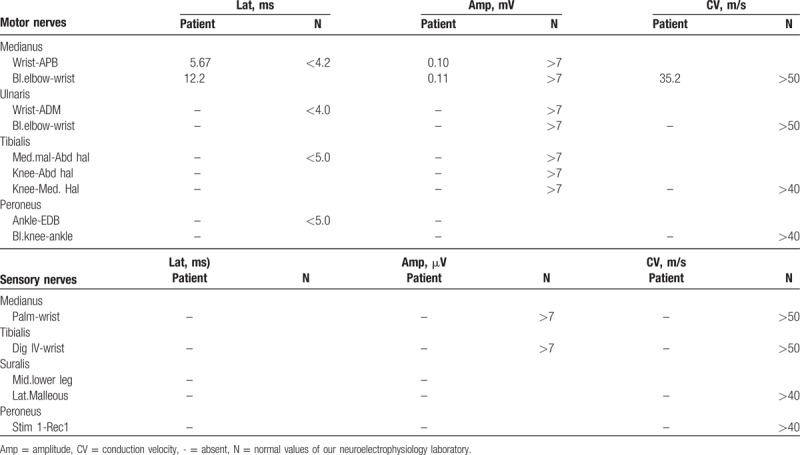
Figure 2.

Axial T2-weighted imaging magnetic resonance showing high signal around lateral ventricle, midbrain aqueduct, and IV ventricle diminished significantly (A, axi-T2WI).
3. Discussion
CIDP is probably the best recognized progressive immune-mediated acquired chronic inflammatory demyelinating peripheral neuropathy, involving the peripheral nerve mainly, and the pathogenesis remains unknown. At present, the diagnosis criteria of CIDP are as follows: the whole course is at least 8 weeks; approximately 50% patients demonstrate typical clinical symptoms, including symmetrical proximal weakness, tendon reflex deficiency, and motor and sensory lesion; laboratory tests show albuminocytologic dissociation in 77% to 95% CIDP patients; EMG shows slow conduction velocity, conduction block, and temporal dispersion; exclude peripheral neuropathy caused by other causes; and effective to glucocorticoids treatment.[4,5]
This case shows complexity in diagnosis. Firstly, because of the critical 8-week and progressive course, subacute instead of chronic disease would be suspected initially. Secondly, the results of brain MRI are quite similar to the NMO, which show obvious abnormities in the AQP4 high expressed area.[2,3] If we just stop here, wrong diagnosis would be made easily. But some similar reports, for example, Lassmann et al[6] found the first CNS and peripheral nervous system (PNS) disease in 1981 and Chinese Professor Li and Yang[7] reported the first 2 CIDP patients’ complicated CNS demyelination in 1994 in China, encouraged us to find out the true answer. And according to his medical history, laboratory, MRI, and EMG results, peripheral neuropathy caused by drugs, tumor, etc could be ruled out. CIDP was diagnosed finally.
The findings of the brain MRI of the patient are as follows: periventricular hemispheric lesions; periventricular and hemispheric lesions; periventricular and brainstem lesions; and periventricular cervical or white matter lesions with nerve roots enhancement.[8–14] From what above all, we can know that the periventricular lesions are the most common change. This patient show not only periventricular demyelination but also bilateral thalamus lesions (Fig. 1), and the lesions are overt, and have not been reported yet.
Although there are reports about CIDP complicated with CNS lesion, there are also reports about existence of peripheral neuropathy in NMO and MS.[15] So whether it is inflammatory demyelinating disease in the PNS and its subsequent extension to the CNS or just on the contrary remains a mystery. For the CNS abnormalities in CIDP patient, Li et al had proposed several conjectures.
They are as follows: the CIDP can develop secondary CNS lesion similar to NMO; maybe this is one kind of unknown syndrome between CIDP and NMO; the PNS lesion maybe a kind of presentation of NMO; and the coexistence of CIDP and NMO.[7] As for This patient, he showed obvious progressive weakness and numbness, which indicated the lesion in the PNS, then the EMG and somatosensory-evoked potential of the lower limbs confirmed the damage of the PNS. He at that time showed no symptoms of the CNS, so we suspected that the CNS lesion was subsequently changed and was not as severe as the PNS. Of course we need to have a longer follow-up of this patient and more similar patients and related researches are needed to find the right answer.
CIDP can combine with central lesions, the pathogenesis is still unclear. On the point of the molecular biology: the myelin consists of inner and outer lipid and in-between myelin sheath. Approximately 15% to 30% of myelin proteins are found in the CNS and the PNS. Up to now, at least 2 common proteins which consist of the myelin have been found, resulting in similar autoimmune disorder in peripheral and central nerve in theory.[16] When the blood-brain and blood-nerve barriers were damaged, the antibodies cross the damaged barrier, resulting in immune reaction in peripheral and central nerve. From what is described above, we can see that if there appears immune-mediated inflammatory response, there is certain molecular basis of the mutual lesion. But the specific mechanism remains unclear.
As we know, the treatment of CIDP includes glucocorticoids, intravenous immunoglobulin, and plasma exchange. Through high dose of corticosteroids, the patient achieved a good prognosis, from which we can know that corticosteroids are effective to the patient who was diagnosed with CIDP and complicated CNS lesion, which was consistent with previous reports.[11–17]
It is a pity that this patient refused to undergo the brain MRI and electrophysiological examinations after discharge. As a result, we are unable to know the brain MRI abnormities and EMG changes in the past year. Simultaneously, because we just report 1 patient, and the number of similar cases is too small, it is hard for us to find out the similarity to eliminate the contingency of the phenomenon that CNS abnormalities in CIDP patient at present. We look forward to further research in the future.
Acknowledgments
The authors thank the patient for cooperation.
Author contributions
BH neurological evaluation of the patient and manuscript composition. YZ, XL, LX, QX, and QL neurological evaluation of the patient and suggestions regarding manuscript composition. XQ and WD neurological evaluation of the patient and manuscript composition. All authors read and approved the final manuscript.
Conceptualization: Xueliang Qi, Weijiang Ding.
Data curation: Bolin Hu, Yibiao Zhou, Xiaoqing Lu.
Formal analysis: Bolin Hu, Xueliang Qi, Weijiang Ding.
Investigation: Bolin Hu, Yibiao Zhou, Xueliang Qi, Weijiang Ding.
Methodology: Yibiao Zhou, Qianqian Xiong and Qing Liu.
Supervision: Xueliang Qi, Weijiang Ding.
Writing – original draft: Bolin Hu.
Writing – review and editing: Xueliang Qi, Weijiang Ding.
Footnotes
Abbreviations: AQP4 = aquaporin-4, CIDP = chronic inflammatory demyelinating polyradiculoneuropathy, CNS = central nervous system, EMG = electromyography, MRI = magnetic resonance imaging, NMO = neuromyelitis optica, PNS = peripheral nervous system.
Declaration: Ethics and Consent to Participate statement: Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the editor of this journal.
Consent to Publish statements: Written informed consent was obtained from the patient for publication of this case report. A copy of the written consent is available for review by the Editor-in-chief of this journal.
Funding: This work was funded by Foundation of the Graduate Innovation Center, Jiangxi province, China (YC2017-S097).
Availability of data and materials statement: Details of the patient are available in the hospital notes for the Editor-in-chief of this journal for review.
Competing Interest statement: The authors declare that they have no competing interests.
References
- [1].Mathey EK, Park SB, Hughes RA, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry 2015;86:973–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wingerchuk DM, Lennon VA, Pittock SJ, et al. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–9. [DOI] [PubMed] [Google Scholar]
- [3].Pittock SJ, Weinshenker BG, Lucchinetti CF, et al. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 2006;63:964–8. [DOI] [PubMed] [Google Scholar]
- [4].De Sousa EA, Chin RL, Sander HW, et al. Demyelinating findings in typical and atypical chronic inflammatory demyelinating polyneuropathy: sensitivity and specificity. J Clin Neuromuscul Dis 2009;10:163–9. [DOI] [PubMed] [Google Scholar]
- [5].Latov N. Diagnosis and treatment of chronic acquired demyelinating polyneuropathies. Nat Rev Neurol 2004;10:435–46. [DOI] [PubMed] [Google Scholar]
- [6].Lassmann H, Budka H, Scnaberth G. Inflammatory demyelinating polyradiculitis in a patient with multiple sclerosis. Arch Neurol 1981;38:99–102. [DOI] [PubMed] [Google Scholar]
- [7].Li XZ, Yang RM. The discussion of 2 cases which show chronic demyelination and central nerves lesion. J Anhui Med Univ 1994;29:53–5. [Google Scholar]
- [8].Di Trapani G, Carnevale A, Cioffi RP, et al. Multiple sclerosis associated with peripheral demyelinating neuropathy. Clin Neuropathol 1996;15:135–8. [PubMed] [Google Scholar]
- [9].Matsuse D, Ochi H, Tashiro K, et al. Exacerbation of chronic inflammatory demyelinating polyradiculopathy during interferon beta-1b therapy in a patient with childhood-onset multiple sclerosis. Int Med 2005;44:68–72. [DOI] [PubMed] [Google Scholar]
- [10].Arias M, Requena I, Pereiro I. Multiple sclerosis and hypertophic demyelinating neuropathy. J Neurol Neurosurg Psychiatry 1992;55:857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wakai S, Watanabe Y, Ichiki T, et al. Childhood multiple sclerosis: MR images and clinical variations in four Japanese cases. Brain Dev 1994;16:52–6. [DOI] [PubMed] [Google Scholar]
- [12].Rubin M, Karpati G, Carpenter S. Combined central and peripheral myelinopathy. Neurology 1987;37:1287–90. [DOI] [PubMed] [Google Scholar]
- [13].Pirko I, Kuntz NL, Patterson M, et al. Contrasting effects of IFNbeta and IVIG in children with central and peripheral demyelination. Neurology 2003;60:1697–9. [DOI] [PubMed] [Google Scholar]
- [14].Quan D, Pelak V, Tanabe J, et al. Spinal and cranial hypertrophic neuropathy in multiple sclerosis. Muscle Nerve 2005;31:772–9. [DOI] [PubMed] [Google Scholar]
- [15].Warabi Y, Yamazaki M, Shimizu T, et al. Abnormal nerve conduction study findings indicating the existence of peripheral neuropathy in multiple sclerosis and neuromyelitis optica. Biomed Res Int 2013;2013:847670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sarova-Pinhas I, Achiron A, Gilad R, et al. Peripheral neuropathy in multiple sclerosis: a clinical and electrophysiologic study. Acta Neurol Scand 1995;91:234–8. [DOI] [PubMed] [Google Scholar]
- [17].Naganuma M, Shima K, Matsumoto A, et al. Chronic multifocal demyelinating neuropathy associated with central nervous system demyelination. Muscle Nerve 1991;14:953–9. [DOI] [PubMed] [Google Scholar]