

Abstract
Objective
Rheumatoid arthritis (RA) is characterized by the activation of B cells that produce anti-citrullinated protein antibodies (ACPA) and rheumatoid factors (RF), but the mechanisms by which tolerance is broken in these B cells remain incompletely understood. Here we investigate whether ACPA and RF B cells break tolerance through distinct molecular mechanisms.
Method
We developed antigen-tetramers to isolate ACPA- and RF-producing B cells and performed single-cell RNA-sequencing on 2,349 B cells from six RA patients to analyze their immunoglobulin repertoires and transcriptional programs. Prominent immunoglobulins were expressed as monoclonal antibodies and tested for autoantigen reactivity.
Results
ACPA and RF B cells were enriched in the peripheral blood of RA patients relative to healthy controls. Characterization of patient-derived monoclonal antibodies confirmed ACPA and RF targeting of tetramer-specific B cells at both antigen-inexperienced and affinity-matured B-cell stages. ACPA B cells utilized more class-switched isotypes and exhibited more somatic hypermutations relative to RF B cells, and these differences were accompanied by the downregulation of CD72 and upregulation of genes that promote class-switching and T cell-dependent responses. In contrast, RF B cells expressed transcriptional programs that stimulate rapid memory reactivation through multiple innate immune pathways. Coexpression analysis revealed that ACPA- and RF- B cell enriched genes belong to distinct transcriptional regulatory networks.
Conclusion
Our findings suggest that ACPA and RF B cells are imprinted with distinct transcriptional programs, suggesting that these autoantibodies associated with increased inflammation in RA arise from two different molecular mechanisms.
Rheumatoid arthritis (RA) is characterized by chronic synovitis and joint destruction. Autoantibodies are a hallmark of RA and include anti-citrullinated protein antibodies (ACPA) and rheumatoid factor (RF) (1). Recent data suggest that ACPA and RF autoantibodies uniquely contribute to systemic inflammation and are associated with increased disease activity scores in RA (2). Further, B cells, which produce autoantibodies, cytokines, and provide T cell help, contribute to pathology as evidenced by the efficacy of B cell-depleting therapies in RA (3).
B cell activation requires coordination of many cell-extrinsic and intrinsic factors (1), and our understanding remains limited of how B cell tolerance is broken in RA. B cell activation is regulated by BCR signaling and costimulatory signals, and dysregulated signaling events can promote the survival and differentiation of autoreactive B cells that would otherwise be deleted or tolerized. RF, which can be present in other autoimmune diseases and chronic inflammation states, can also develop following acute microbial infections (4,5). RF generation has been recapitulated through challenges with LPS or double-stranded DNA in mice, providing evidence that RF B cells become activated in diverse states of inflammation (6,7). In contrast, ACPA are highly specific for RA and promote synovial inflammation and joint destruction in mouse models (8,9). Indeed, ACPA-producing B cells continuously undergo antigen-driven activation throughout disease, as evidenced by the continual regeneration of ACPA plasmablasts in patients (10). Based on these observations, we reasoned that the development and persistence of RF and ACPA may involve differential regulation of B cells at different stages of differentiation and may arise through the loss of tolerance mediated by distinct mechanisms.
Here, we developed antigen-tetramer staining reagents to isolate RF and ACPA B cells from seropositive RA patients and used single-cell-RNA-seq (scRNA-seq) to simultaneously recover the paired BCR variable region and the transcriptional profiles of individual B cells. By using a subset of B cells with assessed surface markers and sequenced immunoglobulins, we developed a computational B cell classifier (“BCellNet”) that used the scRNA-seq profile to organize B cells by subtype and characterize the primary, secondary, and antibody-producing stages of B cell differentiation. We applied these new methods to RA and identified distinct activation programs that distinguish RF from ACPA B cell responses. Our findings suggest that tolerance mediated by two different molecular mechanisms must be broken to activate ACPA and RF B cells to produce autoantibodies and promote synovitis in RA.
Patients and methods
Study design
All samples were collected after obtaining informed consent and according to human subject protocols approved by the Investigational Review Board at Stanford University (Supplementary Table 1). Peripheral blood was obtained from individuals with rheumatoid arthritis (n=6) who (i) met at least 4 of the 7 American College of Rheumatology 1987 classification criteria for RA, (ii) were seropositive for rheumatoid factor and/or cyclic citrullinated peptide, and (iii) did not receive B cell-depleting therapy. Age-matched healthy donors who were negative for HIV or TB were obtained from the Stanford Blood Center (n=5).
Autoreactive B cell isolation
Rheumatoid-factor tetramers were prepared as described in Supplementary Materials. Fourteen citrullinated peptides (400 μM) of highly prevalent citrullinated protein antigens (11) were prepared for citrullinated protein tetramer preparation as described in Supplementary Materials. Live CD3−CD14−CD19+ B cells were sorted using a FACSAriaII flow cytometer (Becton Dickinson) at single cell purity >99%. B cells were considered ACPA+RF− or RF+ACPA− if they stained double-positive for the autoantigen-of-interest and double-negative for the other tetramerized antigen.
Library preparation and next-generation sequencing
Template-switching reverse transcription of mRNA was primed using the STRT-oligo (12), and cDNAs were barcoded using template-switched oligos containing cell and molecular identifiers (Supplementary Table 2). Barcoded cDNA libraries were pooled and amplified using PCR1-FWD and PCR1-REV primers and then sonicated. 5′ fragments of cDNA molecules were purified using MyOne C1 streptavidin dynabeads (Life Technologies) and uncoupled by restriction digest. Libraries were constructed using TruSeq-V2 kits (Illumina) and sequenced using the HiSeq2000 (2×100). Detailed methods are in the Supplementary Materials.
Single-cell RNA-seq pipeline
Custom R scripts extracted cell and molecule barcodes from raw sequencing reads. Reads were aligned to the human genome (hg38) (13) using bowtie2 (v.2.1.0) (14). Duplicate reads generated by PCR were removed and final expression was calculated using RSEM (v.1.2.25) (15). The BCellNet subtype classification was trained as described in Supplementary Materials, allowing us to estimate a classification function that provided the most likely cell type for any cell with an scRNA-seq profile.
De novo assembly of immunoglobulin VH and VL regions; classification of germline V, D, and J regions; and identification of CDR3 clusters were performed as described in Supplementary Materials.
Recombinant monoclonal antibody testing
A preliminary list of candidate antibodies was first generated from autoreactive CDR3 clusters. The top VH-VL pairings from each CDR3 cluster with complete V-region sequence reconstitution were expressed as human IgG1 antibodies with IgL2 or IgK light chains (Lake Pharma). RF reactivity was determined by generating F(ab’)2 fragments, and probing plates coated with human IgG-Fc (Jackson ImmunoResearch). CCP binding was determined using CCP3 ELISA (Inova Diagnostics). The threshold for positivity was 3 SD above the mean of negative control monoclonal antibodies. Autoantigen microarrays were performed as previously described (9). Detailed methods are in the Supplementary Materials.
Differential expression testing
We tested differential expression through a series of linear models including a subset-stratified model, which accounts for the probability of cells belonging to the plamablast (ASC), double negative, memory and naïve subtypes and a global model, which does not account for subtype. Further explanation of the two models is in Supplementary Materials.
Both models were fit in MAST (16) version 0.933 using a two-step procedure to select genes that showed consistent differential expression across patients. Genes were considered to be differentially expressed if they (i) were marginally differentially expressed at 10% FDR and (ii) had a standardized effect size greater than 2. The standardized effect size selects genes most likely to substantially affect transcription in the population of RA patients. Both models also returned a partial residual, which is the log-expression after correcting for nuisance factors. Gene functional categories were defined by organizing the DEGs and 33 genes-of-interest into 16 potentially overlapping groups. Partial residuals were used to calculate functional scores for each cell. Detailed methods are in the Supplementary Materials.
Coexpression analysis
487 genes of interest were tested for coexpression after stratifying by subtype and removing marginal coexpression due to subtype, tetramer specificity, patient, and CDR effects. Pearson correlation matrices were calculated for each pair of genes and the averages of the matrices were compared within the 16 gene sets. Gene pairs were considered differentially expressed if their average correlation differed from the background correlation coefficient at an FDR<5%. Detailed methods are in the Supplementary Materials.
Results
Elevation of naïve, memory, and antibody-secreting autoreactive B cells in the blood of RA patients
In this study, we sampled blood B cells from seropositive RA patients using flow cytometry to examine the frequency of ACPA and RF B cells as well as to isolate these cells for scRNA-seq analysis (Figure 1A).
Figure 1.

Interrogation of autoreactive B cell subsets through tetramer sorting and transcriptomic classification of single cells. (A) Schematic of the experimental workflow for the tetramer-based isolation, single cell-RNA-seq, and simultaneous immunoglobulin repertoire and transcriptomic analyses of B cells. (B) Representative flow cytometry gating approach for the identification of B cells producing RF or ACPA. (C) Comparison of RF and ACPA B cell subtype frequencies in peripheral blood between RA patients and healthy donors. A minimum of 15,000 B cells were analyzed for each sample. * = P<0.05 (D) B cell subtype distribution using the BCellNet classifier for the recovered 2,349 single cells from seven individuals. The ACPA+ and RF+ B cells from subject R01 produced poor quality yields during library preparation and were excluded from analysis. No ACPA+ cells were recovered from subject R04 and HD5 and R04, and no RF+ cells were recovered from subject R48 and HD5.
To isolate RF and ACPA B cells, biotinylated human IgG-Fc as well as 14 citrullinated peptides previously demonstrated to be targeted by RA ACPA were individually coupled to two sets of streptavidin-bound fluorophores (Figure 1B). Autoantigen labeling with dual fluorophores was previously demonstrated to reduce the false-positive detection of fluorophore-reactive B cells, and tetramerization enabled the detection of B cells expressing BCRs with moderate affinity for autoantigen epitopes (10,17). Analysis of the B cell frequencies reveal that RF and ACPA cells are rare in RA blood and virtually undetectable in healthy controls, comprising on average 0.25% and 0.14% of the sampled B cells, respectively (Figure 1C). This small enrichment of RF and ACPA cells in RA patients is significant compared to healthy controls. While the frequency of RF and ACPA B cell subsets varies between patients, autoreactive B cells from the CD27− (naïve, non-affinity matured, and double-negative (DN) memory) and CD27+ (classical memory) compartments are present in all individuals, consistent with previous observations of defective central and peripheral tolerance in RA (18,19). The presence of RF and ACPA ASCs in RA blood reveals that RA patients have active ongoing humoral autoimmunity.
We then performed scRNA-seq on sorted ACPA, RF, and tetramer-negative (Tet−) B cells from RA donors and one healthy donor using a modified STRT-Seq method that enables single molecule quantification and computational reconstitution of the paired BCR for each B cell (Figure 1A) (Supplementary Figure 1). The population of Tet− cells provides a baseline to compare characteristics of the autoreactive populations. After aligning and deduplicating reads, we recovered an average of 4.8E4 molecules per cell with no substantial differences between experimental groups (Supplementary Figure 2).
BCellNet predicts peripheral B cell subsets using scRNA-seq profiles
To evaluate the contribution of various B cell subsets to RA pathology, we organized B cells by subtype using a B cell transcriptomic classifier we developed, termed BCellNet. Predicting subtype with this transcriptomic classification scheme allows us to consider a continuum of differentiation states (20) and assign a subset probability score for each cell, improving upon the quantized subset identifications available in flow cytometric gating. In total, 159 genes (<2.5%) serve as predictors (Supplementary Table 3). Some were previously described in bulk-level studies, such as MS4A1 and TNFRSF13B (21), while others, such as SEC61G and EIF5A, are novel (Supplementary Figure 3).
Applying BCellNet to heterogeneous B cells isolated from RA samples demonstrates positive correlations between subtype fractions assessed by flow cytometry and the classifier (Spearman’s rho of 0.56-0.89), indicating that the classifier recapitulates flow cytometry-identified populations while also parsing subtype-dependent gene effects (Supplementary Figure 4). There are certain discrepancies between the flow fractions and classifier fractions, likely due to sampling variability in the flow sorting and classification error from BCellNet. However, we do not believe these discrepancies significantly altered B cell sub-type classification (see Supplementary Methods). In light of this, rather than using quantized assignments to the most likely subtype, we utilize the continuously-valued posterior probabilities of subtype membership as stratifying covariates for subsequent analysis (Supplementary Table 4).
We observed a general correlation between serological autoantibody levels and the recovery of CIT and RF B cells despite minor variances in the number of cells sorted due to stochastic selection during FACS. We also observed an enrichment in the classification of memory cells through transcriptome analysis (Figure 1D). The BCellNet classifier accounted for much of the variability inherent to scRNA-seq. The remaining bias in classification through this method most likely did not have a significant impact on our further analyses of these cells (see Supplementary Methods).
BCR analysis reveals CDR3 convergence and autoantigen targets
Next, we recovered paired BCR variable-region genes expressed by individual B cells, enabling integrated analysis of affinity maturation between autoreactive populations and functional characterization of recombinant antibodies expressed from reconstituted VH/VL sequences (Supplementary Figure 5).
To investigate how affinity maturation shapes B cell responses against the autoantigens, we clustered RF, ACPA, and Tet− B cells sharing >80% Levenshtein similarities in their CDR3 sequences (Figure 2B, Supplementary Figure 6). This represented the most stringent threshold that preserved cluster membership among cells that exhibit identical germline genes and CDR3 lengths, and therefore putatively derive from the same progenitor (Supplementary Table 6) (22).
Figure 2.
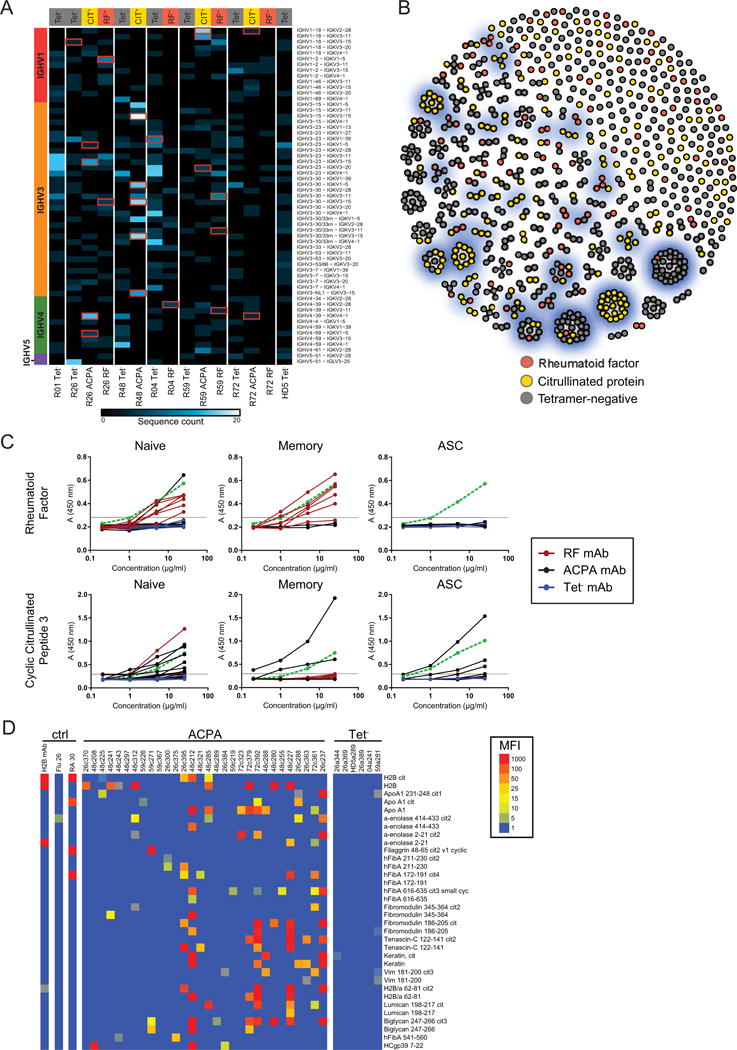
RF and ACPA B cells exhibit distinct immunoglobulin gene usage and CDR3 convergence. (A) Heatmap showing most frequently utilized VH-VL pairs for each patient’s CIT-Tetramer+ (CIT-Tet+), RF-Tet+, and Tet− populations. Column labels denote antigen-specific populations arranged by patient. Row labels denote VH-VL pairs arranged by VH gene family. Red boxes identify prominent VH-VL pairs from which recombinant antibodies were expressed. (B) Greedy, agglomerative clustering of single B cells with H.CDR3 and L.CDR3 sequences sharing >80% homology. Each cell, colored by antigen specificity, is a node. Gray edges connect cells belonging to the same cluster. Prominent clusters from which recombinant antibodies were expressed are highlighted in blue. (C) Representative ELISA results (n=3) against RF and cyclic-citrullinated peptide 3 for all recombinant antibodies, measured using absorbance values, and plotted by B cell subtype as determined by BCellNet. Antibodies expressed from RF, ACPA, and Tet− B cells are represented using red, black, and blue solid lines, respectively. The dotted green line denotes a positive control monoclonal antibody. Threshold for positive reactivity is represented with a horizontal gray line. (D) Top citrullinated autoantigen microarray hits for expressed monoclonal antibodies. Binding intensity is shown for both citrullinated peptides and available corresponding non-citrullinated antigens.
To determine if the size of the CDR3 clusters can measure the magnitude of convergent evolution, we compared the number of cells per tetramer group in each CDR3 cluster to calculate the cluster multiplicity. If each cell from a tetramer group clustered separately, then the multiplicity would be 1, while if all N cells were in a single cluster, the multiplicity would be N. ACPA B cell multiplicities are significantly larger than Tet− multiplicities in 2 of 4 patients (multiplicity differences 1.3 and 0.3 cells/cluster; P<0.002 under hypergeometric sampling), and significantly larger than RF multiplicities in 1 of 3 patients (multiplicity difference 0.17 cells/cluster; P<0.04). No significant differences between RF and Tet− multiplicities are observed. Hence, the elevated frequency of CDR3 convergence suggests that ACPA B cells undergo extensive affinity maturation relative to Tet− and RF populations.
We then expressed 42 representative RF and ACPA BCRs from the most prominent VH-VL pairings and CDR3 clusters as recombinant monoclonal antibodies to identify their autoantigen targets and examine whether autoreactivity is present in antigen-inexperienced and -experienced B cell stages (Figures 2A and B) (Supplementary Table 7). Eleven out of the 12 antibodies cloned from RF-sorted cells bind to human IgG-Fc, whereas only one of the 30 antibodies from ACPA-sorted cells bind to IgG-Fc, and none of the antibodies from Tet− B cells bind to IgG-Fc (Figure 2C). We also find that 13 of the 30 monoclonal antibodies from citrullinated antigen-sorted cells bind to cyclic citrullinated peptide (CCP3), a citrullinated peptide mimic used to diagnose RA. None of the Tet− antibodies recognize CCP3 and only two of the 12 RF antibodies bind CCP3 (Figure 2C). Using the CCP3 ELISA and a protein microarray coated with over 300 RA autoantigens, we identify citrullinated antigen hits for 25 of the 30 citrullinated antigen-sorted antibodies (Figure 2D). We do note that several antibodies cloned from ACPA B cells were cross-reactive with non-citrullinated antigens, as is consistent with previous reports (9). This confirms that the RF and ACPA tetramer reagents enabled isolation of RF and ACPA B cells. Notably, RF and ACPA B cells are in all subsets, including non-affinity matured cells, implicating dysfunctional central and peripheral tolerance in RA.
RF and ACPA B cells differ in class-switch and somatic mutation frequencies
We next asked whether the duration of affinity maturation differs between ACPA and RF B cells, as measured through class-switch recombination (CSR) rates and accumulated somatic mutations on the BCR. Because CSR and mutation rates vary between subtypes, which themselves vary by specificity, we used a mixed-effects logistic regression to measure the unique effect of antigen on CSR and mutation rate adjusted for subtype. We find that ACPA B cells have over 3.8-fold greater odds of being class-switched than Tet− B cells and 3.7-fold greater odds compared to RF B cells but find no difference between Tet− and RF B cells (Figures 3A and B). We also find that RF B cells are 3% less mutated from germline than Tet− B cells, while ACPA B cells non-significantly have 1% higher mutation rates than Tet− B cells (Figures 3C and D). Thus, ACPA B cells undergo more isotype class-switching, have higher mutation rates, and show evidence of increased CDR3 convergence, while RF B cells tend to use unswitched isotypes and have fewer mutations.
Figure 3.
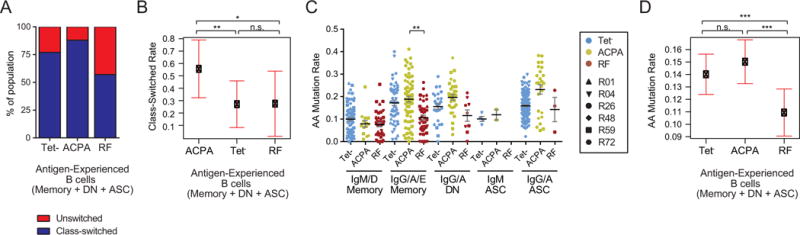
Antigen-experienced RF and ACPA B cells differ in class-switch and somatic hypermutation rates. (A) Proportion of antigen-experienced (i.e. unswitched (IgM/D) and class-switched (IgG/A/E) memory, double negative memory, and antibody-secreting) B cells for each antigen specificity. (B) Class-switch rates for antigen-experienced ACPA, RF, and Tet− B cells in RA patients, adjusting for subset differences and patient variability using a generalized linear mixed model. Error bars indicate 95% confidence intervals. * = P<0.05; ** = P<0.01; n.s. = not significant (C) Somatic mutation rates for antigen-experienced ACPA, RF, and Tet- B cells by subtype. Error bars indicate mean ± SEM. ** = P<0.01 (D) Somatic mutation rates for the immunoglobulins expressed by ACPA, RF, and Tet− B cells adjusting for subset differences and patient-to-patient variability using a linear mixed model. Error bars indicate 95% confidence intervals. *** = P<0.001; n.s. = not significant
Functional differential regulation in the primary and secondary RF and ACPA B-cell responses
To elucidate the transcriptional profiles underlying the BCR differences between RF and ACPA responses, we applied a hurdle model to identify 454 differentially expressed genes (DEGs) between RF/ACPA and Tet− B cell populations for all patients at global (i.e. regressing out the effect of subset on gene expression) and subset-stratified comparisons (Figure 4A) (Supplementary Tables 8 and 9). Performing both global and subset-stratified analyses allowed us to identify a greater amount of DEGs than with either method alone. Both gene sets were relevant to identifying autoreactive B cell signatures. To examine the cumulative functional impact of the DEGs, we organized DEGs into modules based upon previously published literature. The partial residual for each gene, which represents residual expression attributable to antigen specificity, was used to calculate a module score for ACPA, RF and Tet− cells for each subtype (Supplementary Figure 7), pinpointing the developmental stage(s) where differential autoimmune regulation occurs. The most significantly different modules were concentrated within the naïve and memory compartments (Figures 4B and C).
Figure 4.
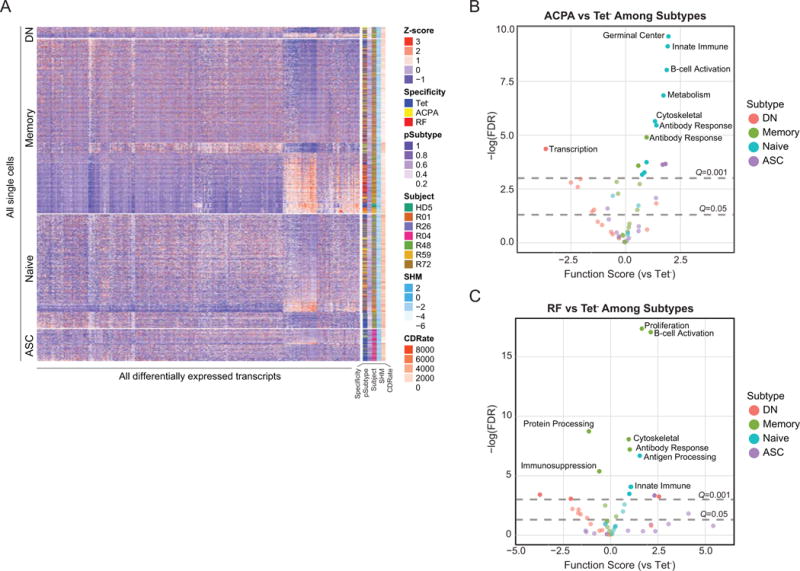
Aberrant primary and secondary autoreactive B cell gene expression signatures in RA. (A) Subset-stratified heatmap showing the Z-scaled partial residual expression of the differentially expressed genes for all 2,349 B cells from patients. Of the 454 differentially expressed genes, 121 genes are known to be immune-related. (B and C) Volcano plots showing differentially expressed functional modules between ACPA versus Tet− (B) and RF versus Tet− (C) for all B cell subtypes. Modules consist of the average partial residuals from genes that share cellular functions. Gray horizontal dotted lines denote FDR q-value cutoffs.
ACPA primary responses initiate class-switch programs and downregulate CD72
In the naïve ACPA population, module scores for antibody response and B cell activation are elevated relative to the RF and Tet− naïve B cell populations (Figures 5A and B). Within these modules, MSH5, JAK3, and EXOC4, which all promote antibody class-switched responses (23–25), are highly elevated in the naïve ACPA compartment and sustained in memory ACPA B cells (Figure 5C). Additionally, CD72, which inhibits the differentiation of resting B cells, is downregulated in naïve and memory ACPA populations but not in RF cells (26). In the B cell activation module, we detect naïve ACPA elevation of adhesion molecules (CD84, JAM2 and ITGB2) which prolong B:T cell interactions and thereby affinity maturation (Figure 5D) (27,28). Together, these signatures suggest that ACPA B cell responses are programmed for class-switching through T cell help and facilitated by CD72 downregulation in the naïve stage.
Figure 5.
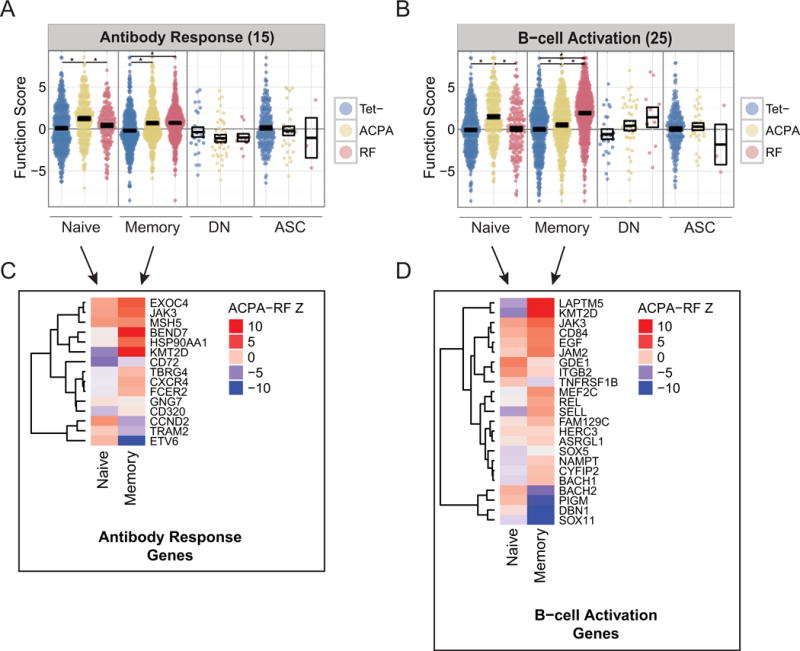
ACPA B cells and RF B cells exhibit distinct transcriptional programs that promote antibody class-switching and B-cell activation. (A) Subset-stratified plot of average partial residual expression in the antibody response module (15 genes). * = P<0.05 (B) Subset-stratified plot of average partial residual expression in the B-cell activation module (25 genes). * = P<0.05 (C) Z-scores testing for differential expression between ACPA and RF B cells in antibody response genes. (D) Z-scores testing for differential expression between ACPA and RF B cells in B-cell activation genes.
RF B cells express transcriptional programs associated with IgM responses and rapid recall responses
Within the B cell activation module, the transcription factor SOX11, which promotes highly proliferative IgM responses and suppresses germinal center transcription factor BCL6, and DBN1, a SOX11-regulated gene, are elevated in memory RF cells relative to memory ACPA Tet− B cells (Figure 5D) (29,30). Transcriptional regulator BACH2 is also significantly upregulated in RF memory relative to ACPA, which is a feature of unswitched B cell responses (Figure 5D) (31,32). The selective upregulation of SOX11 and BACH2 in RF memory B cells provides an explanation for their low class-switch and mutation rates.
Differences in B cell costimulation drive ACPA generation and RF versatility to multiple innate activation pathways
Innate immune mediators provide costimulatory signals that can augment B cell responses (33). In the memory compartment, RF and ACPA cells exhibit differentially expressed subsets of genes despite not having higher average module scores relative to Tet− B cells (Figures 6A and B). RF memory B cells exhibit altered expression of signature genes involved in MyD88 signaling (LRRFIP1), IFN signaling (IFI6, PSMB10), and DAP12 signaling (SIGLEC14), none of which are enriched in ACPA memory B cells (Figure 6B). In contrast, ACPA memory B cells show coordinated upregulation of positive regulators of NF-kB signaling (REL and HSP90AA1) (Figure 6B), which is critical for promoting germinal center responses and survival (34).
Figure 6.
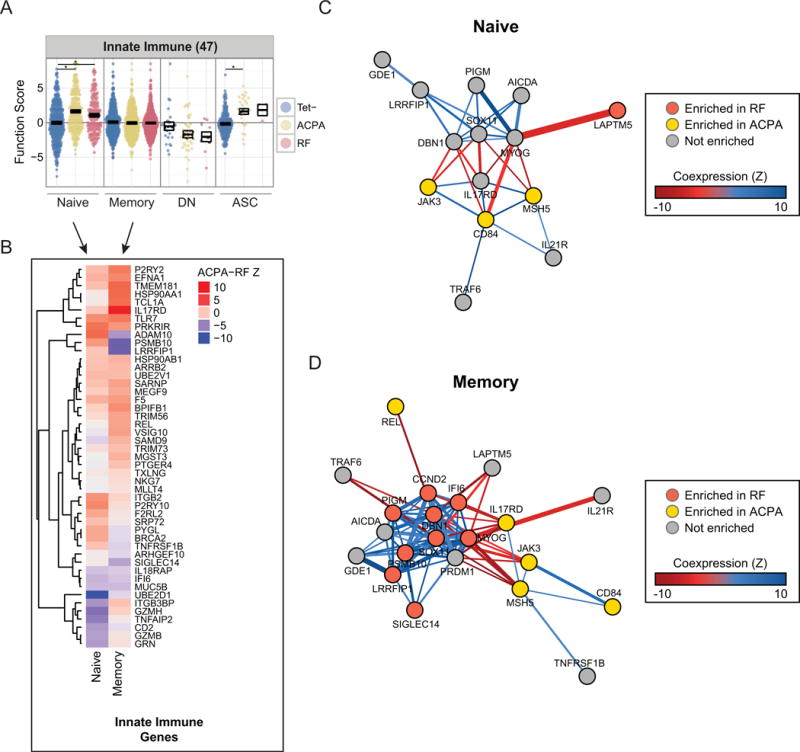
Innate immune mediators enriched in RF B cells and comprise distinct coexpression networks from ACPA-enriched genes. (A) Subset-stratified plot of average partial residual expression in the innate immune module (47 genes). * = P<0.05 (B) Z-scores testing for differential expression between ACPA and RF B cells in innate immune genes. (C and D) Coexpression networks of selected module genes, stratified by subset. Genes enriched in ACPA/RF have differential expression Z-score > 1 relative to RF/ACPA and expression greater than or equal to Tet− population. The color and weight of the edges indicate the Z-score of the Pearson correlation coefficient for pairs of genes. Correlations with FDR-adjusted P-values > 0.05 are omitted. (C) Network displaying the coexpression between B-cell activation and innate immune genes involved in naïve ACPA/RF B cell responses. (D) Network displaying the coexpression between B-cell activation and innate immune genes involved in ACPA/RF memory B cell responses.
Distinct antibody response and innate immune transcriptional regulatory networks characterize ACPA and RF B cells
We analyzed single-cell gene coexpression to identify transcriptional regulatory networks within ACPA and RF DEGs and to provide insights into the mechanisms underlying the loss of tolerance in ACPA as compared to RF B cells (Supplementary Table 10). In both the primary response and memory compartments, ACPA B cells coexpress MSH5, CD84, and JAK3 (Figures 6C and D), revealing a co-regulated transcriptional network specific to ACPA development. We also asked whether upstream effectors of JAK3 signaling were coexpressed with JAK3. Both IL21R and CD40, which receive signals from follicular helper T cells, are coexpressed with the MSH5/CD84/JAK3 network in ACPA memory B cells (Figure 6D) (35,36). Coexpression between these genes suggests that JAK3 links germinal center signals with class-switch functions in both ACPA primary and secondary responses. Thus, antigen-experienced ACPA B cells exhibit a transcriptional program characterized by a robust germinal center response.
We found evidence that RF cells coexpress genes belonging to multiple B cell response pathways. The innate immune genes PSMB10, LRRFIP1, IFI6, and SIGLEC14 are all coexpressed, implying that individual memory RF B cells exhibit coordinate upregulation of genes (Figure 6D). Further, we find that these genes are all significantly coexpressed with PRDM1 and CCND2. The coexpression between these innate immune genes, SOX11, and markers of ASC differentiation, suggests that innate immune genes upregulated in RF memory cells help prime rapid recall responses. Therefore, in contrast to ACPA B cells, RF B cells upregulate transcriptional programs involved in innate immune response pathways and that facilitate rapid recall responses.
Discussion
Accumulating clinical evidence implicates a pathogenic role for B cells in RA (3,37,38), and therapeutic modalities that specifically target dysregulated pathways in autoreactive B cell development will be critical for sustained remission. Here we investigate the differential regulation of B cell tolerance in RA using antigen-tetramers to isolate disease-relevant B cells and scRNA-seq to define RF- and ACPA-specific immunoglobulin usage and transcriptional programs. Our findings demonstrate that ACPA and RF B cells are imprinted with distinct transcriptional programs, suggesting different molecular mechanisms mediate the loss of tolerance in ACPA versus RF B cells.
While self-reactive B cell precursors leak into the periphery at a low frequency in healthy individuals (39), the nearly exclusive detection of CD27−IgM+/IgD+ ACPA B cells and the elevation of CD27−IgM+/IgD+ RF cells in RA patients indicates that ACPA/RF precursors are predominantly deleted in healthy individuals but not in RA patients. This suggests a defect in central tolerance in RA that is likely due to a combination of genetic and environmental factors. Defective regulation through polymorphisms in BCR signaling genes, as well as altered receptor editing have been previously reported and may permit the survival of autoreactive immature B cells that are normally removed (40). Systemic inflammatory mediators can also promote the survival of autoreactive B cells by overwhelming apoptotic or receptor-editing processes (41). While our data indicate that differential regulation of ACPA cells begins at the naïve stage, future studies will be needed to decipher whether this occurs in the bone marrow or the periphery.
The development of ACPA appears to stem from multiple defective tolerance mechanisms that fail to restrain ACPA B cell activation, and from promotion by autoreactive T cell help. In support of this, a study by Makrygiannakis and colleagues found that citrullinated protein levels are increased in both RA and non-RA inflamed tissues; yet only RA patients persistently develop ACPA (42). We find that CD72, an inhibitory co-receptor, is downregulated in naïve ACPA B cells relative to RF and Tet− B cells. This may enable transduction of activating B cell signals that override tolerogenic signals received by ACPA B cells (43). Expression of the co-receptor CD21 was down-regulated between naïve ACPA and Tet− cells, as well, though this effect was not significant in our study.
Once activated, ACPA B cells undergo extensive affinity maturation as evidenced by high mutation rates, class-switching rates, and CDR convergence that are associated with expression of robust transcriptional programs reminiscent of those in germinal centers. The elevation of JAK3 in ACPA B cells suggests dependence on the IL21R/JAK3 pathways for ACPA production and provides a potential explanation for why tofacitinib, which targets JAK3, results in lower remission rates in ACPA+ patients (44). Further emphasizing the importance of T cell help, we find an upregulation in genes that prolong B:T cell interactions (CD84, JAM2, and ITGB2) and molecules that promote antibody class-switching (MSH5). While these transcriptional signatures suggest that affinity maturation of ACPA occurs in germinal centers, future studies will be necessary to determine whether this occurs in conventional germinal center reactions, through ectopic T-cell help, or other mechanisms.
Identification of shared transcriptional programs for ACPA B cells that target diverse citrullinated proteins suggests that exposure to a spectrum of citrullinated proteins rather than a specific citrullinated epitope, drives the ACPA response, in agreement with a previous study that examined ACPA formation during relapse (37). Hence, we propose that differential B cell imprinting, enhanced inflammation, and citrullinated protein availability combinatorially create an environment conducive to ACPA B cell activation and autoantibody production.
In contrast to ACPA, the RF B cell response is characterized by transcriptional programs associated with broad innate immune activation, rapid recall responses, and ASC differentiation. Counter to the extensive affinity maturation driving the ACPA response, the RF response is characterized by B cells that express IgM and exhibit lower mutation rates. The bias toward the IgM isotype, which can bind up to ten IgG molecules in its pentameric form, enables RF responses to aggregate secreted antibodies and immune complexes. The low mutation rates suggest that certain V(D)J combinations may be intrinsically reactive with IgG-Fc. Whereas T cell help causes ACPA to undergo prolonged affinity maturation, RF B cells can differentiate into memory B cells or ASCs with minimal somatic hypermutation.
Our data indicate that RF B cells mount rapid secondary responses with co-stimulation from multiple innate immune pathways (e.g. MyD88, IFN, DAP12), suggesting that RF B cells are primed to expand in response to diverse inflammatory stimuli. Upregulation of these signatures in RF memory B cells correlates with markers of ASC, implying that RF memory generates a reservoir of cells that can mount rapid recall responses.
The etiology behind RF production in multiple inflammatory contexts remains an area of active investigation. Given the presence of RF in other inflammatory diseases and its relatively lower specificity for diagnosing RA (4,5), our results suggest that RF could represent a response to resolve antibody-mediated inflammation by facilitating the clearance of antibodies and immune complexes (45). Thus, RF elevations in RA may represent an expanded pool of naturally occurring B cells for the purpose of engulfing antigen-antibody complexes to enable antigen presentation to T cells. Future studies comparing the gene expression of RF B cells between individuals with and without RA will be important to provide further insights into whether RF in fact results from dysregulated B cell tolerance.”
Our findings reveal that ACPA and RF B cells are imprinted with distinct transcriptional programs at different stages of development, with RF B cells characterized by activation of innate immune activation pathways while ACPA B cells are characterized by loss of tolerance in the primary response followed by robust affinity-matured responses. Future studies will further define how ACPA and RF B cells promote tissue damage in RA, as well as the impact of targeted therapies on ACPA and RF B cell activation.
Supplementary Material
Supplementary Table 1. RA patient and healthy donor demographic and clinical data.
Supplementary Table 2. Cell and molecule identifier oligonucleotide design.
Supplementary Table 3. Genes used for transcriptomic subset prediction.
Supplementary Table 4. B cell subset classifier applied to healthy and RA B cells.
Supplementary Table 5. BCR reconstitution and IG isotype recovery.
Supplementary Table 6. Clustering of antigen-specific B cells by paired HC and LC CDR3 sequences.
Supplementary Table 7. Monoclonal antibody expression and functional characterization.
Supplementary Table 8. Global ACPA and RF B cell differentially expressed transcripts.
Supplementary Table 9. Subsetted ACPA and RF B cell differentially expressed transcripts.
Supplementary Table 10. Subsetted single-cell coexpression testing.
Supplementary Figure 1. Single-cell RNA-seq library preparation and computational de-multiplexing workflow.
Supplementary Figure 2. Distribution of molecule counts (UMIs), pre-deduplicated reads (reads), and genes for each single cell library.
Supplementary Figure 3. Application of the transcriptomic B cell subset classifier to CD19+ B cells from RA patients.
Supplementary Figure 4. Comparison of the distribution of transcriptomic subset classifier frequencies (Classifier Fraction) to flow cytometry subset frequencies (Flow Fraction) from the same samples.
Supplementary Figure 5. Paired immunoglobulin variable-region reconstitution using hidden Markov Model-based assembly.
Supplementary Figure 6. Analysis of multi-specific CDR3 clusters.
Supplementary Figure 7. Differential expression tests for gene modules.
Acknowledgments
We thank S. Elliott, A. Gomez, L. Blum, and Z. Weng for insightful discussions and input.
Funding: This research was supported by NIH \ NIAMS R01 AR063676, U19 AI11049103 and U01 AI101981.
Footnotes
DR. WILLIAM ROBINSON (Orcid ID: 0000-0002-0841-7148)
Competing interests: W.H.R. is a consultant to, owns equity in, and serves on the board of directors of Atreca, Inc.
Author contributions
Study conception and design
Lu, McDavid, Gottardo, Robinson
Acquisition of data
Lu, Kongpachith, Lingampalli, Ju
Analysis and interpretation of data
Lu, McDavid, Robinson, Glanville, Kongpachith, Gottardo, Lingampalli, Ju
Data and materials availability: Raw sequencing data have been deposited in NCBI dbGaP and are accessible through accession number <<submission in process>>.
References
- 1.McInnes IB, Schett G. The Pathogenesis of Rheumatoid Arthritis. N Engl J Med. 2011;365:2205–2219. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 2.Sokolove J, Johnson DS, Lahey LJ, Wagner CA, Cheng D, Thiele GM, et al. Rheumatoid factor as a potentiator of anti-citrullinated protein antibody mediated inflammation in rheumatoid arthritis. Arthritis Rheumatol Hoboken NJ. 2014;66:813–821. doi: 10.1002/art.38307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edwards JCW, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 4.Witte T, Hartung K, Sachse C, Matthias T, Fricke M, Kalden JR, et al. Rheumatoid factors in systemic lupus erythematosus: association with clinical and laboratory parameters SLE study group. Rheumatol Int. 2000;19:107–111. doi: 10.1007/s002960050112. [DOI] [PubMed] [Google Scholar]
- 5.Salonen EM, Vaheri A, Suni J, Wager O. Rheumatoid factor in acute viral infections: interference with determination of IgM, IgG, and IgA antibodies in an enzyme immunoassay. J Infect Dis. 1980;142:250–255. doi: 10.1093/infdis/142.2.250. [DOI] [PubMed] [Google Scholar]
- 6.Izui S, Eisenberg RA, Dixon FJ. IgM rheumatoid factors in mice injected with bacterial lipopolysaccharides. J Immunol Baltim Md 1950. 1979;122:2096–2102. [PubMed] [Google Scholar]
- 7.Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, et al. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci U S A. 2009;106:12061–12066. doi: 10.1073/pnas.0905441106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, et al. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest. 2006;116:961–973. doi: 10.1172/JCI25422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan Y-C, Kongpachith S, Blum LK, Ju C-H, Lahey LJ, Lu DR, et al. Barcode-enabled sequencing of plasmablast antibody repertoires in rheumatoid arthritis. Arthritis Rheumatol Hoboken NJ. 2014;66:2706–2715. doi: 10.1002/art.38754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kerkman PF, Fabre E, van der Voort EIH, Zaldumbide A, Rombouts Y, Rispens T, et al. Identification and characterisation of citrullinated antigen-specific B cells in peripheral blood of patients with rheumatoid arthritis. Ann Rheum Dis. 2016;75:1170–1176. doi: 10.1136/annrheumdis-2014-207182. [DOI] [PubMed] [Google Scholar]
- 11.Sokolove J, Bromberg R, Deane KD, Lahey LJ, Derber LA, Chandra PE, et al. Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PloS One. 2012;7:e35296. doi: 10.1371/journal.pone.0035296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Islam S, Kjällquist U, Moliner A, Zajac P, Fan J-B, Lönnerberg P, et al. Highly multiplexed and strand-specific single-cell RNA 5′ end sequencing. Nat Protoc. 2012;7:813–828. doi: 10.1038/nprot.2012.022. [DOI] [PubMed] [Google Scholar]
- 13.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The Human Genome Browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. 2015 doi: 10.1186/s13059-015-0844-5. Available at: http://dspace.mit.edu/handle/1721.1/100458. Accessed April 24, 2017. [DOI] [PMC free article] [PubMed]
- 17.Doorenspleet ME, Klarenbeek PL, de Hair MJH, van Schaik BDC, Esveldt REE, van AHC Kampen, et al. Rheumatoid arthritis synovial tissue harbours dominant B-cell and plasma-cell clones associated with autoreactivity. Ann Rheum Dis. 2013 doi: 10.1136/annrheumdis-2012-202861. annrheumdis-2012-202861. [DOI] [PubMed] [Google Scholar]
- 18.Samuels J, Ng Y-S, Coupillaud C, Paget D, Meffre E. Impaired early B cell tolerance in patients with rheumatoid arthritis. J Exp Med. 2005;201:1659–1667. doi: 10.1084/jem.20042321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menard L, Samuels J, Ng Y-S, Meffre E. Inflammation-independent defective early B cell tolerance checkpoints in rheumatoid arthritis. Arthritis Rheum. 2011;63:1237–1245. doi: 10.1002/art.30164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bendall SC, Davis KL, Amir ED, Tadmor MD, Simonds EF, Chen TJ, et al. Single-Cell Trajectory Detection Uncovers Progression and Regulatory Coordination in Human B cell Development. Cell. 2014;157:714–725. doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaminski DA, Wei C, Qian Y, Rosenberg AF, Sanz I. Advances in Human B Cell Phenotypic Profiling. Front Immunol. 2012;3 doi: 10.3389/fimmu.2012.00302. Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3467643/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sekine H, Ferreira RC, Pan-Hammarström Q, Graham RR, Ziemba B, de Vries SS, et al. Role for Msh5 in the regulation of Ig class switch recombination. Proc Natl Acad Sci U S A. 2007;104:7193–7198. doi: 10.1073/pnas.0700815104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(Suppl):S121–131. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- 25.Jabara HH, Buckley RH, Roberts JL, Lefranc G, Loiselet J, Khalil G, et al. Role of JAK3 in CD40-Mediated Signaling. Blood. 1998;92:2435–2440. [PubMed] [Google Scholar]
- 26.Yamazaki T, Nagumo H, Hayashi T, Sugane K, Agematsu K. CD72-mediated suppression of human naive B cell differentiation by down-regulating X-box binding protein 1. Eur J Immunol. 2005;35:2325–2334. doi: 10.1002/eji.200425639. [DOI] [PubMed] [Google Scholar]
- 27.Postigo AA, Corbí AL, Sánchez-Madrid F, de Landázuri MO. Regulated expression and function of CD11c/CD18 integrin on human B lymphocytes. Relation between attachment to fibrinogen and triggering of proliferation through CD11c/CD18. J Exp Med. 1991;174:1313–1322. doi: 10.1084/jem.174.6.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong EB, Soni C, Chan AY, Domeier PP, Shwetank, Abraham T, et al. B cell-intrinsic CD84 and Ly108 maintain germinal center B cell tolerance. J Immunol Baltim Md 1950. 2015;194:4130–4143. doi: 10.4049/jimmunol.1403023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Björklund S, Wasik AM, Grandien A, Andersson P, Kimby E, et al. Gene Expression Profiling and Chromatin Immunoprecipitation Identify DBN1, SETMAR and HIG2 as Direct Targets of SOX11 in Mantle Cell Lymphoma. PLOS ONE. 2010;5:e14085. doi: 10.1371/journal.pone.0014085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larbolette O, Wollscheid B, Schweikert J, Nielsen PJ, Wienands J. SH3P7 Is a Cytoskeleton Adapter Protein and Is Coupled to Signal Transduction from Lymphocyte Antigen Receptors. Mol Cell Biol. 1999;19:1539–1546. doi: 10.1128/mcb.19.2.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kometani K, Nakagawa R, Shinnakasu R, Kaji T, Rybouchkin A, Moriyama S, et al. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity. 2013;39:136–147. doi: 10.1016/j.immuni.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 32.Seifert M, Przekopowitz M, Taudien S, Lollies A, Ronge V, Drees B, et al. Functional capacities of human IgM memory B cells in early inflammatory responses and secondary germinal center reactions. Proc Natl Acad Sci. 2015;112:E546–E555. doi: 10.1073/pnas.1416276112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 34.Gerondakis S, Siebenlist U. Roles of the NF-κB Pathway in Lymphocyte Development and Function. Cold Spring Harb Perspect Biol. 2010;2 doi: 10.1101/cshperspect.a000182. Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2857169/. Accessed April 22, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu R, Wu Q, Su D, Che N, Chen H, Geng L, et al. A regulatory effect of IL-21 on T follicular helper-like cell and B cell in rheumatoid arthritis. Arthritis Res Ther. 2012;14:R255. doi: 10.1186/ar4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ouchida R, Kurosaki T, Wang J-Y. A role for lysosomal-associated protein transmembrane 5 in the negative regulation of surface B cell receptor levels and B cell activation. J Immunol Baltim Md 1950. 2010;185:294–301. doi: 10.4049/jimmunol.1000371. [DOI] [PubMed] [Google Scholar]
- 37.Cambridge G, Leandro MJ, Lahey LJ, Fairhead T, Robinson WH, Sokolove J. B cell depletion with rituximab in patients with rheumatoid arthritis: Multiplex bead array reveals the kinetics of IgG and IgA antibodies to citrullinated antigens. J Autoimmun. 2016;70:22–30. doi: 10.1016/j.jaut.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 38.Thurlings RM, Teng O, Vos K, Gerlag DM, Aarden L, Stapel SO, et al. Clinical response, pharmacokinetics, development of human anti-chimaeric antibodies, and synovial tissue response to rituximab treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2010;69:409–412. doi: 10.1136/ard.2009.109041. [DOI] [PubMed] [Google Scholar]
- 39.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant Autoantibody Production by Early Human B Cell Precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 40.Begovich AB, Carlton VEH, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004;75:330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagaoka H, Takahashi Y, Hayashi R, Nakamura T, Ishii K, Matsuda J, et al. Ras Mediates Effector Pathways Responsible for Pre-B Cell Survival, Which Is Essential for the Developmental Progression to the Late Pre-B Cell Stage. J Exp Med. 2000;192:171–182. doi: 10.1084/jem.192.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makrygiannakis D, Klint E af, Lundberg IE, Löfberg R, Ulfgren A-K, Klareskog L, et al. Citrullination is an inflammation-dependent process. Ann Rheum Dis. 2006;65:1219–1222. doi: 10.1136/ard.2005.049403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akatsu C, Shinagawa K, Numoto N, Liu Z, Ucar AK, Aslam M, et al. CD72 negatively regulates B lymphocyte responses to the lupus-related endogenous toll-like receptor 7 ligand Sm/RNP. J Exp Med. 2016 doi: 10.1084/jem.20160560. jem.20160560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bird P, Hall S, Nash P, Connell CA, Kwok K, Witcombe D, Thirunavukkarasu K. Tofacitinib: Treatment Outcomes in Seropositive Versus Seronegative Patients in a Phase 3 RA Population [abstract] 2016 doi: 10.1136/rmdopen-2018-000742. Available at: http://acrabstracts.org/abstract/tofacitinib-treatment-outcomes-in-seropositive-versus-seronegative-patients-in-a-phase-3-ra-population/. Accessed June 9, 2017. [DOI] [PMC free article] [PubMed]
- 45.Roosnek E, Lanzavecchia A. Efficient and selective presentation of antigen-antibody complexes by rheumatoid factor B cells. J Exp Med. 1991;173:487–489. doi: 10.1084/jem.173.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. RA patient and healthy donor demographic and clinical data.
Supplementary Table 2. Cell and molecule identifier oligonucleotide design.
Supplementary Table 3. Genes used for transcriptomic subset prediction.
Supplementary Table 4. B cell subset classifier applied to healthy and RA B cells.
Supplementary Table 5. BCR reconstitution and IG isotype recovery.
Supplementary Table 6. Clustering of antigen-specific B cells by paired HC and LC CDR3 sequences.
Supplementary Table 7. Monoclonal antibody expression and functional characterization.
Supplementary Table 8. Global ACPA and RF B cell differentially expressed transcripts.
Supplementary Table 9. Subsetted ACPA and RF B cell differentially expressed transcripts.
Supplementary Table 10. Subsetted single-cell coexpression testing.
Supplementary Figure 1. Single-cell RNA-seq library preparation and computational de-multiplexing workflow.
Supplementary Figure 2. Distribution of molecule counts (UMIs), pre-deduplicated reads (reads), and genes for each single cell library.
Supplementary Figure 3. Application of the transcriptomic B cell subset classifier to CD19+ B cells from RA patients.
Supplementary Figure 4. Comparison of the distribution of transcriptomic subset classifier frequencies (Classifier Fraction) to flow cytometry subset frequencies (Flow Fraction) from the same samples.
Supplementary Figure 5. Paired immunoglobulin variable-region reconstitution using hidden Markov Model-based assembly.
Supplementary Figure 6. Analysis of multi-specific CDR3 clusters.
Supplementary Figure 7. Differential expression tests for gene modules.