

Abstract
Objective:
To determine the prevalence, spectrum, and prognostic significance of copy number variants of undetermined significance (cnVUS) seen on chromosomal microarray (CMA) in neonates with hypoplastic left heart syndrome (HLHS).
Study design:
Neonates with HLHS who presented between June 2008 and December 2016 to Texas Children’s Hospital were identified. CMA results were abstracted and compared against copy number variations (CNVs) in ostensibly healthy individuals, gathered from the literature. Findings were classified as normal, consistent with a known genetic disorder, or cnVUS. Survival was then compared using Kaplan Meier analysis. Secondary outcomes included tracheostomy, feeding tube at discharge, cardiac arrest, and extracorporeal membrane oxygenation (ECMO).
Results:
Our study included 105 neonates with HLHS; 70 (66.7%) had normal CMA results, 9 (8.6%) had findings consistent with known genetic disorders, and 26 (24.7%) had a cnVUS. Six of the 26 (23.0%) cnVUS patients had a variant that localized to a specific region of the genome seen among the healthy control population. One-year survival in patients with a cnVUS, normal CMA, or known genetic disorders was 84.0%, 68.3% and 33.3%, respectively (P=0.003). There was no significant difference in secondary outcomes, although notably ECMO was utilized in 15.7% of patients with normal CMA and was not used in patients with cnVUS and abnormal results (P=0.038).
Conclusions:
Among children with HLHS, cnVUSs detected on CMA are common. The cnVUSs did not localize to specific regions of the genome and were not associated with worse outcomes compared with children with a normal CMA.
Keywords: copy number variation, genetic test, mutation variant of undetermined significance
Despite surgical palliation and medical management evolving tremendously in the past 40 years, HLHS still carries significant morbidity and mortality.3, 5, 6 The backbone of palliation is the 3-stage surgical intervention comprised of the Norwood, Glenn, and Fontan procedures, respectively. Even with major centers achieving survival rates to discharge of up to 90% following Norwood, these neonates remain at a high risk of death prior to Glenn.7 This so-called interstage mortality can be as high as 24% of children with HLHS.8 At Texas Children’s Hospital (TCH), interstage mortality was 12% from 2002 to 2007, before the single ventricle program (SVP) was established and 8% from 2007 to 2010, with a 12.5% overall hospital discharge mortality following Norwood (2013–2016).9, 10 Further, several clinical factors have been found to be associated with increased mortality, including prematurity, low birth weight, pulmonary venous obstruction, restrictive or intact atrial septum, or children above 1 month of age undergoing the Norwood procedure, and genetic syndrome-associated copy number variation (CNV).11–15
Given the prognostic relevance and high prevalence of CNVs, genetic testing by chromosomal microarray (CMA) has become routine and standard of care.4,16 CMA is a genome-wide screening technique, utilized to detect chromosomal imbalances.4 As CMA testing in neonates with HLHS has increased, so has the detection of copy number variants of unknown/undetermined significance (cnVUS).4 The uncertainty inherent in these findings impairs the ability to adequately predict the potential for morbidity and mortality with consequences for provider management strategies and appropriate counseling of families. To this end, we sought to determine the spectrum, prevalence, and diagnostic relevance of cnVUSs in HLHS.
In this study, we evaluate the association between CMA results and morbidity and mortality in patients with HLHS in order to determine the potential prognostic implications of cnVUS results.
METHODS
In this Institutional Review Board-approved study, we conducted a retrospective review of all patients diagnosed with HLHS at TCH (Houston, Texas, United States) between June 2008 and December 2016. Inclusion criteria were neonates with HLHS who presented to TCH within thirty days of birth, underwent CMA testing through the Baylor Genetics Laboratories (Houston, Texas, United States; previously Baylor College of Medicine Medical Genetics Laboratories). Exclusion criteria were children who received palliative surgical procedures at other centers prior to transfer, and single ventricle lesions not clearly defined as HLHS, including unbalanced atrioventricular septal defects and double outlet right ventricle. Data collected included patient sex, race/ethnicity, age at presentation, gestational age at birth, birthweight, atrial level restriction, echocardiographic characteristics, and CMA testing results. Echocardiograms were analyzed to determine the presence of mitral stenosis (MS)/aortic stenosis (AS), MS/aortic atresia (AA), mitral atresia (MA)/AA, left-sided superior vena cava (LSVC), total anomalous pulmonary venous return/anomalous pulmonary venous return, degree of tricuspid regurgitation, diameter of ascending aorta, and the degree of RV dysfunction.
The patients were studied by V7, V8, V9, V10, or V11 arrays designed by Baylor Medical Genetics Laboratories and manufactured by Agilent Technology (Santa Clara, CA, USA). The V7 array included approximately 105,000 interrogating oligonucleotides, selected from Agilent’s online library (eArray; https://earray.chem.agilent.com/earray/), with backbone coverage of about 30 kb.17, 18 The V8 array included about 180,000 oligonucleotides targeting ~1,714 genes plus 101,644 probes used for SNP analysis for the detection of uniparental disomy (UPD) and absence of heterozygosity (AOH).18 The V9-V11 arrays targeted over 4,800 genes with oligonucleotides including at the exon level and had an average of >4.2 probes per exon for SNP analysis.19 Further details are available at https://www.bcm.edu/geneticlabs/.
We defined structural variation in the genome as variants involving more than 50 base pairs. Our patient cohort was compared with CNV data from a stringent database of CNVs that was later compiled as the Database for Genomic Variants (DGV) genome browser – a database of structural variants of the human genome in healthy patient cohorts20. This database included 72 studies to create a reference cohort including 2,057,386 variants among 2647 subjects across diverse ethnicities with identified structural variations in the genome20. Each CNV locus labeled as a cnVUS was compared withthe healthy reference cohort to discern the frequency of healthy CNV seen at each cnVUS locus. The stringent map variants were utilized to determine control cohort frequency.
The primary outcome studied was overall survival. Secondary outcomes included tracheostomy, nasogastric or gastrostomy tube placement at discharge from initial hospitalization, cardiopulmonary resuscitation (CPR), or extracorporeal membrane oxygenation (ECMO).
Statistical Analyses
Continuous variables were presented as means with standard deviation and medians with interquartile ranges (IQRs). One way ANOVA or Kruskal-Wallis Test were applied to compare the difference among the three groups for data with normal or non-normal distribution, respectively. Categorical variables were expressed as counts with percentages and proportions were compared using the Fisher exact test. Survival was compared using Kaplan-Meier analysis, with comparison by log rank, with birth as time 0, and censoring at death or last follow-up. All statistics were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA).
RESULTS
Given the broad use of CMAs as a diagnostic tool in children with a variety of underlying diseases, we first sought to determine the overall prevalence of abnormal and cnVUS findings in pediatric patients seen at TCH regardless of diagnosis. We identified a total of 6410 CMAs performed on individual subjects. Among these, 4089 (63.2%) individuals were interpreted as “normal”, 1692 (26.2%) as cnVUSs, and 689 (10.6%) as abnormal genetic test results associated with a known genetic syndrome (Figure 1; available at www.jpeds.com). This suggests that a considerable proportion of CMA results, which were cnVUS, did not provide clinicians any clear diagnostic utility.
Figure 1;online only:
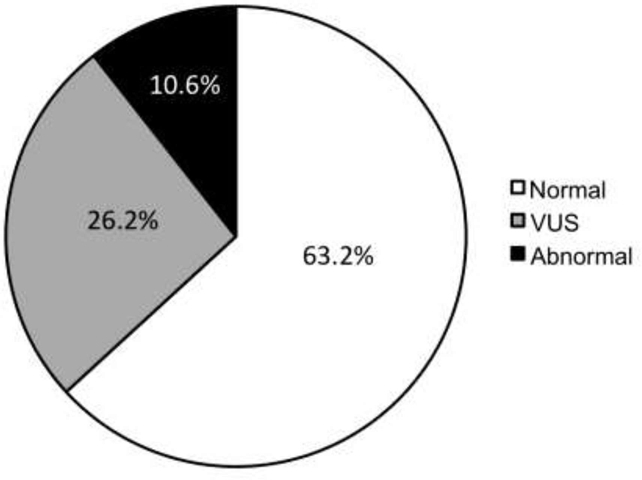
Pie chart of CMA results at Texas Children Hospital’s from June 2008 through December 2016 for any test indication. Test results are divided into those interpreted as abnormal (black fill), cnVUS (gray), and normal (white).
In total, 105 infants met our inclusion and exclusion criteria. Sixty (57%) were male, and the median age of presentation in all groups was the first day of life. These findings, along with birth weight, gestational age, and fronto-occipital circumference are summarized in Table 1 (available at www.jpeds.com). CMAs performed on these children demonstrated no CNV abnormalities in 70 (66.7%), and 26 (24.7%) had cnVUSs, and 9 (8.6%) hosted CNV interpreted as syndromic/pathologic (Table 2; available at www.jpeds.com). The pathologic CNVs identified in patients with HLHS were associated with several medically relevant phenotypes. These include syndromes such as Turner syndrome (N = 4, 67% of abnormal females) and Pallister-Killian syndrome (N = 1). In addition, pathologic CNV was associated with gastrointestinal dysmotility and renal and craniofacial abnormalities. Although loci affected were diverse, four chromosomal rearrangements occurred on the X chromosome. The CNVs were due to copy number losses (50%) and copy number gains (50%). These findings are summarized in Table 3 (available at www.jpeds.com). In comparison, CNV interpreted as cnVUSs were due to copy number gain (59%), loss (37%), and absence of heterozygosity (4%) and were highly variable in their location. These findings are summarized in Table 4 (available at www.jpeds.com).
Table 1; online only.
Summary of clinically relevant findings in subjects with HLHS who underwent CMA testing
| Normal N=70 |
cnVUS N=26 |
Abnormal N=9 |
|||||
|---|---|---|---|---|---|---|---|
| Mean±SD | Median (IQR) | Mean±SD | Median (IQR) | Mean±SD | Median (IQR) | P-value | |
| Age at Presentation (d) | 0.71 ± 2.30 | 0 (0,0) | 1.04 ± 5.09 | 0 (0,0) | 1.00 ± 2.00 | 0 (0,1) | 0.197 |
| Gestational Age | 38.32 ± 1.59 | 38.93 (37.29, 39.43) |
38.92 ± 0.91 | 39.00 (38.71, 39.43) |
37.78 ± 2.00 | 38.29 (36.00, 39.00) |
0.145 |
| Birth Weight (kg) | 3.03 ± 0.55 | 3.08 (2.70, 3.46) |
3.20 ± 0.46 | 3.21 (2.90, 3.46) |
2.56 ± 0.56 | 2.74 (2.23, 2.90) |
0.008 |
| FOC (cm) | 33.28 ± 1.64 | 33.00 (32.50, 34.50) |
33.83 ± 1.95 | 33.70 (32.00, 35.00) |
32.00 ± 1.77 | 32.00 (30.50, 33.50) |
0.103 |
AA, ascending aorta; cm, centimeter; d, days; FOC, fronto-occipital circumference; kg, kilogram.
Table 2; online only.
Summary of clinical demographics for subjects with HLHS who underwent CMA testing
| HLHS Cohort | N (%) |
|---|---|
| Total Cases | 105 |
| Male | 60 (57.0%) |
| Female | 45 (43.0%) |
| CMA cohort | |
| Normal | 70 (66.7%) |
| cnVUS | 26 (24.7%) |
| Abnormal | 9 (8.6%) |
| Race/Ethnicity | |
| Caucasian | 54 (51.0%) |
| Hispanic | 49 (47.0%) |
| African-American | 2 (2%) |
HLHS, hypoplastic left heart syndrome; CMA, chromosomal microarray; cnVUS, copy number variant of unknown significance; d, days.
Table 3; online only:
Summary of copy number variation for subjects with HLHS hosting abnormal/pathogenic CNVs
| CMA Pathologic Variants | |||
|---|---|---|---|
| Subject | CNV | Clinical Presentation or Syndrome | |
| 1 | Xp22.33q28 | Loss | Turner syndrome, renal pyelectasis, cystic kidneys bilaterally |
| 2 | 1p36.33p36.32 | Gain | Gastroparesis and esophageal dysmotility |
| 3 | Xp22.33q28 | Loss | Turner syndrome |
| 4 | 15q11.2, | Loss | Turner Syndrome |
| Xp22.33q28, | Loss | ||
| Yp11.31q11.1 | Gain | ||
| 5 | X, | Loss | Turner Syndrome |
| Yp11.31p11.2 | Gain | ||
| 6 | 12p13.33p11.1 | Gain | Pallister-Killian syndrome |
| 7 | 18q23 | Gain | Partial Trisomy 18q and dysmorphic features |
| 8 | 13q13.1 | Gain | BRCA2 Duplication |
| 9 | 22q11.21 | Loss | Cleft lip/palate (features of DiGeorge Syndrome) |
Table 4; online only:
Summary of copy number variation for subjects with HLHS hosting copy number variants of unknown significance
| Subject | CNV | Genes | ||
|---|---|---|---|---|
| 1 | Xp22.13p22.12 | Gain, Gain |
MTTP, PHKA2, GPR64, PDHA1, MAP3K15 |
|
| 2 | 9p21.3 | Gain | none | |
| 3 | 4q25 | Loss | PAPSS1 | |
| 4 | 8p23.1 | Gain | GATA4, NEIL2, FDFT1, CTSB | |
| 5 | 7q21.11 | Gain | PHTF2, MAGI2, RPL13AP17 | |
| 6 | 22q13.2 | Gain |
BIK, MCAT, TSPO, TTLL12, SCUBE1 |
|
| 7 | 1p36.33 | Gain | PRKCZ, C1orf86, LOC100128003, | |
| SKI | ||||
| 8 | Xq21.31 | Loss | CPXCR1 | |
| 9 | 16q24.2 | Gain | ZNF469, ZFPM1 | |
| 10 | Xq28 | Gain | TMLHE | |
| 11 | 1q21.1q21.2 | Gain |
PRKAB2, PDIA3P, FMO5, CHD1L, LOC100289211, BCL9, ACP6, GJA5, GJA8, GPR89B, GPR89C, PDZK1P1, NBPF11, NBPF24 |
|
| 12 | 15q13.3 | Gain | CHRNA7 | |
| 13 | 20q12q13.11 | Loss | PTPRT | |
| 14 | 15q11.2 | Loss | TUBGCP5, CYFIP1, NIPA2, NIPA1 | |
| 15 | 6p25.3 | Gain | LOC285768 | |
| 16 | 2p21 | Gain | CALM2 | |
| 17 | 1q21.1 | Gain |
SEC22B, NOTCH2NL, NBPF10, HFE2, TXNIP, POLR3GL, RBM8A, GNRHR2, PEX11B, ITGA10, ANKRD34A, LIX1L, ANKRD35, PIAS3, NUDT17, POLR3C, RNF115, CD160, PDZK1 |
|
| 18 | 2p13.1p12 | Gain | SEMA4F, HK2, POLE4, TACR1 | |
| 19 | 17q23.2 | Loss | PPM1D | |
| 20 | 6q23.3 | Loss | AHI1 | |
| 21 | 7q21.13 | Gain | ZNF804B, C7orf62, DPY19L2P4, | |
| 22 | 5p13.2 | Loss | IL7R | |
| 23 | 9p24.3 | Loss | DOCK8 | |
| 24 | 6q25.1 | Loss | PLEKHG1 | |
| 25 | 16q22.1 | Loss | PDXDC2, PDPR | |
| 26 | 6q24.2q25.3 | Absence of | EPM2A, LOC100507557, FBXO30, | |
| Heterozygosity |
SHPRH, GRM1, RAB32, C6orf103, LOC729176, LOC729178, STXBP5, SAMD5, SASH1, UST, LOC100128176, TAB2, SUMO4, ZC3H12D, PPIL4, C6orf72, KATNA1, LATS1, NUP43, PCMT1, LRP11, RAET1E, RAET1G, ULBP2, ULBP1, RAET1K, RAET1L, ULBP3,PPP1R14C, IYD, PLEKHG1, MTHFD1L, AKAP12, ZBTB2, RMND1, C6orf211, C6orf97, ESR1, SYNE1,MYCT1, VIP, FBXO5, MTRF1L, RGS17, OPRM1, IPCEF1, CNKSR3. |
|||
To determine whether HLHS-associated cnVUSs were associated with variation seen in ostensibly healthy individuals, we cross-referenced them with a stringent meta-database of CNV seen in healthy individuals, published in Nature Reviews Genetics. 20 This database compiled information on CNVs regarded as healthy “background” genetic variation. Six of the 26 (23.0%) cnVUS probands with HLHS had a variant that localized to a specific region of the genome identified among the healthy cohort CNV. The remaining 20 (77.0%) had CNV not included in the healthy reference population. In addition, only two of our six patients had loci involved in more than 1% of the healthy patient cohort including the 9p21.3 gain cnVUS which was seen in 12.9% of the cohort and 15q11.2 loss which was seen in 4.16%. These results are summarized in Table 5. Overall, this suggests that cnVUSs do not clearly represent healthy “background” genetic variation and may be a distinct genetic subset with children with HLHS.
Table 5; online only:
Frequency of HLHS-identified cnVUSs within the Stringent Database for Genomic Variants
| Subject | Chromosome | CMA Change | Interval | Genome Map Frequency* |
|---|---|---|---|---|
| 1 | Xp22.13p22.12 | Gain | 100532453–100545071 | - |
| 18936760–19507586 | - | |||
| 2 | 9p21.3 | Gain | 22453166–22808555 | 1513/11732 =12.9% |
| 3 | 4q25 | Loss | 108456682–108806918 | 49/11732=0.42% |
| 4 | 8p23.1 | Gain | 11598718–11843369 | 107/11732=0.912% |
| 5 | 7q21.11 | Gain | 77530094–78018726 | - |
| 6 | 22q13.2 | Gain | 43506447–43722430 | - |
| 7 | 1p36.33 | Gain | 1978815–2201821 | - |
| 8 | Xq21.31 | Loss | 87851064–88148650 | - |
| 9 | 16q24.2 | Gain | 88385967–88530955 | - |
| 10 | Xq28 | Gain | 154741174–154785986 | - |
| 11 | 1q21.1q21.2 | Gain | 146618988–147825678 | - |
| 12 | 15q13.3 | Gain | 32218274–32445252 | 114/11732 = 0.97% |
| 13 | 20q12q13.11 | Loss | 41617589–41737558 | - |
| 14 | 15q11.2 | Loss | 22842145–23086692 | 488/11732= 4.16% |
| 15 | 6p25.3 | Gain | 908246 −1063595 | - |
| 16 | 2p21 | Gain | 47388806–47539278 | - |
| 17 | 1q21.1 | Gain | 145114722–145740657 | - |
| 18 | 2p13.1p12 | Gain | 74908297–75285980 | - |
| 19 | 17q23.2 | Loss | 58740319–58741680 | - |
| 20 | 6q23.3 | Loss | 135715759–135732797 | - |
| 21 | 7q21.13 | Gain | 88424763–89858498 | - |
| 22 | 5p13.2 | Loss | 35903054–35903339 | - |
| 23 | 9p24.3 | Loss | 272815–428641 | - |
| 24 | 6q25.1 | Loss | 151051089–151152761 | 70 /11732 = 0.60% |
| 25 | 16q22.1 | Loss | 68567940 – 68753268 | - |
| 26 | 6q24.2q25.3 | AOH | 145377585–155757781 | - |
No value indicates no CNV observed involving the locus
AOH, Absence of Heterozygosity.
To evaluate the possibility that the remaining cnVUSs not seen in otherwise healthy individuals may involve clinically-relevant genes, we mapped each against a comprehensive list of 935 clinical-relevant genes that are understood to involve CNVs.20 Only a small minority of cnVUS-positive subjects were found to host genes associated with genetic diseases (Table 6). Of the remaining 20 that did not have CNV in the healthy population, only 5 cnVUSs involved loci containing genes in which single gene mutations or CNV have been associated with predisposition to disease. Overall, these genes were associated with developmental delay, autism spectrum disorders, craniofacial abnormalities, macrocephaly and epilepsy. A single cvVUS was associated with congenital heart disease and arrhythmias. The cnVUS with a gain in 1q21.1 contains GJA5, which encodes a gap junction protein, alpha 5 (connexin40) and has been associated with the development of atrial fibrillation, atrial standstill, and cardiac malformations.
Table 6:
CMA-identified cnVUSs in patients with HLHS associated with medically relevant genes/phenotypes.
| Chromosome | CMA Change | Medically Relevant / Phenotype |
|---|---|---|
| Xp22.13p22.12 | Gain, Gain | - |
| 7q21.11 | Gain |
MAGI2: associated with bipolar affective disorder, schizophrenia, and infantile spasms. |
| 22q13.2 | Gain | - |
| 1p36.33 | Gain |
SKI: monosomy 1p36, which is associated with facial clefting anomalies, generalized epilepsy with febrile seizures, cranial suture closure anomalies, and seizures. |
| Xq21.31 | Loss | - |
| 16q24.2 | Gain | - |
| Xq28 | Gain | - |
| 1q21.1q21.2 | Gain |
GJA5: associated with learning disability, autism spectrum disorders, macrocephaly, behavioral features, atrial fibrillation, atrial standstill, and tetralogy of Fallot. GJA8: associated with cataracts. |
| 20q12q13.11 | Loss | - |
| 6p25.3 | Gain | - |
| 2p21 | Gain | - |
| 1q21.1 | Gain |
HFE2: 1q21.1 duplication syndrome (no further info.) |
| 2p13.1p12 | Gain | - |
| 17q23.2 | Loss | - |
| 6q23.3 | Loss | AHI1: haploinsufficient 6q23.3 |
| 7q21.13 | Gain | - |
| 5p13.2 | Loss | - |
| 9p24.3 | Loss | - |
| 16q22.1 | Loss | - |
| 6q24.2q25.3 | Absence of | - |
| Heterozygosity |
The one-year Kaplan-Meier survival in patients with a normal CMA was 68.3%, 84.0% in patients with cnVUS, and 33.3% in those with pathologic CNV (P=0.003 via log rank test). Survival by KM analysis is illustrated in Figure 2. We noted a significantly higher prevalence of extracardiac abnormalities among HLHS infants with abnormal CMAs (55.6%) compared with the much lower prevalence seen in infants with normal (5.7%) and cnVUS (8%) CMA results (P<0.001).
Figure 2:
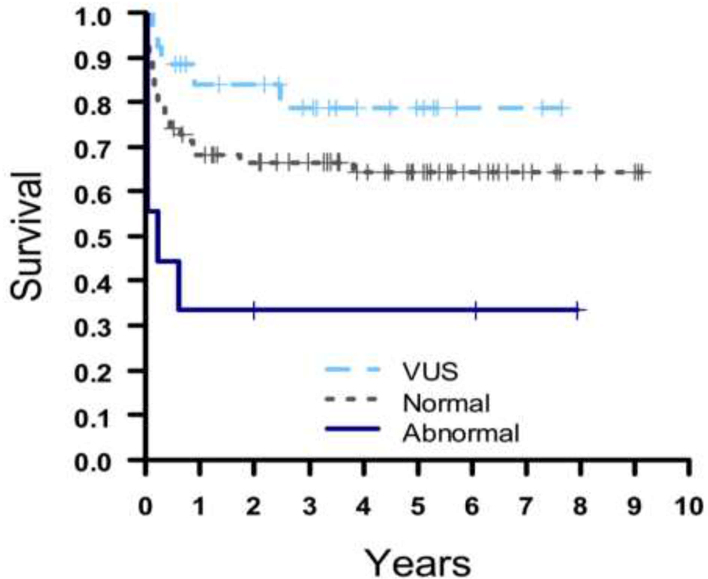
Kaplan-Meier analysis of transplant-free survival among neonates with HLHS by CMA result with normal (gray), cnVUS (light blue), and abnormal (dark blue). Censored observations are indicated as plus signs (+).
RV dysfunction was also more common in our abnormal group (22.2%) as compared with the cnVUS (7.7%) and normal (5.7%) population (P=0.016). There were no significant differences in the HLHS morphologic variation based on CMA class result including prevalence of MS/AS, MS/AA, MA/AA, presence of a LSVC, degree of tricuspid regurgitation, levocardinal vein, abnormal coronary arteries, ascending aorta diameter, and anomalous pulmonary venous return (Table 7).
Table 7:
Clinical characteristics of HLHS cohort by CMA class
| Cardiac Parameters on Presentation | |||||
|---|---|---|---|---|---|
| Normal | cnVUS | Abnormal | |||
| N=70 | N=25 | N=10 | |||
| Parameter | Mean (SD) | Mean (SD) | Mean (SD) | P-value | |
| MS/AS, MS/AA, MA/AA | MA/AA | 32 (46.4) | 17 (65.4) | 3 (33.3) | 0.140 |
| MS/AA | 24 (34.8) | 6 (23.1) | 6 (66.7) | . | |
| MS/AS | 13 (18.8) | 3 (11.5) | 0 | . | |
| Missing | 1 (1.4) | 0 | 0 | . | |
| LSVC | No | 65 (92.9) | 26 (100) | 9 (100) | 0.436 |
| Yes | 5 (7.1) | 0 | 0 | . | |
| PAPVR/TAPVR | No | 68 (97.2) | 24 (92.3) | 9 (100.0) | 0.510 |
| Yes | 2 (2.8) | 2 (7.7) | 0 | . | |
| Degree of TR | Trivial/None | 40 (57.1) | 11 (42.3) | 4 (44.4) | 0.503 |
| Mild | 23 (32.9) | 12 (46.2) | 3 (33.3) | . | |
| Moderate/Severe | 7 (10.0) | 3 (11.5) | 2 (22.2) | . | |
| Mod/Severe TR | No | 64 (91.4) | 23 (88.5) | 7 (77.8) | 0.333 |
| Yes | 6 (8.6) | 3 (11.5) | 2 (22.2) | . | |
| RV function, n (%) | Normal | 62 (89.9) | 20 (80.0) | 5 (55.6) | 0.008 |
| Mildly/Moderately | 7 (10.1) | 5 (20.0) | 4 (44.4) | . | |
| Depressed | |||||
| RV moderate dysfunction, n (%) | No | 69 (98.6) | 24 (92.3) | 7 (77.8) | 0.016 |
| Yes | 1 (1.4) | 2 (7.7) | 2 (22.2) | . | |
| Extra cardiac abnormalities | No | 66 (94.3) | 24 (92.3) | 4 (44.4) | <0.001 |
| Yes | 4 (5.7) | 2 (7.7) | 5 (55.6) | . | |
| Sano, BTS, or Hybrid | BTS | 45 (64.3) | 17 (65.4) | 3 (33.3) | 0.010 |
| Sano | 16 (22.9) | 9 (34.6) | 1 (11.1) | . | |
| Hybrid | 3 (4.3) | 0 | 1 (11.1) | . | |
| None | 6 (8.6) | 0 | 4 (44.4) | ||
| Restrictive ASD, n (%) | No | 54 (77.1) | 22 (84.6) | 7 (77.8) | 0.743 |
| Yes | 16 (22.9) | 4 (15.4) | 2 (22.2) | . | |
| Ascending aorta diameter (cm) | 0.34 ± 0.28 | 0.27 ± 0.15 | 0.24 ± 0.07 | 0.204 | |
AA, aortic atresia; AS, aortic stenosis; ASD, atrial septal defect; BTS, Blalock-Taussig shunt; LSVC, left superior vena cava; MA, mitral atresia; MS, mitral stenosis; RV, right ventricle; TR, tricuspid regurgitation; PAPVR/TAPVR, partial anomalous pulmonary venous return/total anomalous pulmonary venous return.
Given the differences in survival between CMA group, we next evaluated a number of secondary endpoints by CMA class. We identified no significant difference between CMA class and tracheostomy, feeding tube, or CPR (P=1.000, P=1.000P=0.139 respectively, Table 8). Of note, ECMO was not used among probands hosting either cnVUS or pathologic CMA findings, and probands with normal CMAs required ECMO in 11 (15.7%) individuals (P = .038).
Table 8:
Secondary outcome variables divided by CMA class.
| Outcome Variables by CMA Class | |||||
|---|---|---|---|---|---|
| Normal | cnVUS | Abnormal | |||
| Parameter | Mean (SD) | Mean (SD) | Mean (SD) | P-value | |
| n (%) | n (%) | n (%) | |||
| Tracheostomy | No | 67 (95.7) | 25 (96.2) | 9 (100) | 1.000 |
| Yes | 3 (4.3) | 1 (3.8) | 0 | ||
| Feeding tube | No | 53 (75.7) | 20 (76.9) | 7 (77.8) | 1.000 |
| Yes | 17 (24.3) | 6 (23.1) | 2 (22.2) | ||
| CPR | No | 61 (87.1) | 26 (100) | 8 (88.9) | 0.139 |
| Yes | 9 (12.9) | 0 | 1 (11.1) | ||
| ECMO | No | 59 (84.3) | 26 (100) | 9 (100) | 0.038 |
| Yes | 11 (15.7) | 0 | 0 | ||
CPR, cardiopulmonary resuscitation; ECMO, extracorporeal membrane oxygenation
DISCUSSION
CNVs, or unbalanced chromosomal rearrangements, have a wide array of clinical associations. Although there is a broad spectrum of syndromic CNVs, HLHS probands hosting this class of CMA are associated with a worse prognosis when compared withtheir normal counterparts. Multiple studies have found chromosomal abnormalities and genetic syndromes negatively impact inter-stage mortality. One study found one and ten year survival was half for HLHS patients with pathologic genetic syndromes compared withpatients with normal genetics.6, 21–23 Amongst other known syndromes, Turner syndrome, trisomy 18, trisomy 13, and Down syndrome are associated with higher early mortality in HLHS.24, 25 One study demonstrated 50% 1-month survival in patients with HLHS with Turner syndrome, as opposed to 85% survival in non-syndromic patients with HLHS26 , and another exhibited significantly increased cumulative mortality compared withfemales without Turner Syndrome, even with multivariate analysis controlling for low birth weight.27 Another study demonstrated five-year survival of 61% in patients with Down syndrome as opposed to 85% in non-syndromic patients.28 Therefore, an abnormal result detected on CMA is widely considered a predictor of increased mortality in CHD. However, with the rapidly expanded use of clinical CMAs, which are becoming the gold-standard for neonates with HLHS, there is increasing awareness of cnVUS. With 26.2% of all children at our institution referred for CMA testing and 20% of HLHS patients having a cnVUS result, this is a common issue that will likely only increase as genetic testing use increases.
The general consensus amongst most studies is that chromosomal anomaly prevalence in CHD is higher when extracardiac anomalies are also present.29 Hence, both genetic syndromes and extracardiac anomalies, as individual and combined factors, are associated with heightened mortality.30, 31 However, patients with cnVUS results did not exhibit the same characteristics as their abnormal counterpart, achieving significantly higher survival rates and a significantly lower prevalence of extra-cardiac abnormalities.
To determine the genetic significance of cnVUS results, we compared results with loci identified in healthy individuals and determined if there was any overlap with loci of clinical-actionable genes. Studies exploring incidentally identified VUSs associated with cardiomyopathies and channelopathies have suggested that the majority of these variants are likely background genetic noise.32 When comparing our cohort to cnVUS results among ostensibly healthy individuals, fewer than a quarter of patients in our cohort had identical loci identified in healthy individuals. As such, we cannot assume that cnVUS results in HLHS patients are comparable withcnVUS results in healthy individuals and seem genetically distinct. Of the remaining CNVs not included in the cross-referenced database, 5 had clinical-actionable genes involved. Among these, GJA5 has been associated with cardiac disease. Importantly, polymorphisms of GJA5 detected in families with cases of atrial standstill were only clinically manifesting if co-inherited with mutations in the SCN5A gene.33, 34 Further, clinical manifestations of atrial fibrillation were associated with rare, rare, novel missense and nonsense mutations.35–38 Mouse models with limited or absence of expression of GJA5 have demonstrated a higher prevalence of cardiac anomalies usually of conotruncal origin, with one third of the hearts exhibiting tetralogy of Fallot or double outlet right ventricle.39 Although most of these genes have little immediate clinical impact on HLHS patient survival, they may have an impact on patient long-term morbidity that is difficult to determine. Aside from this single cnVUS, associated phenotypes with the other clinical-actionable genes include mainly long-term neurobehavioral/psychiatric conditions. These conditions often cause significant functional limitations, in regards to executive planning, visual-motor integration, and thought-processing.40 Nonetheless, although genetics play a role, neurobehavioral limitations are usually due to a variety of factors, including parental IQ, cardiopulmonary bypass conduct, hemodynamic instability, intraoperative procedures, and perioperative neuroprotection.40–42 Ultimately, cnVUS results were not commonly found among ostensibly healthy individuals nor did loci contain clinically significant genes, suggesting that cnVUSs represent a distinct genetic subclass within HLHS and that there does not appear to be a genetic explanation for the survival seen in individuals hosting cnVUSs
Our finding that patients with HLHS hosting cnVUSs have superior survival to syndromic counterparts suggests the worse prognosis associated with abnormal CMA is restricted to known genetic syndromes. Although we identified a statistically significant greater number of neonates with normal CMAs who received ECMO support compared withpatients with cnVUS and abnormal CMA findings, from our study method we cannot definitively conclude the genetic test results altered patient management. The decision to initiate ECMO support is a multifaceted process. Previous studies have noted the inclusion of known pathologic mutations as part of end of life care discussions with the families of patients, and clinical management has the capacity to be influenced by the presence of abnormal CMA43, 44.
We recognize the limitations of our smaller sample size and the fact that CMA testing of infants with HLHS is not universal. Hence, estimates of CNVs could be amplified in our patient cohort. A better understanding of the effects of cnVUS results on physician/patient family decision-making is needed.
We find that cnVUS are common among children who receive CMAs and among children with HLHS. Although CNV associated with known genetic syndromes carries a negative prognostic association, we find cnVUS-associated survival to be equal, and perhaps superior to, normal CMA class.
Acknowledgments
A.L. receives support from the National Institutes of Health (K08-HL136839 and L40-HL129273), the Pediatric and Congenital Electrophysiology Society Paul C. Gillette Award, pilot grant funding from the Baylor College of Medicine Department of Pediatrics, and the McCrae Foundation. S.M. receives support from the National Institutes of Health (K23-HL127266 and L40-HL124303).
ABBREVIATIONS
- CHD
congenital heart disease
- CMA
chromosomal microarray
- CNV
copy number variation
- cnVUS
copy number variant of undetermined/unknown significance
- CPR
cardiopulmonary resuscitation
- DGV
Database of Genomic Variation
- ECMO
extracorporeal membrane oxygenation
- HLHS
hypoplastic left heart syndrome
- LSVC
left superior vena cava
- MA/AA
mitral atresia and aortic atresia
- MS/AA
mitral stenosis and aortic atresia
- MS/AS
mitral stenosis/aortic stenosis
Footnotes
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- [1].Connor JA, Thiagarajan R. Hypoplastic left heart syndrome. Orphanet J Rare Dis 2007;2:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Park MK. Park’s pediatric cardiology for practitioners Sixth edition. ed. Philadelphia, PA: Elsevier/Saunders,; 2014. [Google Scholar]
- [3].Gilboa SM, Salemi JL, Nembhard WN, Fixler DE, Correa A. Mortality Resulting From Congenital Heart Disease Among Children and Adults in the United States, 1999 to 2006. Circulation 2010;122:2254–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Buckley JR, Kavarana MN, Chowdhury SM, Scheurer MA. Current Practice and Utility of Chromosome Microarray Analysis in Infants Undergoing Cardiac Surgery. Congenital heart disease 2015;10:E131-E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Malik S, Bird TM, Jaquiss RD, Morrow WR, Robbins JM. Comparison of in-hospital and longer-term outcomes of hybrid and Norwood stage 1 palliation of hypoplastic left heart syndrome. J Thorac Cardiovasc Surg 2015;150:474–80.e2. [DOI] [PubMed] [Google Scholar]
- [6].Tabbutt S, Ghanayem N, Ravishankar C, Sleeper LA, Cooper DS, Frank DU, et al. Risk factors for hospital morbidity and mortality after the Norwood procedure: A report from the Pediatric Heart Network Single Ventricle Reconstruction trial. J Thorac Cardiovasc Surg 2012;144:882–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Siehr SL, Norris JK, Bushnell JA, Ramamoorthy C, Reddy VM, Hanley FL, et al. Home monitoring program reduces interstage mortality after the modified Norwood procedure. J Thorac Cardiovasc Surg 2014;147:718–23.e1. [DOI] [PubMed] [Google Scholar]
- [8].Simsic JM, Bradley SM, Stroud MR, Atz AM. Risk factors for interstage death after the Norwood procedure. Pediatr Cardiol 2005;26:400–3. [DOI] [PubMed] [Google Scholar]
- [9].Petit CJ, Fraser CD, Mattamal R, Slesnick TC, Cephus CE, Ocampo EC. The impact of a dedicated single-ventricle home-monitoring program on interstage somatic growth, interstage attrition, and 1-year survival. J Thorac Cardiovasc Surg 2011;142:1358–66. [DOI] [PubMed] [Google Scholar]
- [10].Heart Center Outcomes 2016 Texas Children’s Hospital; 2017. p. 61. [Google Scholar]
- [11].Bove EL, Lloyd TR. Staged reconstruction for hypoplastic left heart syndrome. Contemporary results. Annals of Surgery 1996;224:387–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fixler DE, Nembhard WN, Salemi JL, Ethen MK, Canfield MA. Mortality in First 5 Years in Infants With Functional Single Ventricle Born in Texas, 1996 to 2003. Circulation 2010;121:644–50. [DOI] [PubMed] [Google Scholar]
- [13].Hirsch JC, Copeland G, Donohue JE, Kirby RS, Grigorescu V, Gurney JG. Population-Based Analysis of Survival for Hypoplastic Left Heart Syndrome. J Pediatr 159:57–63. [DOI] [PubMed] [Google Scholar]
- [14].Menon SC, Keenan HT, Weng HY, Lambert LM, Burch PT, Edwards R, et al. Outcome and resource utilization of infants born with hypoplastic left heart syndrome in the Intermountain West. Am J Cardiol 2012;110:720–7. [DOI] [PubMed] [Google Scholar]
- [15].Stasik CN, Gelehrter S, Goldberg CS, Bove EL, Devaney EJ, Ohye RG. Current outcomes and risk factors for the Norwood procedure. J Thorac Cardiovasc Surg 2006;131:412–7. [DOI] [PubMed] [Google Scholar]
- [16].Geng J, Picker J, Zheng Z, Zhang X, Wang J, Hisama F, et al. Chromosome microarray testing for patients with congenital heart defects reveals novel disease causing loci and high diagnostic yield. BMC Genomics 2014;15:1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wiszniewska J, Bi W, Shaw C, Stankiewicz P, Kang SH, Pursley AN, et al. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur J Hum Genet 2014;22:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Boone PM, Bacino CA, Shaw CA, Eng PA, Hixson PM, Pursley AN, et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat 2010;31:1326–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gambin T, Yuan B, Bi W, Liu P, Rosenfeld JA, Coban-Akdemir Z, et al. Identification of novel candidate disease genes from de novo exonic copy number variants. Genome Medicine 2017;9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet 2015;16:172–83. [DOI] [PubMed] [Google Scholar]
- [21].Patel A, Hickey E, Mavroudis C, Jacobs JP, Jacobs ML, Backer CL, et al. Impact of noncardiac congenital and genetic abnormalities on outcomes in hypoplastic left heart syndrome. Ann Thorac Surg 2010;89:1805–13; discussion 13–4. [DOI] [PubMed] [Google Scholar]
- [22].Stasik CN, Goldberg CS, Bove EL, Devaney EJ, Ohye RG. Current outcomes and risk factors for the Norwood procedure. J Thorac Cardiovasc Surg 2006;131:412–7. [DOI] [PubMed] [Google Scholar]
- [23].Gaynor JW, Mahle WT, Cohen MI, Ittenbach RF, DeCampli WM, Steven JM, et al. Risk factors for mortality after the Norwood procedure. Eur J Cardiothorac Surg 2002;22:82–9. [DOI] [PubMed] [Google Scholar]
- [24].Jacobs JP, O’Brien SM, Chai PJ, Morell VO, Lindberg HL, Quintessenza JA. Management of 239 patients with hypoplastic left heart syndrome and related malformations from 1993 to 2007. Ann Thorac Surg 2008;85:1691–6. [DOI] [PubMed] [Google Scholar]
- [25].Siffel C, Riehle-Colarusso T, Oster ME, Correa A. Survival of Children With Hypoplastic Left Heart Syndrome. Pediatrics 2015;136:e864-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Reis PM, Punch MR, Bove EL, van de Ven CJ. Outcome of infants with hypoplastic left heart and Turner syndromes. Obstet Gynecol 1999;93:532–5. [DOI] [PubMed] [Google Scholar]
- [27].Lara DA, Ethen MK, Canfield MA, Nembhard WN, Morris SA. A population-based analysis of mortality in patients with Turner syndrome and hypoplastic left heart syndrome using the Texas Birth Defects Registry. Congenital heart disease 2017;12:105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Colquitt JL, Morris SA, Denfield SW, Fraser CD, Wang Y, Kyle WB. Survival in Children With Down Syndrome Undergoing Single-Ventricle Palliation. Ann Thorac Surg 101:1834–41. [DOI] [PubMed] [Google Scholar]
- [29].Wang Y, Cao L, Liang D, Meng L, Wu Y, Qiao F, et al. Prenatal chromosomal microarray analysis in fetuses with congenital heart disease: a prospective cohort study. Am J Obstet Gynecol 2017. [DOI] [PubMed] [Google Scholar]
- [30].Gaynor JW, Mahle WT, Cohen MI, Ittenbach RF, DeCampli WM, Steven JM, et al. Risk factors for mortality after the Norwood procedure. Eur J Cardiothorac Surg 2002;22:82–9. [DOI] [PubMed] [Google Scholar]
- [31].Allan LD, Apfel HD, Printz BF. Outcome after prenatal diagnosis of the hypoplastic left heart syndrome. Heart 1998;79:371–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Landstrom AP, Dailey-Schwartz AL, Rosenfeld JA, Yang Y, McLean MJ, Miyake CY, et al. Interpreting Incidentally Identified Variants in Genes Associated With Catecholaminergic Polymorphic Ventricular Tachycardia in a Large Cohort of Clinical Whole-Exome Genetic Test Referrals. Circ Arrhythm Electrophysiol 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Groenewegen WA, Firouzi M, Bezzina CR, Vliex S, van Langen IM, Sandkuijl L, et al. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ Res 2003;92:14–22. [DOI] [PubMed] [Google Scholar]
- [34].Makita N, Sasaki K, Groenewegen WA, Yokota T, Yokoshiki H, Murakami T, et al. Congenital atrial standstill associated with coinheritance of a novel SCN5A mutation and connexin 40 polymorphisms. Heart Rhythm 2005;2:1128–34. [DOI] [PubMed] [Google Scholar]
- [35].Gollob MH, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med 2006;354:2677–88. [DOI] [PubMed] [Google Scholar]
- [36].Yang YQ, Zhang XL, Wang XH, Tan HW, Shi HF, Jiang WF, et al. Connexin40 nonsense mutation in familial atrial fibrillation. Int J Mol Med 2010;26:605–10. [DOI] [PubMed] [Google Scholar]
- [37].Yang YQ, Liu X, Zhang XL, Wang XH, Tan HW, Shi HF, et al. Novel connexin40 missense mutations in patients with familial atrial fibrillation. Europace 2010;12:1421–7. [DOI] [PubMed] [Google Scholar]
- [38].Sun Y, Yang YQ, Gong XQ, Wang XH, Li RG, Tan HW, et al. Novel germline GJA5/connexin40 mutations associated with lone atrial fibrillation impair gap junctional intercellular communication. Hum Mutat 2013;34:603–9. [DOI] [PubMed] [Google Scholar]
- [39].Gu H, Smith FC, Taffet SM, Delmar M. High incidence of cardiac malformations in connexin40-deficient mice. Circ Res 2003;93:201–6. [DOI] [PubMed] [Google Scholar]
- [40].Wernovsky G, Newburger J. Neurologic and developmental morbidity in children with complex congenital heart disease. J Pediatr 2003;142:6–8. [DOI] [PubMed] [Google Scholar]
- [41].Sistino JJ, Bonilha HS. Improvements in Survival and Neurodevelopmental Outcomes in Surgical Treatment of Hypoplastic Left Heart Syndrome: A Meta-Analytic Review. J Extra Corpor Technol 2012;44:216–23. [PMC free article] [PubMed] [Google Scholar]
- [42].Mahle WT, Tavani F, Zimmerman RA, Nicolson SC, Galli KK, Gaynor JW, et al. An MRI study of neurological injury before and after congenital heart surgery. Circulation 2002;106:I109-14. [PubMed] [Google Scholar]
- [43].Allen KA. Parental decision-making for medically complex infants and children: An integrated literature review. Int J Nurs Stud 2014;51:1289–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zyblewski SC, Hill EG, Shirali G, Atz A, Forbus G, Gonzalez J, et al. Chromosomal Anomalies Influence Parental Treatment Decisions in Relation to Prenatally Diagnosed Congenital Heart Disease. Pediatr Cardiol 2009;30:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]