

Abstract
Bacterial iron uptake machinery can be hijacked for the targeted delivery of antibiotics into pathogens by attaching antibiotics to siderophores, iron chelators that are employed by bacteria to obtain this essential nutrient. We synthesized and evaluated Ent–SS–Cipro, a siderophore– antibiotic conjugate comprised of the triscatecholate siderophore enterobactin and the fluoroquinolone antibiotic ciprofloxacin that contains a self-immolative disulfide linker. This linker is designed to be cleaved after uptake into the reducing environment of the bacterial cytoplasm. We show that the disulfide bond of Ent–SS–Cipro is cleaved by reducing agents, including the cellular reductant glutathione, which results in release of the unmodified fluoroquinolone antibiotic. Antibacterial activity assays against a panel of Escherichia coli show that Ent–SS–Cipro exhibits activity against some, but not all, E. coli. This work informs the design of siderophore–antibiotic conjugates, particularly those carrying antibiotics with cytoplasmic targets that require release after uptake into bacterial cells, and indicates that disulfide linkers may not be generally applicable for conjugation strategies of antibiotics.
Keywords: Siderophore, antibiotic conjugate, disulfide linker, targeted delivery, iron uptake
Graphical abstract
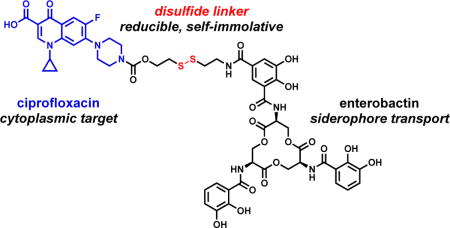
Introduction
The alarming rise of antibiotic-resistant pathogens, the dearth of new antibiotics in the drug pipeline, and the increasing appreciation for the important role of the human microbiota in health and disease necessitate the development of strategies that efficiently deliver and selectively target antibiotics to pathogenic bacteria while sparing non-pathogenic and commensal species [1,2]. One possibility for targeted antibiotic delivery is to hijack bacterial membrane transporters that are required for the uptake of nutrients, such as transition metal ions that bacteria must acquire from the host environment. To starve invading pathogens of these essential nutrients, the human host strongly limits the availability of metal ions at infection sites in an innate immune process termed “nutritional immunity” [3,4]. Nevertheless, high-affinity metal acquisition systems expressed by many bacterial pathogens enable them to thrive in the host despite restricted nutrient access [3,5]. These systems are important virulence factors, and thus are considered to be potential targets and pathogen-selective entry routes for antibiotics.
Siderophores are low-molecular-weight chelators that are biosynthesized and secreted by bacteria for iron acquisition [6–8]. Uptake of the ferric siderophores is mediated by dedicated membrane transporters. Some bacteria also produce siderophores tethered to antibiotics to harm their competitors that express the requisite siderophore receptors [9,10]. These “sideromycins” have inspired the synthesis of various siderophore–drug conjugates to target bacterial siderophore uptake machinery for antibiotic delivery [11–15]. Indeed, conjugation of β-lactam antibiotics to siderophores can significantly increase their antibacterial activity against Gram-negative pathogens that include uropathogenic and enterohemorrhagic Escherichia coli and the opportunistic pathogen Pseudomonas aeruginosa [16–26]. These pathogens are of high clinical interest because their outer membrane serves as a permeability barrier that restricts entry of many small molecules into the cell, and thereby renders these bacteria inherently resistant to many antibiotics in clinical use [27,28]. Siderophore-mediated delivery can overcome this barrier as well as target antibiotics to pathogenic strains when pathogen-associated siderophores are employed [26].
Whereas the antibacterial activity of antibiotics with periplasmic targets, such as β-lactams, can be significantly enhanced by siderophore-mediated delivery, the activity of antibiotics with cytoplasmic targets, such as fluoroquinolones, is often attenuated upon siderophore conjugation [29–42]. One possible explanation for this general observation is that the attachment of siderophores hampers interaction of the antibiotic with its cellular target. Indeed, in the case of the naturally occurring albomycins, peptidase-catalyzed intracellular release of the tRNA synthetase inhibitor from a ferrichrome-like siderophore is essential for the antibacterial activity of these sideromycins [43]. Moreover, we recently reported that an enterobactin (Ent) conjugate carrying the fluoroquinolone antibiotic ciprofloxacin, Ent–Cipro 1 (Scheme 1), exhibits antibacterial activity comparable to unmodified ciprofloxacin when the siderophore moiety is hydrolyzed after uptake into the cytoplasm [44]. This process requires the cytoplasmic Ent esterase IroD, which is encoded by the pathogen-associated iroA gene cluster [45,46]. Thereby, the antibacterial activity of Ent–Cipro 1 is targeted to a subset of E. coli that are predominantly pathogenic.
Scheme 1.
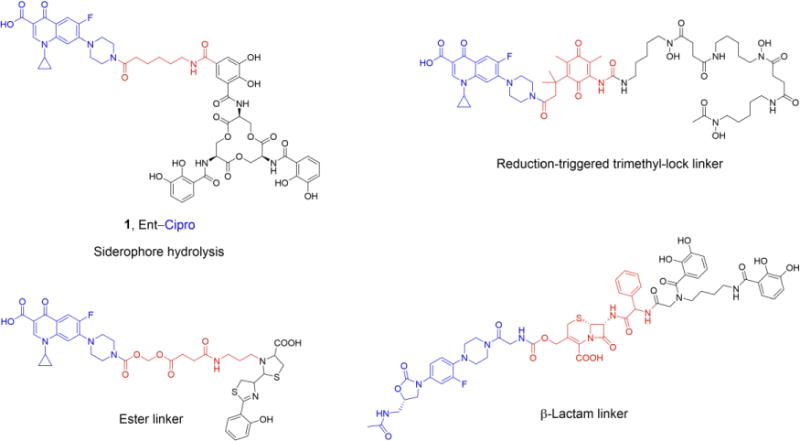
Examples of previously studied siderophore conjugates [33,42,44,50] with cytoplasmic antibiotics (colored blue) and different linkers (colored red)
Intracellular release of antibiotics from synthetic conjugates has been attempted by installing a variety of cleavable linker moieties between the siderophore and antibiotic cargo; however, most prior investigations uncovered limitations. Ester linkers (Scheme 1) were employed to enable release of the antibiotic by intracellular esterase- or acid-catalyzed hydrolysis, but the hydrolytic lability of these linkers resulted in premature cleavage of the conjugates, hence release of the antibiotic in the culture medium [30,31,33,35,41,47–49]. Another approach investigated a trimethyl-lock linker based on a reduction-triggered cleavage mechanism that was designed to be cleaved only in the cytoplasm (Scheme 1), but did not yield a conjugate with high antibacterial activity [42]. Recently, a conjugate in which a β-lactam was used to attach an oxazolidinone antibiotic to the siderophore (Scheme 1) was reported [50]. Oxazolidinones inhibit protein biosynthesis and exhibit antibacterial activity against Gram-positive bacteria. However, these antibiotics are inactive against Gram-negative species due to low uptake into or fast efflux from the bacteria. This linker strategy conferred antibacterial activity to the oxazolidinone against clinical isolates of the Gram-negative pathogen Acinetobacter baumannii. Siderophore-mediated transport across the outer membrane and periplasmic hydrolysis of the β-lactam by a β-lactamase released the antibiotic, presumably followed by translocation into the cytoplasm. The conjugate also exhibited high activity against pathogens that do not express a β-lactamase, likely due to the β lactam remaining intact and inhibiting cell wall biosynthesis. Taken together, all heretofore reported siderophore–antibiotic conjugates with cytoplasmic antibiotic cargo that exhibit high antibacterial activity against Gram-negative bacteria rely on an enzymatic cleavage mechanism for release of the antibiotic.
In this work, we extend our prior studies of siderophore–fluoroquinolone conjugates [44,51] and report the design, synthesis, and evaluation of Ent–SS–Cipro 2 (Structure 1). We use this compound to investigate the applicability of a redox-active disulfide linker for siderophore– antibiotic conjugates. This linker is expected to be stable in the oxidative environmentd of the extracellular space and periplasm, and cleaved in the reducing environment of the cytoplasm, thus preventing premature release of the antibiotic before uptake into the cytoplasm. The linker is designed to release the unmodified fluoroquinolone after reduction of the disulfide and intramolecular attack at the carbamate moiety by the resulting thiol, resulting in elimination of the linker moiety [52]. This self-immolative disulfide linker has previously been studied for conjugates of anticancer agents, including the folate receptor-targeting conjugate vintafolide [53]. Moreover, this linker was used for a dual antibiotic conjugate consisting of kanamycin and an antibacterial cell-penetrating peptide [54]. This peptide delivered the aminoglycoside into mammalian cells, affording clearance of intracellular pathogenic bacteria after cleavage of the disulfide linker and release of both antibiotics. Recently, the self-immolative disulfide linker was employed for a siderophore–fluorophore conjugate and shown to provide intracellular release of the fluorophore in a Gram-negative strain [55]. Herein, we report that the disulfide-linked conjugate Ent–SS–Cipro 2 exhibits antibacterial activity against E. coli independent of intracellular siderophore hydrolysis, which contrasts the requirements for the activity of the structurally similar alkyl-linked conjugate Ent–Cipro 1 (Scheme 1). However, our studies also show that the high antibacterial activity of conjugate 2 is limited to select E. coli strains, indicating that disulfide linkers may not be generally applicable for siderophore-mediated antibiotic delivery strategies.
Structure 1.
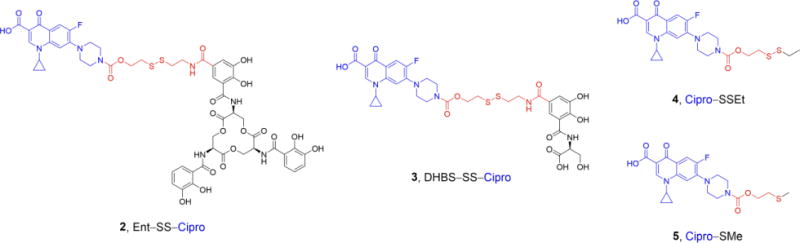
Materials and Methods
General Synthetic Materials and Methods
Details on general synthetic materials and methods, including the syntheses of compounds 1, 6, 10 and 11, as well as NMR spectra, UV/Vis spectra, and analytical HPLC chromatograms of the purified compounds are provided as Supporting Information.
Cipro–STrt (7)
Triphosgene (0.27 g, 0.90 mmol, 3.0 equiv) was dissolved in anhydrous CH2Cl2 (2 mL) and cooled to 0°C. A solution of 6 (0.14 g, 0.45 mmol, 1.5 equiv) and DIPEA (0.31 mL, 1.77 mmol, 6.0 equiv) in anhydrous CH2Cl2 (15 mL) was added slowly, and the yellow solution was stirred for 1 h while slowly warming up to r.t. The solvent and all volatiles (incl. excess of phosgene) were removed in vacuo. The remaining pale yellow solid (dried in vacuo for 1 h) was dissolved in anhydrous CH2Cl2 (5 mL) and the solution was cooled to 0°C. A solution of ciprofloxacin (0.10 g, 0.30 mmol, 1.0 equiv), DIPEA (0.21 mL, 1.2 mmol, 4.0 equiv) and TMSCl (0.12 mL, 0.95 mmol, 3.0 equiv) in anhydrous CH2Cl2 (15 mL) was added slowly, and the reaction mixture was stirred for 7 h at r.t. The orange solution was washed with HCl (1 M) and brine, and the crude product was purified by preparative TLC (hexanes/EtOAc/CH3OH 10:5:3, subsequently CH2Cl2/CH3OH 15:1 with the same TLC plate). Compound 7 was obtained as pale yellow solid (97 mg, 47%). TLC Rf = 0.2 (CH2Cl2/CH3OH 20:1). 1H NMR (CDCl3): δ 14.94 (s, 1H), 8.76 (s, 1H), 8.03 (d, 3JH,H = 13 Hz, 1H), 7.42 (d, 3JH,H = 8 Hz, 6H), 7.36 (br s, 1H), 7.29 (t, 3JH,H = 8 Hz, 6H), 7.23 (t, 3JH,H = 8 Hz, 3H), 3.97 (t, 3JH,H = 6 Hz, 2H), 3.69 (m, 4H), 3.53 (m, 1H), 3.29 (m, 4H), 2.51 (t, 3JH,H = 6 Hz, 2H), 1.38 (m, 2H), 1.20 (m, 2H); 19F NMR (CDCl3): δ –120.9; 13C NMR (CDCl3): δ 176.9, 166.8, 154.8, 153.6 (d, 1JC,F = 250 Hz), 147.4, 145.7 (d, 2JC,F = 11 Hz), 144.6, 139.0, 129.6, 128.0, 126.9, 119.9 (d, 3JC,F = 8 Hz), 112.2 (d, 2JC,F = 23 Hz), 107.9, 105.2, 66.8, 64.1, 49.6, 43.4, 35.4, 31.4, 8.3. HR-MS (ESI): [M+H]+ m/z calcd. 678.2438, found 678.2405.
Cipro–SH (8)
Compound 7 (70 mg, 0.10 mmol) was dissolved in CH2Cl2 (8 mL), and TFA (2 mL) and Et3SiH (0.2 mL) were added. The reaction mixture was stirred for 2 h, and the solvent was removed under reduced pressure. The residue was washed with Et2O, dissolved in CH2Cl2, filtered through cotton, and the solvent was removed under reduced pressure to yield 8 as pale yellow solid (40 mg, 89%). 1H NMR (CDCl3): δ 14.92 (s, 1H), 8.74 (s, 1H), 8.01 (d, 3JH,H = 13 Hz, 1H), 7.37 (d, 3JH,H = 7 Hz, 1H), 4.26 (t, 3JH,H = 7 Hz, 2H), 3.74 (m, 4H), 3.55 (m, 1H), 3.31 (m, 4H), 2.80 (m, 3JH,H = 7 Hz, 2H), 1.47 (t, 3JH,H = 8 Hz, 1H), 1.40 (m, 2H), 1.21 (m, 2H); 19F NMR (CDCl3): δ –121.8; 13C NMR (CDCl3): δ 177.2, 167.0, 154.9, 153.8 (d, 1JC,F = 250 Hz), 147.7, 145.8 (d, 2JC,F = 10 Hz), 139.1, 120.4 (d, 3JC,F = 8 Hz), 112.7 (d, 2JC,F = 24 Hz), 108.3, 105.2, 67.0, 49.7, 43.5, 35.4, 23.9, 8.4. HR-MS (ESI): [M+H]+ m/z calcd. 436.1343, found 436.1327.
Cipro–SSPy (9)
Compound 8 (59 mg, 0.14 mmol, 1.0 equiv) was dissolved in anhydrous CH2Cl2 (7 mL). The solution was slowly added to a solution of aldrithiol (60 mg, 0.27 mmol, 2.0 equiv) in anhydrous CH2Cl2 (3 mL), and the yellow solution was stirred for 2 h. The solvent was removed under reduced pressure and the product was purified by preparative TLC (CH2Cl2/CH3OH 20:1, subsequently CH2Cl2/EtOAc 1:1 with the same TLC plate) to yield 9 as pale yellow solid (37 mg, 50%). TLC Rf = 0.3 (CH2Cl2/CH3OH 20:1). 1H NMR (CDCl3): δ 14.94 (s, 1H), 8.77 (s, 1H), 8.49 (d, 3JH,H = 5 Hz, 1H), 8.04 (d, 3JH,H = 13 Hz, 1H), 7.69 (t, 3JH,H = 8 Hz, 1H), 7.66 (m, 1H), 7.38 (d, 3JH,H = 7 Hz, 1H), 7.12 (t, 3JH,H = 6 Hz, 1H), 4.42 (t, 3JH,H = 6 Hz, 2H), 3.72 (m, 4H), 3.55 (m, 1H), 3.30 (m, 4H), 3.09 (t, 3JH,H = 6 Hz, 2H), 1.41 (m, 2H), 1.21 (m, 2H); 19F NMR (CDCl3): δ –121.9; 13C NMR (CDCl3): δ 177.2, 167.1, 159.7, 154.9, 153.8 (d, 1JC,F = 249 Hz), 149.8, 147.7, 145.8 (d, 2JC,F = 13 Hz), 139.1, 137.3, 121.1, 120.4 (d, 3JC,F = 6 Hz), 112.7 (d, 2JC,F = 23 Hz), 108.3, 105.3, 63.6, 49.8, 43.7, 39.9, 35.5, 29.8, 8.4. HR-MS (ESI): [M+H]+ m/z calcd. 545.1329, found 545.1316.
Bn6Ent–SMmt (12)
Compound 11 (90 mg, 0.07 mmol, 1.0 equiv), HATU (54 mg, 0.14 mmol, 2.0 equiv) and HOAt (20 mg, 0.14 mmol, 2.0 equiv) were dissolved in anhydrous DMF (2 mL). DIPEA (50 μL, 0.29 mmol, 4.0 equiv) was added and the reaction mixture was stirred for 5 min. A solution of 10 (30 mg, 0.09 mmol, 1.2 equiv) and DIPEA (31 μL, 0.18 mmol, 2.4 equiv) in anhydrous DMF (3 mL) was added and the reaction mixture was stirred for 2 h. The solution was diluted with CH2Cl2 and washed with brine. The product was purified by preparative TLC (CH2Cl2/CH3OH 20:1) to yield 12 as pale brown foam (89 mg, 78%). TLC Rf = 0.25 (CH2Cl2/CH3OH 20:1). 1H NMR (CDCl3): δ 8.50 (t, 3JH,H = 8 Hz, 3H), 7.82 (s, 1H), 7.78 (s, 1H), 7.66 (d, 3JH,H = 7 Hz, 2H), 7.44–7.09 (m, 48H), 6.81 (d, 3JH,H = 9 Hz, 2H), 6.32 (m, 1H), 5.21–5.02 (m, 12H), 4.91 (m, 3H), 4.16 (m, 3H), 4.03 (m, 3H), 3.77 (s, 1H), 3.27 (m, 2H), 2.50 (t, 3JH,H = 7 Hz, 2H); 13C NMR (CDCl3): δ 169.2, 169.0, 165.7, 165.1, 165.0, 164.3, 158.2, 152.0, 151.7, 149.3, 147.1, 147.0, 145.0, 136.7, 136.3, 136.1, 136.0, 135.9, 135.5, 130.8, 130.1, 129.5, 129.2, 129.0, 128.9, 128.8, 128.7, 128.6, 128.5, 128.4, 128.3, 128.1, 128.0, 127.9, 127.8, 127.7, 126.8, 126.3, 125.7, 124.4, 123.2, 120.2, 117.6, 116.8, 113.3, 76.5, 76.4, 71.4, 71.3, 66.5, 55.3, 51.7, 51.6, 51.4, 39.0, 31.9. HR-MS (ESI): [M+Na]+ m/z calcd. 1608.5485, found 1608.5067.
Ent–SS–Cipro (2)
Compound 12 (85 mg, 0.05 mmol, 1.0 equiv) was dissolved in CH2Cl2 (7 mL), and TFA (75 μL) and Et3SiH (75 μL) were added. The reaction mixture was stirred for 1 h, and the solvent was removed under reduced pressure to yield a pale yellow foam, which was directly employed in the next synthetic step without further purification.
A solution of the thiol in anhydrous CH2Cl2 (2 mL) was slowly added to a solution of 9 (30 mg, 0.05 mmol, 1.0 equiv) in anhydrous CH2Cl2 (5 mL), and the resulting pale yellow solution was stirred for 3.5 h. The solvent was removed under reduced pressure and the product was purified by preparative TLC (CH2Cl2/CH3OH 20:1, subsequently CH2Cl2/EtOAc 1:1 with the same TLC plate). Benzyl-protected Ent–SS–Cipro was obtained as pale yellow foam (63 mg, 67%). TLC Rf = 0.4 (CH2Cl2/CH3OH 20:1). 1H NMR (CDCl3): δ 8.72 (s, 1H), 8.48 (t, 3JH,H = 7 Hz, 2H), 8.01 (d, 3JH,H = 13 Hz, 1H), 7.98 (s, 1H), 7.83 (s, 1H), 7.61 (m, 2H), 7.44–7.07 (m, 38H), 5.21–5.01 (m, 12H), 4.87 (m, 3H), 4.42 (m, 2H), 4.13 (m, 3H), 4.00 (m, 3H), 3.71 (m, 6H), 3.53 (m, 1H), 3.29 (m, 4H), 2.96 (m, 4H), 1.36 (m, 2H), 1.16 (m, 2H); 19F NMR (CDCl3): δ –121.1. HR-MS (ESI): [M+H]+ m/z calcd. 1747.5572, found 1747.5566.
Benzyl-protected Ent–SS–Cipro (10 mg, 0.01 mmol, 1.0 equiv) and pentamethylbenzene (17 mg, 0.11 mmol, 18 equiv) were dissolved in anhydrous CH2Cl2 (5 mL), and the solution was cooled to −78°C. BCl3 (0.1 mL of 1 M stock in CH2Cl2, 0.10 mmol, 17 equiv) was added and the yellow solution was stirred at –78°C for 1.5 h. The reaction was quenched with DIPEA (50 μL) and CH3OH (1 mL), and the solvent was removed under reduced pressure. The product was purified by semi-preparative HPLC (sample in dioxane/H2O 2:1; gradient: 30–40% over 3 min, 40–55% over 6 min; flow rate 4 mL min−1; elution at 7 min) to yield 2 as pale yellow powder (3.6 mg, 52%). 1H NMR (DMF-d7): δ 15.21 (s, 1H), 12.34 (br s, 1H), 12.05 (br s, 1H), 9.94 (br s, 1H), 9.55 (m, 3H), 9.29 (m, 2H), 8.75 (s, 1H), 8.61 (br s, 1H), 8.17 (br s, 1H), 7.97 (d, 3JH,H = 13 Hz, 1H), 7.70 (d, 3JH,H = 7 Hz, 1H), 7.62 (s, 1H), 7.47 (m, 2H), 7.04 (d, 3JH,H = 7 Hz, 2H), 6.76 (t, 3JH,H = 8 Hz, 2H), 5.10 (m, 3H), 4.78 (m, 3H), 4.61 (m, 3H), 4.39 (t, 3JH,H = 6 Hz, 2H), 3.92 (m, 1H), 3.70 (m, 6H), 3.40 (m, 4H), 3.11 (t, 3JH,H = 6 Hz, 2H), 3.04 (t, 3JH,H = 7 Hz, 2H), 1.44 (m, 2H), 1.32 (m, 2H); 19F NMR (DMF-d7): δ –124.7. HR-MS (ESI): [M+H]+ m/z calcd. 1206.2721, found 1206.2733.
Cipro–SSEt (4)
Ethanethiol (4 μL, 0.06 mmol, 1.0 equiv) was dissolved in anhydrous CH2Cl2 (2.5 mL). The solution was slowly added to a solution of 9 (30 mg, 0.06 mmol, 1.0 equiv) in anhydrous CH2Cl2 (5 mL), and the resulting pale yellow solution was stirred for 3.5 h. The solvent was removed under reduced pressure and the product was purified by preparative TLC (CH2Cl2/CH3OH 20:1) to yield 4 as pale yellow solid (9 mg, 31%). A portion of the compound was further purified by semi-preparative HPLC (sample in dioxane/H2O 2:1; gradient: 40–50% over 3 min, 40–55% over 6 min; flow rate 4 mL min–1; elution at 9 min). TLC Rf = 0.6 (CH2Cl2/CH3OH 20:1). 1H NMR (CDCl3): δ 14.93 (s, 1H), 8.77 (s, 1H), 8.04 (d, 3JH,H = 12 Hz, 1H), 7.37 (s, 1H), 4.39 (t, 3JH,H = 6 Hz, 2H), 3.73 (m, 4H), 3.54 (m, 1H), 3.30 (m, 1H), 2.95 (t, 3JH,H = 6 Hz, 2H), 2.73 (q, 3JH,H = 7 Hz, 2H), 1.40 (m, 2H), 1.34 (t, 3JH,H = 7 Hz, 3H), 1.20 (m, 2H); 19F NMR (CDCl3): δ –121.1; 13C NMR (CDCl3): δ 177.2, 166.9, 154.9, 153.7 (d, 1JC,F = 255 Hz), 147.6, 145.7 (d, 2JC,F = 8 Hz), 139.0, 120.4, 112.7 (d, 2JC,F = 23 Hz), 108.3, 105.1, 63.7, 49.7, 43.4, 37.8, 35.3, 32.9, 14.4, 8.3. HR-MS (ESI): [M+H]+ m/z calcd. 496.1376, found 496.1378.
Cipro–SMe (5)
Triphosgene (0.27 g, 0.90 mmol, 3.0 equiv) was dissolved in anhydrous CH2Cl2 (2 mL) and cooled to 0°C. A solution of 2-(methylthio)ethanol (40 μL, 0.45 mmol, 1.5 equiv) and DIPEA (0.31 mL, 1.77 mmol, 6.0 equiv) in anhydrous CH2Cl2 (15 mL) was added slowly, and the yellow solution was stirred for 1 h while slowly warming up to r.t. The solvent and all volatiles were removed in vacuo. The remaining pale yellow solid was dissolved in anhydrous CH2Cl2 (5 mL) and the solution was cooled to 0°C. A solution of ciprofloxacin (0.10 g, 0.30 mmol, 1.0 equiv), DIPEA (0.21 mL, 1.20 mmol, 4.0 equiv) and TMSCl (0.12 mL, 0.95 mmol, 3.0 equiv) in anhydrous CH2Cl2 (15 mL) was added slowly, and the reaction mixture was stirred for 18 h at r.t. The orange solution was washed with HCl (1 M) and brine, and the product was purified by preparative TLC (hexanes/EtOAc/CH3OH 10:5:3, subsequently CH2Cl2/CH3OH 15:1 with the same TLC plate) to yield 5 as pale yellow solid (65 mg, 48%). A portion of the compound was further purified by semi-preparative HPLC (sample in dioxane/H2O 2:1; gradient: 40–50% over 3 min, 40–55% over 6 min; flow rate 4 mL min–1; elution at 8 min). TLC Rf = 0.4 (CH2Cl2/CH3OH 20:1). 1H NMR (CDCl3): δ 14.90 (s, 1H), 8.71 (s, 1H), 7.97 (d, 3JH,H = 13 Hz, 1H), 7.36 (d, 3JH,H = 7 Hz, 1H), 4.30 (t, 3JH,H = 7 Hz, 2H), 3.73 (m, 4H), 3.55 (m, 1H), 3.31 (m, 4H), 2.77 (t, 3JH,H = 7 Hz, 2H), 2.16 (s, 3H), 1.40 (m, 2H), 1.20 (m, 2H); 19F NMR (CDCl3): δ –121.1; 13C NMR (CDCl3): δ 176.9, 166.8, 155.0, 153.6 (d, 1JC,F = 249 Hz), 147.5, 145.7 (d, 2JC,F = 13 Hz), 139.0, 119.9 (d, 3JC,F = 10 Hz), 112.3 (d, 2JC,F = 24 Hz), 108.0, 105.2, 64.1, 49.6, 43.5, 35.5, 33.1, 15.8, 8.3. HR-MS (ESI): [M+H]+ m/z calcd. 450.1499, found 450.1417.
DHBS–SS–Cipro (3)
A 5-mL solution containing conjugate 2 (500 μM) was prepared in 75 mM Tris-HCl buffer, pH 8.0 and divided into five 1-mL aliquots. The Ent hydrolase IroD (2 μM) was added to each aliquot and the reactions were incubated at r.t. for 5.5 h. Each reaction was quenched by the addition of 6% TFA in Milli-Q water (100 μL per aliquot) and the conjugate was purified by semi-preparative HPLC (20–50% B over 15 min; flow rate 4 mL min−1; elution at 14 min). Compound 3 was obtained as white powder (0.2 mg, 10%). HR-MS (ESI): [M+H]+ m/z calcd. 778.1865, found 778.1838.
Reduction with GSH
To a solution containing conjugate 2 or a ciprofloxacin derivative (100 μM) in 75 mM Tris-HCl buffer, pH 7.4 or 9.0, GSH (1 or 10 mM from a 100 mM stock) was added (final volume: 450 μL). The reaction was incubated at r.t. and aliquots (100 μL) were quenched by adding 6% TFA in Milli-Q water (10 μL) at varying time points (0, 15, 30, 240 min), and analyzed by analytical HPLC.
Reduction with TCEP
To a solution containing conjugate 2 or a ciprofloxacin derivative (100 μM) in 75 mM Tris-HCl buffer, pH 7.4 or 9.0, TCEP (1 mM from a 100 mM stock) was added (final volume: 100 μL); the pH was adjusted by addition of NaHCO3 (1 M stock). The reaction was incubated at r.t. for 30 min, quenched by adding 6% TFA in Milli-Q water (10 μL), and analyzed by analytical HPLC.
General Microbiology Materials and Methods
Bacterial strains were grown in a low-Fe medium (0.6 μM iron content, determined by ICP-MS), modified M9 minimal medium (6.8 g L−1 Na2HPO4, 3 g L−1 KH2PO4, 0.5 g L−1 NaCl, 1 g L−1 NH4Cl, 0.4% glucose, 2 mM MgSO4, 0.1 mM CaCl2, 0.2% casein amino acids, and 16.5 μg mL–1 thiamine) [26]. Stock solutions of Ent and conjugates 1, 2 and 3 were prepared in DMSO; stock solutions of compounds 4 and 5 were prepared in DMF; all stock solutions were aliquoted and stored at – 20°C. The concentrations of the stock solutions were determined by dilution with Milli-Q water and measuring the quinolone absorbance at 279 nm (ε: 12600 M–1 cm–1) [44]. For antimicrobial assays with conjugates 1, 2, 3, and DHBS–Cipro, working dilutions of the stock solutions and ciprofloxacin were prepared in 10% DMSO/H2O; the final cultures contained 1% v/v DMSO. For antimicrobial assays with compounds 4 and 5, working dilutions of the stock solutions and ciprofloxacin were prepared in 10% DMF/H2O; the final cultures contained 1% v/v DMF. For pre-loading of conjugates 1 and 2 with Fe(III), 0.9 equiv of FeCl3 were added to the conjugates and the solutions were incubated for 5 min. Additional details on microbiology materials and methods are provided in the Supporting Information.
Antimicrobial Activity Assays
Overnight cultures were prepared by inoculating 5-mL aliquots of modified M9 medium with single colonies from freezer stocks and incubated at 37°C on a rotating wheel for 16–18 h. The overnight cultures were diluted 1:100 into 5 mL of fresh modified M9 medium and incubated at 37°C on a rotating wheel until an OD600 of approximately 0.6 was reached. The cultures were subsequently diluted to an OD600 of 0.001 with modified M9 medium. Aliquots of the diluted cultures (90 μL) were combined with 10 μL aliquots of 10× solutions of the test compounds in 96-well plates, which were wrapped with plastic (polyvinylidene chloride wrap) and incubated at 30°C with shaking at 150 rpm for 20 h. Bacterial growth was determined by measuring the OD600 using a BioTek Synergy HT plate reader. Each well condition was prepared in duplicate and at least three independent replicates were conducted on different days. The resulting mean OD600 values are reported and the error bars represent the standard deviation.
Results and Discussion
Synthesis of Ent–SS–Cipro 2
Inspired by the synthesis of Ent–Cipro 1 [51], we initially attempted to install the disulfide-containing linker moiety with a terminal amino group at the pendant secondary amine of ciprofloxacin, followed by peptide coupling with acid-derivatized Ent 11 (Scheme S1). However, this procedure provided the benzyl-protected precursor to conjugate 2 in very low yields, and purification of the desired product was hampered by co-elution with an unidentified ciprofloxacin derivative, likely resulting from disulfide scrambling under the basic conditions that were required for the coupling reaction. Thus, Ent–SS–Cipro 2 was synthesized from the thiol-containing building blocks 9 and 12, employing pyridinethiol as a leaving group to prevent formation of symmetric disulfides (Scheme 2). The self-immolative linker was installed at ciprofloxacin by reaction with triphosgene and S-trityl-protected 2-mercaptoethanol 6 to yield carbamate 7. Removal of the trityl protecting group and reaction of the resulting free thiol 8 with dipyridyldisulfide yielded the ciprofloxacin building block 9. An Mmt protecting group was used for the Ent building block 12 to enable thiol deprotection under mild acidic conditions and prevent hydrolysis of the trilactone. Moreover, deprotection of 12 was performed immediately before reaction with 9 to prevent reaction of the free thiol with the trilactone. Final deprotection of the conjugate with BCl3 provided Ent–SS–Cipro 2 in good yield.
Scheme 2.
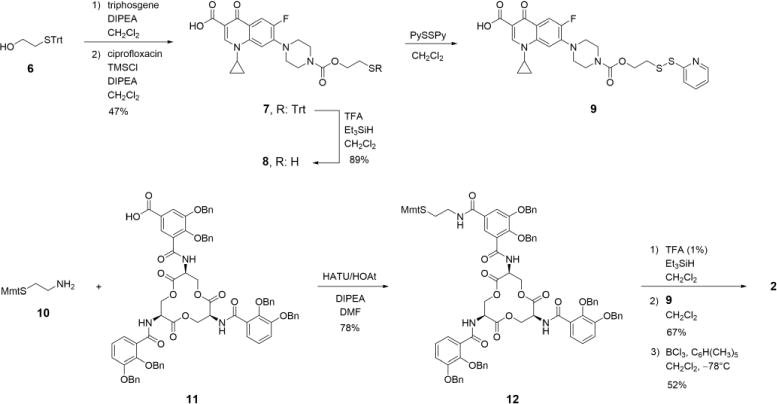
Synthesis of Ent–SS–Cipro 2
Reductive Cleavage of Ent–SS–Cipro
Whereas the extracellular space and the periplasm are oxidative environments, the bacterial cytoplasm is a reducing environment maintained by the glutathione (GSH)/glutathione disulfide (GSSG) redox buffer where disulfides are reduced to the corresponding free thiols [56–58]. The tripeptide GSH is an important cellular antioxidant and present at millimolar concentration in most Gram-negative and some Gram-positive bacteria [57]. We therefore tested the reductive cleavage of Ent–SS– Cipro 2 and release of unmodified ciprofloxacin in the presence of excess GSH (Scheme 3). Incubation of conjugate 2 with GSH results in cleavage of the disulfide bond and release of the free thiols as well as GSH adducts, and eventually release of ciprofloxacin (Fig. 1a,b; Fig. S1,S2). Consistent with the relatively high pKa of the terminal thiol in Cipro–SH 8 (pKa(ethanethiol) = 10.6 [59]), cleavage of the carbamate and release of the unmodified antibiotic proceeds faster at higher pH. The reduction also occurs with other reducing agents, namely tris(2-carboxyethyl)phosphine (TCEP) and 1,4-dithiothreitol (DTT) (Fig. 1c,d; Fig. S3,S4). These results indicate that GSH and likely other cellular reductants can cleave the disulfide bond in Ent–SS–Cipro 2 and mediate release of the unmodified antibiotic.
Scheme 3.
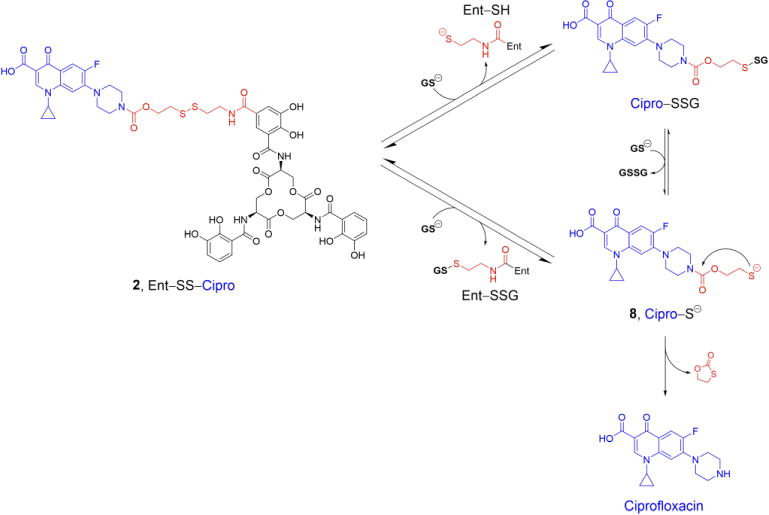
Proposed mechanism for the reductive cleavage of Ent–SS–Cipro 2 by glutathione (GSH) and release of ciprofloxacin
Fig. 1.
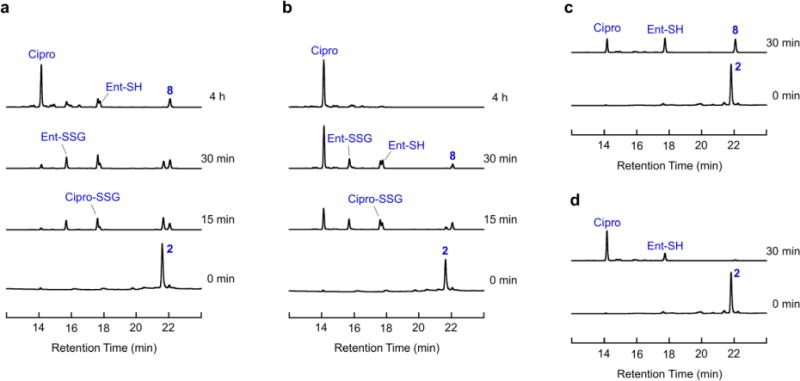
Reductive cleavage of Ent–SS–Cipro 2. (a) Cleavage by GSH at pH 7.4. (b) Cleavage by GSH at pH 9.0. (c) Cleavage by TCEP at pH 7.4. (d) Cleavage by TCEP at pH 9.0. Analytical HPLC traces (316 nm absorption) from reduction assays performed with 100 μM Ent–SS– Cipro 2 and 1 mM GSH or TCEP, respectively, in 75 mM Tris-HCl at the given pH values. Molecular structures of the compounds are presented in Scheme 3. Additional data are presented in Fig. S1–S4
Ent–SS–Cipro Exhibits Antibacterial Activity Independent of Intracellular Siderophore Hydrolysis
We evaluated the antibacterial activity of apo and Fe(III)-bound Ent– SS–Cipro 2 against a panel of non-pathogenic and uropathogenic E. coli strains, and compared its activity with that of the alkyl-linked Ent–Cipro 1, which requires intracellular hydrolysis of the siderophore by IroD to exert growth inhibition. Similar to conjugate 1, Ent–SS–Cipro 2 exhibits no antibacterial activity against the laboratory strain E. coli K-12 (Fig. 2; Fig. S15). We also found that Ent–SS–Cipro 2 does not potently inhibit the growth of IroD-expressing strains that are inhibited by Ent–Cipro 1, namely the uropathogenic strains E. coli UTI89 and CFT073, the probiotic E. coli Nissle 1917, and E. coli K-12(DE3) complemented with iroD (Fig. 2; Fig. S14, S15), although enzymatic activity assays demonstrate that conjugate 2 is hydrolyzed by IroD (Scheme S2; Fig. S19). In contrast to the lack of growth inhibition observed for these E. coli strains, Ent–SS–Cipro 2 exhibits antibacterial activity against the laboratory strains of E. coli B and a commensal isolate from a mouse gut, E. coli JB2. These two strains do not express IroD and are not inhibited by Ent–Cipro 1 (Fig. 2; Fig. S16). Moreover, complementation of the laboratory strain E. coli BL21(DE3) with iroD enhances its susceptibility to Ent–Cipro 1, but the antibacterial activity of Ent–SS–Cipro 2 is not affected by IroD expression (Fig. S17).
Fig. 2.
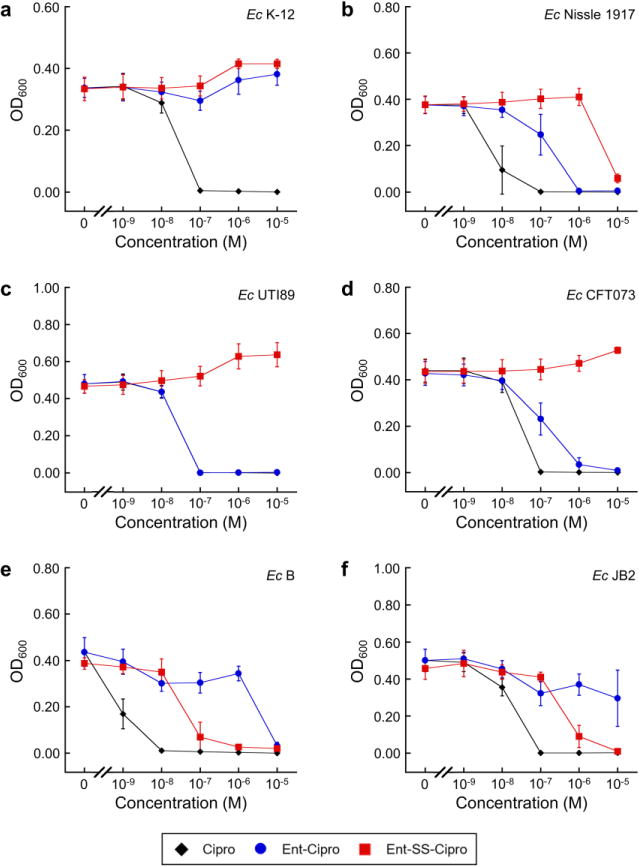
Antibacterial activity of ferric Ent–Cipro 1 and Ent–SS–Cipro 2 against non-pathogenic and uropathogenic E. coli strains. (a) Laboratory strain E. coli K-12. (b) Probiotic strain E. coli Nissle 1917. (c) Uropathogenic E. coli UTI89. (d) Uropathogenic E. coli CFT073. (e) Laboratory strain E. coli B. (f) Commensal strain from mouse gut E. coli JB2. All assays were performed in modified M9 medium (t = 20 h, T = 30°C; mean ± SDM, n = 3). The conjugates were pre-loaded with 0.9 equiv of Fe(III). Additional data are presented in Fig. S14–S16
Treatment of E. coli B or E. coli JB2 with a mixture of Ent and Ent–SS–Cipro 2 (1:1 molar ratio) attenuates the antibacterial activity of the conjugate (Fig. 3; Fig. S18), indicating that it can be outcompeted by the native siderophore for binding at the outer membrane transporter FepA. Similar to the alkyl-linked conjugate 1, the hydrolytic product of the disulfide-linked conjugate, DHBS–SS–Cipro 3 (Structure 1; Scheme S2), exhibits only low growth inhibitory activity (Fig. 3; Fig. S14–S16). Taken together, these results indicate that Ent–SS– Cipro 2 can be delivered into E. coli through the Ent uptake machinery (FepABCDG) and that the intact siderophore is important for uptake. Moreover, the disulfide-linked conjugate 2 exhibits antibacterial activity independent of IroD expression, suggesting that the disulfide linker can be cleaved intracellularly and that hydrolysis of the trilactone is not an important parameter. However, antibacterial activity is observed only against select E. coli strains and not against most IroD-expressing strains. It is possible that reductive cleavage of Ent–SS–Cipro 2 may not proceed efficiently in all strains. Moreover, thiol 8 released upon reduction could form adducts with GSH or other molecules in the cytoplasm, thus preventing release of the unmodified antibiotic and resulting in attenuated inhibition of DNA gyrase, the cytoplasmic target of ciprofloxacin.
Fig. 3.
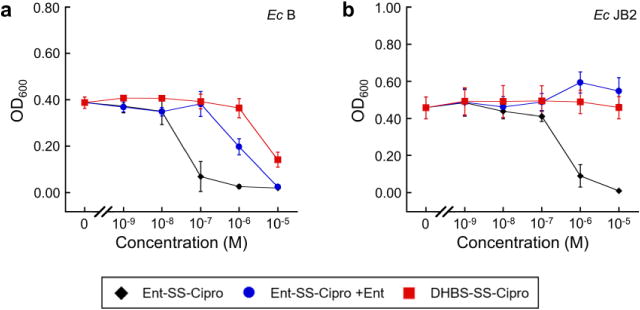
Antibacterial activity of ferric Ent–SS–Cipro 2 in the absence and presence of Ent, and DHBS–SS–Cipro 3 against E. coli strains. (a) E. coli B. (b) E. coli JB2. All assays were performed in modified M9 medium (t = 20 h, T = 30°C; mean ± SDM, n = 3). Ent–SS–Cipro 2 was pre-loaded with 0.9 equiv of Fe(III). For co-treatment, a 1:1 molar ratio of apo Ent and ferric 2 was employed
The Disulfide Linker Attenuates the Antibacterial Activity of Ciprofloxacin
To investigate whether the self-immolative disulfide linker itself could attenuate the antibacterial activity of ciprofloxacin, we synthesized and tested two structurally related ciprofloxacin derivatives; Cipro–SSEt 4 contains the same disulfide linker as Ent–SS–Cipro 2, whereas Cipro– SMe 5 contains a thioether that cannot be cleaved by reducing agents (Structure 1). Consistently, incubation of compound 4 with GSH or TCEP results in release of ciprofloxacin, whereas compound 5 is stable under these conditions (Fig. S5–S10). We tested these compounds against four E. coli strains and observed that both compounds exhibit significantly attenuated antibacterial activity compared to ciprofloxacin (Fig. 4). This behavior may result from reduced transport of the antibiotics into E. coli or from an impaired interaction of ciprofloxacin with DNA gyrase due to the attached linkers. Notably, Cipro–SSEt 4 exhibits lower antibacterial activity against E. coli B and JB2 than Ent–SS–Cipro 2 (Fig. 2), despite the fact that both compounds contain the same linker. This result is reminiscent of the low activity observed for the hydrolytic product, DHBS–SS–Cipro 3 (Fig. 3), and suggests that the intact siderophore facilitates uptake of modified ciprofloxacin. In addition, the non-cleavable derivative Cipro–SMe 5 appears to exhibit overall higher antibacterial activity than the disulfide Cipro–SSEt 4. If both compounds are taken up into the cells, the thioether in 5 prevents release of unmodified ciprofloxacin. In contrast, intracellular reduction of Cipro–SSEt 4 may release ciprofloxacin, but could also result in the formation of larger adducts via the released thiol and thus even lower inhibitory activity. Overall, these observations indicate that the presence of a disulfide linker attenuates the activity of ciprofloxacin.
Fig. 4.
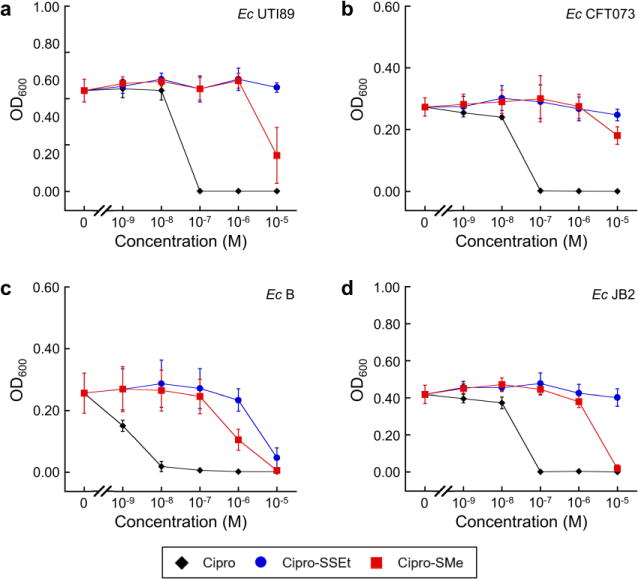
Antibacterial activity of Cipro–SSEt 4 and Cipro–SMe 5 against uropathogenic and non-pathogenic E. coli strains. (a) Uropathogenic E. coli UTI89. (b) Uropathogenic E. coli CFT073. (c) Laboratory strain E. coli B. (d) Commensal strain from mouse gut E. coli JB2. All assays were performed in modified M9 medium (t = 20 h, T = 30°C; mean ± SDM, n = 3)
Conclusions
The strategy of siderophore conjugation holds promise for enhanced delivery of antibiotics into Gram-negative bacteria as well as targeted delivery into pathogenic strains. Despite recent advances, the siderophore-mediated delivery of cytoplasmic antibiotics requires further study and optimization to provide broadly applicable linker strategies that enable release of the active antibiotic cargo after uptake into bacterial cells. We demonstrate that a self-immolative disulfide linker in an Ent–ciprofloxacin conjugate can release the antibiotic upon reaction with cellular reductants and that this linker affords antibacterial activity against some E. coli strains. Our studies also indicate that the siderophore mediates conjugate uptake, and that the intact siderophore is required for activity against the susceptible E. coli strains. However, Ent–SS– Cipro 2 exhibits activity against only a limited number of strains including E. coli B and mouse commensal E. coli JB2. At this point, it is unclear why these particular strains are susceptible to the compound, whereas others are not. Notably, the antimicrobial activity of Ent hydrolase-activated Ent–Cipro 1 against IroD-expressing E. coli is attenuated when the alkyl linker is replaced with the disulfide linker. Throughout this work, we reason that Ent–SS–Cipro 2 enters the cytoplasm of E. coli as we observed for Ent–Cipro 1 and other enterobactin–cargo conjugates; however, we acknowledge that an alternative possibility that warrants evaluation is that the ciprofloxacin cargo is released in the periplasm. Taken together, these results indicate that disulfide linkers are not broadly applicable for siderophore–antibiotic conjugates. Overall, this work, together with previous reports, suggest that enzymatic cleavage of such conjugates may be most effective.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (NIH Grants 1R21AI126465 and 1R01AI114625) for financial support; Lynette Cegelski for providing E. coli UTI89; Manuela Raffatellu for providing E. coli JB2; Ardeypharm GmbH for providing E. coli Nissle 1917. W.N. acknowledges the German National Academy of Sciences Leopoldina for a postdoctoral fellowship (LPDS 2015-08). MS instrumentation maintained by the MIT Center for Environmental Health Sciences (CEHS) is supported by a core center grant from the National Institute of Environmental Health Sciences, National Institutes of Health (NIEHS grant P30-ES002109).
Abbreviations
- DHBS
2,3-dihydroxybenzoyl serine
- DTT
1,4-dithiothreitol
- Ent
enterobactin
- GSH
glutathione
- GSSG
glutathione disulfide
- TCEP
tris(2-carboxyethyl)phosphine
Footnotes
ORCID IDs
Wilma Neumann: 0000-0002-1728-5140
Elizabeth M. Nolan: 0000-0002-6153-8803
Electronic Supplementary Material
The online version of this article contains supplementary material, which is available to authorized users.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Clatworthy AE, Pierson E, Hung DT. Nat Chem Biol. 2007;3:541–548. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 2.Lewis K. Nat Rev Drug Discovery. 2013;12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 3.Hood MI, Skaar EP. Nat Rev Microbiol. 2012;10:525–537. doi: 10.1038/nrmicro2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weinberg ED. JAMA. 1975;231:39–41. doi: 10.1001/jama.231.1.39. [DOI] [PubMed] [Google Scholar]
- 5.Palmer LD, Skaar EP. Annu Rev Genet. 2016;50:67–91. doi: 10.1146/annurev-genet-120215-035146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miethke M, Marahiel MA. Microbiol Mol Biol Rev. 2007;71:413–451. doi: 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hider RC, Kong X. Natural product reports. 2010;27:637–657. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- 8.Chu BC, Garcia-Herrero A, Johanson TH, Krewulak KD, Lau CK, Peacock RS, Slavinskaya Z, Vogel HJ. BioMetals. 2010;23:601–611. doi: 10.1007/s10534-010-9361-x. [DOI] [PubMed] [Google Scholar]
- 9.Wencewicz TA, Miller MJ. Top Med Chem. Springer; Berlin, Heidelberg: pp. 1–33. [Google Scholar]
- 10.Braun V, Pramanik A, Gwinner T, Köberle M, Bohn E. BioMetals. 2009;22:3–13. doi: 10.1007/s10534-008-9199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zahner H, Diddens H, Keller-Schierlein W, Nageli HU. Jpn J Antibiot. 1977;30:S201–206. [PubMed] [Google Scholar]
- 12.Diarra MS, Lavoie MC, Jacques M, Darwish I, Dolence EK, Dolence JA, Ghosh A, Ghosh M, Miller MJ, Malouin F. Antimicrob Agents Chemother. 1996;40:2610–2617. doi: 10.1128/aac.40.11.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ji C, Juárez-Hernández RE, Miller MJ. Future Med Chem. 2012;4:297–313. doi: 10.4155/fmc.11.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Page MGP. Ann N Y Acad Sci. 2013;1277:115–126. doi: 10.1111/nyas.12024. [DOI] [PubMed] [Google Scholar]
- 15.Tillotson GS. Infect Dis (Auckl) 2016;9:45–52. doi: 10.4137/IDRT.S31567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watanabe N-A, Nagasu T, Katsu K, Kitoh K. Antimicrob Agents Chemother. 1987;31:497–504. doi: 10.1128/aac.31.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curtis NAC, Eisenstadt RL, East SJ, Cornford RJ, Walker LA, White AJ. Antimicrob Agents Chemother. 1988;32:1879–1886. doi: 10.1128/aac.32.12.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silley P, Griffiths JW, Monsey D, Harris AM. Antimicrob Agents Chemother. 1990;34:1806–1808. doi: 10.1128/aac.34.9.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hashizume T, Sanada M, Nakagawa S, Tanaka N. J Antibiot (Tokyo) 1990;43:1617–1620. doi: 10.7164/antibiotics.43.1617. [DOI] [PubMed] [Google Scholar]
- 20.Nikaido H, Rosenberg EY. J Bacteriol. 1990;172:1361–1367. doi: 10.1128/jb.172.3.1361-1367.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKee JA, Sharma SK, Miller MJ. Bioconjugate Chem. 1991;2:281–291. doi: 10.1021/bc00010a013. [DOI] [PubMed] [Google Scholar]
- 22.Dolence EK, Minnick AA, Lin C-E, Miller MJ, Payne SM. J Med Chem. 1991;34:968–978. doi: 10.1021/jm00107a014. [DOI] [PubMed] [Google Scholar]
- 23.Ji C, Miller PA, Miller MJ. J Am Chem Soc. 2012;134:9898–9901. doi: 10.1021/ja303446w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng T, Nolan EM. J Am Chem Soc. 2014;136:9677–9691. doi: 10.1021/ja503911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohira N, West J, Ito A, Ito-Horiyama T, Nakamura R, Sato T, Rittenhouse S, Tsuji M, Yamano Y. Antimicrob Agents Chemother. 2016;60:729–734. doi: 10.1128/AAC.01695-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chairatana P, Zheng T, Nolan EM. Chem Sci. 2015;6:4458–4471. doi: 10.1039/c5sc00962f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruiz N, Kahne D, Silhavy TJ. Nat Rev Microbiol. 2006;4:57–66. doi: 10.1038/nrmicro1322. [DOI] [PubMed] [Google Scholar]
- 28.Silhavy TJ, Kahne D, Walker S. Cold Spring Harb Perspect Biol. 2010;2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hennard C, Truong QC, Desnottes J-F, Paris J-M, Moreau NJ, Abdallah MA. J Med Chem. 2001;44:2139–2151. doi: 10.1021/jm990508g. [DOI] [PubMed] [Google Scholar]
- 30.Rivault F, Liébert C, Burger A, Hoegy F, Abdallah MA, Schalk IJ, Mislin GLA. Bioorg Med Chem Lett. 2007;17:640–644. doi: 10.1016/j.bmcl.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 31.Wencewicz TA, Möllmann U, Long TE, Miller MJ. BioMetals. 2009;22:633–648. doi: 10.1007/s10534-009-9218-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Md-Saleh SR, Chilvers EC, Kerr KG, Milner SJ, Snelling AM, Weber JP, Thomas GH, Duhme-Klair A-K, Routledge A. Bioorg Med Chem Lett. 2009;19:1496–1498. doi: 10.1016/j.bmcl.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 33.Noël S, Gasser V, Pesset B, Hoegy F, Rognan D, Schalk IJ, Mislin GLA. Org Biomol Chem. 2011;9:8288–8300. doi: 10.1039/c1ob06250f. [DOI] [PubMed] [Google Scholar]
- 34.Juárez-Hernández RE, Miller PA, Miller MJ. ACS Med Chem Lett. 2012;3:799–803. doi: 10.1021/ml300150y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ji C, Miller MJ. Bioorg Med Chem. 2012;20:3828–3836. doi: 10.1016/j.bmc.2012.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wencewicz TA, Miller MJ. J Med Chem. 2013;56:4044–4052. doi: 10.1021/jm400265k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wencewicz TA, Long TE, Möllmann U, Miller MJ. Bioconjugate Chem. 2013;24:473–486. doi: 10.1021/bc300610f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milner SJ, Seve A, Snelling AM, Thomas GH, Kerr KG, Routledge A, Duhme-Klair AK. Org Biomol Chem. 2013;11:3461–3468. doi: 10.1039/c3ob40162f. [DOI] [PubMed] [Google Scholar]
- 39.Souto A, Montaos MA, Balado M, Osorio CR, Rodríguez J, Lemos ML, Jiménez C. Bioorg Med C. 2013;21:295–302. doi: 10.1016/j.bmc.2012.10.028. [DOI] [PubMed] [Google Scholar]
- 40.Milner SJ, Snelling AM, Kerr KG, Abd-El-Aziz A, Thomas GH, Hubbard RE, Routledge A, Duhme-Klair A-K. Bioorg Med Chem. 2014;22:4499–4505. doi: 10.1016/j.bmc.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 41.Fardeau S, Dassonville-Klimpt A, Audic N, Sasaki A, Pillon M, Baudrin E, Mullié C, Sonnet P. Bioorg Med Chem. 2014;22:4049–4060. doi: 10.1016/j.bmc.2014.05.067. [DOI] [PubMed] [Google Scholar]
- 42.Ji C, Miller MJ. BioMetals. 2015;28:541–551. doi: 10.1007/s10534-015-9830-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Braun V, Günthner K, Hantke K, Zimmermann L. J Bacteriol. 1983;156:308–315. doi: 10.1128/jb.156.1.308-315.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neumann W, Sassone-Corsi M, Raffatellu M, Nolan EM. J Am Chem Soc. 2018;140:5193–5201. doi: 10.1021/jacs.8b01042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin H, Fischbach MA, Liu DR, Walsh CT. J Am Chem Soc. 2005;127:11075–11084. doi: 10.1021/ja0522027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu M, Valdebenito M, Winkelmann G, Hantke K. Microbiology. 2005;151:2363–2372. doi: 10.1099/mic.0.27888-0. [DOI] [PubMed] [Google Scholar]
- 47.Zheng T, Nolan EM. Bioorg Med Chem Lett. 2015;25:4987–4991. doi: 10.1016/j.bmcl.2015.02.034. [DOI] [PubMed] [Google Scholar]
- 48.Paulen A, Gasser V, Hoegy F, Perraud Q, Pesset B, Schalk IJ, Mislin GLA. Org Biomol Chem. 2015;13:11567–11579. doi: 10.1039/c5ob01859e. [DOI] [PubMed] [Google Scholar]
- 49.Paulen A, Hoegy F, Roche B, Schalk IJ, Mislin GLA. Bioorg Med Chem Lett. 2017;27:4867–4870. doi: 10.1016/j.bmcl.2017.09.039. [DOI] [PubMed] [Google Scholar]
- 50.Liu R, Miller PA, Vakulenko SB, Stewart NK, Boggess WC, Miller MJ. J Med Chem. 2018;61:3845–3854. doi: 10.1021/acs.jmedchem.8b00218. [DOI] [PubMed] [Google Scholar]
- 51.Zheng T, Bullock JL, Nolan EM. J Am Chem Soc. 2012;134:18388–18400. doi: 10.1021/ja3077268. [DOI] [PubMed] [Google Scholar]
- 52.Vrudhula VM, MacMaster JF, Li Z, Kerr DE, Senter PD. Bioorg Med Chem Lett. 2002;12:3591–3594. doi: 10.1016/s0960-894x(02)00784-9. [DOI] [PubMed] [Google Scholar]
- 53.Vlahov IR, Leamon CP. Bioconjugate Chem. 2012;23:1357–1369. doi: 10.1021/bc2005522. [DOI] [PubMed] [Google Scholar]
- 54.Brezden A, Mohamed MF, Nepal M, Harwood JS, Kuriakose J, Seleem MN, Chmielewski J. J Am Chem Soc. 2016;138:10945–10949. doi: 10.1021/jacs.6b04831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim HS, Song WY, Kim HJ. Org Biomol Chem. 2015;13:73–76. doi: 10.1039/c4ob01810a. [DOI] [PubMed] [Google Scholar]
- 56.Ritz D, Beckwith J. Annu Rev Microbiol. 2001;55:21–48. doi: 10.1146/annurev.micro.55.1.21. [DOI] [PubMed] [Google Scholar]
- 57.Masip L, Veeravalli K, Georgiou G. Antioxid Redox Signaling. 2006;8:753–762. doi: 10.1089/ars.2006.8.753. [DOI] [PubMed] [Google Scholar]
- 58.Van Loi V, Rossius M, Antelmann H. Front Microbiol. 2015;6:187. doi: 10.3389/fmicb.2015.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Serjeant EP, Dempsey B, International Union of Pure and Applied Chemistry (IUPAC) IUPAC Chemical Data Series, No 23. Pergamon Press; Oxford, New York: 1979. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.